

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancer. It has an excessive desmoplastic stroma that can limit the intratumoral delivery of chemotherapy drugs, and protect tumor cells against radiotherapy. Therefore, both stromal and tumor compartments need to be addressed in order to effectively treat PDAC. We hereby co-deliver a sonic hedgehog inhibitor, cyclopamine (CPA), and a cytotoxic chemotherapy drug paclitaxel (PTX) with a polymeric micelle formulation (M-CPA/PTX). CPA can deplete the stroma-producing cancer-associated fibroblasts (CAFs), while PTX can inhibit tumor proliferation. Here we show that in clinically relevant PDAC models, M-CPA effectively modulates stroma by increasing microvessel density, alleviating hypoxia, reducing matrix stiffness while maintaining the tumor-restraining function of extracellular matrix. M-CPA/PTX also significantly extends animal survival by suppressing tumor growth and lowering the percentages of poorly to moderately differentiated tumor phenotypes. Our study suggests that using multifunctional nanoparticles to simultaneously target stromal and tumor compartments is a promising strategy for PDAC therapy.
Keywords: polymeric micelles, sonic hedgehog signaling, pancreatic cancer, stromal modulation, cancer-associated fibroblast
Introduction
Pancreatic ductal adenocarcinoma (PDAC) belongs to one of the most lethal types of cancer, with an overall 5-year survival rate of patients below 5% [1]. Therefore, there is an unmet medical need to develop an effective treatment for this disease. PDAC is distinguished from other cancers by its extensive desmoplastic stroma, a fibrotic deposition produced by cancer-associated fibroblasts (CAFs) [2]. The exact role of stroma during PDAC progression has been under debate. Conventionally, stroma was thought to protect tumor cells against radiation, restrict the delivery of chemotherapy drugs into tumors, and promote tumor progression and metastasis [3]. The stroma-producing CAFs also induce drug resistance in tumor cells [4]. Therefore, it was once considered beneficial to ablate stroma and CAFs. Sonic hedgehog (SHH) pathway is a key regulator for CAF activation and stromal deposition. It also contributes to the initiation and progression of PDAC, and maintenance of tumor-initiating cells [5]. As a result, SHH inhibitors were tested as a promising strategy for pharmacological depletion of CAFs [6].
However, SHH inhibitors produced disappointing results in clinical trials on PDAC patients, giving no survival benefit over standard-of-care therapies [7–10]. Further studies on genetically engineered mouse models of PDAC showed that genetic or pharmacologic inhibition of SHH signaling only resulted in shortened median survival of mice [11, 12]. In these studies, high doses of SHH inhibitors (up to 100 mg/kg daily) were administered over an extended time to ablate the alpha smooth muscle actin-positive (α-SMA+) CAFs (~80% reduction) and type I collagen in the tumor extracellular matrix (ECM). Surprisingly, the loss of collagenous ECM induced the epithelial-mesenchymal transition (EMT) in tumor cells, which led to more invasive and metastatic tumor phenotypes, and subsequently treatment failure [12]. These results highlighted the importance of preserving the tumor-restraining function of collagenous matrix during PDAC therapy. Because SHH inhibitors used at high doses might have caused significant systemic toxicity and possible off-target effects, we argued that the benefits of SHH inhibition in PDAC treatment should not be completely ruled out [13]. Numerous preclinical studies had shown that treatment with SHH inhibitors, e.g., cyclopamine (CPA) and IPA-926 (saridegib, a semisynthetic derivative of CPA), could increase tumor vasculature [14], sensitize tumor cells to radiotherapy [15], and deplete cancer stem cells [16].
We hereby propose an alternative approach with two key features. First, the extent of stroma reduction might be controlled by lowering the dose of SHH inhibitors. In this way, we can avoid the catastrophic consequences of stroma ablation, while retaining the benefits of SHH inhibition. Second, a cytotoxic agent, e.g. paclitaxel (PTX), might be co-administered to suppress the proliferation of invasive tumor cells. To test this approach, we developed a polymeric micelle-based nanoformulation (termed M-CPA/PTX) to co-deliver CPA and PTX, and examined its performance in three orthotopic PDAC models: human PDAC MiaPaca-2-luciferase xenograft model, patient-derived xenograft model, and an genetically engineered mouse (GEM) model of PDAC (KPC-Luc model, KrasG12D; p53T172H; Pdx1-Cre; Luciferase). The preclinical results reported here demonstrate that M-CPA/PTX successfully modulated stroma without ablating the collagenous matrix. Remarkably, M-CPA/PTX treatment significantly prolonged the survival of KPC-Luc mice compared to gemcitabine and abraxane, and significantly lowered the percentiles of poorly to moderately differentiated phenotypes. Taken together, multifunctional nanoparticles targeting multiple cellular populations is a promising approach for PADC therapy.
Materials and Methods
Experimental design
The in vivo studies were designed to evaluate the antitumor efficacy of M-CPA/PTX in preclinical tumor models. Sample size of 5 to 10 is standard for xenograft studies with frequent measurements of tumor burden via bioluminescence or T2-MRI and was sufficient for detection of significant differences between control and treatment groups. Sample size of 8 to 15 has been used for survival studies in spontaneous tumor models and was sufficient for detection of significant differences between the untreated control and M-CPA/PTX groups. The late-treatment M-CPA/PTX group had only 7 mice because 1 mouse had to be euthanized owing to severe fight wounds and was censored. Cohorts were randomized by a technician, and pathological analyses were completed by a licensed pathologist; both were blinded to the treatment history. Study groups were followed until mice were moribund or until the predetermined time of study completion. Animal morbidity was independently examined by in-house veterinarians, and mice were euthanized upon their recommendation. All animal studies were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center and were carried out in accordance with institutional guidelines.
Orthotopic MiaPaca-2-luc pancreatic xenograft model
Orthotopic MiaPaca-2-luc pancreatic tumor xenografts were established in female NCR nude mice (Taconic, Hudson, NY). A small incision was made in the left abdomen of each mouse. Directly into the pancreas was injected 50 µL of cell suspension (1 × 106 MiaPaca-2-luc cells and 3 × 106 HPSCs) in a 1:1 volume mixture of Hank’s balanced salt solution and Matrigel. The incision was closed with absorbable sutures, and the tumors were allowed to grow for 4 weeks before treatment. Mice were randomly assigned to 4 different groups consisting of 10 mice per group: (1) untreated (control); (2) M-CPA; (3) M-PTX; and (4) M-CPA/PTX. Each micellar formation was injected intravenously at a dosage of 10 mg/kg/drug/injection, 3 injections per week. Luciferase activity was monitored weekly by intraperitoneal injection of D-luciferin (150 mg/kg) according to the manufacturer’s instructions. The luminescence signals were recorded up to 20 min after D-luciferase injection and the peak value was recorded using a Xenogen IVIS-200 optical system (PerkinElmer, Waltham, MA).
Orthotopic PDX models
Orthotopic PDX models were established in female J:Nu nude mice (Jackson Lab, Bar Harbor, ME) according to published procedures [17]. A small incision was made in the left abdomen of each mouse. PDX tumors (PAXT#69 and #102) less than 5 passages were cut into 1-mm3 pieces and sutured to the pancreas. The incision was closed with absorbable sutures. MRI images were acquired on a Biospec USR70/30 imaging system (Bruker Biospin MRI, Billerica, MA) equipped with a 7T magnet using micro-imaging gradient and a purpose-built small animal coil [18].
Tumor size was monitored using respiration-gated T2-MRI following standard protocols. The following parameters were used: TE/TR = 38/2000 ms; BW = 101010.10 Hz; Rare = 8; averages = 3; matrix size = 256 × 192; field of view = 4 cm × 3 cm; slick thickness = 0.75 mm; slice gap = 0.25 mm. Images were processed using Bruker Biospin software. Tumor size was measured at the largest tumor cross-section of axial images and reported as the mean of the length and width of tumor at this point. Mice were randomly assigned to the untreated control or M-CPA/PTX treatment group, 5 mice per group, once tumor size reached 6 to 8 mm. M-CPA/PTX was injected intravenously at 5 mg/kg/drug/injection, 3 injections per week, for the first 2 weeks and then intraperitoneally at the same schedule for up to 8 weeks.
For DCE-MRI, mice were anesthetized with isoflurane, and a catheter fitted with a heparin-flushed 27-gauge needle was securely inserted into the tail vein. Three-dimensional (3D), fast spoiled gradient recalled (3D-fSPGR) images were acquired before and sequentially for 15 minutes after intravenous bolus injection of the biotin-BSA-Gd-DTPA. The following parameters were used: TE/TR = 1.3/40 ms; BW = 79365.10 Hz; averages = 1; matrix size = 128 × 96; field of view = 3 cm × 3 cm; slick thickness = 2 mm; slice gap = 0.00 mm. Ktrans values were calculated according to published protocols[19].
