

Abstract
Background
Post-malaria neurological syndrome (PMNS) is a debated entity, defined by neurological complications following a post-malaria symptom-free period and a negative blood smear. Four cases of PMNS are hereby reported and a review the literature performed to clarify the nosological framework of this syndrome.
Methods
A French teaching hospital infectious diseases database was investigated for all PMNS cases occurring between 1999 and 2016 and the PubMed database for cases reported by other institutions after 1997. A case was defined by the de novo appearance of neurological signs following a post-malaria symptom-free period, a negative blood smear, and no bacterial or viral differential diagnoses.
Results
Four patients from the database and 48 from PubMed, including 4 following Plasmodium vivax infection were found matching the definition. In the institution, the estimated PMNS incidence rate was 1.7 per 1000 malaria cases overall. Of the 52 patients (mean age 33 years), 65% were men. Malaria was severe in 85% of cases, showed neurological involvement in 53%, and treated with quinine in 60%, mefloquine in 46%, artemisinin derivatives in 41%, antifolic drugs in 30%, doxycycline in 8% and other types in 8%. The mean symptom-free period was 15 days. PMNS signs were confusion (72%), fever (46%), seizures (35%), cerebellar impairment (28%), psychosis (26%), and motor disorders (13%). Cerebrospinal fluid analyses showed high protein levels in 77% (mean 1.88 g/L) and lymphocytic meningitis in 59.5% (mean 48 WBC/mm3) of cases. Electroencephalograms were pathological in 93% (14/15) of cases, and brain MRIs showed abnormalities in 43% (9/21) of cases with white matter involvement in 100%. Fourteen patients were treated with steroids. The 18 patients with follow-up data showed no sequelae. The mean time to recovery was 17.4 days.
Conclusion
PMNS is a rare entity englobing neurological signs after severe or non-severe malaria. It appears after a symptom-free period. PMNS occurred following treatment of malaria with a wide range of anti-malarials. The disease is self-limiting and associated with good outcome. MRI patterns underline a possible link with acute disseminated encephalomyelitis (ADEM) or auto-immune encephalitis. Plasmodium falciparum and Plasmodium vivax should be added to the list of pathogens causing ADEM.
Background
Falciparum malaria remains a common cause of morbidity and mortality, with an estimated 212 million cases and 429,000 deaths in 2015 [1]. The disease causes neurological impairment during its acute phase and cerebral malaria can provoke neurological sequelae. Additionally, a return of neurological signs after cure has been reported since 1987 [2].
A work by Senanayake et al. [3] published in 1994 may be the first large report on this phenomenon. In it, the authors described 74 Sri Lankan patients, who, during a large epidemic in the 1980s, presented an isolated, self-limiting, delayed cerebellar ataxia (DCA) syndrome 3–41 days after the onset of fever due to falciparum malaria. However, it was difficult to establish a clear relationship with a post-malaria phenomenon as the onset of neurological signs occurred while almost half (34/74) of the patients still had positive blood smears, and 11 of them had not received any anti-malarial treatment.
In 1997, Nguyen et al. [4] provided a first definition of post-malaria neurological syndrome (PMNS) in a report on 22 Vietnamese patients who presented encephalitic signs after a symptom-free period (median time of 96 h) after malaria cure, and additionally had negative blood smears for Plasmodium falciparum and negative etiological investigations. In their study, the use of mefloquine for severe malaria was associated with PMNS (relative risk 7.4). Since then, a number of case reports and series have been published but a clear definition and pathophysiological hypotheses for this syndrome are still lacking. PMNS may be a part of acute disseminated encephalomyelitis (ADEM) or acute post-infectious encephalitis but this too remains controversial. Finally, the medical community knows little about the condition’s underlying pathophysiology, the duration of its symptom-free period, or its outcome and prognosis, and furthermore has little in the way of diagnostic tools or treatment options. Four new cases of PMNS are herein reported and the characteristics of the cases reported since 1997 discussed further with the goal of contributing to a better understanding of this rare entity.
Methods
Case definition of malaria
Malaria was defined as the association of compatible clinical signs with a positive blood smear and/or antigens for a Plasmodium spp. The disease of patients with imported malaria in France were classified as severe or non-severe using the French 2007 classification [5].
Case definition of PMNS
PMNS was defined as the occurrence of de novo neurological signs after a symptom-free period following acute malaria (whatever the Plasmodium species, i.e., P. falciparum Plasmodium vivax, Plasmodium ovale, Plasmodium malariae or Plasmodium knowlesi), associated with a negative blood smear and no retainable differential diagnosis. The symptom-free period was a crucial criterion.
Case origin
All the cases fitting the case definition and seen in the department between 1999 and 2016 were included. PubMed was also investigated for neurological conditions following malaria using the following Mesh terms date-limited to ≥ 1997: “post-malaria neurological syndrome”, “post-malaria and ADEM”, “Plasmodium/falciparum/vivax/ovale/malariae/knowlesi” and “neurological” and “encephalitis” and “ADEM” and “cerebellar”.
In the resulting patient population (hospital patients plus those screened from the literature), the following data was assessed: age, gender, malaria severity, malaria treatment, symptom-free period duration, fever presence, neurological signs (confusion, cerebellar and motor impairment, seizures, psychosis), C-reactive protein (CRP), cerebral spinal fluid (CSF) characteristics, electroencephalography (EEG) signs, brain magnetic resonance imaging (MRI) results, treatment and outcome when available.
Statistical analysis
Rates and means were calculated on the available data. Continuous variables were expressed as medians with minimum–maximum values (min–max) and means with standard deviations (SD). Qualitative variables were expressed as percentages. 95% confidence intervals (95% CI) for sample proportions were calculated using Wilson’s method. Statistical analyses were performed using STATA1 software, version 12 for Windows. Analysis was restricted to the P. falciparum group.
Results
Four cases of PMNS after imported malaria (Table 1)
Table 1.
