

Abstract
Programmed death one homolog (PD-1H) is a cell surface molecule of the B7/CD28 immune modulatory gene family. Although PD-1H has been shown to function as a coinhibitory receptor on T cells to limit naive T-cell activation and proliferation, its role in the regulation of the T-cell response to allergens is unknown. We report here that genetic ablation or blockade of PD-1H drastically promotes pulmonary inflammation with massive accumulation of eosinophils in a mouse model of experimental asthma, indicating a suppressive function of PD-1H in allergic inflammation. The loss of PD-1H led to elevated production of both innate cytokines (IL-6, MCP-1 and TNFα) and Th2 cytokines (IL-5 and IL-13) in the lung, indicating a critical role of PD-1H in suppressing the production of airway inflammatory cytokines. In addition, the loss of PD-1H also impaired the expansion of systemic and pulmonary regulatory T cells during asthma induction. These findings support a critical role of intrinsic PD-1H in the regulation of inflammatory responses to allergens. Finally, we showed that treatment with a PD-1H agonistic monoclonal antibody reduced the severity of asthma, which was accompanied by suppressed lung inflammation. Our findings support PD-1H as a potential target and suggest a possible strategy for the treatment of allergic asthma in humans.
Keywords: Asthma, PD-1H, Th2, VISTA
Introduction
PD-1H (also called Gi24, VISTA, DD1α or Dies1) is a cell surface molecule of the B7/CD28 immune modulatory gene family.1 Alignment of the PD-1H Immunoglobulin V region with CD28 members shows the highest identity with the programmed death one (PD-1) protein. PD-1H is constitutively expressed on most hematopoietic cells, including both lymphoid cells (except B cells) and myeloid cells.2 Several lines of evidence have supported the hypothesis that PD-1H functions as a coinhibitory receptor on T cells to limit naive T-cell activation, whereas PD-1H expressed on antigen-presenting cells interacts with an unknown receptor on T cells to suppress T-cell responses.3,4 A recent study has shown that the PD-1H/PD-1H interaction between different cells (homophilic interaction) could promote macrophage-mediated clearance of dead cells and T-cell suppression.5 PD-1H knockout (KO) mice were shown to develop more severe inflammation and autoimmune diseases in several mouse models,6,7 indicating a role of PD-1H in the suppression of T-cell immunity. By contrast, agonistic monoclonal antibodies (mAbs) to mouse PD-1H were shown to suppress T-cell-mediated acute hepatitis and prevent acute graft-versus-host disease (GVHD) in semi- and fully allogeneic murine models, leading to full chimerism following treatment. It appears that the effect of PD-1H on the suppression of the T-cell response could be divided into two stages with an early event in arresting allo-reactive donor T cells from activation and a later event in promoting donor Treg expansion.
Although PD-1H could suppress the CD4+ T-cell response to antigens, its role in Th2-like responses, especially under pathogenic conditions, is not yet known. Asthma is a common, chronic inflammatory disease of the airways, and CD4+ Th2 cells have been shown to have a critical role in disease induction, pathogenesis and progression. The hallmarks of Th2-type responses in asthma are eosinophilic airway inflammation with mucus secretion, airway remodeling, and hyper-reactivity.8,9 IL-5 and IL-13 are critical for the pathophysiology of asthma. IL-5 has multifaceted roles, including the direct activation of eosinophils, influencing adhesion and inducing chemotaxis and inflammatory mediator synthesis. IL-13 has a major impact on influencing bronchial hyper-reactivity, inflammation, and airway remodeling.10 Moreover, IL-13 drives epithelial cell maturation and mucus production, the synthesis of extracellular matrix proteins and enhances contractility of airway smooth muscle cells. The mechanisms of the regulation of cytokine production during asthma pathogenesis, however, are yet to be elucidated. Recent therapeutic efforts have focused on blockade of the interaction of these cytokines to their receptors as an approach for asthma treatment.