KPC-Luc transgenic mouse model
KPC-Luc mice were generated from breeding LSL-KrasG12D/+; LSL-p53T172H/+; Pdx1-Cre (KPC) mice with ROSA26-pGAGGs-LSL-Luciferase mice[20]. PDX-1-Cre, which drives KrasG12D, also drives an inducible luciferase in pancreatic acinar cells, and the resultant tumors are luciferase positive. Bioluminescence imaging (Xenogen IVIS-200 optical system, PerkinElmer) was used to monitor tumor presence in KPC-Luc mice between the ages of 90 and 140 days. For the early-treatment protocol, mice with positive bioluminescence signals (5 × 108 to 20 × 108 photons/second) were recruited at ages 95 to 100 days and randomly assigned to the untreated control (N = 15), gemcitabine (N = 11), abraxane (N = 10) or M-CPA/PTX treatment groups (N = 10). M-CPA/PTX was injected intravenously at 5 mg/kg/drug/injection at 3 injections/week for the first 2 weeks and then intraperitoneally at the same schedule until mice became moribund or until 90 days after treatment initiation. Gemcitabine was intraperitoneally injected at 100mg/kg twice per week until mice died. Abraxane was given at 5 mg/kg/injection with the same schedule of M-CPA/PTX. The last surviving mouse was sacrificed at the age of 280 days. For the late-treatment protocol, mice with palpable tumors were enrolled at ages 120 to 130 days and randomly assigned to the untreated control or 1 of 4 treatment groups: untreated control (N = 8), gemcitabine (N = 8), abraxane (N = 8), M-CPA + M-PTX (N = 8), M-CPA/PTX (N = 8). Each micellar formulation was injected intravenously at 5 mg/kg/drug/injection at 3 injections/week for the first 2 weeks and then intraperitoneally at the same schedule until mice became moribund or until 60 days after treatment initiation, when all the surviving mice were sacrificed. Gemcitabine was intraperitoneally injected at 100 mg/kg twice per week until mice died. Abraxane was given at 5 mg/kg/injection with the same schedule of M-CPA/PTX.
UPLC-MS/MS assay for simultaneous quantification of PTX and CPA
For quantification of PTX and CPA, chromatographic separation was achieved on Hypersil GOLD C18 Selectivity LC Columns (1.9 µm, 50 × 2.1 mm, Thermo Fisher Scientific, Madison, WI) with a gradient elution using mobile phase A of 0.1% formic acid in water and mobile phase B of 0.1% formic acid in acetonitrile. The detection was performed with a triple quadrupole mass spectrometer (API 5500, Applied Biosystems/MDS SCIEX, Foster City, CA) in the positive ionization using APCI source and multiple reaction monitoring mode, using docetaxel as an internal standard for quantification. Linearity of the assay was demonstrated over the range of 0.5 ng/ml to 2,000 ng/ml, with the lower limit of quantification of 0.5 ng/ml for both PTX and CPA, in mouse blood and pancreatic tumor. The assay was accurate and precise with bias and percentage in coefficient of variation of less than 15% and extraction recovery of 85% to 104% for PTX and 85% to 89% for CPA.
Statistical analysis
Values are expressed as mean ± SEM. Data were evaluated using Student’s t test or 1-way analysis of variance followed by post hoc Tukey multiple comparisons. The log-rank test was used in Kaplan-Meier survival analyses. A p value of less than 0.05 was considered statistically significant.
Results
Polymeric micelles with CPA and PTX co-payloads displayed optimal physicochemical properties
The polymeric micelles were formulated from an anionic block copolymer (Fig. 1A) and a cationic block copolymer (Fig. 1B). The anionic copolymer consisted of a poly(methacrylate) backbone with one block grafted with poly(ethylene glycol) (PEG) and a second block grafted with biodegradable poly(ε-caprolactone). Each poly(ε-caprolactone) side chain was end-capped with a succinic acid monoester. The residual carboxylic acid of succinic acid reversibly interacted with the secondary amine of CPA, which stabilized CPA-encapsulated micelles. The cationic polymer had the brush-like PEG-grafted poly(methacrylate) block and a second block with pendent quaternary ammonium cations. The numbers of repeating units in each block of the copolymers were calculated from proton nuclear magnetic resonance spectra (Supplementary Figs. S1–5). Upon addition of drug payloads, these two oppositely charged polymers attracted each other through ionic and hydrophobic interaction to form the micelles (Fig. 1C). These features enabled co-encapsulation of both hydrophobic and positively charged compounds with high stability.
Figure 1. Formulation and characterization of CPA- and/or PTX-loaded polymeric micelles.
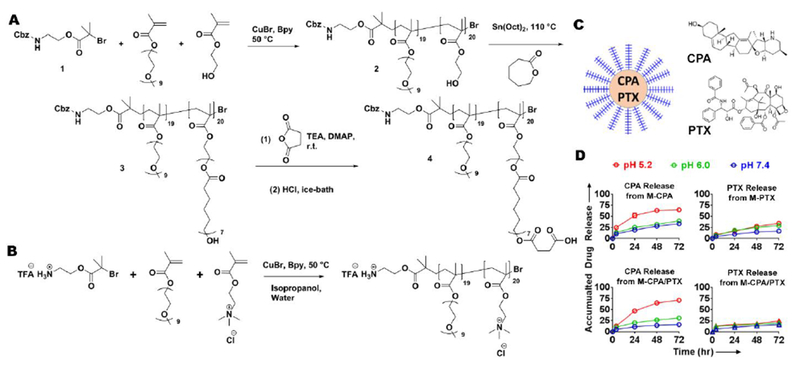
(A) Synthesis route of anionic block copolymer. (B) Synthesis route of cationic block copolymer. (C) Schematic illustration of polymeric micelles. (D) Drug release profiles at 37°C under pH 5.2, 6.0, and 7.4. N = 3 in each group.
We prepared micelle formulations with different drug loading and CPA-to-PTX ratio in order to minimize the micelle size (Table 1). Because PDAC tumors are hypovascular, smaller nanoparticles have better a chance to penetrate the tumor bed [21]. We therefore used both size and drug payload as a criteria for the selection of optimal formulation for further studies. On the basis of data summarized in Table 1, we chose polymeric micelles with a total drug loading of 5% by weight and a CPA-to-PTX weight ratio of 50%:50%, and denoted this formulation as M-CPA/PTX. CPA release from M-CPA/PTX was pH-dependent. The cumulative release of CPA after 72-hour incubation at 37 °C was 71.0 ± 2.4% at pH 5.2, and 16.0 ± 1.8% at pH 7.4. PTX release from M-CPA/PTX was less dependent on pH. The cumulative release of PTX after 72-hour incubation at 37 °C was 24.6 ± 1.3% at pH 5.2, and 16.1 ± 0.9% at pH 7.4 (Fig. 1D).Similar release profiles were observed for CPA from M-CPA, and PTX from M-PTX. M-CPA/PTX showed little burst release of CPA under physiological conditions (11.2 ± 2.0% release at pH 7.4 after 24-hour incubation) (Fig. 1D). The pH-responsive release of CPA should prevent its premature release during blood circulation and allow its faster release in the acidic microenvironment of pancreatic tumor [22]. M-CPA/PTX could be stored as frozen aqueous solutions over a period of 6 months without apparent changes in drug payload and particle size (data not shown).
Table 1.
Size and size distribution of drug-loaded micelles measured by dynamic light scattering1
| Number Average Diameter (nm) | Polydispersity Index | |
|---|---|---|
| Effect of CPA:PTX weight ratio (Total drug loading = 5% by weight) | ||
| CPA only | 74.7 ± 1.5 | 0.148 |
| CPA:PTX = 67%:33% | 53.9 ± 5.6 | 0.131 |
| CPA:PTX = 50%:50% | 45.2 ± 4.5 | 0.114 |
| CPA:PTX = 33%:67% | 77.3 ± 11.7 | 0.175 |
| PTX only | 121.3 ± 2.1 | 0.124 |
| Effect of total drug loading (CPA:PTX = 50%:50% by weight) | ||
| 2.5% by weight | 85.5 ± 10.6 | 0.113 |
| 5% by weight | 45.2 ± 4.5 | 0.114 |
| 10% by weight | 65.4 ± 3.1 | 0.110 |
Data are presented as mean ± standard error of mean (n = 3).
M-CPA maintained the biological activity of free CPA
We then examined whether M-CPA functioned as an SHH inhibitor. M-CPA/PTX was not used in this study because at the half maximal inhibitory concentration (IC50) of CPA, M-CPA/PTX would have all cells killed due to cytotoxicity of PTX. During the canonical activation of SHH signaling, the SHH ligand binds to the cell surface protein Patched (PTCH1), and releases another membrane receptor Smoothened (SMO). SMO then moves to cell nucleus to activate the GLI transcription factors(Fig. 2A) [23]. CPA inhibits SHH pathway by irreversibly binding to SMO, therefore prevents its translocation into cell nucleus. Figure 2B shows that M-CPA inhibited the binding of CPA-BODIPY, a fluorescent derivative of CPA with similar binding property, to SMO receptors on the surface of MiaPaca-2 pancreatic cancer cells. The half maximal effective M-CPA concentration required to block 50% of CPA-BODIPY’s binding to SMO (EC50) was 0.35 µM to 1.3 µM in 3 different PDAC cell lines (Table 2). Downstream of SMO, M-CPA reduced the protein expression of GLI1, SHH, and PTCH1 in 2 CPA-responsive cell lines (MiaPaca-2 and L3.6pl) and in immortalized human pancreatic stellate cells (HPSCs) but not in CPA-resistant Panc-1 cells (Fig. 2C). Panc-1 cells express another member of the GLI family transcription factors GLI3, which is believed to mediate cell survival and sensitivity to CPA [24]. These data suggest that the CPA in polymeric micelles retained the biological activity of free CPA.
Figure 2. Evaluation of drug-loaded polymeric micelles in human pancreatic cancer cell lines.
