

Abstract
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) has been implicated in the regulation of inflammation in rheumatoid arthritis (RA), primarily due to its ability to promote apoptosis in synoviocytes and infiltrating lymphocytes. The aim of this study was to investigate the immunomodulatory mechanism and role of TRAIL in inflammatory arthritis. We created an animal model of inflammatory arthritis and demonstrated that TRAIL significantly inhibited joint inflammation and reduced the severity of arthritis. The suppression of joint inflammation was not due to the TRAIL-mediated induction of apoptosis in T cells, macrophages or synovial fibroblasts. In contrast, TRAIL directly inhibited T-cell proliferation and suppressed the production of cytokines, which indicated that TRAIL exerted its anti-inflammatory effects by direct inhibition of T-cell activation. Moreover, TRAIL receptor (TRAIL-R)-knockout mice developed more severe disease, and the protective effects of TRAIL were abolished in the experimental arthritis model in TRAIL-R knockout mice. From these results, we conclude that TRAIL suppresses joint inflammation via an apoptosis-independent pathway and directly inhibits T-cell activation. Our results provide a novel apoptosis-independent, immune regulatory role for TRAIL in suppressing inflammatory arthritis and shed light on the development of effective new therapies for autoimmune inflammatory diseases.
Keywords: collagen-induced arthritis, T-cell activation, TRAIL
INTRODUCTION
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) belongs to the TNF superfamily, the expression of which has been demonstrated in a wide range of tissues.1 TRAIL binds to five receptors, TRAIL-R1 (death receptor (DR)4), TRAIL-R2 (DR5), TRAIL-R3 (decoy receptor (DcR)1), TRAIL-R4 (DcR2), and a soluble decoy receptor, osteoprotegerin (OPG).2,3,4,5 In mice, there is only one TRAIL receptor, mTRAIL-R, which has an apoptosis-inducing activity.6 Interaction of TRAIL with the TRAIL receptor induces oligomerization of the receptor and recruitment of the FAS-associated protein with the death domain (FADD), which recruits pro-caspase-8. The resulting complex, death-inducing signaling complex (DISC), in turn activates the caspase cascade and eventually leads to cell death.7,8,9,10 Although TRAIL induces apoptosis in many tumor cell lines, almost no normal cells are sensitive to TRAIL-induced cell death.11,12 The biological role of TRAIL remains to be elucidated. Recently, accumulating evidence has focused on an emerging role of TRAIL in modulating immune responses. TRAIL administration induces anti-inflammatory effects in several autoimmune animal models.13,14,15 Treatment with TRAIL significantly reduces the antigen-specific antibody production in the autoimmune-prone lymphoproliferative strain of mice, C3H/HeJgld/gld.15 Furthermore, in mice with autoimmune encephalomyelitis (EAE), TRAIL decreases the disease score, inhibits the inflammation in the central nervous system and prevents the activation of autoreactive T cells.16 In addition, TRAIL inhibits the development of autoimmune diabetes in non-obese diabetic (NOD) mice by inhibiting the proliferation of NOD diabetogenic T cells.14 Patients with inflammation associated with rheumatoid arthritis (RA) demonstrate an increased level of the TRAIL receptor that is associated with the T-cell subsets and clinical scores,17,18 and treatment with TRAIL reduces the severity of joint inflammation in an animal model of inflammatory arthritis,19 which suggests that TRAIL may have a role in RA. However, the mechanism by which TRAIL regulates inflammatory arthritis is still unclear. TRAIL has long been regarded as a pro-apoptotic signal transducer, and most of its regulatory role has been believed to be due to the promotion of apoptosis in the synoviocytes and/or infiltrating lymphocytes. Recently, evidence has accumulated that suggested that TRAIL may have an apoptosis-independent role in immune regulation.13,20,21,22,23 TRAIL was demonstrated to be able to transduce a reverse signal and induce T-cell costimulation.20,21,22 In contrast, in addition to triggering apoptosis, TRAIL itself can directly inhibit T-cell activation via interaction with the TRAIL receptor.23 These results suggest that TRAIL has an apoptosis-independent role in the immune system and that it may have potential therapeutic implications in inflammatory autoimmune diseases.
In this study, we investigated the role of TRAIL in regulating inflammation and its potential therapeutic applications in inflammatory arthritis. We demonstrate herein that TRAIL strongly suppressed joint inflammation via an apoptosis-independent pathway and directly inhibited T-cell activation. Our study suggests a novel role for TRAIL in suppressing autoimmune inflammatory diseases and sheds light on possible therapeutic applications in inflammatory arthritis and autoimmune diseases.
MATERIALS AND METHODS
Animals
Sprague–Dawley (SD) rats (male, 6–8 weeks old) and C57BL/6 mice (male, 6–8 weeks old) were housed under specific pathogen-free conditions and provided with standard food and water. TRAIL-receptor knockout (TRAIL-R KO) mice (C57BL/6 background) were obtained from Henning Walczak (UCL Cancer Institute, University College London, United Kingdom). All animal work in the study was conducted following the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care. All animal experiments were approved by the Animal Ethics Committee of the National Taiwan University Medical Center.
Induction of collagen-induced arthritis and collagen antibody-induced arthritis
To induce collagen-induced arthritis (CIA), bovine collagen type II (bCII) was solubilized at 2 mg/ml in 0.05 m acetic acid, and then emulsified in an equal volume of complete Freund’s adjuvant (CFA) containing 4 mg/ml of heat-killed Mycobacterium tuberculosis (Arthrogen-CIA; Chondrex, Redmond, WA, USA) in an ice-cold water bath. Male SD rats were first immunized (day 0) subcutaneously at the base of the tail with 0.2 ml of this emulsion. On day 7, the rats were given a booster injection of 0.2 ml of the emulsion.
Collagen antibody-induced arthritis (CAIA) was induced in wild-type (WT) C57BL/6 and TRAIL-R KO mice using an intravenous injection of 0.2 mg/g body weight of a cocktail of 5 anti-type II collagen monoclonal antibodies (Chondex) on day 0. On day 3, all mice received an intraperitoneal injection of 2 μg/g body weight lipopolysaccharide (LPS; E. coli 0111:B4; Chondrex).