In this study, we evaluate the role of PD-1H in chicken ovalbumin (OVA)-induced allergic airway inflammation by employing PD-1H KO mice and soluble PD-1H fusion protein. Our findings show that PD-1H is a critical regulator of Th2 T-cell responses. Furthermore, our studies support the development of agonists of PD-1H as potential therapeutic agents for the treatment of human asthma.
Materials and methods
Mice
All experiments were carried out in accordance with the guidelines of Sun Yat-sen University on animal care and the ethical guidelines for the investigation of experimental animals. BALB/c mice were purchased from the Experimental Animal Centre of Sun Yat-sen University (Guangzhou, China). PD-1H KO mice were described previously and were backcrossed with BALB/c mice to generate the H-2d/PD-1H KO strain. Eight- to ten-week-old mice were used for all experiments and were kept under specific pathogen-free conditions.
Generation of PD-1H mAb
Full-length mouse PD-1H-pcDNA3.1 was stably transfected into Chinese hamster ovary cells (CHO cells) by lipofection, and the stably transfected PD-1H+ CHO cells were confirmed by flow cytometry using a mam82 mAb. The PD-1H-Ig fusion protein was produced and purified as described previously.11 PD-1H KO mice were immunized with mouse PD-1H-Ig, and the generation of hybridomas secreting PD-1H mAb was performed as described previously.12 The specificity of the mAb was validated by ELISA and flow cytometry using PD-1H+ CHO cells. A clone of PD-1H mAb, 4C11, was selected for further experiments, and was produced and purified as previously described.
Mouse model of experimental asthma
Mice were sensitized by intraperitoneal (i.p.) injection of 10 μg OVA (Sigma-Aldrich, St Louis, MO, USA) with 4 mg aluminum hydroxide (Thermo Fisher, Waltham, MA, USA) gel on days 0 and 5, followed by challenge with 15 or 25 ml of 1% aerosolized OVA for 20 or 40 min on days 12, 13 and 14. The aerosol was generated by a nebulizer (NE-U07; Yuyue, Suzhou, China). Mice were killed for analyses on day 15. For PD-1H-Ig treatment, groups of mice received hydrodynamic injections13 of 20 μg PD-1H plasmid in 2 ml PBS intravenously (i.v.) on days −1, 4 and 11, and the Flag plasmid was the control. The levels of PD-1HIg fusion protein in the sera were detected by a specific sandwich ELISA method (Supplementary Figure 1). For mAb treatment, groups of mice received i.p. injections of 200 μg anti-mouse PD-1H mAb (4C11) on days 0, 3, 6, 9 and 12, or control mouse immunoglobulin G (mIgG) (Rockland, Gilbertsville, PA, USA).
Assays of bronchoalveolar lavage fluid and sera
Mice were anesthetized with a lethal dose of pentobarbital, and the lungs were gently lavaged with 0.5 ml PBS three times (1.5 ml of total bronchoalveolar lavage fluid (BALF)) via a tracheal cannula. Samples were centrifuged at 2000 r.p.m. for 5 min. Mouse IL-4, IL-5 and IL-13 in BALF were quantified using ELISA kits (eBioscience, San Diego, CA, USA) according to the manufacturer’s protocols, and MCP-1, IL-6, TNFα and IFN-γ were measured using the CBA kit (BD Bioscience, San Jose, CA, USA). The total counts of cells in BALF were determined using a microscope. To identify BALF differential cell counts, BALF cells were spun onto microscope slides by CytoFuge and were stained with Diff-Quick (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Differential cell counts were performed by counting 400 cells per slide using a high-magnification microscope. Sera from experimental mice was collected, and OVA-specific IgE was examined by specific sandwich ELISA.
Histology
After the bronchial lavage procedure, lungs were exsanguinated and fixed by intratracheal instillation with 1 ml 10% formalin and subsequently removed from mice. After successive dehydration, the lungs were embedded in paraffin and sectioned and subsequently stained with H&E and periodic acid-Schiff (PAS).