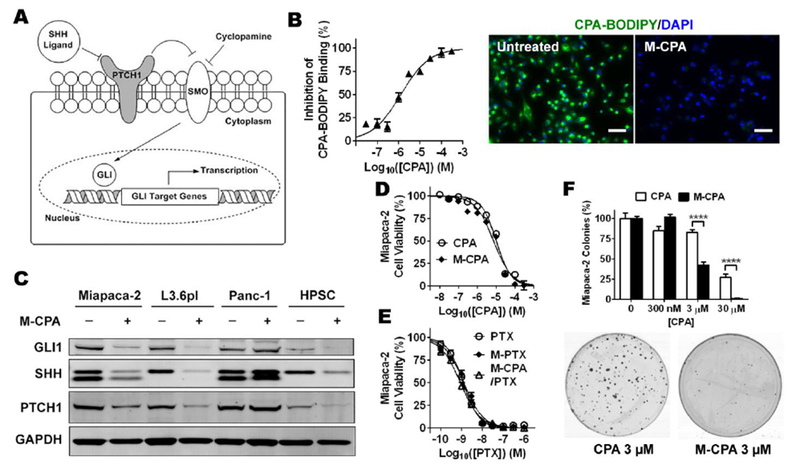
(A) Schemes of canonical SHH signaling pathway. (B) M-CPA inhibited binding of CPA-BODIPY to SMO receptors on MiaPaca-2 cells after 24-h incubation. Left: percentage inhibition as a function of equivalent CPA concentration curve. Right: representative photomicrography showing binding of CPA-BODIPY to cancer cells in the absence and presence of M-CPA. Scale bars = 50 µm. (C) Western blots of representative SHH pathway proteins in MiaPaca-2, L3.6pl, and Panc-1 pancreatic cancer cells and immortalized HPSCs after 48 h of incubation with and without 10 µM M-CPA. (D) MiaPaca-2 cell viability after 72-h incubation with free CPA (dissolved in DMSO) or M-CPA measured by MTS assay. N = 6 for each data point. (E) MiaPaca-2 cell viability after 72-h incubation with free PTX (dissolved in DMSO), M-PTX, or M-CPA/PTX measured by MTS assay. N = 6 for each data point. (F) Normalized number of MiaPaca-2 colonies formed after 10-day incubation with CPA or M-CPA, with representative photographs of colonies. N = 3 for each group.
Table 2.
SMO binding property and cytotoxicity for drug-loaded polymeric micelles1
| MiaPaca-2 | L3.6pl | Panc-1 | HPSC | |
|---|---|---|---|---|
| EC50 for inhibition of CPA-BODIPY’s binding to SMO | ||||
| M-CPA (µM) | 1.3 ± 0.3 | 0.9 ± 0.2 | 0.35 ± 0.05 | N/A |
| IC50 for cell proliferation | ||||
| CPA (µM) | 10.2 ± 0.1 | 16.0 ± 1.5 | ~ 163 | 13.3 ± 1.2 |
| M-CPA2 (µM) | 7.3 ± 1.4 | 6.2 ± 0.6 | ~ 249 | 12.7 ± 1.1 |
| PTX (nM) | 1.2 ± 0.1 | 1.4 ± 0.1 | 2.4 ± 0.4 | 3.6 ± 0.3 |
| M-PTX3 (nM) | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.6 ± 0.2 | 4.4 ± 0.2 |
| M-CPA/PTX4 | 0.9 ± 0.1 | 1.1 ± 0.2 | 1.2 ± 0.5 | 3.3 ± 0.1 |
The concentrations are equivalent CPX or equivalent PTX concentration. Data are presented as mean ±standard deviation (n = 3). EC50, half maximal effective concentration; IC50, half maximal inhibitory concentration.
CPA loading = 5% by weight;
PTX loading = 5% by weight;
CPA and PTX were equally loaded in micelles at 2.5% by weight.
M-CPA and free CPA were cytotoxic only to the two CPA-responsive PDAC cell lines and HPSCs, with IC50 values in the lower micromolar range (Fig. 2D, Table 2). In contrast, M-PTX and free PTX (Fig. 2E, Table 2) exhibited potent though nonselective cytotoxicity against all 3 tested PDAC cell lines and HPSCs with sub-nanomolar half-maximal inhibitory concentrations (IC50). The cytotoxicity of M-CPA/PTX was of the same order of magnitude as the cytotoxicity of M-PTX, suggesting that CPA in M-CPA/PTX did not potentiate the cytotoxicity of PTX. Colony formation assay in MiaPaca-2 cells showed that M-CPA inhibited colony formation more effectively than CPA did at micromolar concentrations (Fig. 2F). Compared to un-treated CTL, 3 µM CPA reduced the number of colonies by 18.2 ± 1.3%, and 3 µM M-CPA reduced the number of colonies by 57.7 ± 2.3%. Based on the results, we concluded that although there was no difference between CPA or M-CPA after 72 hours of incubation (Fig. 2D), M-CPA was significantly more potent than free CPA in inhibiting colony formation during a longer term incubation period (i.e., 10 days). Because the release profiles of CPA from M-CPA and M-CPA/PTX were similar, these data indicate that M-CPA and M-CPA/PTX maintained the same biological activity of free CPA, and that M-CPA/PTX was as potent as free PTX.
M-CPA/PTX was more effective than M-CPA, M-PTX, or a mixture of M-CPA and M-PTX in suppressing tumor growth
We next investigated whether M-CPA/PTX was more effective than M-CPA or M-PTX in suppressing tumor growth. We used an orthotopic human PDAC xenograft model generated from MiaPaca-2-luciferase cells and HPSCs. Studies have shown that co-inoculation with HPSCs promotes tumorigenesis and fibrosis in xenograft PDAC models [25]. Nude mice bearing established MiaPaca-2-luciferase tumors were intravenously injected with each micellar drug every other day for 6 days (3 injections) at 10 mg/kg/drug/injection (all doses from here onward are equivalent CPA or PTX doses) or left untreated (CTL), and tumor growth was monitored by bioluminescence imaging. At 35 days after the initial treatment, mean tumor burden normalized to the initial mean tumor burden was 19.1 ± 2.2 fold for CTL, 5.2 ± 2.3 fold for M-CPA, 1.2 ± 0.2 fold for M-PTX, and 0.4 ± 0.1 fold for M-CPA/PTX (p < 0.01). The tumor growth curves show that M-CPA alone did not effectively suppress tumor growth. Although both M-PTX and M-CPA/PTX suppressed tumor growth for 28 days, a significant tumor relapse was observed in M-PTX-treated tumors on day 35 (Fig. 3A). No significant loss of body weight was observed in any treatment groups (Supplementary Fig. S6A).
Figure 3. M-CPA/PTX had better antitumor efficacy than M-CPA or M-PTX in an orthotopic human PDAC xenograft model.
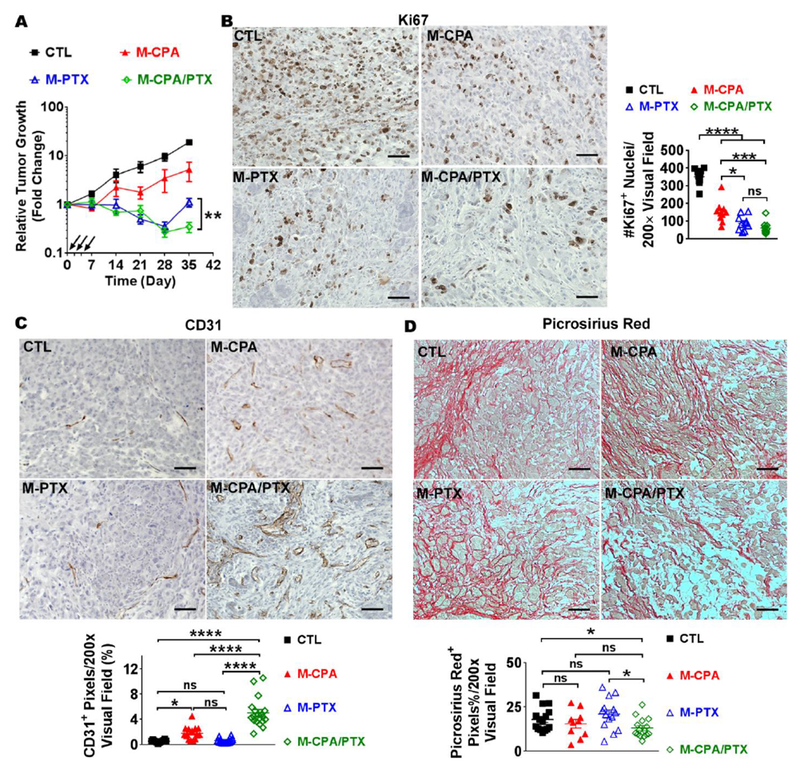
(A) Relative tumor growth curves of M-CPA, M-PTX, or M-CPA/PTX-treated tumors in MiaPaca-2-luciferase orthotopic xenograft mouse model. Tumor growth was monitored by bioluminescence imaging. Injections are marked by black arrows. Untreated mice were used as control (CTL). **p < 0.01, N = 10 in each group. (B-D) Representative micrographs of Ki67 (B), CD31 (C), and Picrosirius red (D) IHC staining and corresponding quantifications. N = 10 in each group. Scale bars = 50 µm. Data are presented as mean ± SEM. Significance was determined using 1-way ANOVA followed by Tukey post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant.