Main clinical characteristics for cases of post-malaria neurological syndrome following P. falciparum (A), P. vivax (B) and mixed (C) infections
| N | Age | Gender | Cerebral malaria | Severe malaria | Treatment of malaria | Symptom-free period (days) | Fever | Confusion | Seizures | Psychosis | Cerebellar involvement | Motor deficit | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A—PMNS post P. falciparum | |||||||||||||
| Case 1 | 1 | 33 | M | No | Yes | AD/AP | 10 | Yes | Yes | No | Yes | Yes | No |
| Case 2 | 1 | 29 | M | No | No | AP/Q/AD | 13 | Yes | Yes | Yes | Yes | No | Yes |
| Case 3 | 1 | 36 | F | No | No | AP | 15 | Yes | Yes | No | Yes | Yes | No |
| Case 4 | 1 | 43 | F | No | Yes | Q/MF | 15 | No | No | No | No | Yes | No |
| Nguyen [4] | 22 | 29a | 15 M | Yes 13 No 9 |
Yes 21 No 1 |
Q46% MF77% O18% AD59% | 1–60 (median 4) | Yes 9 No 13 |
Yes 15 No 7 |
Yes 8 No 14 |
Yes 6 No 16 |
Yes 1 No 21 |
Yes 0 No 22 |
| O’Brien [6] | 1 | 42 | M | Yes | Yes | Q/D | 45 | No | Yes | No | No | No | No |
| Zambito [7] | 1 | 60 | M | Yes | Yes | Q/D | 11 | No | Yes | No | No | Yes | Yes |
| Mizuno [8] | 1 | 54 | M | Yes | Yes | AD/MF | 21 | Yes | Yes | No | No | No | No |
| Nayak [9] | 1 | 23 | M | Yes | Yes | AD/O | 7 | No | No | No | No | No | Yes |
| Prendki [10] | 1 | 19 | M | Yes | Yes | Q | 46 | Yes | Yes | Yes | Yes | No | No |
| Prendki [10] | 1 | 19 | M | Yes | Yes | Q | 7 | Yes | Yes | Yes | No | No | No |
| Falchook [11] | 1 | 50 | F | No | Yes | Q/D/AP | 11 | No | Yes | No | Yes | Yes | No |
| Matias [12] | 1 | 61 | M | No | Yes | Q/D | 2 | Yes | Yes | No | Yes | Yes | Yes |
| Markley [13] | 1 | 42 | M | Yes | Yes | MF/Q/D/AP | 19 | No | Yes | No | No | Yes | No |
| Forestier [14] | 1 | 50 | M | No | No | AP | 21 | No | Yes | No | No | No | No |
| Rakoto [15] | 1 | 16 | M | Yes | No | Q/AD/O | 13 | No | Yes | Yes | No | Yes | No |
| Pace [16] | 1 | 48 | F | No | No | AD/O | 2 | No | No | No | No | No | Yes |
| Caetano [17] | 1 | 60 | M | No | No | NA | NA | No | Yes | Yes | NA | Yes | No |
| Mohsen [18] | 1 | 30 | F | No | Yes | Q/D | 35 | No | Yes | Yes | No | No | No |
| Schnorf [19] | 1 | 34 | F | Yes | Yes | Q/O | 17 | Yes | No | Yes | No | Yes | No |
| Schnorf [19] | 1 | 61 | M | Yes | Yes | Q/MF | 10 | Yes | No | No | No | Yes | No |
| Agrawal [20] | 1 | 12 | F | Yes | Yes | Q/D | 16 | No | Yes | Yes | No | No | No |
| Rachita [21] | 1 | 4 | F | No | No | Q | 7 | Yes | No | No | No | No | Yes |
| Lawn [22] | 1 | 44 | M | No | Yes | Q/AF | 7 | Yes | Yes | No | No | No | No |
| Lawn [22] | 1 | 22 | F | No | Yes | Q/AF | 3 | Yes | Yes | No | No | Yes | No |
| Total | 46 | 30 M | 24 | 38 | Q28, MF21, AD19, AF14, D7, O2 | 21 | 33 | 16 | 12 | 13 | 6 | ||
| % [95% CI] | 65.2 [50.7–77.3] | 52.2 [38.1–65.9] | 82.6 [69.3–90.9] | Q 60, MF 46, AD 41, AF 30, D 18, O 8 | 45.6 [32.1–59.8] | 71.7 [57.4–82.7] | 34.8 [22.7–49.2] | 26.1 [15.6–40.3] | 28.3 [17.3–42.6] | 13.0 [6.1–25.7] | |||
| Mean (SD) | 33.3 (12.7) | 15.4b (12.0) | |||||||||||
| Median (min–max) | 29 (4–61) | 13b (2–46) | |||||||||||
| B—PMNS post P. vivax | |||||||||||||
| Goyal [23] | 1 | 1.5 | F | Yes | Yes | AD/Q | 7 | Yes | Yes | No | No | No | Yes |
| Sidhu [24] | 1 | 8 | F | Yes | Yes | AD | 7 | No | No | No | No | No | No |
| Kochar [25] | 1 | 55 | M | No | No | CQ | 14 | No | No | No | No | No | Yes |
| Kasundra [26] | 1 | 14 | F | No | NA | NA | 14 | No | No | No | No | Yes | Yes |
| C—PMNS post mixed infections (P. falciparum/P. vivax) | |||||||||||||
| Koibichi [27] | 1 | 24 | M | No | Yes | Q | 26 | Yes | Yes | No | No | No | No |
| Mani [28] | 1 | Adult | F | NA | NA | D/AD | NA | No | No | No | No | No | Yes |
M male, F female, D days, NA not available, Q quinin, AD artemisinin derivatives, MF mefloquine, AF anti-folic, O other, SD standard deviation, [95% CI] 95% confidence interval
aMean on 22 patients
bCalculated on 23 available figured data
Four of 2314 patients treated in the hospital for imported malaria during the study period fit the case definition of PMNS. Therefore, the estimated PMNS incidence rate for the hospital was 1.7 per 1000 malaria cases overall (95% CI 0.7–4.0 per 1000).