Assessment of arthritis severity
To assess the arthritis severity, measurements and scoring for the manifestation of arthritic symptoms were performed every day. For the rats with CIA, the clinical signs of arthritis in each hind paw were assessed under blinded conditions every day. The hind paw thickness was measured with Vernier calipers. The arthritis severity in each hind paw was scored and monitored based on the following criteria: 0=no evidence of swelling or erythema; 1=mild swelling and erythema confined to the tarsals or ankle joint; 2=mild swelling and erythema extending from the ankle to the tarsals; 3=moderate swelling and erythema extending from the ankle to the metatarsal joints; and 4=severe swelling and erythema encompassing the foot, ankle and digits. Each hind paw was graded and an average score of ⩾3 was defined as severe arthritis. For the CAIA mice, the hind paw thicknesses were measured with Vernier calipers and the clinical scores were provided as the sum of the arthritis scores of four limbs.
Reagents
Recombinant murine TRAIL was from PeproTech (Rocky Hill, NJ, USA). Subcutaneous injection of TRAIL was administered at a dose of 1.5 μg/kg in each rat, and intraperitoneal injection of TRAIL was given at a dose of 50 μg/mouse per day. The soluble TNF-α receptor, etanercept (Enbrel), was purchased from Pfizer (New York City, NY, USA) and used for subcutaneous injection at a dose of 6 mg/kg in each rat every 3 days. Mouse immunoglobulin G (IgG) was obtained from R&D Systems (Minneapolis, MN, USA) and administered by subcutaneous injection at a dose of 200 ng/rat per day. The pan-caspase inhibitor, z-VAD-fmk, was purchased from a commercial source (BioVision, Milpitas, CA, USA) and administered by subcutaneous injection at a dose of 1.5 mg/kg in the rats or by intraperitoneal injection at a dose of 1.5 μg/g per day in the mice.
Histological analysis
The rats were killed at 28 days after the treatment, and the hind paws were fixed in 4% formalin for 12 h and then decalcified in 20% ethylenediaminetetraacetic acid for 14 days at room temperature. For the assessment of synovial inflammation and bone erosion, serial paraffin sections (5 μm) of the hind paws were stained with hematoxylin and eosin (HE).
Measurement of cytokines and chemokines
Serum samples were obtained from SD rats on day 28 after the collagen immunization. The serum levels of cytokines including TNF-α (Biolegend, San Diego, CA, USA), interleukin (IL)-1-β (Peprotech) and IL-6 (Peprotech) as well as chemokines including CCL2, CCL5 and CXCL8 (all from Peprotech) were determined using ELISA kits according to the manufacturer’s instructions.
To measure the T cell-derived cytokines, 105 primary CD4+ T cells from WT C57BL/6 or TRAIL-R KO mice were enriched (Stem Cell Technology, Vancouver, BC, Canada) and cultured in a 96-well plate that had been precoated with 1 μg/ml of anti-CD3 and 1 μg/ml of anti-CD28 for 24 h with or without TRAIL. The supernatants were collected, and the levels of IL-2 (BioLegend), interferon (IFN)-γ (BioLegend), TNF-α (BioLegend) and IL-17 (BioLegend) were determined using ELISA kits.
Terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling staining
To evaluate the apoptotic cell death within the inflamed joints, three unstained sections corresponding to the HE-stained synovial slides were deparaffinized, and the synovium was stained for apoptosis using a TACS TdT-DAB In Situ Cell Death Detection Kit (Trevigen, Gaithersburg, MD, USA) following the manufacturer’s instructions.
Analysis of cell apoptosis by flow cytometry
Enriched primary CD4+ T cells from C57BL/6 mice (Stem Cell Technology) were cultured in a 96-well plate precoated with 1 μg/ml of anti-CD3 and 1 μg/ml of anti-CD28 in the presence of 100, 200 or 400 ng/ml TRAIL, and Jurkat cells were treated with 400 ng/ml TRAIL for 48 h. After 48 h, 105 cells were collected and stained with FITC-conjugated annexin and PE-conjugated propidium iodide (PI) in fluorescence-activated cell sorter (FACS) buffer, then analyzed on a FACSCanto II flow cytometer (BD Biosciences, San Diego, CA, USA) using Cell Quest software (Becton Dickinson, Franklin Lakes, NJ, USA). In this analysis, Annexin V+ PI− cells indicated early apoptotic cells, and annexin V+ PI+ cells indicated late apoptotic and necrotic cells.
Carboxyfluorescein succinimidyl ester staining
Mouse CD4+ T cells from the WT C57BL/6 or TRAIL-R KO mice were enriched (Stem Cell Technologies), labeled with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, Paisley, UK) and cultured in 96-well culture plates precoated with 1 μg/ml of anti-CD3 and 1 μg/ml of anti-CD28 in the presence or absence of 10 μg/ml of TRAIL for 5 days. After 5 days, 105 cells were collected in FACS buffer and analyzed on a FACSCanto II flow cytometer (BD Biosciences) using Cell Quest software (Becton Dickinson) for the CFSE-dilution patterns.
In vitro apoptosis detection
To detect apoptotic cells, the cytoplasmic histone-associated DNA fragments were measured using the Cell Death Detection ELISA PLUS system according to the manufacturer’s protocol (Roche Mannheim Biochemicals, Mannheim, Germany). Briefly, enriched CD4+ T cells (Stem Cell Technology), bone marrow-derived macrophages (BMMs) and synovial fibroblasts isolated from WT C57BL/6 or TRAIL-R KO mice as well as Jurkat cells were cultured for 24 h in a 96-well plate (104 cells per well) precoated with 1 μg/ml of anti-CD3 and 1 μg/ml of anti-CD28 in the presence or absence of TRAIL. The cells were lysed, and the lysates were collected and mixed with the immunoreagent for 2 h, following by reaction with the substrate solution in the dark until development of the color. The reaction was quantified using spectrophotometry at 405 nm. The percentage of apoptotic cells were defined as absorbance at 405 nm of cells from quadruplicate samples of the indicated stimulated culture/absorbance at 405 nm of the positive control.
Statistical analysis
Student’s t-test was used to compare the different parameters in two mice groups. All P-values in the study refer to two-tailed tests, and P-values <0.05 were defined as statistically significant. SPSS software, version 16.0 (SPSS, Chicago, IL, USA), was used for the analyses.