Hematoxylin-eosin staining showed extensive reduction in cellular density in the M-CPA/PTX-treated tumors compared with control (Supplementary Fig. S6B). Immunohistochemical (IHC) staining showed that the population of Ki67+ (proliferating) cells was in the order of CTL > M-CPA > M-PTX ~ M-CPA/PTX (Fig. 3B). There was no significant difference between the tumors treated with M-PTX or M-CPA/PTX, suggesting that CPA did not potentiate the cytotoxicity of PTX in M-CPA/PTX in vivo. Surprisingly, microvessel density (MVD, expressed as percent of CD31+ pixels per visual field) was significantly higher in M-CPA/PTX treated tumors (5.0 ± 0.5%) than untreated group (0.5 ± 0.2%) and both monotherapy-treated groups (p < 0.0001). Compared to CTL, MVD was also increased by treatment with M-CPA (1.8 ± 0.2%, p < 0.05), but not with M-PTX (0.6 ± 0.1%) (Fig. 3C). M-CPA/PTX moderately depleted the deposition of collagen (27% decrease); whereas monotherapies had lesser impact on collagen content than M-CPA/PTX did (14% decrease with M-CPA; 17% increase with M-PTX) (Fig. 3D). Treatment with all three formulations reduced the population of α-SMA+ CAFs compared to CTL (p < 0.001, Supplementary Fig. S6C). The tumors treated with M-CPA/PTX had similar number of α-SMA+ CAFs with those treated with M-CPA, but fewer than those treated with M-PTX (p < 0.001); suggesting that CPA was the major component in M-CPA/PTX contributing to the depletion of α-SMA+ CAFs.
Because cell line-derived xenograft models such as MiaPaca-2 produce tumors of reduced complexity and stroma elements [14], and may fail to accurately model the tumor microenvironment, we next used a GEM model of PDAC to investigate the antitumor efficacy of M-CPA/PTX in comparison with a physical mixture of M-CPA and M-PTX (Supplementary Fig. S7). M-CPA/PTX at a dose of 5 mg/kg/drug/dose led to a significantly longer survival of KPC-Luc mice with established tumor (median survival 58 days after treatment initiation) than the mixture of M-CPA and M-PTX (median survival 31.5 days after treatment initiation, p = 0.0036) administered at the same doses (in CPA and PTX equivalence) and schedules. This result suggested that simultaneous delivery of CPA and PTX, both temporally and spatially, was an indispensable attribute to the superior efficacy of M-CPA/PTX.
M-CPA/PTX increases lifespan of GEM KPC-Luc mice with both early-treatment and late-treatment protocols in favor of suppression of invasive phenotypes
GEM models can faithfully recapitulate some of human PDAC genetics and phenotype [26]. KPC mice are generated by the concomitant expression of oncogenic KrasG12D and of Trp53 with a point mutation (Trp53R172H), both driven by a pancreas-specific Cre [26]. The KPC model is a common GEM model of PDAC used to test the efficacy of antitumor therapies. Previous studies showed that stroma ablation created invasive tumor phenotypes, and reduced animal survivals in KPC mice [11, 12]. However, because substantially high doses of Hh inhibitors were used in these studies (i.e., 100 mg/kg/dose), possible systemic toxicity to poor-performance tumor-bearing mice, or off-target effects, cannot be ruled out. We therefore used M-CPA/PTX to address the question of whether stroma modulation mediated by low drug dose is effective in prolonging animal survival. To facilitate monitoring of tumor growth, we used KPC-Luc mice generated by crossing LSL-KrasG12D/+; LSL-p53T172H/+; Pdx1-Cre (KPC) mice with ROSA26-pGAGGs-LSL-Luciferase mice [20]. The tumors in the resultant mice are luciferase positive. Similar to the original KPC mice, KPC-Luc mice develop spontaneous pancreatic tumors at the age of 100 to 140 days, and tumors in the KPC-Luc model are highly stromal with dense desmoplasia [26]. Untreated KPC-Luc mice succumbed to extensive local invasion and metastasis, as shown by the bioluminescence image of an untreated control mouse acquired at age 135 days (Supplementary Fig. S8A&B).
M-CPA/PTX was injected at 5 mg/kg/drug/injection, 3 injections week, which was only 2% to 5% the dose of hedgehog inhibitors used in other studies (40 to 100 mg/kg daily [11, 12]). Gemcitabine and abraxane were included as the standard-of-care therapy controls. The dose of abraxane was adjusted to 5 mg/kg/injection in PTX equivalent, which was the same as the M-CPA/PTX regimen. In the early-treatment protocol, M-CPA/PTX was initiated at ages of 95 to 100 days, when bioluminescence flux reached ~ 5×108 to 20×108 photons/second. At this time point, pancreases exhibited severe inflammation, acinar ductal metaplasia, and pancreatic intraepithelial neoplasia but had not transformed to PDAC. Mice with early M-CPA/PTX treatment showed localized bioluminescence signals throughout an 8-week monitoring period (Supplementary Fig. S8C). The median survival in the untreated control, gemcitabine, and abraxane groups were 139, 167, and 155 days, respectively. Remarkably, in the M-CPA/PTX group, the first death occurred at 167 days of age, and the median survival was 236.5 days, 69 days longer than in the gemcitabine group (p = 0.0001, log-rank test) and 81 days longer than in the abraxane group (p = 0.0018, log-rank test) (Fig. 4A).
Figure 4. M-CPA/PTX prolonged survival of KPC-Luc mice and alleviated hypoxia.
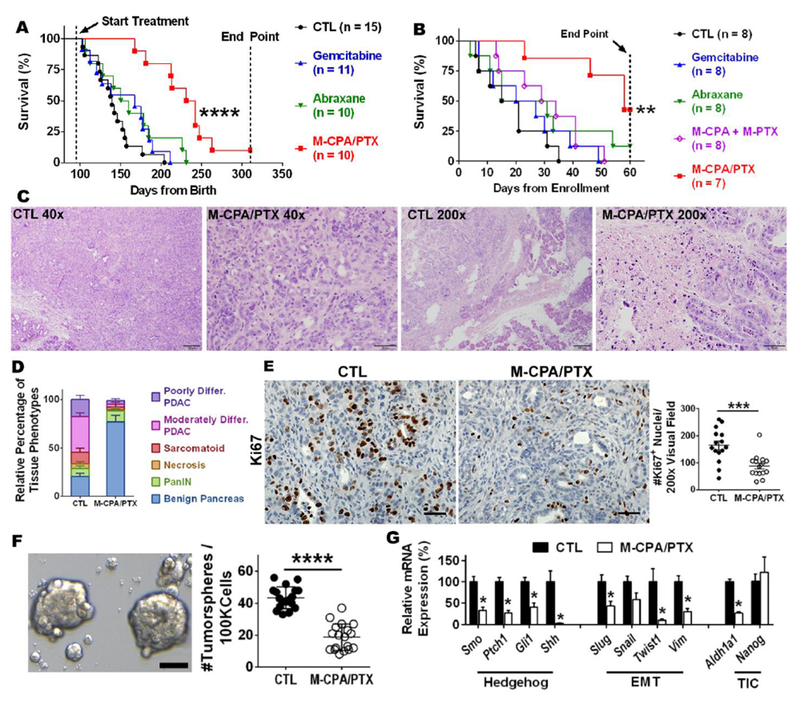
(A) Kaplan-Meier survival analysis for KPC-Luc mice with early treatment. ****p < 0.0001, log-rank test. (B) Kaplan-Meier survival analysis for KPC-Luc mice with treatment started when tumors became palpable. ***p = 0.0033, log-rank test. (C) Representative H&E-stained sections of late-stage tumors after 2 weeks of treatment with M-CPA/PTX. Untreated tumors were used as control (CTL). Scale bars = 200 µm for 40× images and 50 µm for 200x images. (D) Distribution of histological phenotypes of late-stage tumors after 2 weeks of treatment with M-CPA/PTX (N = 10). Untreated tumors were used as CTL (N = 8). The M-CPA/PTX-treated tumor group had a significantly lower proportion of poorly differentiated PDAC (p < 0.05) or moderately differentiated PDAC (p < 0.0001) and a significantly higher proportion of benign pancreas (p < 0.0001). Significance of differences was determined using 2-way ANOVA followed by Sidak’s multiple comparison test. differ. = differentiated. (E) Representative micrographs of Ki67 IHC staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (F) Representative micrographs of tumor spheres and corresponding quantifications. N = 18 for each group. Arrows indicate eligible tumor spheres (>50 µm). Scale bars = 50 µm. (G) Expression levels of mRNA for selected genes from CTL and M-CPA/PTX-treated tumors. Results are mean ± SEM of 4 mice in each group. RT-PCR was performed in technical duplicates, and values were normalized to 18S. Significance of differences between CTL and M-CPA/PTX groups was determined using Student’s unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant.
PDAC patients usually present with late-stage disease at the time of diagnosis. We therefore designed an additional study to evaluate efficacy on well-established tumors. In the late-treatment protocol, M-CPA/PTX was initiated once the tumor burden was confirmed by palpation and bioluminescence imaging (~125 days of age). Gemcitabine or abraxane only marginally increased median survival by 5 days compared to untreated control. The median survival times after enrollment were 18 days for untreated control mice, 23.5 days for gemcitabine, and 23 days for abraxane (Fig. 4B). Vismodegib (GDC-0449), a synthetic SHH inhibitor that has been approved by the FDA for treatment of advanced basal cell carcinoma, shortened the animal survival by 3 days compared to untreated control (Supplementary Fig. S9). In contrast, the first death of M-CPA/PTX group occurred at 23 days after enrollment, and the median survival was 58 days and significantly longer than the other groups (p = 0.0033, log-rank test). These data indicated that M-CPA/PTX was highly efficacious against PDAC at substantially reduced dose compared to SHH inhibitors used in previous studies [9, 12].