Case 1 was a 33-year-old Caucasian male. He was a pilot and flew routes between France, Guinea and the Republic of the Congo. In September 2016 he presented with fever, headaches and vomiting, and thereafter received treatment in Paris for severe malaria (positive thick drop for P. falciparum with 5 parasites/2 μL, positive HRP2 antigen test) with hepatic impairment (SGOT/SGPT 92/105 U/L and hyperbilirubinaemia (93 µmol/L, normal range < 25 µmol/L) but no neurologic involvement or any other severity criteria. The treatment regimen included intravenous artesunate (2.4 mg/kg, 5 doses for 3 days) then atovaquone/proguanil (1000/400 mg per day for 3 days), and the patient improved quickly, both clinically and biologically (blood smear negative for P. falciparum on day 3). On day 7, he presented headaches and fever (38 °C) and on day 8 abdominal pain, nausea and vomiting. The renewed blood smear was negative. On day 10, the patient showed confusion, ataxia, tremor, and dysarthria, and his fever increased to 39 °C. On day 11, he was given ceftriaxone for presumed enteric fever. On day 12, he remained confused and started having visual hallucinations and urine incontinence. CSF analysis showed lymphocytic meningitis (Table 2), MRI was normal and EEG revealed asymmetric (right) frontal slowing. Laboratory results showed no inflammation, a slight hyperbilirubinaemia that diminished over the first days and a weak positive titre of anti-nuclear factor (1/80) with no positivity for anti-DNA. Thereafter, he was treated with cefotaxime and acyclovir from day 12–21 (until a second CSF analysis showed no viral or bacterial infection), and corticosteroids from day 15–30 (methylprednisolone 500 mg/od for 3 days then prednisone 1 mg/kg/od), with clinical improvement on day 19. The patient was discharged with only a slight residual cerebellar ataxia on day 29 and had fully recovered on day 60.
Table 2.
Biological and radiological features of PMNS post P. falciparum, P. vivax or mixed infection
| CSF (WBC/%L) | CSF protein (g/L) | CRP (mg/L) | EEG | MRI | MRI matching ADEM or AIE | |
|---|---|---|---|---|---|---|
| P. falciparum infection | ||||||
| Case 1 | 32/90 | 1.05 | N | Abnormal | N | – |
| Case 2 | 82/87 | 2.41 | N | Abnormal | Limbic and hippocampal hypersignal | ADEM plausible |
| Case 3 | 173/89 | 1.88 | 40 | Abnormal | N | – |
| Case 4 | NA | NA | N | NA | NA | |
| Nguyen [4] (N = 22) | > 5 in 8/lymphocytic predominance | > 0.5 in 13 | NA | NA | NA | |
| O’Brien [6] | NA | NA | NA | NA | WM lesions in CH, brainstem, cerebellum, thalamus and basal ganglia | ADEM plausible |
| Zambito [7] | 20/100 | 0.86 | NA | Abnormal | N | – |
| Mizuno [8] | 10/100 | 0.83 | 27 | Abnormal | N | – |
| Nayak [9] | N/N | 0.66 | NA | NA | NA | |
| Prendki [10] | 76/100 | 0.52 | 163 | Abnormal | N | – |
| Prendki [10] | 26/91 | 1.88 | 9 | Abnormal | N | – |
| Falchook [11] | NA | NA | N | NA | Pons, posterior internal capsule, thalamus, corona radiata, and periventricular hypersignal | ADEM unlikely |
| Matias [12] | N/N | 1.83 | N | Abnormal | Extensive demyelinating lesions (subcortical WM and cerebellum) | ADEM or dysimmune plausible |
| Markley [13] | 20/100 | 0.92 | NA | Abnormal | N | – |
| Forestier [14] | 43/95 | 1.2 | N | Abnormal | N | – |
| Rakoto.[15] | 31/98 | 2 | N | NA | NA | |
| Pace [16] | NA | NA | 8 | NA | Brainstem and spinal cord high signal and swelling | ADEM plausible |
| Caetano [17] | 123/100 | 1.88 | NA | Normal | N | |
| Mohsen [18] | 22/100 | 1.4 | NA | Abnormal | Subcortical unilateral frontal and temporal, and cerebellar hypersignal with gadolinium enhancement | ADEM unlikely but not impossible |
| Schnorf [19] | 10/95 | 0.6 | NA | Abnormal | Peri and supraventricular and cerebellar hypersignal | ADEM plausible |
| Schnorf [19] | 80/87 | 1.8 | NA | Abnormal | N | |
| Agrawal [20] | 5/100 | 1.12 | NA | NA | Asymmetric supraventricular, semi-ovale center, genu of corpus callosum WM hypersignal | NA |
| Rachita [21] | 7/100 | 1.25 | NA | NA | Multifocal asymmetric diffuse WM hypersignal with small mass effect | ADEM |
| Lawn [22] | N/N | 0.89 | N | NA | N | – |
| Lawn [22] | 59/100 | 2.89 | N | Abnormal | N | – |
| Total abnormal | 25/42 | 33/42 | 5/14 | 14/15 | 9/21 | |
| % abnormal [95% CI] | 59.5 [44.5–72.9] | 78.6 [64.1–88.3] | 35.7 [16.3–61.3] | 93.3 [70.2–98.8] | 42.8 [24.5–63.5] | |
| Mean WBC/%L (SD) | 48a/96a (46)/(5.1) | 1.4b (0.6) | 49.4c (64.9) | |||
| Median WBC/%L (min–max) | 31a/100a (5–173)/(87–100) | 1.2b (0.5–2.9) | 27c (8–163) | |||
| P. vivax infection | ||||||
| Goyal [23] | 70/NA | 0.5 | NA | NA | Diffuse periventricular, deep and subcortical WM hypersignal | |
| Sidhu [24] | NA | NA | NA | NA | Subcortical, cortical, left parietal periventricular regions and pons hypersignal | |
| Kochar [25] | NA | NA | NA | NA | NA | |
| Kasundra [26] | 10/100 | 0.65 | NA | NA | T1-weighted isointense and T2 and fluid-attenuated inversion recovery high signal in bilateral cerebellar hemispheres including vermis | |
| Mixed infection | ||||||
| Koibuchi [27] | 30/NA | 0.46 | 52 | NA | Asymmetric spotty mottled cortical and subcortical lesions | |
| Mani [28] | NA | NA | NA | Multifocal confluent areas of demyelination in the corpus callosum and periventricular region, myelitis | ||
Meningitis is defined in the CSF by CSF WBC ≥ 5/mL. CSF Protein ≥ 0.5 g/L is considered abnormal. CRP normal value ≤ 5 mg/L
CSF cerebrospinal fluid, WBC white blood count, %L proportion of lymphocytes, CRP c-reactive protein, WM white matter, NA not available, N normal, ADEM acute disseminated encephalomyelitis, AIE autoimmune encephalitis, LP lumbar puncture, SD standard deviation, [95% CI] 95% confidence interval
aCalculated on abnormal and available figured data, n = 17
bCalculated on abnormal and available figured data, n = 20
cCalculated on abnormal and available figured data, n = 5
Case 2 was a 29-year-old Caucasian male who worked in Ivory Coast. He presented a first episode of acute falciparum malaria without severity criteria in July 2013. That episode was treated with a 3-day course of atovaquone/proguanil (standard treatment). The patient experienced a second symptomatic episode with a positive blood smear (0.18%) 3 weeks later and received a 3-day course of artemether/lumefantrine with good outcome. He then consulted on day 37 for vomiting and a 40 °C fever for which intravenous quinine was initiated despite a negative blood smear. On day 38, he presented convulsions and a severe alteration of consciousness requiring sedation and ventilation. He was evacuated to Paris where his anti-malarial treatment was changed to artesunate despite a still negative blood smear. On day 42, sedation was stopped but the patient presented visual hallucinations and generalized convulsions and consequently had to be re-sedated. The blood test was again negative for malaria but an HRP2 antigen test was positive. T2 and T1 gadolinium-enhanced MRI sequences on day 48 showed hippocampal lesions (Fig. 1), EEG diffuse slowing, and CSF analysis lymphocytic meningitis (Table 2a). Differential diagnoses such as infectious or inflammatory/immunological diseases were ruled out (tests for all of the following were negative or normal: HSV, EBV, VZV, HIV, VHB, VHC, HHV6, adenovirus, dengue fever, Chikungunya, Rift Valley fever virus, West-Nile virus, Borrelia, Coxiella, Brucella, Bartonella, Tropheryma whipplei, gram-negative bacilli, TPHA-VDRL, African trypanosomiasis, cysticercosis, toxocariasis, anti-nuclear factor (ANF), anti-neutrophil cytoplasmic antibodies (ANCA), complement, anti-phospholipid (APL) antibodies, anti-neuronal antibodies including NMDA-receptor in CSF, rheumatoid factor, anti-CCP, angiotensin conversion enzyme and 16S RNA on CSF). The patient improved around day 54 without specific treatment. MRI on day 63 had returned to normal. On day 73, the patient was free of neurological symptoms.