Flow cytometric analysis
Cells (0.5–1 × 106) were first incubated with unlabeled anti-FcR mAb to block nonspecific binding of mAbs to FcR and then were incubated with labeled mAbs. The mAbs against H-2Kd, H-2Kb, mIgG, CD3, CD4, CD8, Siglec-F, IL-4, IFN-γ and Foxp3 were purchased from eBioscience. For intracellular Foxp3 staining, lymphocytes from lungs and spleens were isolated as described previously,14 and the cells were subsequently fixed, permeabilized and stained following the manufacturer’s protocol of the Cytofix/Cytoperm Plus kit (BD Biosciences). For intracellular IL-4 and IFN-γ staining, isolated lymphocytes were stimulated by 10 ng/ml PMA (Sigma-Aldrich), 1 μg/ml ionomycin (Sigma-Aldrich) and Golgi plug (BD Biosciences) for 4 h and then were stained using the Cytofix/Cytoperm Plus kit (BD Biosciences).
Statistical analysis
Data were analyzed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Unpaired student’s t-test was used to determine the statistical significance between groups, with P<0.05 being considered significant.
Results
PD-1H is required to suppress airway inflammation in an experimental asthma model
Wild-type (WT) and PD-1H KO mice were immunized and challenged by OVA to induce asthma (Figure 1a). Mice were killed at day 15, and differential cell counts were performed in collected BALF. The challenge by the OVA induced pulmonary infiltration of inflammatory cells in WT mice and inflammatory cells was dominated by eosinophils as well as with increased lymphocytes and macrophages. This prominent eosinophilic response is highly characteristic of allergic asthma in this model. Interestingly, PD-1H KO mice had significantly higher total numbers of inflammatory cells in BALF than WT mice, with significant increase in eosinophils. Infiltrating lymphocytes were found in small numbers, and macrophages were negligible (Figures 1b and c). These findings indicate that PD-1H is required to inhibit the recruitment of inflammatory cells to the lung in this model.
Figure 1.
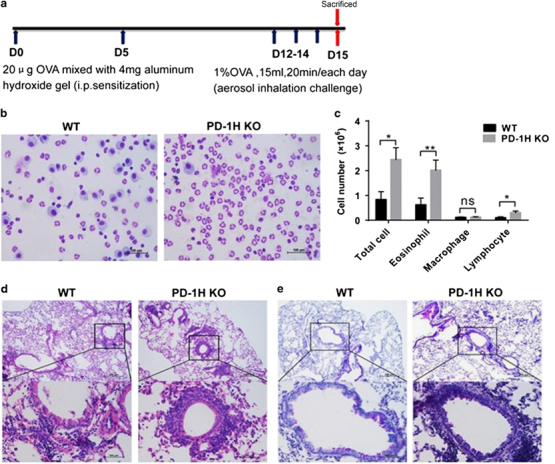
PD-1H KO mice developed severe lung inflammation and mucus secretion in the OVA-induced asthma model. (a) Experimental protocol to induce experimental asthma: WT and PD-1H KO mice were immunized by i.p. injection of 20 μg OVA mixed with 4 mg aluminum hydroxide gel on days 0 and 5, followed by challenge with 1% OVA (15 ml/20 min/day) on three consecutive days (12–14) by aerosol instillation. The mice were killed for analyses on day 15. (b, c) BALF was collected and stained for leukocyte counts. (d) H&E staining of lung paraffin sections (e) PAS staining of lung paraffin sections for mucus-secreting goblet cells. The scale bars in b, d and e represent 100 μm. *P<0.05 and **P<0.01 (two-tailed student’s t-test). All values are expressed as means±s.e.m. N=8 per group. All experiments were repeated at least three times. BALF, bronchoalveolar lavage fluid; i.p., intraperitoneal; KO, knockout; OVA, ovalbumin.
