

Abstract
The discovery of functional RNAs critical for normal and disease physiology continues to expand at a break-neck pace. Many RNA functions are controlled by the formation of specific structures; an understanding of each structural component is necessary to elucidate its function. Measuring solvent accessibility intracellularly with experimental ease is an unmet need in the field. Here, we present a novel method for probing nucleobase solvent accessibility, Light Activated Structural Examination of RNA (LASER). LASER depends on light activation of a small molecule, nicotinoyl azide (NAz), to measure solvent accessibility of purine nucleobases. In vitro, this technique accurately monitors solvent accessibility and identifies rapid structural changes due to ligand binding in a metabolite-responsive RNA. LASER probing can further identify cellular RNA-protein interactions and unique intracellular RNA structures. Our photo-activation technique provides an adaptable framework to structurally characterize solvent accessibility of RNA in a myriad of environments.
Introduction.
RNA molecules can perform a myriad of functions to control nearly every step of gene regulation1–3. This is due to the unique ability of RNA molecules to fold into complex structures. Biochemical methods that can interrogate structural elements in RNA can impart specific molecular details on their biological activity. Chemical methods such as dimethylsulfate (DMS) alkylation are used to determine base pairing for adenosine and cytosine residues4. Another method is selective hydroxyl acylation (SHAPE), which interrogates RNA structural flexibility5. SHAPE and DMS excel at monitoring changes in Watson-Crick pairing, but only reveal limited information where solvent accessibility is the major alteration due to structural transitions or protein binding6.
Hydroxyl radical probing is arguably the gold standard when measuring solvent accessibility of RNA. Due to its fast rate constant (t1/2 ~ 1 us), the technique is especially powerful for monitoring the exposed backbone of RNA, and is frequently used to footprint RNA structural transitions and RNA-protein interactions7. Despite its wide-spread use in vitro, adoption of hydroxyl radical probing in cells is limited because of the experimental challenges with using synchrotron radiation8. Assaying solvent accessibility intracellularly can be powerful for footprinting sensitive changes in RNA structure and identifying RNA-protein interactions6,9. Development of more accessible techniques to assay RNA solvent accessibility in cells will provide additional experimental methods for deriving comprehensive models relating RNA structure, RNA interactions, and function.
A potential complement to hydroxyl radical probing is to measure solvent accessibility of the major and minor groove surrounding the nucleobases. Many RNA structures or RNA-protein complexes rely on major groove interactions to fold into their complex states and to control affinity and selectivity10–13. DMS can alkylate the N-7 position of guanine in a structure-dependent manner, yet these approaches often utilize additional chemical conditions, which could compromise the integrity of the modified RNA, resulting in many unavoidable false positives14. Expanding upon the reactivity on the major groove to assay solvent accessibility could reveal similar structural transitions observed by hydroxyl radical probing. The growing catalogue of RNA functions implores such an extension of chemical approaches to interrogate RNA structure.
The major challenge to adopting a novel method to measure RNA solvent accessibility is that it should recapitulate many of the positive aspects of hydroxyl radical probing, but be easy to implement experimentally. For use in the cellular context, such reagents would ideally have the following characteristics: (1) to be highly soluble for use in cells15, (2) to be readily converted from an inert to active state, as to prevent quenching during incubation before reaction with RNA, (3) to have fast half-lives to sensitively measure RNA structure transitions, (4) to be self-quenching as to obviate the need for a quencher, and lastly (5) to be amenable to conventional reverse-transcription protocols which would enable widespread adoption of the procedure by labs interested in RNA structure and function.
One potential solution is to utilize the ability of light to transform an inert compound into a highly reactive species: this approach is a well-established paradigm for controlling reactivity in organic chemistry and has been used in cells to “uncage” metabolic precursors and complex biomolecules16–18. Nevertheless, its use for monitoring the structure of biomolecules has yet to be demonstrated.
Within this study, we present a novel chemical approach to probe the solvent accessibility of purine nucleobases in RNA, Light Activated Structural Examination of RNA (LASER). We demonstrate the utility of this novel method to footprint RNA structure and solvent accessibility. LASER is highly sensitive and can detect very subtle changes in structure due to ligand binding in vitro. LASER identifies similar sites of solvent accessibility as hydroxyl radical probing, and reveals rich structural transitions not observed by either DMS or SHAPE. Lastly, we demonstrate that LASER can measure unique structural states of RNA such as footprinting RNA-protein interactions inside cells. Our approach provides the community with a novel approach to characterize RNA structure both inside and outside the cellular environments.
Results.
Aroyl azides as a photo-inducible probe for RNA structure
Developing such an approach to chemical probing of RNA implored us to understand how other molecules perturb solvent accessible regions of nucleobases. Carcinogenic aryl nitrenium ions are highly reactive electrophiles, which have been demonstrated to react with solvent accessible regions of adenosine and guanosine in DNA to form C8 adducts on picosecond time scales19–22. While such chemicals could be highly useful for measuring RNA structure, the rate of enzymatic nitrene formation is slow as it relies on complicated activation pathways that convert inert pro-carcinogens into reactive nitrene intermediates23. Lastly, rearrangements of aryl nitrenes to form benzazirine and ring-expanded ketenimine structures would complicate the structural studies because these additional intermediates alter their sites of reactivity on nucleobases20.
Aroyl nitrenes are another class of reactive nitrogen species. Photo-excited aroyl azides can form single-site amidation products with electron-rich heteroarenes, similar in structure to the electron-rich 5-membered rings in purines24. In these reactions, the formed aroyl nitrene is protonated by solvent to form an aroyl nitrenium ion, which is a strong electrophile24,25. Aroyl nitrenes, in contrast to the aryl nitrenes, are exquisitely selective in forming single addition products with heteroarenes containing a wide array of other functional groups24. A dominant competing Curtius rearrangement results in the formation of an aryl isocyanate (Fig. 1A), which is later quenched to reveal amino aryl rings. We hypothesized that taking advantage of the exquisite selectivity and rates of such reactions could permit a similarly sensitive measurement of RNA structure and solvent accessibility.
Figure 1: Characterizing the reactivity of NAz.