Fig. 1.
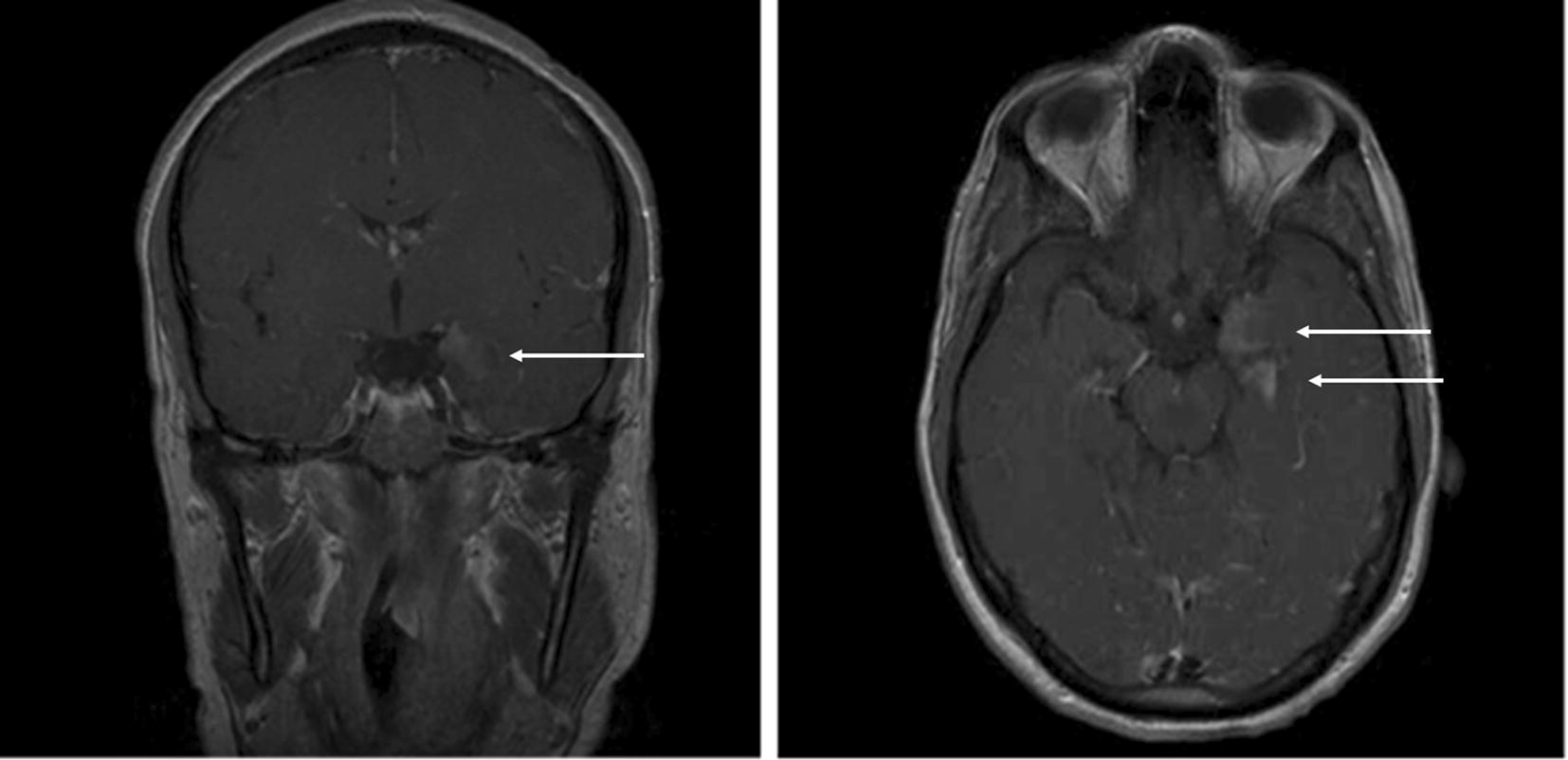
T1 gadolinium-enhanced MRI sequences at day 48 (case 2). Left limbic and hippocampal hypersignals are indicated with white arrows
Case 3 was a 36-year-old Caucasian female who lived in France but had visited friends in Mali in November 2009. On 17 November, she complained of fever, diarrhoea and vomiting and on the 18th returned to Paris. She was diagnosed with non-severe (2% parasitaemia) falciparum malaria on 28 November and treated with a 3-day course of atovaquone/proguanil with good outcome. Thirteen days later (day 15) she became confused, aphasic and anosognosic. CSF analysis showed lymphocytic meningitis (Table 2). EEG showed frontal bilateral slowing and MRI no abnormalities. Infectious investigations were all negative (including HSV, VZV and enterovirus, HIV, Chikungunya, dengue, West Nile and Rift Valley fevers, yellow fever and African trypanosomiasis). Intravenous acyclovir was stopped after 2 days following the negative result for HSV PCR in CSF. On day 19, she was discharged with partial recovery. On day 22, she was re-admitted for confusion, aphasia, ataxia, delirium, and fever (38 °C). MRI remained unremarkable but EEG showed major frontal slowing and spike-and-wave discharges. Treatment with acyclovir and levetiracetam was re-initiated for 3 weeks. The patient’s clinical status gradually improved allowing for her discharge on day 41. She had fully recovered on day 87.