We next compared the lung histology of OVA-challenged WT and KO mice. Compared with WT mice, OVA-challenged KO mice showed substantially more inflammatory cell infiltration mainly around bronchi (Figure 1d). At the same time, OVA-challenged KO mice showed significantly larger and more PAS+ mucin-producing goblet cells lining the bronchial air spaces (Figure 1e), accompanied by profound goblet cell hyperplasia and metaplasia, indicating primarily bronchial involvement and extensive airway remodeling during inflammation. These results demonstrate substantially more severe histopathologic changes characteristic of inflammatory asthma in OVA-challenged lungs in KO than in WT mice. Taken together, our results indicate that endogenous PD-1H suppresses the development of severe inflammatory cell infiltration and airway remodeling following OVA challenge.
To validate our findings, we also tested the effect of PD-1HIg, a fusion protein of the extracellular domain of PD-1H and the IgG2a Fc portion. In our preliminary experiments, we found that high pressure injection of the PD-1HIg plasmid (hydrodynamic injection) led to high-level expression of the PD-1HIg fusion protein (Supplementary Figure S1) as detected in mouse sera and that this method had a more profound effect than the infusion of purified recombinant PD-1HIg fusion protein (Liu et al., unpublished data). At the beginning of the OVA-induced asthma model, the mice were injected i.v. with PD-1H-Ig or Flag-Ig control plasmid via the tail vein under high pressure on days −1, 4 and 11. The mice were killed at day 15, and the BALF was collected. Similar to PD-1H KO mice, PD-1H-Ig-treated mice showed significantly increased eosinophil counts compared to mice treated with the Flag-Ig plasmid control (Figures 2a and b), indicating that PD-1H-Ig may block PD-1H’s interaction with its counter-receptor to promote airway inflammation in OVA-induced asthma. Thus, our results further support a suppressive role of endogenous PD-1H in the development of lung inflammation during the induction of experimental asthma.
Figure 2.

PD-1H-Ig promotes airway inflammation in OVA-induced asthma. Allergic asthma was induced by OVA as described in Figure 1a. WT mice were i.v. injected with 20 μg PD-1H-Ig plasmid in 2 ml PBS through the tail vein on days −1, 4 and 11. Flag-Ig plasmid was used as the control. BALF was collected after the last OVA challenge and stained (a) for leukocyte counts (b). The scale bar in a represents 100 μm. *P<0.05 and ***P<0.001 (two-tailed student’s t-test). All values are expressed as means±s.e.m. N=8 per group. All experiments were repeated at least three times. BALF, bronchoalveolar lavage fluid; i.v., intravenously; OVA, ovalbumin; WT, wild-type.
Mechanisms of PD-1H-mediated suppression of airway inflammation
We first determined the levels of cytokines in the BALF in OVA-challenged WT and PD-1H KO mice. The KO mice challenged with OVA had significantly higher levels of IL-5, IL-13, IL-6, MCP-1 and TNFα in the BALF than WT mice, whereas the changes in IL-4 and IFN-γ were insignificant (Figure 3a). The OVA-specific IgE in sera was measured as well, but no difference was observed (Figure 3b). These results are consistent with the cell counts and histopathological findings, showing increased lung inflammation biased to eosinophil recruitment in the KO mice. Similar results were also obtained in the WT mice treated by hydrodynamic inoculation of the PD-1H-Ig plasmid, with significantly higher levels of IL-5 and IL-13 than those in the mice treated with the control plasmid (Figure 3c). Interestingly, treatment with PD-1H-Ig led to significantly increased IL-4 in BALF (Figure 3d). Our results indicate that endogenous PD-1H inhibits the production of innate (MCP-1, TNFα, IL-6) and Th2-like cytokines (IL-5, IL-13) during the induction of airway inflammation.
Figure 3.