(A) Proposed mechanism of NAz activation and adduct formation with purines. (B) fs TRIR of the singlet state after photoexcitation of NAz with 273 nm light in CHCl3. The asterisk denotes the S1 state of the azide. (C) Nitrene formation from higher singlet excited state (Sn, where n≥2) in CCl4. The asterisk denotes the nitrene. (D) Formation of isocyanate after NAz photoexcitation with 273 nm light in CHCl3. The asterisk denotes the isocyanate. (E) Reaction of NAz with guanosine to yield 8AG. (F) HPLC analysis of the reaction depicted in E.
Activation of NAz forms 8-adduct with guanosine
We designed and synthesized nicotinoyl azide (NAz, (1), Supplementary Note 1), with a similar pyridinoyl scaffold from previous highly soluble RNA structure probes (Fig. 1A)26. Extensive density functional theory (DFT) calculations were performed to characterize the reactive species formed by photo-irradiation and to analyze the reaction mechanism (Online Methods; Supplementary Note 2). Consistent with previous reports of excited aroyl azides25, NAz gives rise to the nitrene species with a thermally accessible singlet nitrene, which will be protonated in protic solvent to form a nitrenium cation in picoseconds (Supplementary Figs. 1 & 2; Supplementary Dataset 1; Supplementary Note 2; Supplementary Tables 1–3)22. The formed nitrenium ions are computed to react with groove face of guanosine via an almost barrierless electrophilic aromatic substitution reaction. We calculated that this should occur in the singlet potential energy surface (PES) to form C8-N bond directly (see Supplementary Fig. 2).
To experimentally characterize the excitation pathway of NAz, we performed time-resolved infrared spectroscopy (TRIR). Following light-excitation of NAz, a new band around 2105 cm−1 was born immediately (< 1 ps) after the laser pulse corresponding to the singlet excited state (S1) of NAz (Fig. 1B, Supplementary Note 2). A weak, broad, band centered around 1734 cm−1 was observed in carbon tetrachloride and is assigned to the singlet nitrene (calculations predicted singlet nitrene to be around 1752 cm−1). The singlet nitrene (1734 cm−1) was immediately formed after the laser pulse indicating initial formation of singlet nitrene from higher singlet excited state (Sn, where n≥2) of NAz (Fig. 1C). The S1 state’s kinetic behavior is biphasic with an ultrafast growth component of 19 ps and a longer-lived decay component of 247 ps. Another positive band was observed in the range of 2310–2185 cm−1 (Fig. 1D) with a lifetime of 37 ps, and this band is readily assigned to the isocyanate. Consistent with the known reactivity of aroyl azides, isocyanate formation is the dominant product (Supplementary Figs. 3–6; ; Supplementary Dataset 2; Supplementary Note 2; Supplementary Tables 4–8). Steady-state photolysis in acetonitrile (a singlet nitrene quencher) resulted in the formation of oxadiazole products27 (Supplementary Note 2), further demonstrating that NAz forms a singlet nitrene upon photoirradiation. Quenching of the aroyl nitrene (by nucleophiles) must occur on a picosecond time scale, making such reaction diffusion controlled (Fig. 1B & C). These results demonstrate that NAz is capable of forming high energy intermediates known to form C-8 adducts with adenosine and guanosine19–22.
Following the known reactivity of aroyl azides with heteroarenes24, we predicted that light activation on NAz with guanosine would result in the formation of an 8-amido-guanosine (8AG. (2)) adduct (Fig. 1D). We focused on guanosine, as compared to adenosine, as it has been demonstrated that the yield of C-8 products with guanosine is much higher due the higher HOMO energy (−8.1 eV for G), in comparison to other nucleobases (−8.4 eV for A, −9.2 eV for C, and −9.5 eV for U)28,29,1H NMR experiments demonstrated a lack of reactivity between guanosine and NAz without light (Supplementary Fig. 7). We next incubated NAz with guanosine under 310 nm light from a conventional handheld UV lamp (As depicted in Fig. 1, D). HPLC-MS analysis revealed the presence of a single new peak with a mass consistent with 8AG.
We synthesize 8AG (10 steps from commercially available guanosine, (Supplementary Note 1), and compared it to the reaction sample by HPLC-MS. Perfect overlap was observed between 8AG and the new peak (Fig. 1E). Other dominant peaks in the spectrum were matched with the known products and intermediates of the Curtius rearrangement. Masses in negative control are matched to the azo products due to different reaction pathways from nitrene30. In addition, a peak with same retention time was detected in both samples of incubation of guanosine with NAz in the presence and absence of UV. Further HPLC analysis demonstrates this is from acylation of NAz with 5’OH; no peak was detected for incubation of 5’GMP with NAz (Supplementary Fig. 7). We note that this product would not be observed in structure probing experiments. Incubation of guanosine with the predicted isocyanate from the Curtius rearrangement yielded no reaction (Supplementary Fig. 8). These results overall demonstrate that simple light activation of NAz results in the formation of a C-8 adduct with guanosine. The selectivity of product formation is consistent with carcinogenic aryl nitrenes that form C-8 adducts with guanosine and adenosine19,28.
NAz probes purine solvent accessibility in folded RNA
We next investigated NAz as an RNA structure probe. As our control RNA, we utilized the SAM-I riboswitch, an RNA that binds S-adenosylmethionine (SAM). SAM-I RNA has been well characterized by both solution and X-Ray crystallographic methods31,32. The SAM-I aptamer core is highly conserved and comprised of four helical regions (P1–P4) centered on a four-way junction with three joining regions (J1/2, J3/4, and J4/1). The helices form two coaxial stacks (P1/P4 and P2/P3) that are organized through a set of tertiary interactions involving a pseudoknot (PK) between L2 and J3/4, a base triple tying together L2, J3/4 and J4/1, and several long-range interactions involving base triples between J1/2 and J3/4 and paired regions 27,28 (Supplementary Fig. 9). The key characteristic of SAM-I is that the structure mostly exists in a pre-formed state. Upon binding to SAM, structural changes can occur on the microsecond time-scale to compact the overall structure of the RNA through an induced-fit mechanism33,34. As such, SAM-I RNA is a powerful control for determining whether NAz is able to measure nucleobase accessibility and structural transitions.