RESULTS
TRAIL significantly reduced the severity and incidence of the joint inflammation and paw swelling in rats with CIA
To address the crucial role of TRAIL in the development and pathogenesis of inflammatory arthritis, TRAIL was administered to rats that had been immunized with collagen, and the therapeutic effects of TRAIL were compared with those of the soluble TNF receptor, etanercept, in this animal model of CIA. As illustrated in Figure 1, TRAIL, etanercept or the TRAIL plus etanercept combination was administered subcutaneously from day 11 after rats were immunized with collagen (day 4 after the booster injection). Prompt paw swelling developed on approximately day 12 after the collagen injection in both the PBS and IgG groups, and the frequency and severity of the paw erythema and swelling increased in a time-dependent manner. In contrast, in the CIA rats treated with TRAIL, etanercept or TRAIL plus etanercept, very little paw swelling developed over the entire 28-day experimental course. Markedly reduced paw thickness and decreased clinical scores in the TRAIL group were observed from day 16 after the collagen injection compared with the control group. In addition, the rats with CIA that were treated with either TRAIL, etanercept or TRAIL plus etanercept exhibited similar impacts on paw swelling and clinical scores, which indicated that TRAIL had effects on joint inflammation comparable to those of etanercept. Moreover, TRAIL significantly prevented the incidence of severe arthritis. The histopathological analysis demonstrated that TRAIL significantly abrogated the synovial tissue inflammation and significantly reduced the inflammatory cell infiltration. Taken together, our results indicate that TRAIL treatment significantly reduced the severity and incidence of joint inflammation and paw swelling in rats with CIA and this effect was as potent as that of TNF-α blocking agent. The combination of TRAIL and TNF-α blocking agent led to better therapeutic effects.
Figure 1.
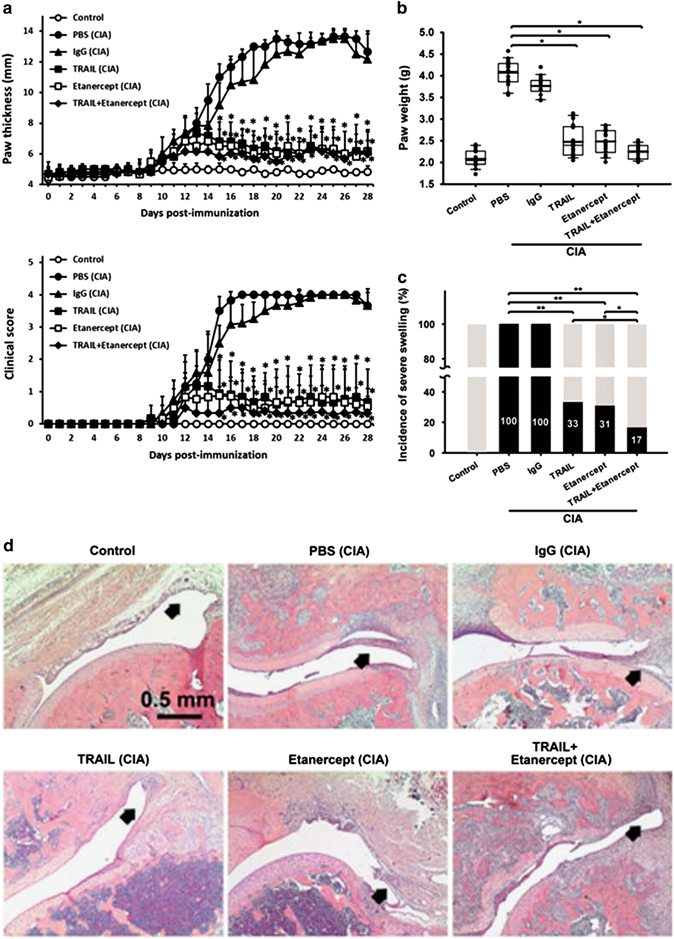
TRAIL reduced the severity and incidence of joint inflammation in rats with CIA. SD rats were immunized with bovine collagen type II (bCII; 100 μg/rat, sc) in CFA. On day 7 after the collagen injection, the rats were rechallenged with an injection of bCII in CFA. Beginning on day 11 post-collagen injection, the rats were treated with PBS (200 μl/rat/day, sc), IgG (200 ng/rat/3 days, sc), TRAIL (1.5 μg/kg/day, sc), etanercept (6 mg/kg/3 days, sc) or TRAIL plus etanercept. (a) The hind paw thicknesses and clinical scores of the groups were measured at the indicated time points (n=12 in each group). *P<0.05 by Student’s t-test, compared with the PBS group of rats with CIA. (b) Hind paw swelling among the groups was measured as the weights using a water displacement method on day 28 after the collagen injection (n=12 in each group). *P<0.05 by Student’s t-test. (c) The incidence of severe hind paw swelling (defined as a severity score of ⩾3) among the groups was measured on day 28 after the collagen injection (n=12 in each group). *P<0.05; **P<0.01 by Student’s t-test. (d) H&E staining of the ankle joints among the groups on day 28 after the collagen injection is shown. The arrow indicates the synovium of the inflamed joints in each group. CIA, collagen-induced arthritis; CFA, complete Freund’s adjuvant; H&E, hematoxylin and eosin; PBS, phosphate-buffered saline; SD, Sprague–Dawley.
TRAIL significantly inhibited the production of proinflammatory cytokines and chemokines in inflammatory arthritis
To further investigate whether TRAIL modulates the inflammatory process by regulating the production of proinflammatory cytokines and chemokines, the serum proinflammatory cytokines and chemokines were measured in the rats with CIA under different treatments on day 28 after the collagen injection. As illustrated in Figure 2, treatment of rats with CIA with TRAIL significantly decreased the production of proinflammatory cytokines including TNF-α, IL-1β and IL-6. These results were comparable to those in the rats with CIA that were treated with etanercept. In particular, the combination of TRAIL and etanercept demonstrated more profound effects in decreasing the systemic inflammatory cytokine production. Similar results were observed for proinflammatory chemokines including CCL2 (MCP-1), CCL5 (RANTES) and CXCL8 (IL-8; Figure 2). Altogether, these results showed that TRAIL significantly reduced the systemic proinflammatory cytokine and chemokine production in inflammatory arthritis.
Figure 2.