Case 4 was a 43-year-old Caucasian female who presented with severe falciparum malaria (4.5% parasitaemia) in July 1999 after a trip to Ivory Coast. She was initially treated with intravenous quinine (25 mg/kg/day) and on day 5 was given an oral dose of quinine (500 mg tid) after which she developed hypoacusis. On day 6, she was treated with mefloquine (6 × 250 mg for 24 h). She was discharged after recovery but on day 18 she experienced dizziness, limb weakness, gait impairment, nausea, an increase in pre-existing headaches and an episode where she was unable to read. Tremor and ataxia were observed during a resulting physical examination. On day 21, the level of mefloquine in her blood was high (5 µg/L, HPLC method, normal value < 1.5 µg/L). On day 27, her clinical status improved spontaneously and she was discharged without any specific treatment. Follow-up blood examinations showed that the half-life of mefloquine elimination for this patient was 9 days. On day 41, the patient had a normal clinical status and no sequelae.
Literature review (Table 1)
The systematic review of the literature found 48 PMNS patient cases of which 42 involved P. falciparum alone, 4 P. vivax alone, and 2 P. falciparum and P. vivax together. Of these 48 patients, 16 were travellers (15 P. falciparum and 1 P. vivax) who acquired malaria while abroad and 32 were inhabitants of countries endemic for the infection (27 P. falciparum and 5 P. vivax). No cases were described with P. ovale, P. malariae or P. knowlesi.
Matching the results of the local cases and those of the literature
The main clinical findings are summarized in Table 1. Four patients presented PMNS after P. vivax infection alone [23–26] and 2 after mixed malaria infections [27, 28]. Three were children, all after P. vivax infection [23, 24, 26]. One PMNS patient presented bilateral facial palsy that recovered spontaneously [25]. Table 2 summarizes CSF, CRP, EEG and MRI findings. In the P. falciparum group, of the 42 CSF analyses available, 25 (60%) showed lymphocytic meningitis (mean of 48 cells/mm3), and 33 (78%) high protein levels (mean of 1.4 g/L) without low glucose levels. When abnormal, EEG showed diffuse or frontal slowing. Computed tomography was never abnormal when performed (9/9). MRI revealed brain abnormalities in 9 of 21 P. falciparum patients (43%), showing hypersignals, sometimes with gadolinium enhancement, in different localizations (periventricular, sub-cortical, corpus callosum, brainstem, cerebellar), usually with little extension. One child showed clinical and MRI characteristics of severe ADEM [20]. One case presented extensive myelitis. Four and two patients with P. falciparum and P. vivax infection, respectively, showed grey matter hypersignals.
Treatment and outcome
In the P. falciparum group, corticosteroids were used to treat 10 patients, among whom 6 had MRI abnormalities. Six of these 10 patients received intravenous high-dose methylprednisolone (with oral tapering). Of the 4 patients in the P. vivax group, 3 received corticosteroids (missing data for one) and one in the mixed infection group. None of the 22 patients with follow-up data had sequelae. The mean time to recovery in the 21 patients with that datum was 17.4 days (12.3), (min–max, 2–45 days). The characteristics of PMNS are summarized in Table 3.
Table 3.
Summary of PMNS due to Plasmodium falciparum
| Plasmodium falciparum PMNS features (determined on 46 published cases) | |
|---|---|
| Mean age | 33 years |
| Sex ratio (% male) | 66% |
| Previous severe malaria | 85% |
| Previous cerebral malaria | 50% |
| Malaria species | Plasmodium falciparum (also P. vivaxa) |
| Previous treatment for malaria | No effect |
| Traveler or local population | Described in both populations |
| Symptom-free period since malaria (mean) | 15 days |
| Fever | 46% |
| Mental confusion | 72% |
| Seizures | 35% |
| Psychosis | 26% |
| Cerebellar disorders | 28% |
| Motor deficit | 13% |
| MRI (abnormal) | 43% mostly white matter lesions |
| CSF | 52% lymphocytic meningitis, 77% high protein level but normal glucose level |
| EEG (abnormal) | 94% (encephalopathy) |
| Treatment | Corticosteroids advised in severe forms (lack of specific recommendation) |
| Prognosis | Excellent (100% ad integrum recovery) |
aSee text for further details
Discussion
For the present work, the literature’s 48 cases of PMNS published since 1997 were assessed and 4 local imported cases added thereto. From the database of the hospital, an incidence of 1.7 per 1000 was determined, comparable with that of 1.2 per 1000 reported in a previous study [4].
In the studied patient population, PMNS developed mostly in adults (mean age of 33) after severe falciparum malaria (85%), with cerebral malaria in 50% of the cases. It was seen most frequently in people who live in (two-thirds of reported cases), but also in those who travel to, malaria-endemic areas. PMNS was also seen after vivax malaria, notably in children in 50% of the cases [23, 24, 26]. There was a mean symptom-free period of 15 days before the onset of PMNS symptoms. The most common clinical feature was confusion, followed by fever, seizures, psychosis, cerebellar involvement, and motor deficits. More rarely, cranial nerve palsy [25], visual impairment [26, 27], sphincter disorders [16], and headaches were seen. When performed, CSF analyses were pathological in 60% of cases, showing high protein levels (1.4 g/L), and lymphocytic meningitis (96% lymphocytes). White matter abnormalities were always present in abnormal (43% of the cases) MRI studies. However, clinical cerebellar impairment did not appear to be associated with the presence of cerebellar lesions in MRI (only 2 patients with cerebellar hypersignals). MRI studies in post-P. vivax PMNS, appeared to show the same types of lesions (noting however that there were only 3 available [23, 24, 26]). EEGs, when realized, usually showed signs of encephalopathy.