We first determined if NAz probing would be amenable to conventional reverse transcription assays. Since our data demonstrated the installation of the large bulky group at the C-8 position of guanosine, we reasoned this would stall reverse transcription through a trans to cis isomerization of the glycosidic bond on adenosine and guanosine. Incubation of NAz with SAM-I RNA only in the presence of light from the handheld UV lamp (310 nm) resulted in cDNA truncation products (Fig. 2A). Increased light exposure resulted in more adducts (Supplementary Fig. 10). High signal-to-noise ratios can be observed in as little as one minute time of light exposure. Due to the fast rate of nitrene quenching, removal of light is sufficient to stop NAz reactivity (Supplementary Fig. 11). cDNA truncations mapped almost exclusively to guanosine and adenosine residues. Guanosine residues tend to have higher reactivity, consistent with guanosine being more electron-rich than adenosine28. We note that NAz has a different modification profile compared to other chemical methods (DMS and SHAPE), since guanosine and adenosine form adducts on both single and double stranded regions.
Figure 2. Reactivity of NAz with SAM-I RNA.

(A) Denaturing gel electrophoresis demonstrating the formation of adducts only in the presence of NAz and light. (B) NAz probing of SAM-I RNA with increasing amounts of SAM. (C) Differential reactivity of B mapped onto the SAM-I RNA secondary structure. (D) Differential reactivity of B mapped onto the SAM-I RNA crystal structure PDB: 2GIS. A and G residues are represented in stick. (E) Differential reactivity of NAz (+/−) SAM. A and G residues are represented on the X-axis (F) Correlation between LASER and C-8 solvent accessibility, comparing the +SAM lane of B. A and G residues are represented on the X-axis.
To better understand the mode of NAz reactivity in LASER, we subjected SAM-I RNA to a titration of SAM (Fig. 2B). Residues with differential reactivity were mapped onto the SAM-I secondary structure and a recent crystal structure (Fig. 2 C-E)35. Sites modified by LASER are clustered around the SAM binding site. Consistent with what others have observed, peripheral loop regions of SAM-I undergo very little structural changes due to ligand binding33–35. Sites of differential reactivity can be found within J1/2, PK, P2a, P3, and J4/1. These sites are known to undergo aptamer domain collapse to stabilize the pseudoknot and associated tertiary contacts remote from the SAM-binding site. Nearly all residues within the pseudoknot (A24, G27–28) have decreased reactivity, consistent with the collapsed and stabilized bound state33–35. Notably, LASER detected changes in reactivity upon SAM binding for A24, which is a critical lynchpin residue that is part of the base triple connecting L2 with joining regions J4/1 and J3/433.
In the bound state, SAM forms specific hydrogen bonding interactions with residues 45 and 46 in P3 and helices P1 and P2 (at J1/2). SAM interactions with J1/2 are major contributors to binding as these interactions help to fix J1/2 into a configuration that is favorable for P1 helix formation. This interaction has been proposed to be the kinetic barrier to the induced-fit mechanism employed for anti-anti-terminator formation and control of gene expression34. Differential probing is observed at residues A45 and A46, the residues directly interacting with SAM. Nearly every residue in J1/2 (the critical residues for induced fit and SAM binding) displayed differential LASER reactivity in the SAM-bound state. For example, A9 and A14, which are splayed out into the solvent in the SAM-bound state, are now highly reactive to LASER. In contrast, G11 and G58 which form H-bonding interactions with SAM, are tucked back into the structure resulting in decreased LASER reactivity. Residues such as A12 is not making any structural contacts with SAM or other residues in the RNA that would control its solvent accessibility; thus, A12 was observed to have a lack of change in LASER reactivity. Overall, these results demonstrate that LASER is capable of detecting structural differences important for RNA function.
To gain further insight into the mode of reactivity, we compared the C-8 solvent accessibility of several SAM-I crystal structures with SAM bound to the modification profile of LASER probing. LASER probing displayed a linear relationship with solvent accessibility (r = 0.82, Fig. 2F. We note that this value rivals hydroxyl radical probing which have reported r values of ~0.636,37. Two residues make direct contact with SAM and have been shown to be critical for ligand binding, A46 and A45. A46 shows high LASER reactivity in the bound state, whereas A45 displays low reactivity. This finding also correlates with their solvent accessibility of the N7-C8 bond. G58, which is stacked directly above SAM, has a reduction in reactivity with ligand binding which correlates to a solvent inaccessible N7-C8 bond in the structure. The residues 49–51 are reactive with NAz whose N7-C8 bond is also observed to be solvent exposed in the crystal structures. Lack of reactivity on residues 52 and 53 N7-C8 bond can also be explained by these structures since these N7-C8 bonds are protected from solvent (Supplementary Fig. 12). One major outlier we identified was G68, a residue that has high LASER reactivity, but has its N7-C8 bond protected (Supplementary Fig. 12). Inspection of this position in the structure shows the lack of strong contacts with the RNA, where G68 is in a non-coplanar W-C pair with C25, implies that the specific position may be dynamic in solution and could have sampled conformations that are solvent exposed. This analysis of SAM-I supports the notion that LASER profiling is measuring C-8 solvent accessibility of purine nucleobases.
We put our results into context by comparing NAz reactivity to other methods of chemical probing. We interrogated other chemical probing techniques, while focusing on G and A residues. We first compared LASER probing (Fig. 3A) to SHAPE and DMS. As shown in Fig. 3B, published differences in DMS probing were very scarce in comparison to LASER, although DMS successfully monitored residues A46 and A45 due to direct interactions with SAM33. When compared to SHAPE (Fig. 3C) and observed that SHAPE probing misses many of the J1/2 residues, while still being capable of identifying flexibility changes in residues A46 and A45 (Fig. 3C & Supplementary Fig. 13). This is consistent with what others have observed with SHAPE probing on the same SAM-I construct35. Because LASER, SHAPE, and DMS all perform different reactions, these striking differences may be due to a combination of the fast rate constant of LASER reactivity and its unique specificity for the C-8 position of adenosine and guanosine. These two characteristics may make LASER incredibly sensitive to structural changes. Lastly, we compared our data to published hydroxyl radical probing33. As shown in Fig. 3D, hydroxyl radical probing also identified the key sites of J1/2 (and residues 45/46) for changes in structure due to the induced fit mechanism of SAM-I. The residues, which have lower reactivity, also aligned well with P4 and P1 (similar residues that have lower LASER probing), further supporting the overall compact structural transition that SAM-I undergoes due to ligand binding. We also note that there are some residues undergoing reactivity changes that are missed by LASER; however, these are mostly U and C residues, which are not reactive to LASER (Supplementary Fig. 14). These results support the notion that LASER probing can reveal similar structural changes to hydroxyl radical probing and is sensitive to changes in solvent accessibility.