Tumor response was studied in the mice of late-treatment setting after 6 intravenous injections of M-CPA/PTX over 2 weeks at 5 mg/kg/drug/injection. Extensive poorly or moderately differentiated PDAC was observed in the untreated CTL, whereas focal residual moderately differentiated PDAC with extensive necrosis was observed in the M-CPA/PTX-treated tumors (Fig. 4C). Compared to CTL, M-CPA/PTX reduced the area fraction of poorly differentiated PDAC by 78% (p = 0.037) and that of moderately differentiated PDAC by 92% (p < 0.0001) (Fig. 4D). Remarkably, M-CPA/PTX increased the area fraction of benign pancreas by 270% (p < 0.0001). In terms of tumor proliferation, M-CPA/PTX reduced the density of Ki67+ cells by 52% (Fig. 4E, p < 0.001). These results demonstrate that M-CPA/PTX could prevent the formation of undifferentiated aggressive tumor phenotypes. This was in sharp contrast to earlier studies that had shown induction of more invasive phenotypes after prolonged, high-dose SHH inhibitor vismodegib [11].
To gain mechanistic insight into reduced invasive phenotypes with M-CPA/PTX treatment, we performed tumorsphere assay to examined the effect of M-CPA/PTX on potency of tumor-initiating cells (TICs). The number of spheres formed per isolated 100,000 tumor cells was significantly lower from M-CPA/PTX-treated tumors than those from untreated CTL (19 ± 8 vs. 43 ± 7 spheres per 100,000 cells, p < 0.0001, Fig. 4F). RT-PCR showed that M-CPA/PTX-treated tumors had significantly lower mRNA levels of genes involved in Hh signaling (Smo, Ptch1, Gli1, and Shh), EMT (Slug, Snail1, Twist1, and Vim), and TICs (Aldh1a1) (Fig. 4G). SHH (p < 0.05) and PTCH1 (p = 0.059) were also reduced at the protein level (Supplementary Fig. S10). These data suggest that M-CPA/PTX simultaneous inhibited SHH signaling, eliminated proliferating tumor cells, and suppressed EMT and potency of TICs in favor of tumor control.
M-CPA/PTX enhances angiogenesis and reduces hypoxia in KPC-Luc stroma without depleting collagen
To address the extent to which M-CPA/PTX treatment impacted PDAC stroma, we analyzed tumors from the KPC mice with the late treatment protocol. Atomic force microscopy revealed that M-CPA/PTX therapy reduced the elastic modulus of tumor by 55% (Fig. 5A, p < 0.01). IHC staining showed that M-CPA/PTX reduced α-SMA+ CAFs by 28% (Fig. 5B, p < 0.01) and FAP-α+ CAFs by 56% (Fig. 5C, p < 0.01), but did not affect the Picrosirius red–stained collagen (Fig. 5D). Hyaluronic acid (HA) binding protein (HABP1), a marker for total HA content, was decreased by 51% (Fig. 5E, p < 0.05); and lysyl oxidase, a collagen crosslinking enzyme, was reduced by 57% (Fig. 5F, p < 0.0001). IHC staining also showed that M-CPA/PTX doubled the number of CD31+ vessels (Fig. 5G, p < 0.0001) and reduced the area fraction of hypoxia to one third of CTLs (Fig. 5H, p < 0.001). These data suggest that M-CPA/PTX created a softer tumor matrix unfavorable for invasive transformation and metastasis [27]. The mRNA expression levels of hypoxia markers (Ca9, Glut1, Hif1a, and Vegfa) were significantly lower in the treated tumors than in CTLs. In terms of fibrosis markers, the mRNA expression levels of Fap (encoding fibroblast activation protein alpha [FAP-α]) and Acta2 (encoding α-SMA) were significantly down regulated by M-CPA/PTX, while Col1a1 (encoding type I collagen) was not affected (Fig. 5I).
Figure 5. M-CPA/PTX modulated stromal components in KPC-Luc tumor.
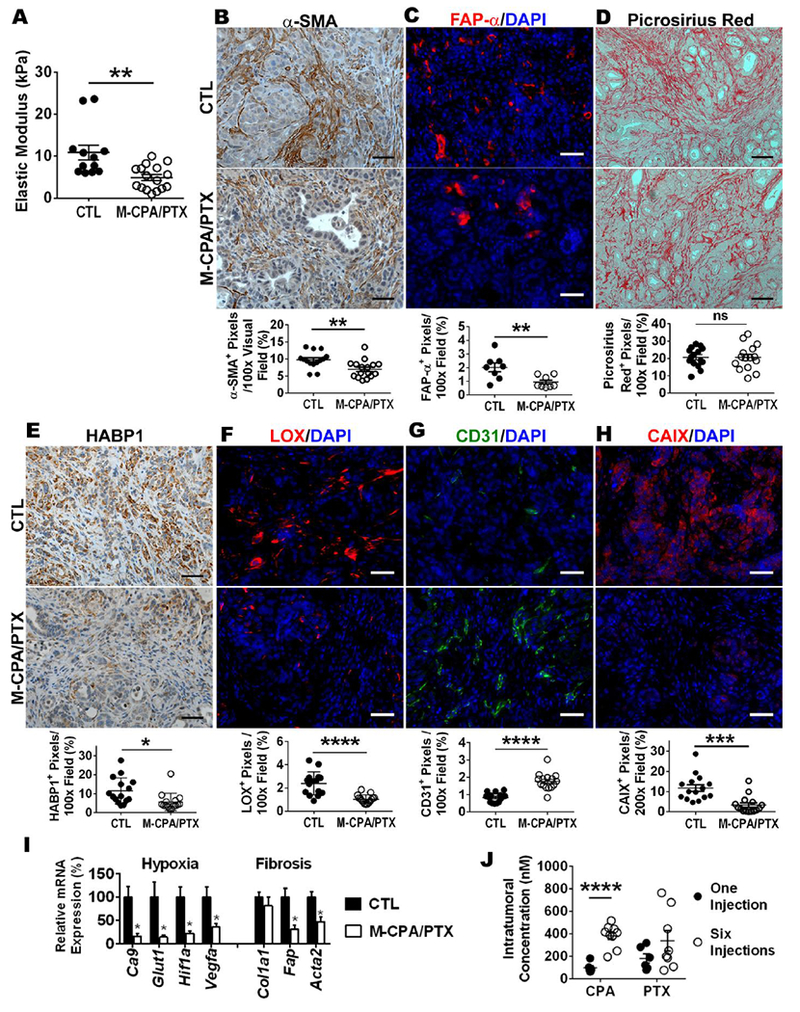
(A) Quantification of the elastic modulus of tumors. Results are representative of 3 mice in each group. Five randomly selected locations were measured on each tumor. (B) Representative micrographs of α-SMA IHC staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (C) Representative micrographs of FAP-α (red) and DAPI (blue) dual-immunofluorescence staining and corresponding quantifications. N = 8 in each group. Scale bars = 50 µm. (D) Representative micrographs of Picrosirius red IHC staining and corresponding quantifications. N = 15 in each group. Scale bars = 100 µm. (E) Representative micrographs of HABP1 IHC staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (F Representative micrographs of LOX (red) and DAPI (blue) dual-immunofluorescence staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (G) Representative micrographs of CD31 IHC staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (H) Representative micrographs of CAIX (red) and DAPI (blue) dual-immunofluorescence staining and corresponding quantifications. N = 15 in each group. Scale bars = 50 µm. (I) Relative mRNA expression for selected genes from CTL and M-CPA/PTX-treated tumors. Results are representative of 4 mice in each group. RT-PCR was performed in technical duplicates, and values were normalized to 18S. (J) Intratumoral drug concentrations of late-stage tumor at 24 h after 1 or 6 intravenous injections of M-CPA/PTX. The dose for each injection was 5 mg/kg/drug. Six injections were completed over 2 weeks. The CPA concentration was significantly higher after 6 injections than after 1 injection (p < 0.0001). N = 6 for 1-injection group; N = 9 for 6-injection group.
Repeated injections of M-CPA/PTX enhances drug delivery to KPC-Luc tumor without significant systemic toxicity
Lastly, we investigated whether pharmacologically relevant concentrations of CPA and PTX were delivered to the KPC-Luc tumors. The concentrations of CPA and PTX were measured using quantitative liquid chromatography–tandem mass spectrometry (LC-MS-MS) from tumors harvested at 24 hours after the last dose of M-CPA/PTX. The CPA intratumoral concentration after 6 injections was 4 times that after 1 injection (383.4 ± 33.8 nM vs 95.9 ± 18.0 nM, p < 0.0001) (Fig. 5J), both of which were much lower than the IC50 values of CPA against tumor cells or immortalized HPSCs (~10 µM, Fig. 1, Table 2). Although CPA at this low concentration was not expected to kill most tumor cells or CAFs, the sustained release of CPA from M-CPA/PTX still down-regulated SHH signaling in the tumor (Fig. 4G, Supplementary Fig. S10). The PTX concentration after 6 injections was approximately twice that after 1 injection (305.9 ± 83.9 nM vs 163.2 ± 37.1 nM, p = 0.19), both of which were remarkably higher than the IC50 values of PTX in cell culture (Fig. 2, Table 2). This high PTX concentration was in correlation with the substantial reduction of Ki67+ proliferating cells in M-CPA/PTX-treated tumors (Fig. 4E).
After the 6 doses of M-CPA/PTX (30 mg/kg/drug total dose), there was no significant differences in body weight among the age-matched wild-type mice, tumor-bearing control mice, and tumor-bearing M-CPA/PTX-treated mice (Supplementary Fig. S11A). No observable toxic effects were found in major organs (Supplementary Fig. S11B). In a separate long-term study, there was no change in the histopathologic appearance of the intestine and Ki67+ cells (Supplementary Fig. S11C) after injecting M-CPA/PTX at a total dose of 120 mg/kg/drug over 8 weeks. The absence of systemic toxicity was consistent with the low CPA and PTX accumulations in the blood (< 3 nM, 24 hours after 1 and 6 injections) and relatively low accumulation of PTX in liver or spleen after 1 and 6 injections (Supplementary Fig. S12).