A study from Vietnam had concluded that mefloquine was a risk factor for PMNS after severe malaria (relative risk of 7.4; 95% CI 2.5–22) [4]. However, only 3 of the 19 patients published after that study, and only one of the local patients, had been treated with mefloquine. Also, half of the P. falciparum patients reported had received a range of treatments (including artemisinin combination therapy). Moreover, PMNS occurred after P. vivax infection for which mefloquine had not used. Considered together, these observations cast doubt on an association or causative effect for mefloquine and PMNS.
Cerebellar signs can be seen not only in severe malaria, where they respond to anti-malarial treatment [29–31], but also in PMNS, where the inefficacy of anti-malarials and the absence of parasites argue for a different, perhaps immune, mechanism. Overall, 30% of the studied PMNS cases presented cerebellar involvement and various neurological signs and symptoms (Table 1). Therefore, PMNS should include DCA as they share the same prognosis and outcome [4, 19].
PMNS shares many features with CNS post-infectious diseases such as ADEM and auto-immune encephalitis. ADEM [32–37] is a rare, acute, post-infectious (bacterial or viral) or post-vaccinal syndrome. It is a monophasic disease characterized by multiple inflammatory demyelination lesions. Its signs classically appear 2–30 days after the infectious trigger. The clinical picture may include fever, headaches, encephalopathy, seizures, sensorimotor focal deficits, acute cerebellar ataxia, cranial nerve palsy, myelitis, and optic neuritis. CSF analyses only show lymphocytic pleiocytosis and high protein content. CT scans are pathological in up to 30% of cases. MRI show diffuse, asymmetric signs of white matter demyelination and more rarely grey matter involvement in the cortex, the thalamus, basal ganglia [32, 37–39] and the spinal cord. With corticosteroid treatment, the outcome is usually good and free of sequelae [32]. Multiple pathophysiological hypotheses have been proposed, including immunization against some cerebral antigens after a neurotropic infection, mediated by molecular mimicry and T cell-activated cerebral aggression [35, 40]. Although P. falciparum and P. vivax have not been identified as causative agents for ADEM, the great number of characteristics shared between this latter and PMNS suggest that the relationship is nonetheless highly plausible.
In 2007, a new group of neurological disorders mediated by neuronal antibodies (against ions channels and synapses) appeared in the literature under the term auto-immune encephalitis (AIE) [41]. AIE causes neurological and psychiatric impairment usually in a setting of malignancy or post-viral infection (herpes virus, for example) [42]. Various antibodies have been described, some leading to encephalitis or cerebellar ataxia, i.e., N-methyl-d-aspartate-receptor antibody-NMDAR or P/Q type voltage-gated-calcium-channel antibody. The NMDAR antibody was described in 12 (24.5%) of 49 patients 3 months after HSV encephalitis [43]. No neuronal auto-antibodies have been found to be related with malaria but new auto-antigens are being discovered at a pace of about one per year [35]. In AIE as in PMNS, MRIs are normal in about 50% of cases, improvement is observed after corticosteroids and sequelae are largely absent.
The clinical features of PMNS are compatible with post-infectious encephalitis, either ADEM or AIE. Symptom-free latency and negative extensive screening of possible infectious or systemic causes argue for a post-infectious immunologically mediated cerebral aggression. CSF analyses, inflammation characteristics and EEG are all compatible with post-infectious encephalitis (Table 2). For ADEM, MRI is a key point for diagnosis as it is almost always abnormal, to the point where a normal MRI is a commonly-accepted criteria against it [35, 44]. It is important to note however that an MRI may be normal in ADEM when performed too early in the course of the disease [38, 45]. In AIE, MRI shows no abnormalities in 50% of cases and non-specific abnormalities in the other 50%. In PMNS, less than half of MRIs are pathological. Therefore, cases of PMNS with normal MRIs, could involve AIE.
The mechanisms underlying PMNS are poorly understood. The delayed onset, negativity of blood smear, association with fever, and sometimes elevated CRP may suggest an inflammatory pathway, probably involving cross presentation like in AIE, but no demonstration of that has been made to date. The authors of the only study on inflammatory pathways of malarial post infectious neurological complications reported increased blood and CSF pro-inflammatory cytokines, like TNF, IL-6 and IL-2, in 12 patients with DCA (compared to 8 patients without it) [46]. PMNS has not been investigated in that manner. The approximately 15-day symptom-free period is not without interest, since it corresponds roughly to the 2–3 weeks needed for the production of specific antibodies. The cerebral microvasculature could be the locus of this immunization process, since parasites and pigments are known to be sequestered there due to cytoadherence; this is the case even after P. falciparum clearance and even in non-neurological malaria [47]. The fact that cytoadherence is less significant, or at least less frequent, [48, 49] in P. vivax infection could explain why most PMNS cases follow P. falciparum infections. The first case presented non-specific anti-nuclear factors and the second had neither anti-nuclear factors nor anti-neuronal antibodies. Recently, Sahuguet et al. [50] reported a case of AIE with anti-voltage-gated-potassium-channel antibodies in the setting of PMNS, and in so doing opened a field of pathophysiological possibilities and made autoimmune investigations useful. The cases of PMNS with normal MRIs should accordingly be investigated as AIE. It appears that PMNS is a post-infectious syndrome probably due to various pathophysiological mechanisms (Fig. 2).
Fig. 2.

Nosological framework for post-malaria neurological syndrome. DCA is difficult to classify in PMNS because in most of the published cases, data on parasite clearance was lacking. However, other elements (delay, lack of sensitivity to antimalarials) advocate for at least a relationship between them. Delayed post-infectious cerebellar involvement was described in a number of the confirmed PMNS cases; MRIs were normal for some and ADEM-compatible for others. ADEM: acute diffuse encephalomyelitis. MRI magnetic resonance imaging, DCA diffuse cerebellar ataxia, AIE autoimmune encephalitis, PMNS post-malaria neurological syndrome
The use of steroids in PMNS is another topic worth discussing. According to the follow-up data available, all patients fully recovered in a mean delay of 17.2 days (2–45 days; delay to recovery could not be determined for post-P. vivax patients) and most without steroid treatment. However, steroids were given to the 14 most-severe cases (including 11 with MRI abnormalities). This observation casts a shadow on the ‘self-limiting course’ [13] of PMNS, forwarded by most of the reports in the literature, and raises questions as to whether or not steroids contribute to the cure of severe cases, reduce sequelae or shorten time to recovery. An interventional study would be needed to respond to these questions. In analogy with AIE treatment, corticosteroids do nonetheless appear to be a first-line treatment for PMNS, at least in severe cases and at least for now. The prognosis of PMNS seems to be good and corticosteroid treatment should be discussed for the most severe cases. If corticosteroids are not effective, first the diagnosis of PMNS should be reconsidered, and thereafter intravenous immunoglobulin or plasma exchange discussed.