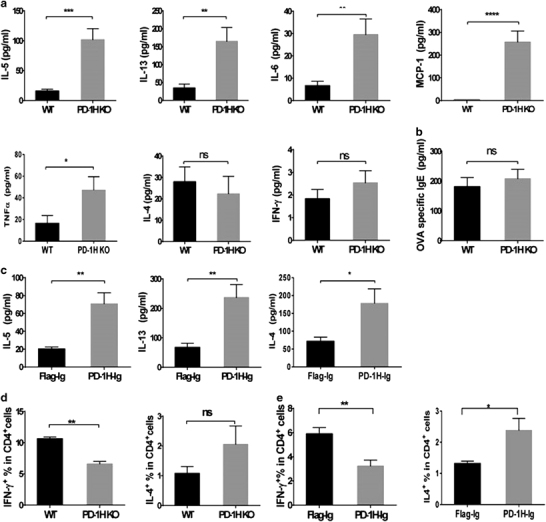
Blockade of PD-1H enhanced innate and Th2-type cytokine production in OVA-induced asthma. (a) BALF from WT and PD-1H KO mice was collected, and the indicated cytokines were assessed by ELISA or CBA (N=8 per group). (b) Sera from the WT and KO mice was collected, and OVA-specific IgE was measured by ELISA (N=8 per group). (c) BALF from mice treated with the PD-1H-Ig or flag plasmid was collected, and the indicated cytokines were assessed by ELISA (N=8 per group). (d) Percentage of IL-4-producing and IFN-γ-producing CD4+ T cells in WT and KO mice in OVA-induced asthma. Lung lymphocytes isolated after the last OVA challenge were stimulated with PMA and ionomycin for 4 h in the presence of GolgiStop with brefeldin A. IL-4 and IFN-γ were measured by intracellular staining (N=8 per group). (e) The percentages of IL-4-producing and IFN-γ-producing CD4+T cells in wild-type mice treated with PD-1H-Ig or Flag-Ig plasmid in OVA-induced asthma were analyzed (N=8 per group). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 (two-tailed student’s t-test). All values are expressed as means±s.e.m. All experiments were repeated at least three times. BALF, bronchoalveolar lavage fluid; KO, knockout; OVA, ovalbumin; WT, wild-type.
We also determined CD4+ T-helper-cell types in lung lymphocytes after brief PMA/ionomycin stimulation in vitro by intracellular staining of cytokines.15 CD4+IL-4+ Th2-type cells were significantly higher in KO than in WT mice in the lung during the asthma induction. By contrast, the Th1-type cells (CD4+IFN-γ+) were significantly fewer in KO than in WT mice (Figure 3e). Similar results were also observed in WT mice treated with PD-1H-Ig versus control Ig plasmids (Figure 3f). These results of intracellular cytokine detection further validate previous findings of extracellular cytokine measurement in the BALF and support the role of endogenous PD-1H in the suppression of CD4+Th2-like responses in the experimental asthma model.
We showed previously that PD-1H promoted Treg expansion in the induction of GVHD in several mouse models. We next evaluated whether there was impaired generation of Tregs during the induction of the airway inflammatory response in the absence of PD-1H. CD4+Foxp3+ Tregs in the lungs and spleens were evaluated by flow cytometry after last OVA challenge of WT and KO mice. The percentage of Tregs was significantly lower in both the lungs (Figure 4a) and spleens (Figure 4b) of KO mice than in those of WT mice. Our results suggest a possible contribution of Tregs in the PD-1H-mediated suppression of airway inflammation. Therefore, PD-1H may operate via multiple mechanisms to suppress airway inflammation.
Figure 4.

Decreased Treg levels during experimental asthma induction in PD-1H KO mice. (a) Lung lymphocytes and (b) spleens from WT and KO mice were collected after the last OVA inhalation, and the percentages of Foxp3+CD4+ T cells were analyzed by intracellular staining. **P<0.01 and ****P<0.0001 (two-tailed student’s t-test). All values are expressed as means±s.e.m. N=8 per group. All experiments were repeated at least three times. KO, knockout; OVA, ovalbumin; WT, wild-type.