Figure 3. Differential structure probing mapped onto the SAM-I crystal structure.

(A) Mapped with NAz. (B) Mapped with DMS. (C) Mapped with SHAPE. (D) Mapped with hydroxyl radical. A and G residues are represented in stick. C and U nucleobases are not represented in stick. PDB for SAM-I riboswitch structures: 2GIS. The figure legends here are very terse. Could you provide some additional text clarifying what the orange and blue colors are?
LASER probes purine solvent accessibility in living cells
To test LASER reactivity in cells15,38, we first focused on probing ribosomal RNA (rRNA). We chose rRNA as it is highly abundant and available high-resolution cryo-EM structures (2.9 Å)39. This permits a molecular level analysis of LASER probing in comparison to structural states representative of the cellular environment. We first focused on H15 and H16, because the majority of the helices are hidden from solvent, due to extensive interactions with other sections of 18S rRNA and ribosomal proteins S9, S24, and S30. The section of interest in H4 has extensive G-C base pairing and a large portion of the junction between H15 and H16 is connected by a long stretch of single-stranded adenosine residues. In vitro, LASER modified many of the residues connecting H15 and H16, in the adenosine-rich sequence (Fig. 4A & Supplementary Fig. 15). However, in stark contrast, these residues did not show LASER reactivity in cells. When mapping the interaction of these residues back onto the cryo-EM model, analysis revealed that many of them are in direct contact with the three rRNA binding proteins. The C-8 position of these residues are packed tightly against ribosomal protein S9 (Fig. 4B).
Figure 4. LASER probing inside living cells.
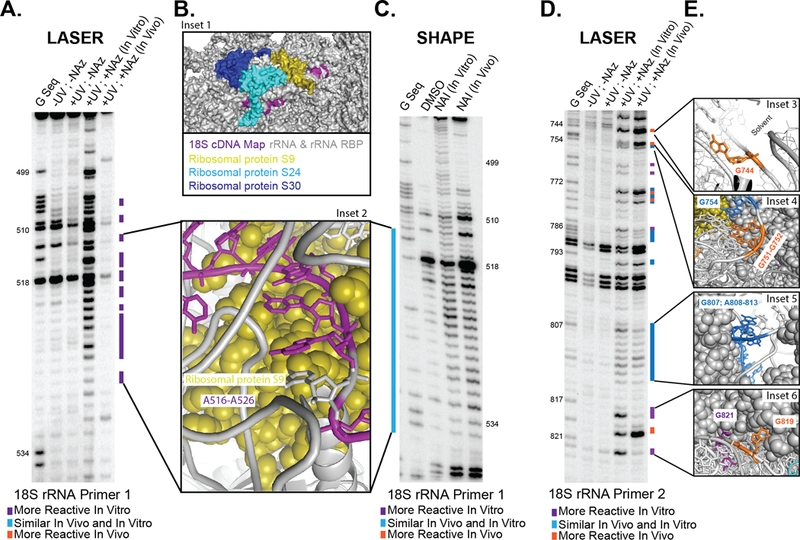
(A) Denaturing gel electrophoresis of LASER on section 1 of 18S rRNA. (B) Space filling model of 18S rRNA and close up of A-stretch showing interactions with proteins and RNA blowing solvent. (C) Denaturing gel electrophoresis of SHAPE on section 1 of 18S rRNA. (D) Denaturing gel electrophoresis of LASER on section 2 of 18S rRNA. (E) Close up of residues, which have differential NAz probing. PDB for ribosomal structures: 4V6X.
Many residues within H15 and H16 in the cryo-EM structure are single stranded or are in non-optimal weak base pairs. As such, we predicted that these residues would have similar reactivity in and outside of cells to SHAPE probing, even though they have extensive interactions with proteins. Consistent with this prediction, there was no difference in SHAPE reactivity in the long single-stranded stretch of residues G513 and A526 (Fig. 4C). As such, LASER probing is a good complement to SHAPE and is very powerful at identifying solvent protected residues.
We next sought to extend our analysis onto a second segment of 18S rRNA, which has a more complex intracellular environment (Fig. 4 D-E & Supplementary Fig. 16). Residue 819 is highly reactive in cells, where it sits completely solvent exposed, residues 817 and 821 are non-reactive with their nucleobases are solvent protected. Residues 807–813 is part of a long A-rich bulge that has similar reactivity in cells and in vitro. Inspection of the crystal structure shows that these residues have high solvent accessibility and as such, would not have significant differences in NAz reactivity. Guanosine residues near the end of the helix displayed differential NAz reactivity. G751/752 both have their C-8 positions completely solvent exposed in the crystal structure. G744 is part of an S-turn in rRNA and the N7-C8 side of the ring is pointing out into the solvent, completely solvent exposed in the crystal structure. These sites of differential probing are depicted in Fig. 4E. These results, combined with the 18S rRNA probing above demonstrate that NAz is capable of reading RNA structure inside cells and can accurately report on solvent accessibility of complex RNA structures in native environments.
LASER Probing of the U1 snRNP in cells
Lastly, we further tested the limitations of LASER probing on a small nuclear ribonucleo proteins (snRNP). The U1 snRNP participates in the recognition of 5’ splice sites via base pairing between the 5’ splice site and the 5’ end of U1 snRNA13,40,41. Recent low-resolution crystal structures of the U1-and 70K-RNA (Fig. 5A) have been solved (5.5 Å)42 and comparison of these structures to analysis in cells would further support their findings.
Figure 5. LASER probing of the U1 snRNP inside living cells.

(A) Secondary structure of the U1 snRNP. (B) Denaturing gel electrophoresis of LASER on U1 snRNP. (C) Close up of residues, which have differential NAz probing and their relationship to the U1 and 70K proteins. PDB for both structures: 4PKD.