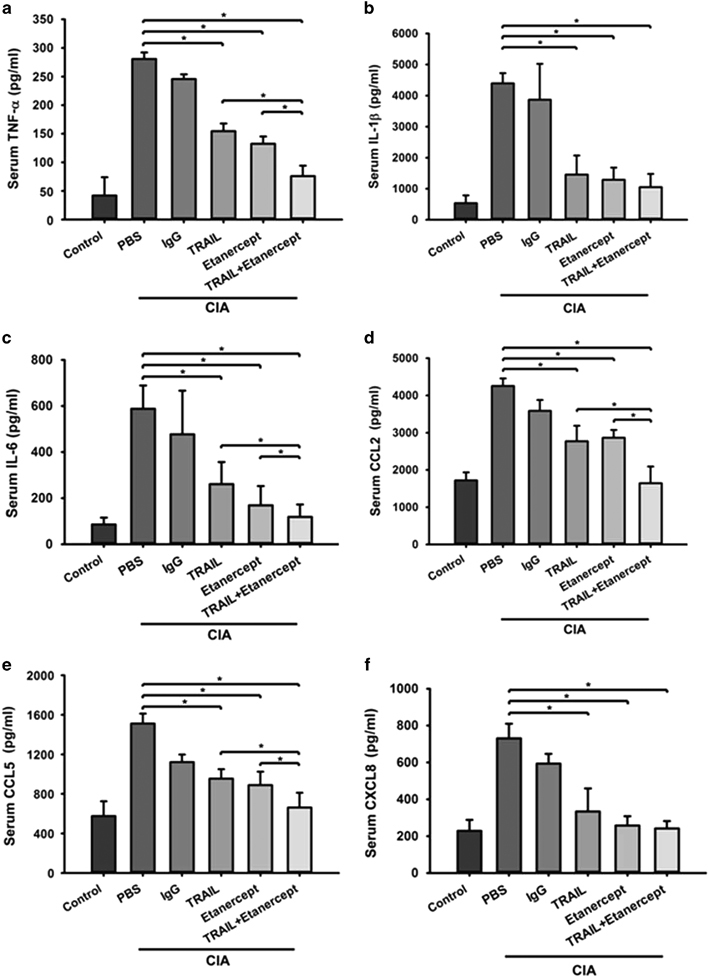
TRAIL reduced the production of systemic inflammatory cytokines and chemokines in rats with CIA. SD rats were immunized with bovine collagen type II (bCII) (100 μg/rat, sc) in CFA. On day 7 after the collagen injection, the rats were rechallenged with an injection of bCII in CFA. Beginning on day 11 after the collagen injection, the rats were treated with PBS (200 μl/rat/day, sc), IgG (200 ng/rat/3 days, sc), TRAIL (1.5 μg/kg/day, sc), etanercept (6 mg/kg/3 days, sc) or TRAIL plus etanercept. The serum levels of inflammatory cytokines including (a) TNF-α, (b) interleukin (IL)-1β and (c) IL-6 as well as inflammatory chemokines including (d) CCL2 (MCP-1), (e) CCL5 (RANTES) and (f) CXCL8 (IL-8) were assayed in each group on day 28 after the collagen injection. *P<0.05 by Student’s t-test. The data are representative of at least three independent experiments in each group. CIA, collagen-induced arthritis; CFA, complete Freund’s adjuvant; PBS, phosphate-buffered saline; SD, Sprague–Dawley; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Suppression of joint inflammation by TRAIL was not due to the induction inflammatory cell apoptosis
TRAIL induces cell death by binding to its death receptor through which it induces a caspase-mediated signaling cascade. To determine whether the effects of suppression of joint inflammation by TRAIL involved the promotion of apoptosis, we further evaluated the apoptosis in the joint tissues of rats with CIA in the presence and absence of TRAIL treatment. As shown in Figure 3, in the rats with CIA, no differences in the numbers of apoptotic cells within the synovial tissue were detected in the comparison between the control and TRAIL-treated group, which indicated that the pro-apoptotic effect of TRAIL on the synovial tissues is not easily detected in experimental arthritis. We further examined whether TRAIL-induced apoptosis in the primary cells within joints, including the T cells, BMMs and synovial fibroblasts. The results in Figure 3 demonstrated that there was very little cell apoptosis among these primary cells when treated with TRAIL. In contrast, TRAIL induced a significant amount of apoptosis in the human T-cell line, Jurkat, in a dose-dependent manner. Similarly, on the basis of the annexin V and PI apoptotic staining, TRAIL did not promote apoptosis in activated primary T cells. All these results indicate that treatment with TRAIL did not enhance the cellular apoptosis in inflammatory arthritis.
Figure 3.
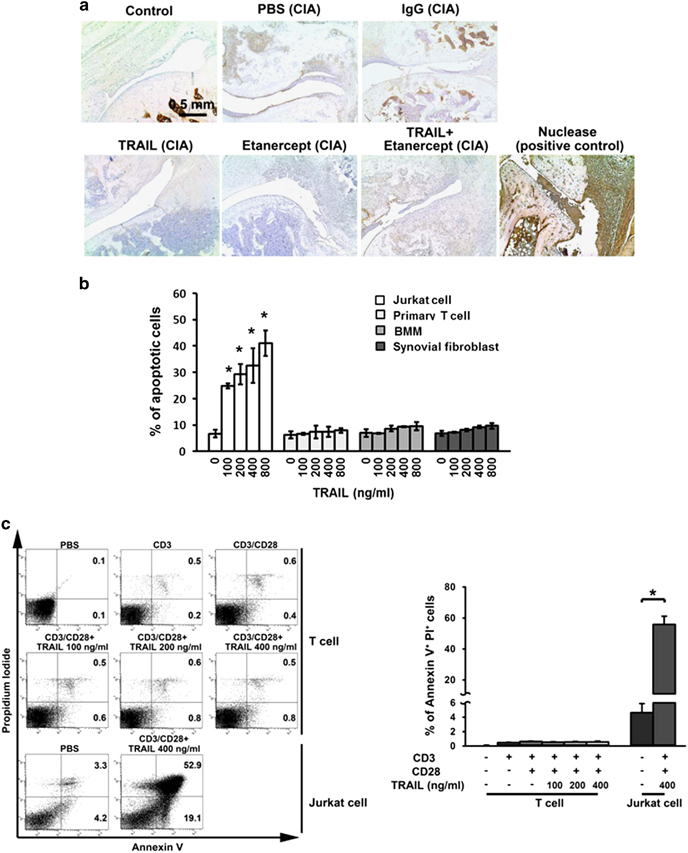
TRAIL did not promote cell apoptosis within the inflamed joints in inflammatory arthritis. (a) SD rats were immunized with bovine collagen type II (bCII) (100 μg/rat, sc) in CFA. On day 7 after the collagen injection, the rats were rechallenged with an injection of bCII in CFA. Beginning on day 11 after the collagen injection, the rats were treated with PBS (200 μl/rat/day, sc), IgG (200 ng/rat/3 days, sc), TRAIL (1.5 μg/kg/day, sc), etanercept (6 mg/kg/3 days, sc) or TRAIL plus etanercept. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining of the ankle joints in each group was carried out on day 28 after the collagen injection. Scale bar, 0.5 mm. (b) Activated primary CD3+ T cells, BMMs and synovial fibroblasts from C57BL/6 mice as well as the Jurkat cell line were cultured in a 96-well plate (104 cells per well) and treated with various concentrations of TRAIL for 24 h. The culture supernatants were collected and quantified using apoptotic assays. *P<0.05 by Student’s t-test, compared with Jurkat cells without TRAIL treatment. (c) Annexin V and PI staining in anti-CD3/CD28-activated primary T cells from C57BL/6 mice and the Jurkat cell line after being cultured in a 96-well plate (1 × 106 cells per well) and treatment with various concentrations of TRAIL for 24 h. Representative figures for each group are shown (left panel). The percentages of annexin V+ PI+ cells were quantified (right panel). *P<0.05 by Student’s t-test. The data are representative of at least three independent experiments in each group. BMMS, bone marrow-derived macrophages; CIA, collagen-induced arthritis; CFA, complete Freund’s adjuvant; PBS, phosphate-buffered saline; PI, propidium iodide; SD, Sprague–Dawley; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
To confirm that the anti-inflammatory effects of TRAIL were independent of its pro-apoptotic effect, we further evaluated TRAIL-induced anti-inflammatory effects in rats with CIA and in mice with CAIA in the presence and absence of the pan-caspase inhibitor, z-VAD-fmk. The results in Figure 4 and Supplementary Figure S1 demonstrated that the pan-caspase inhibitor, z-VAD-fmk, was not able to reverse the TRAIL-mediated suppression of joint inflammation. Taken together, these results indicated that the inhibition of joint inflammation by TRAIL occurred via an apoptosis-independent pathway.