Conclusion
PMNS is a rare entity englobing a range of neurological disorders following malaria cure. There appears to be no direct role for the parasite but rather a putative immune-mediated aggression of the CNS. Most of its characteristics can fit into the diagnosis of post-infectious encephalitis, ADEM or AIE. The addition of malaria due to P. falciparum and P. vivax parasites to the list of pathogens causing ADEM should be highly considered. Further studies on this rare syndrome should include immune investigations (i.e., auto-antibodies).
Authors’ contributions
YT made the research and wrote the manuscript. SD, EC contributed to the draft manuscript. SJ conceived the research, the design of the study and performed the statistical analysis and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. Thellier for our fruitful discussions and the local epidemiology on imported malaria in our hospital, and K. Erwin for his editorial assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Please contact author for data requests.
Consent for publication
All patients were informed and consent for publication of their story.
Ethics approval and consent to participate
Data collection was approved by the French National Commission for Data Protection and Liberties (CNIL) under the number 2063450 v 0 for retrospective and non-interventional studies without consent.
Funding
None.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. World malaria report 2016. Geneva: World Health Organization. 2016. http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/.
- 2.Senanayake N. Delayed cerebellar ataxia: a new complication of falciparum malaria? Br Med J. 1987;294:1253. doi: 10.1136/bmj.294.6582.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senanayake N, de Silva HJ. Delayed cerebellar ataxia complicating falciparum malaria: a clinical study of 74 patients. J Neurol. 1994;241:456–459. doi: 10.1007/BF00900965. [DOI] [PubMed] [Google Scholar]
- 4.Mai NTH, Day NP, Chuong LV, Waller D, Phu NH, Bethell DB, et al. Post-malaria neurological syndrome. Lancet. 1996;348:917–921. doi: 10.1016/S0140-6736(96)01409-2. [DOI] [PubMed] [Google Scholar]
- 5.Prise en charge et prévention du paludisme d’importation à Plasmodium falciparum: recommandations pour la pratique clinique 2007 (Revisions de la Conference de consensus 1999). Texte long—ScienceDirect. http://www.sciencedirect.com.gate2.inist.fr/science/article/pii/S0399077X07003940?_rdoc=1&_fmt=high&_origin=gateway&_docanchor=&md5=b8429449ccfc9c30159a5f9aeaa92ffb&ccp=y. Accessed 5 June 2017. [DOI] [PubMed]
- 6.O’Brien MD, Jagathesan T. Lesson of the month 1: post-malaria neurological syndromes. Clin Med. 2016;16:292–293. doi: 10.7861/clinmedicine.16-3-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zambito Marsala S, Ferracci F, Cecotti L, Gentile M, Conte F, Candeago RM, et al. Post-malaria neurological syndrome: clinical and laboratory findings in one patient. Neurol Sci. 2006;27:442–444. doi: 10.1007/s10072-006-0728-2. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno Y, Kato Y, Kanagawa S, Kudo K, Hashimoto M, Kunimoto M, et al. A case of postmalaria neurological syndrome in Japan. J Infect Chemother. 2006;12:399–401. doi: 10.1016/S1341-321X(06)70902-3. [DOI] [PubMed] [Google Scholar]
- 9.Nayak R. Post-malaria neurological syndrome: a rare manifestation of common disease. Trop Dr. 2013;43:86–87. doi: 10.1177/0049475513486643. [DOI] [PubMed] [Google Scholar]
- 10.Prendki V, Elzière C, Durand R, Hamdi A, Cohen Y, Onnen I, et al. Post-malaria neurological syndrome—two cases in patients of African origin. Am J Trop Med Hyg. 2008;78:699–701. doi: 10.4269/ajtmh.2008.78.699. [DOI] [PubMed] [Google Scholar]
- 11.Falchook GS, Malone CM, Upton S, Shandera WX. Postmalaria neurological syndrome after treatment of Plasmodium falciparum malaria in the United States. Clin Infect Dis. 2003;37:e22–e24. doi: 10.1086/375269. [DOI] [PubMed] [Google Scholar]
- 12.Matias G, Canas N, Antunes I, Vale J. Post-malaria neurologic syndrome. Acta Médica Port. 2008;21:387–390. [PubMed] [Google Scholar]
- 13.Markley JD, Edmond MB. Post-malaria neurological syndrome: a case report and review of the literature. J Travel Med. 2009;16:424–430. doi: 10.1111/j.1708-8305.2009.00349.x. [DOI] [PubMed] [Google Scholar]
- 14.Forestier E, Labe A, Raffenot D, Lecomte C, Rogeaux O. Post-malaria neurological syndrome complicating a relapse of Plasmodium falciparum malaria after atovaquone-proguanil treatment. Med Mal Infect. 2011;41:41–43. doi: 10.1016/j.medmal.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Rakotoarivelo RA, Razafimahefa SH, Andrianasolo R, Fandresena FH, Razanamparany MMO, Randria MJD, et al. Post-malaria neurological syndrome complicating a Plasmodium falciparum malaria in Madagascar. Bull Soc Pathol Exot. 1990;2012(105):199–201. doi: 10.1007/s13149-012-0208-7. [DOI] [PubMed] [Google Scholar]
- 16.Pace AA, Edwards S, Weatherby S. A new clinical variant of the post-malaria neurological syndrome. J Neurol Sci. 2013;334:183–185. doi: 10.1016/j.jns.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Caetano A, Mendonça MD, Ferreira NR, Alves L. Post-malaria neurological syndrome or viral encephalitis? BMJ Case Rep. 2016 doi: 10.1136/bcr-2015-213591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohsen AH, McKendrick MW, Schmid ML, Green ST, Hadjivassiliou M, Romanowski C. Postmalaria neurological syndrome: a case of acute disseminated encephalomyelitis? J Neurol Neurosurg Psychiatry. 2000;68:388–389. doi: 10.1136/jnnp.68.3.388a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnorf H, Diserens K, Schnyder H, Chofflon M, Loutan L, Chaves V, et al. Corticosteroid-responsive postmalaria encephalopathy characterized by motor aphasia, myoclonus, and postural tremor. Arch Neurol. 1998;55:417–420. doi: 10.1001/archneur.55.3.417. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal A, Goyal S. Acute demyelinating encephalomyelitis in a child following malaria. Indian Pediatr. 2012;49:922–923. doi: 10.1007/s13312-012-0198-y. [DOI] [PubMed] [Google Scholar]