Amelioration of experimental asthma using a new agonistic PD-1H antibody
In the context of the roles of PD-1H in the suppression of airway inflammation, we explored whether PD-1H could be targeted to suppress asthma in the experimental model. We reported several agonistic mAbs (clones MH5A, mam82) with suppressive function in GVHD in mouse models. In our preliminary studies, these mAbs, however, are less consistent in suppressing airway inflammation in the OVA-induced asthma model (Liu et al., data not shown). We generated additional mAbs through the immunization of PD-1H KO mice with PD-1H-Ig. One mAb, designated 4C11, was selected based on its specificity and high affinity for murine PD-1H. The mAb 4C11 reacted strongly with PD-1H+ CHO cells in a similar manner to clone mam82 (Figure 5a), and this binding could be blocked by the inclusion of PD-1H-Ig (data not shown).
Figure 5.

4C11 monoclonal antibody and its role in suppressing airway inflammation. (a) PD-1H+ CHO cells were stained with mIgG, mam82 and 4C11 and were analyzed by flow cytometry. (b, c) Airway inflammation and asthma were induced as described in Figure 1a, but the mice were challenged with 1% OVA (25 ml/40 min/day) on three consecutive days (12–14). Meanwhile, mice were treated by i.p. injection of 200 μg 4C11 or control mIgG in 200 μl PBS on days 0, 3, 6, 9 and 12. BALF was collected after the last challenge and was stained for leukocyte counts. (d) H&E staining of lung paraffin sections. (e) PAS staining of lung paraffin sections for mucus-secreting goblet cells. The scale bars in b, d and e represent 100 μm). *P<0.05 and **P<0.01 (two-tailed student’s t-test). All values are expressed as means±s.e.m. N=8 per group. All experiments were repeated at least three times. BALF, bronchoalveolar lavage fluid; i.p., intraperitoneal; mIgG, mouse immunoglobulin G; PAS, periodic acid-Schiff.
To test the effect of 4C11, mice were administered 4C11 or control mIgG on days 0, 3, 6, 9 and 12 during the induction of OVA-induced asthma. Mice treated with control IgG developed typical accumulation of eosinophils in BALF fluid (Figures 5b and c). The histopathological examination showed massive inflammatory cell infiltration around the bronchi (Figure 5d) and mucus overproduction into the bronchi (Figure 5e). By contrast, the accumulation of eosinophils in BALF was greatly reduced in the 4C11-treated mice (Figures 5b and c). Meanwhile, the infiltration of inflammatory cells around the bronchi (Figure 5d) and overproduction of mucus (Figure 5e) were also decreased significantly. These results indicate that agonistic PD-1H mAb 4C11 can suppress airway inflammation and asthma development and imply that PD-1H could be a potential therapeutic target for asthma treatment.
Discussion
Using a PD-1H-deficient mouse strain and recombinant PD-1H-Ig fusion protein, we showed that PD-1H deficiency or blocking PD-1H engagement with its ligand via soluble PD-1H led to vastly increased accumulation of infiltrating inflammation in the airways with eosinophils as the major cell component. PD-1H deficiency or blocking mice show elevated levels of Th2-type cytokines, including IL-5 and IL-13, and innate inflammatory cytokines MCP-1 and IL-6, as well as increased Th2-like CD4+ T cells and decreased Treg, supporting the possibility that PD-1H could function as a negative regulator in the control of airway inflammation in experimental asthma progression.