Figure 4.
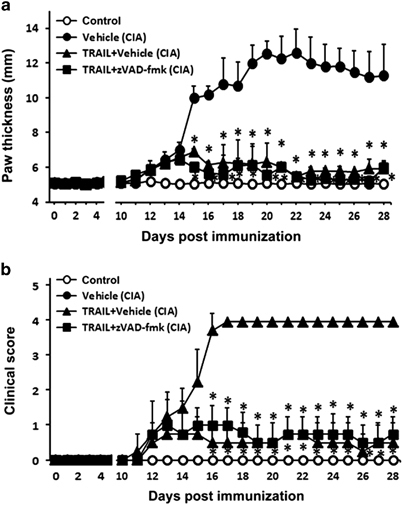
A pan-caspase inhibitor did not reverse the TRAIL-mediated inhibition of joint inflammation. SD rats were immunized with bovine collagen type II (CII) (100 μg/rat, sc) in CFA. On day 7 after the collagen injection, the rats were rechallenged with an injection of bovine CII in CFA. Beginning on day 11 after the collagen injection, the rats were treated with the vehicle, TRAIL (1.5 μg/kg/day, sc) or TRAIL plus z-VAD-fmk (1.5 μg/kg/day, sc). (a) The hind paw thicknesses and (b) clinical scores among the groups were measured at the indicated time points (n=10 in each group). *P<0.05 by Student’s t-test compared with CIA with vehicle group. CIA, collagen-induced arthritis; CFA, complete Freund’s adjuvant; SD, Sprague–Dawley; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Suppression of joint inflammation by TRAIL is via TRAIL/TRAIL–receptor interaction
To confirm that the anti-inflammatory effects of TRAIL were dependent on TRAIL/TRAIL–receptor interaction, we further evaluated the TRAIL-induced anti-inflammatory effects in the CAIA model using TRAIL–receptor knockout (TRAIL-R KO) mice. The results in Figure 5 demonstrated the enhanced joint inflammation and disease severity score in the TRAIL-R KO mice with CAIA compared with these parameters in WT controls. Moreover, the anti-inflammatory effects of treatment with recombinant TRAIL in mice with CAIA were abolished in the TRAIL-R KO mice. In addition, treatment of the CAIA mice with TRAIL significantly decreased the production of TNF-α and T cell-derived cytokines including IL-17 and IFN-γ. These effects were also abrogated in the TRAIL-R KO mice. All these results indicated the TRAIL-mediated inhibition of joint inflammation occurred via an interaction with the TRAIL receptor.
Figure 5.
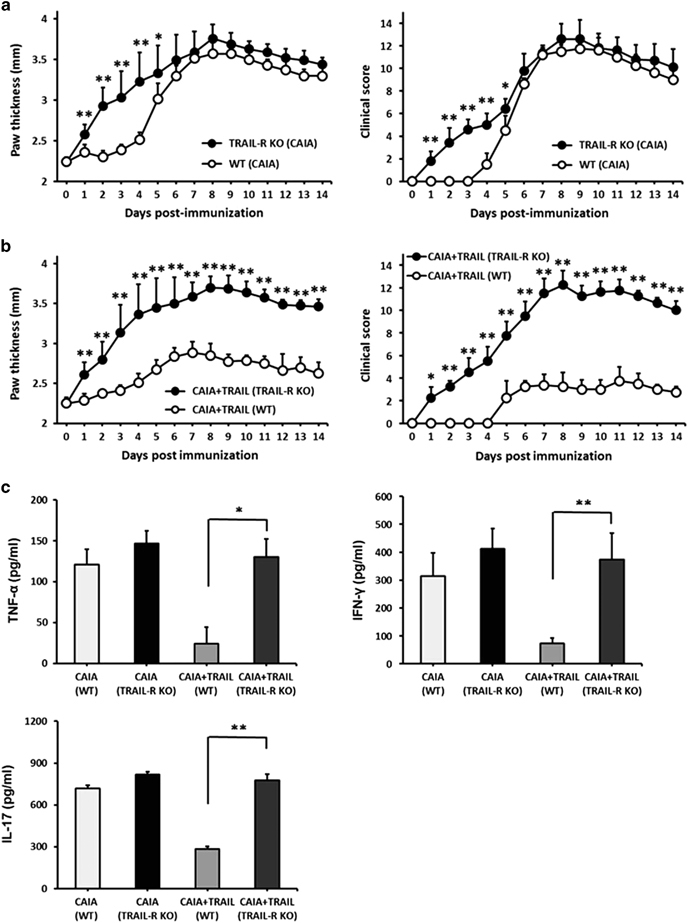
TRAIL-mediated inhibition of joint inflammation occurred via the TRAIL–receptor. (a) C57BL/6 mice and TRAIL-receptor knockout (TRAIL-R KO) mice were immunized with a cocktail of five monoclonal antibodies to type II collagen (3 mg/mouse, IV) on day 0 followed by LPS on day 3 (50 μg/mouse, IP). The hind paw thicknesses (left panel) and clinical scores (right panel) among groups were measured at the indicated time points (n=10 in each group). *P<0.05; **P<0.01 by Student’s t-test. (b) C57BL/6 mice and TRAIL-R KO mice were immunized with a cocktail of five monoclonal antibodies to type II collagen (3 mg/mouse, IV) on day 0 followed by LPS on day 3 (50 μg/mouse, IP). Beginning on day 3 after the immunization, the mice were treated daily with TRAIL (50 μg/mouse, IP). The hind paw thicknesses (left panel) and clinical scores (right panel) among groups were measured at the indicated time points (n=12 in each group). *P<0.05; **P<0.01 by Student’s t-test. (c) The serum T cell-derived cytokines were assayed in each group on day 14 after the immunization. *P<0.05; **P<0.01 by Student’s t-test. The data are representative of at least three independent experiments in each group. LPS, lipopolysaccharide; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
TRAIL directly inhibits T-cell activation by suppressing the T-cell proliferation and cytokine production
To further investigate the possible mechanisms by which TRAIL inhibited joint inflammation and to explore the role of TRAIL in T-cell activation in inflammatory arthritis, we examined the effects of TRAIL on T-cell proliferation and cytokine production. As indicated by the results shown in Figure 6, the proliferation of primary T cells following activation with anti-CD3/CD28 was significantly inhibited by TRAIL in a dose-dependent manner. Notably, this effect was abolished in the T cells from TRAIL-R KO mice. In addition, TRAIL directly inhibited the IL-2 production in the primary T cells following stimulation with anti-CD3/CD28. However, this effect was abrogated in the T cells from the TRAIL-R KO mice. The TRAIL-induced inhibitory effects on the T cells were not due to cellular apoptosis or the pro-apoptotic effects of TRAIL (Supplementary Figure S2) but instead involved the suppression of the downstream T-cell receptor (TCR) signaling pathway (Supplementary Figure S3). Taken together, these results indicated that instead of inducing apoptosis, TRAIL directly inhibited the T-cell activation, thus suppressing the T-cell proliferation and cytokine production.