NAz probing in cells identified the U1A-RNA interface. Residues 76–75, 70, and 68 all displayed significant differences in their NAz profiles (Fig. 5B - C). Residues 76 and 75 have their C-8 positions pushed back against the bottom of the U1 protein (direct contacts with Arg52 prevent NAz reactivity) where they are now solvent protected. Their W-C faces are involved base pair interactions with cytosine residues. Residue A70 also showed lower reactivity and its N7-C8 bond is protected by pi-stacking with Phe56. This result is revealing because A70 is known to be a critical residue for U1A-RNA particle assembly43. Lastly, G68 is in direct contact U67, which combines with Gln54 and Asn15 on U1 to protect the nucleobase from LASER reactivity.
LASER probing also identified key residues in the 70K-U1 snRNA interface. As shown in Fig. 5C, 70K binding provides a large portion of surface interaction to prevent NAz reactivity. Many of the residues in Loop I are observed to be protected. The entire 3’-end of loop I has lower NAz reactivity in cells, which is consistent with the interactions between the major groove of such residues and the binding face of 70K.
LASER reactivity remained unchanged in Loop III, as this loop is known to lack of any known protein binding factors inside cells. However, LASER reactivity in cells was much higher in the H-helix, which is consistent with the concept that LASER is probing the engaged U1 snRNP, which opens up its H-helix when bound to the target mRNA for regulation of splicing13. Thus, we have demonstrated the capability of our technique to profile RNA protein interactions of lowler expressed RNAs inside living system.
Discussion
Herein we have introduced a novel method for measuring RNA solvent accessibility: Light Activated Structural Examination of RNA (LASER). LASER works through light activation of aroyl azides to form aroyl nitrenes. We demonstrate that our new method forms adducts with the solvent-exposed region of purine residues. LASER works with exquisite sensitivity and can identify sites of structural change due to ligand binding in the SAM-I riboswitch. Importantly, LASER can identify changes in structure that are missed by other conventional probing methods. LASER and hydroxyl radical probing yield very similar results. Due to the sensitivity of LASER probing, it enables the ability for understanding RNA structural transitions at faster time scales. Furthermore, our method can be easily implemented in living cells. This allows us to measure unique structural motifs of RNA in its native environment and also have the ability to identify RNA-protein interactions.
We anticipate that LASER will be an invaluable addition to the methods used to probe RNA structure. NAz reactivity can be mapped back to both single and double stranded regions in RNA. This reactivity makes this reagent unique because other canonical structure probing reagents (DMS and SHAPE reagents) work by identifying single stranded regions. As such, NAz reactivity (solvent accessibility) can complement DMS and SHAPE probing. Thus, we anticipate it could reveal aspects of RNA structure that may be missed by conventional methods. Furthermore, it should be extremely useful for footprinting RNA-protein interactions for proteins that recognize structured regions in RNA and do not necessarily bind to single-stranded residues. Tracking solvent accessibility may prove more feasible for characterizing such binding events. We expect that such experimental interrogations will become more important as the catalog of noncoding RNA molecules10 and functional motifs in coding transcripts15 continues to expand.
Online Methods
Synthesis of NAz.
Synthesis of NAz is reported in Supplementary Note 1.
Synthesis of 8AG.
Synthesis and spectral characterization of 8AG is reported in Supplementary Note 1.
Characterization of NAz reactivity with guanosine.
Experimental methods detailing the reactivity between NAz and guanosine are reported in the Online Methods, Supplementary Note 2, and Supplementary Information.
General Biological Methods
Buffer salts and NTPs were purchased from commercial sources. All chemical reagents were purchased from commercial sources. Superscript III was purchased from Life technologies. 32P was purchased from Perkin Elmer. SAM-I gene and primers were purchased from Integrated DNA Technologies (IDT). T7 RNA polymerase was graciously donated by the Luptak Lab. Gels were imaged on the Typhoon Imager GE Healthcare. Optikinase was purchased from Affymetrix. TURBO DNase was purchased from Thermo Fisher Scientific. Zilla Desert UVB 50 fluorescent coil bulb was purchased from Amazon.
SAM-I construct
A 94 nucleotide construct consisting of the sequence for the SAM-I riboswitch from the metF-metH2 operon of T. tencongensis was designed into a plasmid with IDT31. SAM-I plasmid was transformed into One Shot Top 10 chemically competent cells and plated on lysogeny broth (LB) supplemented with ampicillin (100 mg/mL) agar plates. A single colony was selected in 3 mL culture and grown overnight. The resulting plasmid was isolated according to conditions using QIAprep Miniprep. Transcription template was prepared by PCR using primers directed against the T7 promoter (5’, TAATACGACTCACTATAGGG, 3’) and an adaptor sequence for reverse transcription (5’, ATTTAGGTGACACTATAGTT, 3’). RNA was transcribed in 40 μL reaction containing 40 mM Tris-HCl (pH 8.0), 20 mM DTT, 0.01% Triton X-100, 2 mM spermidine, 1 mM each NTP, 25 mM MgCl2, 200 ng template DNA and 1 U of T7 RNA polymerase provided by the Luptak Lab. The transcription reaction was allowed to proceed for four hours at 37 °C. Subsequent samples were treated with 1 μL of TURBO DNase and incubated for 30 min at 37 °C. The resulting RNA was ethanol precipitated at −80 °C for 30 minutes. The resulting RNA was resuspended in 50 μL of RNase-free water. An aliquot of RNA was run on a denaturing PAGE gel (15% polyacrylamide, 0.5X TBE, 7 M urea) alongside a 100 bp RNA ladder. The band of interest was visualized through SYBR-gold (1x) in water for 15 minutes. Resulting concentrations of RNA was quantified by integrating intensity of the ladder with the RNA band of interest.
32P End labeling for reverse transcription
200 pmol of primer DNA was phosphorylated according to manufacturer’s conditions by Affymetrix. The reaction was allowed to proceed for two hours at 37 °C. Reactions were stopped by the addition of equal amounts of Gel Loading Buffer II (Ambion, Inc.). The reactions were loaded onto a 15% denaturing PAGE gel. The band of interest was visualized by a phosphorimager (Typhoon, GE healthcare). The resulting band was excised and eluted overnight in 300 mM KCl. Resulting solution was EtOH precipitated and dissolved to 8,000 counts per minute (cpm)/μL for further use in reverse transcription.