M-CPA/PTX eliminated proliferating cells, enhanced blood perfusion, and reduced hypoxia without depleting collagen in patient-derived orthotopic (PDX) model
The finding that M-CPA/PTX increased tumor MVD in human PDAC xenografts and the KPC-Luc mice prompted us to further investigated the role of M-CPA/PTX in modulating tumor stroma using PDX models. PDX models mirror the cellular diversity within tumor and genetic heterogeneity among patients, therefore are suitable preclinical models to study treatment response [28]. Mice were randomized into untreated control (CTL) or M-CPA/PTX-treated groups. T2-weighted magnetic resonance imaging (T2-MRI) revealed that tumor growth was suppressed in 4 out of the 5 treated mice, while 4 out of the 5 CTL tumors progressed (Fig. 6A). To gain insight on the impact of treatment on stroma, we measured tumor blood permeability using dynamic contrast-enhanced MRI (DCE-MRI) and expressed the data as Ktrans [29]. Both untreated CTL and treated tumors had similar Ktrans at the start of study (0.53 ± 0.14 vs. 0.62 vs. 0.25 min−1, Fig. 6B). At the end of study (8 weeks), the Ktrans value of treated tumors was 4.2 times higher than that of CTL (0.55 ± 0.11 vs. 0.13 ± 0.03 min−1, p < 0.01). The intratumoral distribution of Ktrans values are illustrated using heat maps on two size-matched CTL and treated tumors (Fig. 6C). The treated tumor had > 0.4 min−1 Ktrans values throughout the cross-section, while the CTL tumor had lower Ktrans at the center (~ 0.1 min−1) than its peripheral rim (~ 0.3 min−1).
Figure 6: M-CPA/PTX was effective against orthotopic PDX models.
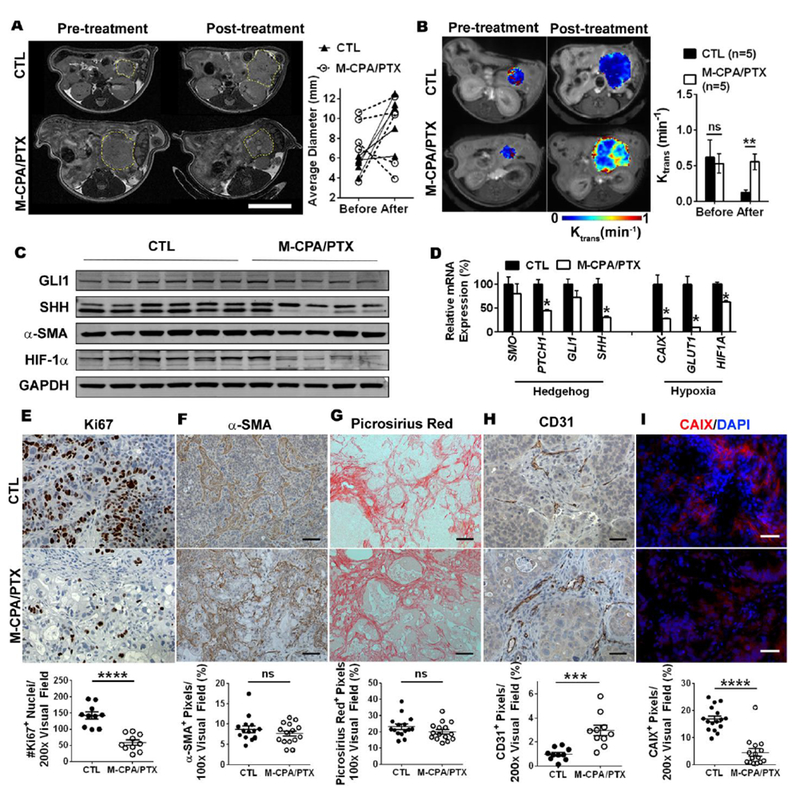
(A) Representative T2-weighted axial magnetic resonance images and individual sizes of CTL and M-CPA/PTX-treated tumors at the start and end of the study. Tumor margin is outlined by yellow circle. Scale bars = 10 mm. (B) Representative T1-dyanmic contrast enhanced MRI of size-matched CTL and M-CPA/PTX-treated tumors, and corresponding quantification of Ktrans values. (C) Western blot images of selected SHH and hypoxia proteins. Tumor lysates were prepared from 6 CTL mice and 5 M-CPA/PTX-treated mice. (D) Relative mRNA expression of selected genes from CTL and M-CPA/PTX-treated tumors. Results are mean ± SEM of 4 mice in each group. RT-PCR was performed in technical duplicates, and values were normalized to HPRT1. (E) Representative Ki67 staining and corresponding quantification in PDX models (N = 15 in each group). (F&G) Representative micrographs of α-SMA IHC staining (F) and Picrosirius red–stained collagen IHC staining (G) and corresponding quantifications. N = 15 in each group. Scale bars = 100 µm. (H) Representative micrographs of CD31 and corresponding quantifications. N = 10 in each group. Scale bars = 50 µm. (I) Representative micrographs of CAIX (red)/DAPI (blue) dual-immunofluorescence staining and corresponding quantifications. N = 15 in each group. Scale bar = 50 µm. Data are presented as mean ± SEM. Significance of differences between CTL and M-CPA/PTX groups was determined using Student’s unpaired t test. ***p < 0.001, ****p < 0.0001, ns = not significant.
Western blots (Fig. 6c and Supplementary Fig. S13) and RT-PCR (Fig. 6D) confirmed that M-CPA/PTX inhibited the Hh signaling pathway and relieved hypoxia. However, the expression of αSMA was not significantly affected by treatments (p>0.05). SHH was significantly reduced at both protein and mRNA levels (p < 0.05), while PTCH1 was significantly reduced at mRNA level (p < 0.05). The expression of hypoxia biomarkers was also significantly decreased: HIF-1α at both protein and mRNA levels (p < 0.05), and CAIX and SLC2A1 (GLUT1) at mRNA level (p < 0.05).
IHC staining showed that M-CPA/PTX significantly reduced the number of Ki67+ proliferating cells in PDX tumor (Fig. 6E, p < 0.0001), which was consistent with the slower growth of treated tumors from T2-MRI imaging. Although M-CPA/PTX did not deplete α-SMA+ CAFs (Fig. 6F) or Picrosirius red–stained collagen (Fig. 6G), it tripled the percentage of CD31+ pixels per visual field (Fig. 6H, p < 0.001) and reduced hypoxia area fraction to 27% that of the CTL (Fig. 6I, p < 0.0001). Taken together, we have demonstrated that M-CPA/PTX replenished tumor blood vessels in PDX model, alleviated hypoxia, and eliminated proliferating tumor cells without depleting the collagenous matrix.
Discussion
In this study we have developed a dual-functional nano-formulation, M-CPA/PTX, to simultaneous modulate PDAC stroma and suppress tumor growth. In the three mouse models of this study, M-CPA/PTX restored tumor blood vessels, alleviated hypoxia and exhibited prominent anti-tumor efficacy. Specifically, the improvements of animal survival in KPC-Luc model, i.e. 6 weeks in late-stage setting and more than 3 months in early-stage setting, were comparable to the best survival data reported in the literature. Indeed, among the preclinical studies using GEM models of PDAC, the extension of survival ranged from 2 weeks in late-stage settings to about 2 months in early-stage settings [14, 30–35]. Therefore our strategy of simultaneous treating the stromal and tumor compartments is a promising and effective method to enhance PDAC therapy. The importance of a simultaneous delivery of CPA and PTX was underscored by the survival difference between the M-CPA/PTX group (58 days) and M-CPA + M-PTX mixture group (31.5 days) (Fig. 4B). For the mixture group, the two micelle formulations (M-CPA and M-PTX) may have different level and kinetics of tumor uptake with mismatched drug release profiles, compromising their anti-tumor efficacy.
Previous studies have showed that the collagenous matrix in PDAC stroma could restrain the rapid tumor growth and limit local tumor invasion [11]. In support of this viewpoint, genetic depletion of α-SMA+ CAFs (83% reduction) caused extensive ablation of collagen (46 ~ 66%), and subsequently created an aggressive phenotype of PDAC tumor that rendered gemcitabine ineffective [9]. The α-SMA+ and FAP-α+ CAFs have different roles during PDAC progression. Alpha-SMA+ CAFs produce collagen. Clinical studies have established that a high α-SMA expression in PDAC correlated with a high collagen deposition and stromal density [36]. In both preclinical and clinical studies, a high collagen deposition corresponded to longer survival [37]. On the other hand, FAP-α upregulation predicts an aggressive remodeling of tumor matrix, and consequently shorter survival of PDAC patients [38]. In our study, M-CPA/PTX caused moderate depletion of α-SMA+ CAFs ranging from 28% in the KPC-Luc model to 12% in the PDX models, although in the latter case the difference between untreated control and M-CPA/PTX treated tumors were not statistically significant. M-CPA/PTX did not affect the collagen deposition in either KPC-Luc (Fig. 5D) or PDX (Fig. 6G) models. The collagen reduction in MiaPaca-2/HPSC model by M-CPA/PTX (Fig. 3D) can be attributed to the limitations of this xenograft model (i.e., the need to co-inject fibroblasts together with tumor cells to promote stroma formation), and underlined the importance of using more clinically relevant PDAC models to study treatments directed at tumor stroma.