Figure 6.
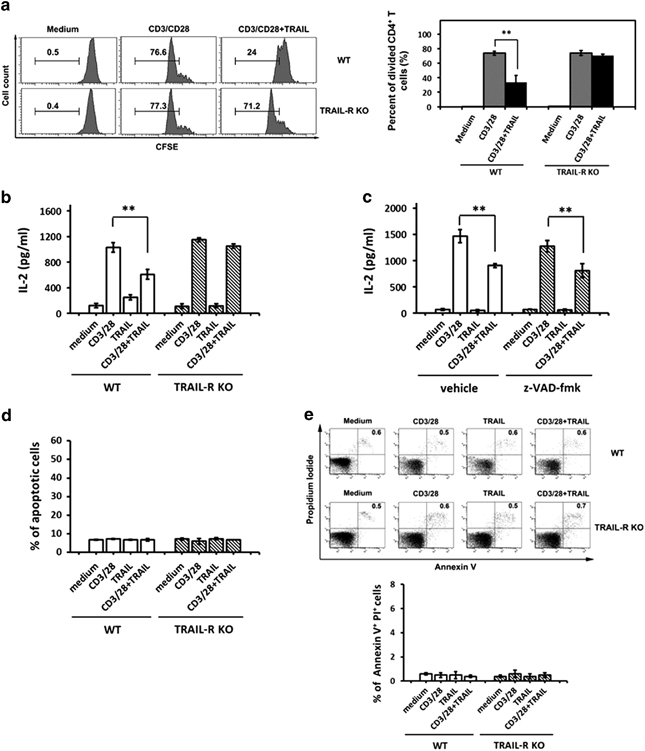
TRAIL inhibited the proliferation of activated T cells and the production of cytokine via the TRAIL receptor in an apoptosis-independent manner. (a) A total of 2 × 105 primary CD4+ T cells from C57BL/6 mice and TRAIL-R KO mice were labeled with CFSE and cultured in 96-well flat-bottomed microtiter plates coated with medium or anti-CD3 Ab (1 μg/ml) and anti-CD28 Ab (1 μg/ml) in the presence or absence of TRAIL (10 μg/ml) for 5 days. Representative figures of the CFSE staining (left panel) and quantified data (right panel) are shown. **P<0.01 by Student’s t-test. The data are representative of at least three independent experiments in each group. (b) Overall, 2 × 106 primary CD4+ T cells from C57BL/6 mice or TRAIL-R KO mice were cultured for 24 h in 96-well flat-bottomed plates precoated with medium, anti-CD3 mAb and anti-CD28, TRAIL or a combination of anti-CD3/anti-CD28 and TRAIL. The supernatants were collected and assayed for IL-2 production by ELISA. The data are shown as the mean±s.d. of triplicate samples. **P<0.01 by Student’s t-test. (c) Overall, 2 × 106 primary CD4+ T cells from C57BL/6 mice were cultured for 24 h in 96-well flat-bottomed plates precoated with medium, anti-CD3 mAb (1 μg/ml) and anti-CD28 (1 μg/ml), TRAIL (10 μg/ml) or the combination of anti-CD3/anti-CD28 and TRAIL in the presence or absence of a pan-caspase inhibitor, z-VAD-fmk (2 μm). The supernatants were collected and assayed for IL-2 production by ELISA. **P<0.01 by Student’s t-test. (d) Overall, 2 × 106 primary CD4+ T cells from C57BL/6 mice or TRAIL-R KO mice were cultured for 24 h in 96-well flat-bottomed plates precoated with medium, anti-CD3 mAb and anti-CD28, TRAIL or the combination of anti-CD3/anti-CD28 and TRAIL. The supernatants were collected and quantified using apoptotic assays. *P<0.05 by Student’s t-test. All data are shown as the mean±s.d. of triplicate samples. (e) Overall, 2 × 106 primary CD4+ T cells from C57BL/6 mice or TRAIL-R KO mice were cultured for 24 h in 96-well flat-bottomed plates precoated with medium, anti-CD3 mAb and anti-CD28, TRAIL or the combination of anti-CD3/anti-CD28 and TRAIL, followed by Annexin V and PI staining. Representative figures of each group are shown (upper panel). The percentages of annexin V+ PI+ cells were quantified (lower panel). *P<0.05 by Student’s t-test. The data are representative of at least three independent experiments in each group. CFSE, carboxyfluorescein succinimidyl ester; CIA, collagen-induced arthritis; PI, propidium iodide; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
DISCUSSION
In this study, we demonstrated that TRAIL significantly inhibited joint inflammation and reduced the severity of arthritis in animal models of inflammatory arthritis. TRAIL profoundly suppressed the joint inflammation in rats with CIA. In addition, TRAIL-R knockout mice developed more severe joint inflammation and were resistant to the TRAIL treatment in the CAIA animal model. Previous studies have demonstrated that treatment of mice with inflammatory arthritis using recombinant TRAIL protein reduced the joint inflammation and the severity of arthritis.19 In agreement with those observations, chronic TRAIL blockade in mice exacerbated the disease severity in autoimmune arthritis and intraarticular administration of TRAIL gene ameliorated the disease.13,24 However, most of these previous results attributed the protective effects against arthritis to the pro-apoptotic activity of TRAIL and its ability to trigger apoptosis of the inflammatory and synovial cells19,24,25 or to blocking cell-cycle progression.13 Although some evidence has indicated that TRAIL promotes synoviocyte proliferation in RA,26,27,28 the effects of TRAIL on the proliferation of inflammatory cells and synovial cells has remained controversial.25,29 In this study, we clearly demonstrated that the suppression of the joint inflammation by TRAIL was not due to the induction of apoptosis in T cells, macrophages or synoviocytes (Figure 3; Supplementary Figure S4). In contrast, TRAIL exerted its anti-inflammatory effects by directly inhibiting the T-cell activation via a TRAIL/TRAIL–R interaction. In addition, it had been demonstrated that RA synovial cells are resistant to apoptotic cell death,26,30 which suggested that an apoptosis-independent pathway was involved in the TRAIL-induced suppression of inflammatory arthritis. These results suggested a novel role for TRAIL in modulating inflammatory arthritis and autoimmune diseases.