In the OVA-induced experimental asthma model, PD-1H appeared to mainly suppress IL-5 and IL-13 production, whereas IL-4 was less altered (Figure 3). These findings are consistent with the observation that most of the accumulated leukocytes in the airways are eosinophils but not lymphocytes. IL-5 was originally defined as a T-cell-derived cytokine and is now appreciated as a major cytokine that affects many eosinophil-related aspects, including maturation, differentiation, migration and survival. IL-13 can be produced by various immune and non-immune cells including T cells and eosinophils16 and could induce airway eosinophilia, airway hyper-responsiveness, and mucus overproduction.17 Although IL-4 appears to be less affected by the loss of PD-1H, the effect of IL-4 may be replaced by a high level of IL-13 because IL-13 and IL-4 share a common α chain receptor subunit as their receptors18,19 and have overlapping biological functions.20 PD-1H is constitutively found on the surface of naive T lymphocytes, whereas its expression on eosinophils has not yet been reported. In our experiments, however, inflammatory eosinophils isolated from BALF do not express PD-1H (Liu et al., unpublished result). Because both IL-5 and IL-13 could be produced by T cells, it is thus possible that PD-1H on T cells may directly mediate the suppression of cytokine production. On the other hand, IL-13 could be produced by various hematopoietic cells, whereas PD-1H is also broadly expressed on most hematopoietic cells. It is thus tempting to speculate that PD-1H may have a broader suppressive function beyond T cells for IL-13 production from hematopoietic cells in addition to T cells and eosinophils.
Both naturally occurring thymus-derived CD4+CD25+Foxp3+ Tregs and an inducible population of Tregs suppress the development of allergies, including allergic asthma, through multiple mechanisms21,22 and the inhibition of other effector Th1, Th2 and Th17 cells, eosinophils, basophils, mast cells, inflammatory DCs and inflammatory cell migration to tissues.23 We showed previously that the agonistic mAb MH5A for mouse PD-1H selectively promotes Treg cell expansion in murine GVHD models. Our results showed insignificant differences in the percentage of CD4+ and CD8+T cells between WT and KO mice in both the lungs and spleens during experimental asthma induction (Supplementary Figure 2). However, PD-1H deficiency led to a markedly reduced percentage of CD4+ Foxp3+ Tregs in the lungs compared with WT mice, and the proportion of Tregs in the spleens was reduced as well during airway inflammation (Figure 4). These findings may partially explain the more severe asthma pathogenesis in PD-1H KO mice.
We reported previously that administration of several PD-1H agonistic mAbs could suppress acute hepatitis and GVHD in animal models. Using a newly generated mAb 4C11, the treatment could remarkably decrease the number of inflammatory cells in BALF, especially the eosinophils and lymphocytes, as well as pulmonary inflammation and mucus production (Figure 5). Our results thus implicate the possibility of targeting PD-1H for the treatment of allergic diseases. In our early attempt to treat the experimental asthma model with MH5A and mam82 agonistic mAb, however, the effect was inconsistent among different experiments (Liu et al., unpublished observation). Currently, the reason behind the different effects of these PD-1H agonistic mAbs has yet to be investigated. It is possible that these mAbs may interact with PD-1H at different functional domains, subsequently triggering signals that are different in quantity or in quality, leading to different biological outcomes. Nevertheless, our results suggest a possible strategy for the treatment of allergic asthma using an agonistic mAb against PD-1H.
Electronic supplementary material
Acknowledgements
This work is supported in part by the 985 project grant from Sun Yat-Sen University, Guangdong Province Innovative Research Program Project 2011Y035, PRC; National Institutes of Health grants P50 CA196530 and P30 CA016359; and an endowment from the United Technologies Corporation, USA.
Author contributions
HL and LC developed the concept, designed the experiments and wrote the manuscript. XL, LH and LL advised, and/or performed the experiments.
Conflict of interest
LC serves as an advisor/board member for Pfizer, AstraZeneca, NextCure, GenomiCare and Vcanbio, and receives research support from Boehringer Ingelheim, Pfizer and NextCure. LC also serves as an uncompensated adjunct faculty member of Sun Yat-Sen University. The remaining authors declare no conflict of interest.