Primers used for Reverse transcription
SAM-I (5’, ATTTAGGTGACACTATAGTT, 3’)
HeLa 18s Primer 1 (5’, CCAATTACAGGGCCTCGAAA, 3’)
HeLa 18s Primer 2 (5’, TCCAGGCGGCTCGGGCCT, 3’)
U1 Primer (5’, CCCACTACCACAAATTATGCAG, 3’)
Characterization with NAz structure probe with RNA
Modification of RNA with NAz structure probe in vitro.
In a typical experiment, 10 pmol of in vitro transcribed RNA or 5 μg of total RNA (isolated from HeLa cells) was heated in 6 μL metal free water at 95° C for two minutes, then the RNA was snap cooled on ice. Resulting RNA was incubated in 1× SHAPE buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, and 333 mM NaCl), and the RNA was allowed to equilibrate at 37 °C for three minutes. To this mixture, 1 μL of 10x NAz stock (3 M in DMSO) was added. In a typical light exposure experiment, RNA with NAz are laid flat in a 1.7 mL microcentrifuge tube. A 20 W UVB lamp is positioned around 5 cm above the samples. Exposure time could be reduced if a stronger light source is used. However, care should be taken over potential health concerns with UV damage. The reaction was exposed to a handheld lamp (20 W; λmax ~ 310 nm) for three minutes unless otherwise noted. 1 μL of 10× 2-methylnicotinic acid imidazolide (1 M in DMSO) (NAI) was incubated for 15 minutes in place of NAz for comparison. Modified RNA was quenched with the addition of 90 μL water, and precipitated with 10 μL of 3 M sodium acetate buffer (pH 5.2), and 1 μL of glycogen (20 μL/uL). Pellets were washed twice with 70% cold ethanol and resuspended in 5 μL of RNase-free water.
Modification of RNA with NAz structure probe in vitro with SAM ligand.
RNA modification protocol was altered with the following changes. 10 pmol of in vitro transcribed SAM-I RNA was heated in 5 μL metal free water at 95° C for two minutes, then the RNA was snap cooled on ice. Resulting RNA was incubated in 1x SHAPE buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, and 333 mM NaCl), and the RNA was allowed to equilibrate at 37 °C for three minutes. Starting with 1 mM final concentration, the RNA was incubated for 2 minutes with SAM ligand with a 10-fold dilution for each subsequent sample down to 1 nM final concentration. RNA modification proceeded with NAz reagent as indicated above.
Modification of 18s rRNA or U1 snoRNA with NAz structure probe in vivo.
HeLa cells were grown in DMEM (high glucose) culture medium supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were washed two times with DPBS (Dulbecco’s phosphate-buffered saline) and then scraped and spun down at 1000 r.p.m. for 5 minutes. Cells (3–6×107) were resuspended in DPBS and DMSO (10% final concentration), or NAz (300 mM final concentration) was added to the desired final concentration. Cell suspensions were exposed to a handheld lamp (20 W; λmax ~ 310 nm) for 5 minutes. 1 mL of TRIzol was added to the resulting mixture, and then 200 μL of chloroform was added. RNA was precipitated following the TRIzol LS manufacturer’s instructions. RNA was resuspended with 5 μL of RNase-free water.
Reverse transcription of modified RNA (in vitro and in vivo).
32P-end-labeled DNA primers were annealed to modified RNA by incubating 95 °C for two minutes, then at 25 °C for two minutes, and finally 4 °C for two minutes. To the reaction, first strand buffer, DTT, and dNTP’s were added. The reaction was pre-incubated at 52 °C for 1 minute, then superscript III (2 units/μL final concentration) was added. Extensions were performed for 15 minutes. To quench the reaction, 1 μL of 4 M sodium hydroxide was added and allowed to react at 95 °C for five minutes. The resulting complementary DNA (cDNA) was snapped cooled on ice, and ethanol precipitated according to above procedures. Purified cDNA was resuspended in 2 μL of nuclease-free water and 2 μL of Gel Loading Buffer II was added. cDNA products were resolved on 10% denaturing PAGE gel.
Characterization of Reverse transcription stops.
cDNA extensions were visualized by a phosphorimager (Typhoon, GE healthcare). cDNA bands were integrated with SAFA. NAz reactivities were normalized to the average of the top 92–98 percentile. Solvent accessibility calculations for SAM-I unbound (PDB ID: 3IQN)35 and bound(PDB ID: 2GIS)31 RNA was performed with Crystallography & NMR System (CNS)_ENREF_50 using a probe size of 5 Å. PDBs used to calculate the variation in solvent accessibility were: 5FJC, 5FK1, 5FK2, 5FK3, 5FK4, 5FK5, 5FK6, 5FKG, 5FKH, 4KQY, 3IQN, 3IQP, 3IQR, 3GX2, 3GX3, 3GX5, 3GX6, 3GX7, 2GIS.
Structural modeling
Figure graphics were generated in PyMOL (http://www.delanoscientific.com/).
Computational methods
All structures were initially optimized in the gas phase using the TPSS46 functional and polarized triple-ζ valence (def2-TZVP47–51) basis sets in combination with the zero-damped D3-dispersion correction (denoted –D3 below)52. Final single-point energies were obtained using polarized quadruple-ζ valence (def2-QZVP47–51) basis sets yielding energies close to the basis set limit in DFT calculations. All computations employed spin-unrestricted Kohn-Sham references; open-shell singlet states were allowed to spin-polarize using converged triplet orbitals as initial guess. Solvation effects were included in the final single-point energies using the COSMO solvation model for aqueous solution with a dielectric constant of 80.153. Chemical potentials (c.p.) for obtaining the Gibbs free energies at 298.15 K (G = E(0) + c.p.) were computed from the harmonic vibrational frequencies using the quasi-RRHO approach where the vibrational entropy is replaced with the free-rotor entropy for all modes with frequencies less than 100 cm−1; the latter reduces the error in the vibrational entropy caused by deficiencies of the harmonic approximation54. The β-N9-glycosidic bond of guanosine was replaced with N9-CH3, see Figure S1.