In recent years, accumulating evidence has demonstrated that TRAIL modulates the immune responses in autoimmune diseases.13,14,15,16 Administration of TRAIL led to a significant reduction in the antigen-specific antibody production in the autoimmune-prone lymphoproliferative strain of mice, C3H/HeJgld/gld.15 Chronic blockade of TRAIL exacerbated the disease severity in experimental autoimmune EAE16 and autoimmune arthritis 13 in mice. In addition, TRAIL inhibited development of autoimmune diabetes in NOD mice.14 All of these studies and our results emphasize the regulatory role of the TRAIL in modulating autoimmune immune responses and suggest that there are potential implications for TRAIL in therapy for autoimmune diseases.
RA is a prototype of autoimmune arthritis which presents as repeated polyarthritis and causes severe joint damage, osteoporosis and an impaired quality of life. The intense inflammation in RA is mediated by over-reactions of the infiltrating mononuclear phagocytes, lymphocytes, and synovial fibroblasts in the inflamed synovium, and osteoclastogenesis. Proinflammatory cytokines and chemokines act as important mediators of the joint inflammation, acceleration of pannus formation, and subsequent joint destruction and bone loss during the pathogenesis of RA.31,32 In this study, our results demonstrated the potent anti-inflammatory effects of TRAIL in animal models of inflammatory arthritis with effects comparable to the TNF-α blocking agent. In addition, the combination of TRAIL with etanercept synergistically suppressed the joint inflammation and joint destruction and inhibited the production of proinflammatory cytokines and chemokines in inflammatory arthritis. Moreover, TRAIL was demonstrated to strongly suppress the receptor activator of nuclear factor kappa-B ligand (RANKL)-induced osteoclastic differentiation.33 Therefore, TRAIL may regulate osteoclast differentiation during inflammation and has an important role in osteoimmunology. These results suggest that TRAIL is not only potent in inhibiting joint inflammation but is also efficacious in protecting against the joint erosion and bone loss in inflammatory arthritis. Notably, the combination of TRAIL and etanercept synergistically suppressed the joint inflammation in inflammatory arthritis. All of these results suggest that TRAIL has potential as a therapeutic target in RA.
T cells have important roles in the pathogenesis and development of RA.34 Increased expression of TRAIL and its receptors on the peripheral blood T cells in RA patients have been reported to be associated with the RA disease activity.18 In addition to triggering apoptosis, recent evidence has suggested that TRAIL may have a role in T-cell homeostasis and the regulation of immune responses to viral infections and autoimmune diseases.15,35 The inhibitory effect of TRAIL on T-cell activation was also observed in T cells, particularly the CD8+ population, that had infiltrated synovial fluid of RA patients.36 Furthermore, a recent study demonstrated that TRAIL-R costimulation inhibited proximal TCR signaling and suppressed human T-cell activation.23 In support of this, Song et al. 13 demonstrated that blockade of TRAIL led to enhanced proliferation of synovial cells and lymphocytes and increased the production of cytokines in autoimmune arthritis. Our results also demonstrated that TRAIL efficiently suppressed the T-cell proliferation and that the deficiency of TRAIL-R abolished this inhibitory effect. Furthermore, TRAIL effectively inhibited the production of the T cell-derived cytokines (IL-2, IFN-γ and IL-4), which indicated that TRAIL directly inhibited T-cell activation and thus suppressed the T-cell proliferation and cytokine production. Our results describe a novel apoptosis-independent role for TRAIL in suppressing inflammatory arthritis, and shed light on the development of new effective therapies for autoimmune inflammatory diseases.
Electronic supplementary material
Acknowledgements
We thank the core laboratory and Department of Medical Research of National Taiwan University Hospital for facility support. This work was supported by grants from the National Science Council, Taiwan (NSC 101-2321-B-002-008 and 104-2314-B-281-002-; MOST 105-2320-B-002-034- and 105-2320-B-038-065-).