Footnotes
The original online version of this article was revised: In the Acknowledgement section of this article, NIH grants numbered P50 CA196530 and P30 CA016359 were incorrectly attributed to author Lieping Chen and should not have been cited. The original article has been corrected.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.16
References
- 1.Flies DB, Higuchi T, Chen L. Mechanistic assessment of PD-1H coinhibitory receptor-induced T cell tolerance to allogeneic antigens. J Immunol. 2015;194:5294–5304. doi: 10.4049/jimmunol.1402648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flies DB, Wang S, Xu H, Chen L. Cutting edge: a monoclonal antibody specific for the programmed death one homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187:1537–1541. doi: 10.4049/jimmunol.1100660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J Clin Invest. 2014;124:1966–1975. doi: 10.1172/JCI74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015;349:1261669. doi: 10.1126/science.1261669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceeraz S, Sergent PA, Plummer SF, Schned AR, Pechenick D, Burns CM et al. VISTA deficiency accelerates the development of fatal murine lupus nephritis. Arthritis Rheumatol, 2016 epubdate ahead of print. [DOI] [PMC free article] [PubMed]
- 7.Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci USA. 2014;111:14846–14851. doi: 10.1073/pnas.1407447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. 2014;134:499–507. doi: 10.1016/j.jaci.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 9.Veres TZ, Kopcsanyi T, van Panhuys N, Gerner MY, Liu Z, Rantakari P, et al. Allergen-induced CD4+ T cell cytokine production within airway mucosal dendritic cell-T cell clusters drives the local recruitment of myeloid effector cells. J Immunol. 2017;198:895–907. doi: 10.4049/jimmunol.1601448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.May RD, Fung M. Strategies targeting the IL-4/IL-13 axes in disease. Cytokine. 2015;75:89–116. doi: 10.1016/j.cyto.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Ogasawara K, Yoshinaga SK, Lanier LL. Inducible costimulator costimulates cytotoxic activity and IFN-gamma production in activated murine NK cells. J Immunol. 2002;169:3676–3685. doi: 10.4049/jimmunol.169.7.3676. [DOI] [PubMed] [Google Scholar]
- 12.Paczesny S, Choi SW, Ferrara JL. Acute graft-versus-host disease:new treatment strategies. Curr Opin Hematol. 2009;16:427–436. doi: 10.1097/MOH.0b013e3283319a6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azuma T, Zhu G, Xu H, Rietz AC, Drake CG, Matteson EL, et al. Potential role of decoy B7-H4 in the pathogenesis of rheumatoid arthritis: a mouse model informed by clinical data. PLoS Med. 2009;6:e1000166. doi: 10.1371/journal.pmed.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Dong Z, Zhou R, Luo D, Wei H, Tian Z. Isolation of lymphocytes and their innate immune characterizations from liver, intestine, lung and uterus. Cell Mol Immunol. 2005;2:271–280. [PubMed] [Google Scholar]
- 15.Miller RA, Garcia G, Kirk CJ, Witkowski JM. Early activation defects in T lymphocytes from aged mice. Immunol Rev. 1997;160:79–90. doi: 10.1111/j.1600-065X.1997.tb01029.x. [DOI] [PubMed] [Google Scholar]
- 16.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 19.Zurawski SM, Chomarat P, Djossou O, Bidaud C, McKenzie AN, Miossec P, et al. The primary binding subunit of the human interleukin-4 receptor is also a component of the interleukin-13 receptor. J Biol Chem. 1995;270:13869–13878. doi: 10.1074/jbc.270.23.13869. [DOI] [PubMed] [Google Scholar]
- 20.Chomarat P, Banchereau J. Interleukin-4 and interleukin-13: their similarities and discrepancies. Int Rev Immunol. 1998;17:1–52. doi: 10.3109/08830189809084486. [DOI] [PubMed] [Google Scholar]
- 21.Duvernelle C, Freund V, Frossard N. Transforming growth factor-beta and its role in asthma. Pulm Pharmacol Ther. 2003;16:181–196. doi: 10.1016/S1094-5539(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 22.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. RegulatoryT cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Palomares O, Yaman G, Azkur AK, Akkoc T, Akdis M, Akdis CA. Role of Treg in immune regulation of allergic diseases. Eur J Immunol. 2010;40:1232–1240. doi: 10.1002/eji.200940045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.