All computations were performed using the Turbomole 7.155 program package (http://www.turbomole.com/) with default settings with the following exceptions: Finer DFT integration grids m4 were used and automatic level shifting was employed if E(HOMO-LUMO) was less than 0.5 eV. The multipole-accelerated resolution-of-the-identity approximation for the Coulomb energy (MARI-J)56 with the corresponding auxiliary basis sets57 was employed. The optimized structures were visualized using Cylview (http://www.cylview.org/). The CASSCF and B3LYP/6–31+G** were performed by JJ, WHC, and CMH using Gaussian09.
Spin states of nitrene and nitrenium ion.
The ground state multiplicities of the neutral nitrene and the nitrenium ion have important implications for the mechanism and serve as a benchmark for the computational methodology. The singlet nitrene is computed to be a closed shell species with significant N-O interaction in accordance with previous studies, see Figure S158,59. The energetic ordering of the singlet and triplet states is sensitive to the electronic structure method, basis sets, and solvation model: Double-ζ basis sets overstabilize the triplet state by several kcal/mol compared to triple- and quadruple-ζ basis sets. Additionally, the amount of exact exchange (EXX) affects the spin splitting: At the TPSS-D3/def2-QZVP (0% of EXX) level, the singlet and triplet states are almost isoenergetic, while at the TPSSh-D360/def2-QZVP (10% of EXX) level, the triplet state is 4 kcal/mol more stable than the singlet state. CASSCF(12e,11o)/6–31G* calculations also slightly favor the triplet state by 1.8 kcal/mol. On the other hand, solvation stabilizes the singlet state by approximately 1 kcal/mol. Thus, our best estimate is that the singlet and triplet states are isoenergetic within a few kcal/mol, and the singlet state may be slightly favored in solution. This is consistent with spectroscopic evidence for the singlet state presented below.
HPLC conditions:
All samples were run using DIONEX UltiMate 3000 analytic HPLC instrument. A Supelco analytical column (Discovery® C18 (Cat. No. 569223-U) 10cmX4.6mm, 5 micrometer). 0.1 M TEA/Acetic Acid in DI water and MeCN solvents were used as eluents. MeCN gradient was set from 0 (2 min) to 30% (38 min). The HPLC was run at room temperature. Flow rate: 1.00 mL/min. 260 nm UV lamp was used for detection.
Preparation of HPLC samples:
NAz+G+UV: NAz (15 mg, 0.101 mmol) and guanosine (30 mg, 0.106 mmol) was dissolved in 1 mL wet DMSO and the samples were illuminated by UV lamp (20 W, 300–320 nm) for over 1 h. Then 10 microliter sample was taken and resuspended in 1 mL water for HPLC analysis.
NAz-G+UV: A solution of NAz (25 mg) in wet DMSO (1 mL) was illuminated by UV lamp (20 W, 300–320 nm) over 1 h, then 10 microliter sample was taken and resuspended in 1 mL water for HPLC analysis.
NAz+G-UV: NAz (15 mg, 0.101 mmol) and guanosine (30 mg, 0.106 mmol) was dissolved in 1 mL wet DMSO. After 1 h in dark, 10 microliter sample was taken and resuspended in 1 mL water for HPLC analysis.
Supplementary Material
Acknowledgements
We thank members of the Spitale lab for their careful reading and critique of the manuscript. Spitale lab is supported by start up funds from the University of California, Irvine, and the NIH (1DP2GM119164 RCS) and 1RO1MH109588 (RCS). F.F. and the computational work is supported by the U.S. Department of Energy under Award DE-SC0008694. CMH acknowledges financial support from the National Science Foundation (DMR-1212842 and CHE-1609889) as well as generous allocations of computational resources at the Ohio Supercomputer Center. Femtosecond TRIR experiments were performed at The Ohio State University’s Center for Chemical and Biophysical Dynamics.
Footnotes
Competing Financial Interest Statement
The authors declare no competing financial interest.
Data Availability
The data that support the findings of this study are available from the corresponding authors on reasonable request. A Life Sciences Reporting Summary for this paper is available.
Additional Information
Supplementary information and chemical compound information is available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should R.C.S.
References
- 1.Mattick JS The functional genomics of noncoding RNA. Science 309, 1527–1528 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Chappell J et al. The centrality of RNA for engineering gene expression. Biotechnol. J 8, 1379–1395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharp PA The centrality of RNA. Cell 136, 577–580 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Tijerina P, Mohr S & Russell R DMS footprinting of structured RNAs and RNA-protein complexes. Nat. Protoc 2, 2608–2623 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson KA, Merino EJ & Weeks KM Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc 1, 1610–1616 (2006). [DOI] [PubMed] [Google Scholar]
- 6.McGinnis JL, Duncan CD & Weeks KM High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol 468, 67–89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tullius TD & Greenbaum JA Mapping nucleic acid structure by hydroxyl radical cleavage. Curr. Opin. Chem. Biol 9, 127–134 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Adilakshmi T, Lease RA & Woodson SA Hydroxyl radical footprinting in vivo: mapping macromolecular structures with synchrotron radiation. Nucleic Acids Res 34, e64 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramaswamy P & Woodson SA S16 throws a conformational switch during assembly of 30S 5’ domain. Nat. Struct. Mol. Biol 16, 438–445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batey RT, Rambo RP & Doudna JA Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed 38, 2326–2343 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Moras D & Poterszman A Getting into the major groove. Protein-RNA interactions. Curr. Biol 6, 530–532 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Varani L & Spillantini MG Structural basis for recognition of the RNA major groove in the tau exon 10 splicing regulatory element by aminoglycoside antibiotics. Nucleic Acids Res 28, 710–719 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L & Frankel AD A peptide interaction in the major groove of RNA resembles protein interactions in the minor groove of DNA. Proc. Natl. Acad. Sci. USA 92, 5077–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawley PD & Brookes P Further Studies on the Alkylation of Nucleic Acids and Their Constituent Nucleotides. Biochem. J 89, 127–138 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee B et al. Comparison of SHAPE reagents for mapping RNA structures inside living cells. RNA 23, 169–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klan P et al. Photoremovable protecting groups in chemistry and biology: reaction mechanisms and efficacy. Chem. Rev 113, 119–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker AS & Deiters A Optical control of protein function through unnatural amino acid mutagenesis and other optogenetic approaches. ACS Chem. Biol 9, 1398–1407 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Song C-X & He C Bioorthogonal Labeling of 5-Hydroxymethylcytosine in Genomic DNA and Diazirine-Based DNA Photo-Cross-Linking