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.2
References
- 1.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 2.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- 3.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 5.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 6.Falschlehner C, Schaefer U, Walczak H. Following TRAIL's path in the immune system. Immunology. 2009;127:145–154. doi: 10.1111/j.1365-2567.2009.03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122–2133. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- 8.Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/S1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- 9.Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–620. doi: 10.1016/S1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 10.Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 11.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 12.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song K, Chen Y, Goke R, Wilmen A, Seidel C, Goke A, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J Exp Med. 2000;191:1095–1104. doi: 10.1084/jem.191.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mi QS, Ly D, Lamhamedi-Cherradi SE, Salojin KV, Zhou L, Grattan M, et al. Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes. 2003;52:1967–1975. doi: 10.2337/diabetes.52.8.1967. [DOI] [PubMed] [Google Scholar]
- 15.Kayagaki N, Yamaguchi N, Abe M, Hirose S, Shirai T, Okumura K, et al. Suppression of antibody production by TNF-related apoptosis-inducing ligand (TRAIL) Cell Immunol. 2002;219:82–91. doi: 10.1016/S0008-8749(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 16.Hilliard B, Wilmen A, Seidel C, Liu TS, Goke R, Chen Y. Roles of TNF-related apoptosis-inducing ligand in experimental autoimmune encephalomyelitis. J Immunol. 2001;166:1314–1319. doi: 10.4049/jimmunol.166.2.1314. [DOI] [PubMed] [Google Scholar]
- 17.Xiao H, Wang S, Miao R, Kan W. TRAIL is associated with impaired regulation of CD4+CD25- T cells by regulatory T cells in patients with rheumatoid arthritis. J Clin Immunol. 2011;31:1112–1119. doi: 10.1007/s10875-011-9559-x. [DOI] [PubMed] [Google Scholar]
- 18.Bisgin A, Terzioglu E, Aydin C, Yoldas B, Yazisiz V, Balci N, et al. TRAIL death receptor-4, decoy receptor-1 and decoy receptor-2 expression on CD8+ T cells correlate with the disease severity in patients with rheumatoid arthritis. BMC Musculoskelet Disord. 2010;11:192. doi: 10.1186/1471-2474-11-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin CH, Chae SY, Kim TH, Yang HK, Lee EY, Song YW, et al. Effect of tumor necrosis factor-related apoptosis-inducing ligand on the reduction of joint inflammation in experimental rheumatoid arthritis. J Pharmacol Exp Ther. 2010;332:858–865. doi: 10.1124/jpet.109.159517. [DOI] [PubMed] [Google Scholar]
- 20.Tsai HF, Lai JJ, Chou AH, Wang TF, Wu CS, Hsu PN. Induction of costimulation of human CD4 T cells by tumor necrosis factor-related apoptosis-inducing ligand: possible role in T cell activation in systemic lupus erythematosus. Arthritis Rheum. 2004;50:629–639. doi: 10.1002/art.20038. [DOI] [PubMed] [Google Scholar]
- 21.Huang SC, Tsai HF, Tzeng HT, Liao HJ, Hsu PN. Lipid raft assembly and Lck recruitment in TRAIL costimulation mediates NF-kappaB activation and T cell proliferation. J Immunol. 2011;186:931–939. doi: 10.4049/jimmunol.1001092. [DOI] [PubMed] [Google Scholar]
- 22.Chou AH, Tsai HF, Lin LL, Hsieh SL, Hsu PI, Hsu PN. Enhanced proliferation and increased IFN-gamma production in T cells by signal transduced through TNF-related apoptosis-inducing ligand. J Immunol. 2001;167:1347–1352. doi: 10.4049/jimmunol.167.3.1347. [DOI] [PubMed] [Google Scholar]
- 23.Lehnert C, Weiswange M, Jeremias I, Bayer C, Grunert M, Debatin KM, et al. TRAIL-receptor costimulation inhibits proximal TCR signaling and suppresses human T cell activation and proliferation. J Immunol. 2014;193:4021–4031. doi: 10.4049/jimmunol.1303242. [DOI] [PubMed] [Google Scholar]
- 24.Yao Q, Seol DW, Mi Z, Robbins PD. Intra-articular injection of recombinant TRAIL induces synovial apoptosis and reduces inflammation in a rabbit knee model of arthritis. Arthritis Res Ther. 2006;8:R16. doi: 10.1186/ar1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichikawa K, Liu W, Fleck M, Zhang H, Zhao L, Ohtsuka T, et al. TRAIL-R2 (DR5) mediates apoptosis of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2003;171:1061–1069. doi: 10.4049/jimmunol.171.2.1061. [DOI] [PubMed] [Google Scholar]
- 26.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–15718. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 27.Audo R, Combe B, Coulet B, Morel J, Hahne M. The pleiotropic effect of TRAIL on tumor-like synovial fibroblasts from rheumatoid arthritis patients is mediated by caspases. Cell Death Differ. 2009;16:1227–1237. doi: 10.1038/cdd.2009.38. [DOI] [PubMed] [Google Scholar]
- 28.Audo R, Calmon-Hamaty F, Baeten D, Bruyer A, Combe B, Hahne M, et al. Mechanisms and clinical relevance of TRAIL-triggered responses in the synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheum. 2011;63:904–913. doi: 10.1002/art.30181. [DOI] [PubMed] [Google Scholar]
- 29.Terzioglu E, Bisgin A, Sanlioglu AD, Ulker M, Yazisiz V, Tuzuner S, et al. Concurrent gene therapy strategies effectively destroy synoviocytes of patients with rheumatoid arthritis. Rheumatology. 2007;46:783–789. doi: 10.1093/rheumatology/kel448. [DOI] [PubMed] [Google Scholar]
- 30.Perlman H, Nguyen N, Liu H, Eslick J, Esser S, Walsh K, et al. Rheumatoid arthritis synovial fluid macrophages express decreased tumor necrosis factor-related apoptosis-inducing ligand R2 and increased decoy receptor tumor necrosis factor-related apoptosis-inducing ligand R3. Arthritis Rheum. 2003;48:3096–3101. doi: 10.1002/art.11302. [DOI] [PubMed] [Google Scholar]
- 31.Smolen JS, Tohidast-Akrad M, Gal A, Kunaver M, Eberl G, Zenz P, et al. The role of T-lymphocytes and cytokines in rheumatoid arthritis. Scand J Rheumatol. 1996;25:1–4. doi: 10.3109/03009749609082660. [DOI] [PubMed] [Google Scholar]
- 32.Lunemann JD, Waiczies S, Ehrlich S, Wendling U, Seeger B, Kamradt T, et al. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J Immunol. 2002;168:4881–4888. doi: 10.4049/jimmunol.168.10.4881. [DOI] [PubMed] [Google Scholar]
- 33.Zauli G, Rimondi E, Nicolin V, Melloni E, Celeghini C, Secchiero P. TNF-related apoptosis-inducing ligand (TRAIL) blocks osteoclastic differentiation induced by RANKL plus M-CSF. Blood. 2004;104:2044–2050. doi: 10.1182/blood-2004-03-1196. [DOI] [PubMed] [Google Scholar]
- 34.Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology. 2012;51:v3–11. doi: 10.1093/rheumatology/kes113. [DOI] [PubMed] [Google Scholar]
- 35.Gyurkovska V, Ivanovska N. Distinct roles of TNF-related apoptosis-inducing ligand (TRAIL) in viral and bacterial infections: from pathogenesis to pathogen clearance. Inflamm Res. 2016;65:427–437. doi: 10.1007/s00011-016-0934-1. [DOI] [PubMed] [Google Scholar]
- 36.Martinez-Lorenzo MJ, Anel A, Saez-Gutierrez B, Royo-Canas M, Bosque A, Alava MA, et al. Rheumatoid synovial fluid T cells are sensitive to APO2L/TRAIL. Clin Immunol. 2007;122:28–40. doi: 10.1016/j.clim.2006.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.