In contrast to its mild or non-impact on α-SMA+ CAFs, M-CPA/PTX significantly reduced the expression of FAP-α, HA, and lysyl oxidase (LOX) (Figs. 5C, E &F), all of which are validated targets in PDAC therapy [35, 39, 40]. FAP-α+ CAFs can promote tumor growth by suppressing antitumor immune reactions in the stroma [39]. HA contributes to the stiffness and physical stress of solid tumors that can cause the collapse of microvessels [35]. LOX is an extracellular enzyme that catalyzes collagen and elastin crosslinking and mediates stromal stiffness [40]. The stroma modulation of M-CPA/PTX may arise from the inhibition of sonic hedgehog signaling (Fig. 4G & Supplementary Fig. S10), because a sustained activation of SHH pathway is known to cause an excessive deposition of fibrotic stroma. PTX from M-CPA/PTX may also contribute to the stroma modulation. Awasthi et al. [41] found in AsPC-1 human pancreatic cancer cell line-derived xenograft models that abraxane reduced α-SMA expression and collagen deposition, which is consistent with our findings in the MiaPaca-2/HPSC model (Supplementary Fig. S6C).
M-CPA/PTX increased the tumor blood vessels in all three PDAC models: 9 folds in the MiaPaca-2/HPSC model, 3 folds in the PDX models, and 2 folds in the KPC-Luc model. Moreover, DCE-MRI analyses of the PDX tumor revealed that M-CPA/PTX increased tumor permeability compared to CTL, indicating a normalization of vascular functions (Fig. 6B). This finding agrees with the accruing evidences that stroma modulation can affect tumor angiogenic activity [11, 12]. The vasculature normalization can be attributed to the depletion of CAFs [11, 12], degradation of HA, and reduction of lysyl oxidase. HA compress tumor microvessels by increasing the stiffness and physical stress of solid tumors. In a mouse model of colorectal-to-liver cancer metastasis [42], deposition of HA occurred following the treatment with anti-vascular endothelial growth factor, and subsequently induced collapse of blood vessels and tumor hypoxia. Other studies also showed that HA impaired vascular function and drug delivery in preclinical PDAC models while enzymatic depletion of HA increased drug delivery to tumor [43, 44]. LOX inhibition increased intratumoral vasculatures [45]. Taken together, both HA and LOX can increase tumor stiffness and play important roles in suppressing the development of tumor vasculature and limiting intratumoral delivery of chemotherapy drugs. By reducing HA and LOX contents, M-CPA/PTX significantly reduced the interstitial barriers to drug delivery within the tumor microenvironment. This interpretation is supported by the increased concentrations of CPA and PTX after 2 weeks of treatment, as compared to those after single injection (Fig. 5J).
In addition to drug delivery, the normalized microvessels also alleviated tumor hypoxia, which is known to cause chemo-resistance, induce immunosuppression, and increase tumor aggressiveness[46–48]. Glucose transporter-1 protein (GLUT1) and carbonic anhydrase IX (CAIX) are regulated by hypoxia inducible factor-1 (HIF-1) and have been used as molecular markers to indicate the degree of hypoxia [49]. M-CPA/PTX significantly reduced the mRNA levels of Caix, Glut1, and Hif1a (Fig. 5I). IHC staining for CA-IX showed that the fraction of CAIX-positive pixels in M-CPA/PTX-treated tumors was only 28.5% that in untreated KPC-Luc PDAC (p = 0.0012) (Fig. 5H).
Conclusions
We propose the following model for M-CPA/PTX’s mechanisms of action (Fig. 7). M-CPA/PTX nanoparticles are initially delivered to PDAC via blood circulation. Because of their small size (< 150 nm) and the leakiness of the intratumoral vasculature [50], M-CPA/PTX extravasate into the extravascular and extracellular fluid space, where CPA and PTX are released and exert therapeutic effects on both tumor cells and CAFs. In the tumor cell compartment, while PTX eliminates proliferating cells and enriches TICs, CPA ablates TICs. In the stromal compartment, CPA and PTX act in concert to disrupt tumor cell–CAF communication and remodel the ECM (Fig. 7A). The remodeling of the ECM is a highly dynamic process. The “togetherness” of CPA and PTX enabled by M-CPA/PTX ensures a positive feedback loop to enhance drug delivery and increase blood perfusion in subsequent injections (Fig. 7B). M-CPA/PTX, by directing CPA and PTX to proliferating tumor cells, TICs, and CAFs, efficiently disrupts tumor cell–CAF communication and breaks the vicious cycle of tumor cell proliferation, induction of tumor-supporting stroma, and stimulation of tumor invasion and metastasis.
Figure 7. Proposed model of mechanisms of action for M-CPA/PTX.
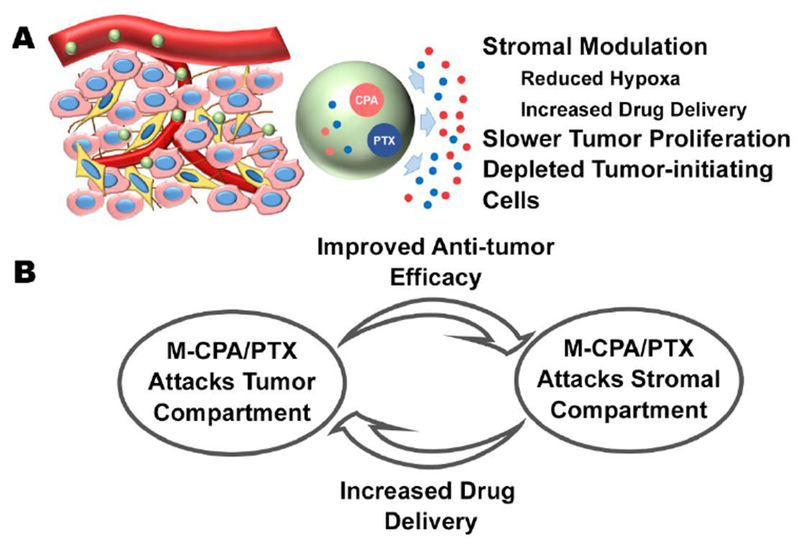
(A) CPA and PTX released from M-CPA/PTX act on different compartments of PDAC. While PTX enriches TICs, CPA nullifies such an effect. Furthermore, CPA and PTX work together to disrupt tumor cell–CAFs communication, resulting in remodeling instead of depletion of PDAC stroma. (B) Multiple injections of M-CPA/PTX generate a positive feedback loop to enhance its efficacy. M-CPA/PTX modulates the stromal compartment to increase the drug delivery from ensuing injections. More efficient drug delivery increases the intratumoral concentration of drugs and thereby improves the antitumor efficacy.
Our study has several limitations. First, the exact mechanism remains unclear as to why the collagenous matrix was preserved while HA and LOX were reduced by M-CPA/PTX. Future studies need to address the complex interactions between CAFs and other components in tumor microenvironment, as well as the different contributions from respective CAF phenotypes. Second, the dose of M-CPA/PTX and dosing schedules need further optimization to minimize systemic toxicity and maximize therapeutic outcomes. Finally, given that M-CPA/PTX depleted FAPα+ CAFs, further combination with immunotherapies may provide additional therapeutic benefits. In summary, we have demonstrated that M-CPA/PTX provided a promising alternative treatment for PDAC through the combination of stromal modulation with cytotoxic chemotherapy. Our results suggest that using multifunctional nanoparticles to target multiple cellular targets is a viable strategy to enhance the efficacy of PDAC therapy.
Supplementary Material
Significance Statement.
Recent studies have shown that high doses of Hedgehog pathway inhibitors may disrupt the tumor-restraining collagenous network of pancreatic ductal adenocarcinoma (PDAC), and therefore promoted tumor progression and metastases. Here, we find in three distinct mouse models of PDAC, that by using a nanoformulation to co-delivery cyclopamine (a sonic hedgehog inhibitor) and paclitaxel (a cytotoxic chemotherapy drug), the animal survival is significantly extended along with lower percentages of poorly to moderately differentiated tumor phenotypes. Stromal analyses reveal that the nanoformulation increases microvessel density, alleviates hypoxia, and reduces matrix stiffness while maintaining the tumor-restraining collagenous matrix. Our study suggests that multifunctional nanoparticles simultaneously targeting stromal and tumor compartments are promising tools for PDAC therapy.
Acknowledgments:
We thank Stephanie Deming for editing the manuscript. The HPSCs cells were kindly provided by Dr. Rosa Hwang (MD Anderson Cancer Center). This work was supported in part by the Skip Viragh Foundation, John S. Dunn Foundation, and Gillson Longenbaugh Foundation. The Research Animal Support Facility, High Resolution Electron Microscopy Facility, Nuclear Magnetic Resonance Facility, and Small Animal Imaging Facility are supported by a Cancer Center Support Grant from the National Institutes of Health (P30CA016672).