Probes. Acc. Chem. Res 44, 709–717 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wild D A novel pathway to the ultimate mutagens of aromatic amino and nitro compounds. Environ. Health Perspect 88, 27–31 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue J, Du L, Zhu R, Huang J & Phillips DL Direct time-resolved spectroscopic observation of arylnitrenium ion reactions with guanine-containing DNA oligomers. J. Org. Chem 79, 3610–3614 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Kuska MS et al. Structural influence of C8-phenoxy-guanine in the NarI recognition DNA sequence. Chem. Res. Toxicol 26, 1397–1408 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Voskresenska V et al. Photoaffinity labeling via nitrenium ion chemistry: protonation of the nitrene derived from 4-amino-3-nitrophenyl azide to afford reactive nitrenium ion pairs. J. Am. Chem. Soc 131, 11535–11547 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loeb LA & Harris CC Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res 68, 6863–6872 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brachet E, Ghosh T, Ghosh I & Konig B Visible light C-H amidation of heteroarenes with benzoyl azides. Chem. Sci 6, 987–992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubicki J et al. Direct Observation of Acyl Azide Excited States and Their Decay Processes by Ultrafast Time Resolved Infrared Spectroscopy. J. Am. Chem. Soc 131, 4212–4213 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Spitale RC et al. RNA SHAPE analysis in living cells. Nat. Chem. Biol 9, 18–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desikan V, Liu YL, Toscano JP & Jenks WS Photochemistry of sulfilimine-based nitrene precursors: Generation of both singlet and triplet benzoylnitrene. J. Org. Chem 72, 6848–6859 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Humphreys WG, Kadlubar FF & Guengerich FP Mechanism of C8 Alkylation of Guanine Residues by Activated Arylamines - Evidence for Initial Adduct Formation at the N7 Position. Proc. Natl. Acad. Sci. USA 89, 8278–8282 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reha D et al. Intercalators. 1. Nature of stacking interactions between intercalators (ethidium, daunomycin, ellipticine, and 4’,6-diaminide-2-phenylindole) and DNA base pairs. Ab initio quantum chemical, density functional theory, and empirical potential study. J. Am. Chem. Soc 124, 3366–3376 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Wentrup C, Reisinger A & Kvaskoff D 4-Pyridylnitrene and 2-pyrazinylcarbene. Beilstein J. Org. Chem 9, 754–760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montange RK & Batey RT Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature 441, 1172–1175 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Winkler WC, Nahvi A, Sudarsan N, Barrick JE & Breaker RR An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol 10, 701–707 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Hennelly SP & Sanbonmatsu KY Tertiary contacts control switching of the SAM-I riboswitch. Nucleic Acids Res 39, 2416–2431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heppell B et al. Molecular insights into the ligand-controlled organization of the SAM-I riboswitch. Nat. Chem. Biol 7, 384–392 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Stoddard CD et al. Free State Conformational Sampling of the SAM-I Riboswitch Aptamer Domain. Structure 18, 787–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mortimer SA, Johnson JS & Weeks KM Quantitative analysis of RNA solvent accessibility by N-silylation of guanosine. Biochemistry 48, 2109–2114 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Adams PL et al. Crystal structure of a group I intron splicing intermediate. RNA 10, 1867–1887 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kubota M, Tran C & Spitale RC Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Chem. Biol 11, 933–941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khatter H, Myasnikov AG, Natchiar SK & Klaholz BP Structure of the human 80S ribosome. Nature 520, 640–645 (2015). [DOI] [PubMed] [Google Scholar]
- 40.So BR et al. A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat. Struct. Mol. Biol 23, 225–230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du H & Rosbash M The U1 snRNP protein U1C recognizes the 5’ splice site in the absence of base pairing. Nature 419, 86–90 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Kondo Y, Oubridge C, van Roon AM & Nagai K Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5’ splice site recognition. eLife 4, e04986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McConnell TS, Lokken RP & Steitz JA Assembly of the U1 snRNP involves interactions with the backbone of the terminal stem of U1 snRNA. RNA 9, 193–201 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das R, Laederach A, Pearlman SM, Herschlag D & Altman RB SAFA: Semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11, 344–354 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunger AT et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Tao J, Perdew JP, Staroverov VN & Scuseria GE Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett 91, 146401 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Weigend F & Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 7, 3297–3305 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Schäfer A, Horn H & Ahlrichs R Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys 97, 2571–2577 (1992). [Google Scholar]
- 49.Schäfer A, Huber C & Ahlrichs R Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys 100, 5829–5835 (1994). [Google Scholar]
- 50.Eichkorn K, Weigend F, Treutler O & Ahlrichs R Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc 97, 119–124 (1997). [Google Scholar]
- 51.Weigend F, Furche F & Ahlrichs R Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys 119, 12753–12762 (2003). [Google Scholar]
- 52.Grimme S, Antony J, Ehrlich S & Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Schäfer A, Klamt A, Sattel D, Lohrenz JC & Eckert F COSMO Implementation in TURBOMOLE: Extension of an efficient quantum chemical code towards liquid systems. Phys. Chem. Chem. Phys 2, 2187–2193 (2000). [Google Scholar]
- 54.Grimme S Supramolecular binding thermodynamics by dispersion‐corrected density functional theory. Chem. Eur. J 18, 9955–9964 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Furche F et al. Turbomole. WIREs Comput. Mol. Sci 4, 91–100 (2014). [Google Scholar]
- 56.Sierka M, Hogekamp A & Ahlrichs R Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys 118, 9136–9148 (2003). [Google Scholar]
- 57.Weigend F Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys 8, 1057–1065 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Gritsan NP & Pritchina EA Are aroylnitrenes species with a singlet ground state? Mendeleev Commun 11, 94–95 (2001). [Google Scholar]
- 59.Pritchina EA et al. Matrix isolation, time-resolved IR, and computational study of the photochemistry of benzoyl azide. Phys. Chem. Chem. Phys 5, 1010–1018 (2003). [Google Scholar]
- 60.Staroverov VN, Scuseria GE, Tao J & Perdew JP Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys 119, 12129–12137 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.