References
- [1].Tempero MA, Berlin J, Ducreux M, Haller D, Harper P, Khayat D, Schmoll HJ, Sobrero A, Van Cutsem E, Pancreatic cancer treatment and research: an international expert panel discussion, Ann. Oncol. 22(7) (2011) 1500–6. [DOI] [PubMed] [Google Scholar]
- [2].Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA, The pancreas cancer microenvironment, Clin. Cancer Res. 18(16) (2012) 4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rucki AA, Zheng L, Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies, World J. Gastroenterol. 20(9) (2014) 2237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kalluri R, The biology and function of fibroblasts in cancer, Nat. Rev. Cancer 16(9) (2016) 582–98. [DOI] [PubMed] [Google Scholar]
- [5].Onishi H, Katano M, Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer, World J. Gastroenterol. 20(9) (2014) 2335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, Ramachandran V, Arumugam T, Ji B, Logsdon CD, Brown JL, Godin R, Inhibition of the Hedgehog Pathway Targets the Tumor-Associated Stroma in Pancreatic Cancer, Mol. Cancer Res. 10(9) (2012) 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, Chang WC, Kantoff E, Vannier MW, Catenacci DV, Venook AP, Kindler HL, A phase I study of FOLFIRINOX plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma, Pancreas 45(3) (2016) 370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].De Jesus-Acosta A, O’Dwyer PJ, Ramanathan RK, Von Hoff DD, Maitra A, Rasheed Z, Zheng L, Rajeshkumar NV, Le DT, Hoering A, Bolejack V, Yabuuchi S, Laheru DA, A phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients (pts) with untreated metastatic pancreatic ductal adenocarcinoma (PDA). J. Clin. Oncol. 32 (Suppl 3; Abstr 257) (2014). [Google Scholar]
- [9].Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R, Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival, Cancer Cell 25(6) (2014) 719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, Cohen D, Wade J, Sleckman B, Lenz HJ, Stiff P, Kumar P, Xu P, Henderson L, Takebe N, Salgia R, Wang X, Stadler WM, de Sauvage FJ, Kindler HL, Randomized phase IB/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer, J. Clin. Oncol. 33(36) (2015) 4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, Bardeesy N, Beachy PA, Stromal response to Hedgehog signaling restrains pancreatic cancer progression, Proc. Natl. Acad. Sci. USA 111(30) (2014) E3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ, Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma, Cancer Cell 25(6) (2014) 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, Klimstra DS, Zhu LJ, Simin K, Xu L, McMahon AP, Lewis BC, Mao J, The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis, Proc. Natl. Acad. Sci. USA 109(17) (2012) E1038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA, Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer, Science 324(5933) (2009) 1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao J, Wu C, Abbruzzese J, Hwang RF, Li C, Cyclopamine-loaded core-cross-linked polymeric micelles enhance radiation response in pancreatic cancer and pancreatic stellate cells, Mol. Pharm. 12(6) (2015) 2093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mueller MT, Hermann PC, Witthauer J, Rubio–Viqueira B, Leicht SF, Huber S, Ellwart JW, Mustafa M, Bartenstein P, D’Haese JG, Schoenberg MH, Berger F, Jauch KW, Hidalgo M, Heeschen C, Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer, Gastroenterology 137(3) (2009) 1102–1113. [DOI] [PubMed] [Google Scholar]
- [17].Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE, Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice, Nat. Protocols 4(11) (2009) 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, Sood AK, Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma, Nat. Med. 12(8) (2006) 939–944. [DOI] [PubMed] [Google Scholar]
- [19].Dafni H, Kim SJ, Bankson JA, Sankaranarayanapillai M, Ronen SM, Macromolecular dynamic contrast-enhanced (DCE)-MRI detects reduced vascular permeability in a prostate cancer bone metastasis model following anti-platelet-derived growth factor receptor (PDGFR) therapy, indicating a drop in vascular endothelial growth factor receptor (VEGFR) activation, Magn. Reson. Med. 60(4) (2008) 822–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, Kaelin WG Jr., Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination, Mol. Imaging 2(4) (2003) 297–302. [DOI] [PubMed] [Google Scholar]
- [21].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat. Nanotechnol. 6(12) (2011) 815–23. [DOI] [PubMed] [Google Scholar]
- [22].Kimbrough CW, Khanal A, Zeiderman M, Khanal BR, Burton NC, McMasters KM, Vickers SM, Grizzle WE, McNally LR, Targeting Acidity in Pancreatic Adenocarcinoma: Multispectral Optoacoustic Tomography Detects pH-Low Insertion Peptide Probes In Vivo, Clin. Cancer Res. 21(20) (2015) 4576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Varjosalo M, Taipale J, Hedgehog: functions and mechanisms, Genes Dev. 22(18) (2008) 2454–2472. [DOI] [PubMed] [Google Scholar]
- [24].Steg A, Amm HM, Novak Z, Frost AR, Johnson MR, Gli3 mediates cell survival and sensitivity to cyclopamine in pancreatic cancer, Cancer Biol. Ther. 10(9) (2010) 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD, Cancer-associated stromal fibroblasts promote pancreatic tumor progression, Cancer Res. 68(3) (2008) 918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L, Mouse models of pancreatic cancer, World J. Gastroenterol. 18(12) (2012) 1286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, Del Rio Hernandez A, Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells, Oncogenesis 6(7) (2017) e352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Holloway S, Davis M, Jaber R, Fleming J, A clinically relevant model of human pancreatic adenocarcinoma identifies patterns of metastasis associated with alterations of the TGF-beta/Smad4 signaling pathway, Int. J. Gastrointest. Cancer 33(1) (2003) 61–9. [DOI] [PubMed] [Google Scholar]
- [29].Kim H, Folks KD, Guo L, Sellers JC, Fineberg NS, Stockard CR, Grizzle WE, Buchsbaum DJ, Morgan DE, George JF, Zinn KR, Early therapy evaluation of combined cetuximab and irinotecan in orthotopic pancreatic tumor xenografts by dynamic contrast-enhanced magnetic resonance imaging, Mol. Imaging 10(3) (2011) 153–67. [PMC free article] [PubMed] [Google Scholar]
- [30].Stellas D, Szabolcs M, Koul S, Li Z, Polyzos A, Anagnostopoulos C, Cournia Z, Tamvakopoulos C, Klinakis A, Efstratiadis A, Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer, J. Natl. Cancer Inst. 106(12) (2014). [DOI] [PubMed] [Google Scholar]
- [31].Banerjee S, Modi S, McGinn O, Zhao X, Dudeja V, Ramakrishnan S, Saluja AK, Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer, Clin. Cancer Res. 22(2) (2016) 415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI, Tuveson DA, Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma, J. Exp. Med. 209(3) (2012) 437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saloman JL, Albers KM, Li D, Hartman DJ, Crawford HC, Muha EA, Rhim AD, Davis BM, Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer, Proc. Natl. Acad. Sci. USA 113(11) (2016) 3078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM, Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy, Cell 159(1) (2014) 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR, Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma, Cancer Cell 21(3) (2012) 418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sinn M, Denkert C, Striefler JK, Pelzer U, Stieler JM, Bahra M, Lohneis P, Dorken B, Oettle H, Riess H, Sinn BV, alpha-Smooth muscle actin expression and desmoplastic stromal reaction in pancreatic cancer: results from the CONKO-001 study, Br. J. Cancer 111(10) (2014) 1917-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J, The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma, Clin. Gastroenterol. Hepatol. 6(10) (2008) 1155–61. [DOI] [PubMed] [Google Scholar]
- [38].Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC, Tumor-associated stromal cells as key contributors to the tumor microenvironment, Breast Cancer Res 18(1) (2016) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT, Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer, Proc. Natl. Acad. Sci. USA 110(50) (2013) 20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Le Calve B, Griveau A, Vindrieux D, Marechal R, Wiel C, Svrcek M, Gout J, Azzi L, Payen L, Cros J, de la Fouchardiere C, Dubus P, Guitton J, Bartholin L, Bachet JB, Bernard D, Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution, Oncotarget 7(22) (2016) 32100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Awasthi N, Zhang C, Schwarz AM, Hinz S, Wang C, Williams NS, Schwarz MA, Schwarz RE, Comparative benefits of Nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer, Carcinogenesis 34(10) (2013) 2361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, Edrich CM, Jung K, Daubriac J, Chen I, Heishi T, Martin JD, Huang Y, Maimon N, Reissfelder C, Weitz J, Boucher Y, Clark JW, Grodzinsky AJ, Duda DG, Jain RK, Fukumura D , Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases, Sci. Transl. Med. 8(360) (2016) 360ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chauhan VP, Boucher Y, Ferrone CR, Roberge S, Martin JD, Stylianopoulos T, Bardeesy N, DePinho RA, Padera TP, Munn LL, Jain RK, Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure, Cancer Cell 26(1) (2014) 14–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Lolkema MP, Jiang P, Kultti A, Thompson CB, Maneval DC, Jodrell DI, Frost GI, Shepard HM, Skepper JN, Tuveson DA, Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer, Gut 62(1) (2013) 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, Bailey P, Chang D, Oien K, Karim S, Au A, Steele C, Carter CR, McKay C, Anderson K, Evans TR, Marais R, Springer C, Biankin A, Erler JT, Sansom OJ, Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy, EMBO Mol. Med. 7(8) (2015) 1063–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Casanovas O, Hicklin DJ, Bergers G, Hanahan D, Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors, Cancer Cell 8(4) (2005) 299–309. [DOI] [PubMed] [Google Scholar]
- [47].Lee C-T, Mace T, Repasky EA, Hypoxia-driven immunosuppression: A new reason to use thermal therapy in the treatment of cancer?, Int. J. Hyperthermia 26(3) (2010) 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Guillaumond F, Leca J, Olivares O, Lavaut M-N, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini O, Iovanna JL, Tomasini R, Vasseur S, Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma, Proc. Natl. Acad. Sci. USA 110(10) (2013) 3919–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hoskin PJ, Sibtain A, Daley FM, Wilson GD, GLUT1 and CAIX as intrinsic markers of hypoxia in bladder cancer: relationship with vascularity and proliferation as predictors of outcome of ARCON, Br. J. Cancer 89(7) (2003) 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rosler A, Vandermeulen GW, Klok HA, Advanced drug delivery devices via self-assembly of amphiphilic block copolymers, Adv. Drug Deliv. Rev. 53(1) (2001) 95–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.