- 21.Rachita S, Satyasundar M, Mrutunjaya D, Birakishore R. Acute disseminated encephalomyelitis (ADEM)—a rare complication of falciparum malaria. Indian J Pediatr. 2013;80:499–501. doi: 10.1007/s12098-012-0814-9. [DOI] [PubMed] [Google Scholar]
- 22.Lawn SD, Flanagan KL, Wright SG, Doherty TF, Godfrey-Faussett P. Postmalaria neurological syndrome: two cases from the Gambia. Clin Infect Dis. 2003;36:e29–e31. doi: 10.1086/344774. [DOI] [PubMed] [Google Scholar]
- 23.Goyal JP, Shah VB, Parmar S. Acute disseminated encephalomyelitis after treatment of Plasmodium vivax malaria. J Vector Borne Dis. 2012;49:119–121. [PubMed] [Google Scholar]
- 24.Sidhu J, Maheshwari A, Gupta R, Devgan V. Acute disseminated encephalomyelitis after Plasmodium vivax infection: case report and review of literature. Pediatr Rep. 2015;7:5859. doi: 10.4081/pr.2015.5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kochar DK, Sirohi P, Kochar SK, Bindal D, Kochar A, Jhajharia A, et al. Post-malaria neurological syndrome–a case of bilateral facial palsy after Plasmodium vivax malaria. J Vector Borne Dis. 2007;44:227–229. [PubMed] [Google Scholar]
- 26.Kasundra GM, Bhargava AN, Bhushan B, Shubhakaran K, Sood I. Post-Plasmodium vivax malaria cerebellar ataxia and optic neuritis: a new form of delayed cerebellar ataxia or cerebellar variant of acute disseminated encephalomyelitis? J Pediatr Neurosci. 2015;10:58. doi: 10.4103/1817-1745.154354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koibuchi T, Nakamura T, Miura T, Endo T, Nakamura H, Takahashi T, et al. Acute disseminated encephalomyelitis following Plasmodium vivax malaria. J Infect Chemother. 2003;9:254–256. doi: 10.1007/s10156-003-0244-8. [DOI] [PubMed] [Google Scholar]
- 28.Mani S, Mondal SS, Guha G, Gangopadhyay S, Pani A, Das Baksi S, et al. Acute disseminated encephalomyelitis after mixed malaria infection (Plasmodium falciparum and Plasmodium vivax) with MRI closely simulating multiple sclerosis. Neurologist. 2011;17:276–278. doi: 10.1097/NRL.0b013e3182173668. [DOI] [PubMed] [Google Scholar]
- 29.Sakaria A, Mahajan S, Desai R, Shah K. Delayed cerebellar ataxia: a rare self limiting complication of Plasmodium falciparum malaria. Adv Biomed Res. 2013;2:27. doi: 10.4103/2277-9175.107997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhandari K, Bhandari P. Cerebellar syndrome in malaria. Indian Pediatr. 1989;26:1037–1038. [PubMed] [Google Scholar]
- 31.Chitkara AJ, Anand NK, Saini L. Cerebellar syndrome in malaria. Indian Pediatr. 1984;21:908–910. [PubMed] [Google Scholar]
- 32.Sonneville R, Klein I, de Broucker T, Wolff M. Post-infectious encephalitis in adults: diagnosis and management. J Infect. 2009;58:321–328. doi: 10.1016/j.jinf.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bester M, Petracca M, Inglese M. Neuroimaging of multiple sclerosis, acute disseminated encephalomyelitis, and other demyelinating diseases. Semin Roentgenol. 2014;49:76–85. doi: 10.1053/j.ro.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Pohl D, Alper G, Van Haren K, Kornberg AJ, Lucchinetti CF, Tenembaum S, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology. 2016;87:S38–S45. doi: 10.1212/WNL.0000000000002825. [DOI] [PubMed] [Google Scholar]
- 35.Menge T, Hemmer B, Nessler S, Wiendl H, Neuhaus O, Hartung H-P, et al. Acute disseminated encephalomyelitis: an update. Arch Neurol. 2005;62:1673–1680. doi: 10.1001/archneur.62.11.1673. [DOI] [PubMed] [Google Scholar]
- 36.Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016;15:967–981. doi: 10.1016/S1474-4422(16)30043-6. [DOI] [PubMed] [Google Scholar]
- 37.Steiner I, Kennedy PGE. Acute disseminated encephalomyelitis: current knowledge and open questions. J Neurovirol. 2015;21:473–479. doi: 10.1007/s13365-015-0353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tenembaum S, Chitnis T, Ness J, Hahn JS, International Pediatric MS Study Group Acute disseminated encephalomyelitis. Neurology. 2007;68:S23–S36. doi: 10.1212/01.wnl.0000259404.51352.7f. [DOI] [PubMed] [Google Scholar]
- 39.Tenembaum SN. Acute disseminated encephalomyelitis. Handb Clin Neurol. 2013;112:1253–1262. doi: 10.1016/B978-0-444-52910-7.00048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esposito S, Di Pietro GM, Madini B, Mastrolia MV, Rigante D. A spectrum of inflammation and demyelination in acute disseminated encephalomyelitis (ADEM) of children. Autoimmun Rev. 2015;14:923–929. doi: 10.1016/j.autrev.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: the 2016 Cotzias Lecture. Neurology. 2016;87:2471–2482. doi: 10.1212/WNL.0000000000003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mailles A, Stahl J-P, Bloch KC. Update and new insights in encephalitis. Clin Microbiol Infect. 2017;23:607–613. doi: 10.1016/j.cmi.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Westman G, Studahl M, Ahlm C, Eriksson BM, Persson B, Rönnelid J, et al. N-methyl-d-aspartate receptor autoimmunity affects cognitive performance in herpes simplex encephalitis. Clin Microbiol Infect. 2016;22:934–940. doi: 10.1016/j.cmi.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 44.Sonneville R, Demeret S, Klein I, Bouadma L, Mourvillier B, Audibert J, et al. Acute disseminated encephalomyelitisin the intensive care unit: clinical features and outcome of 20 adults. Intensive Care Med. 2008;34:528–532. doi: 10.1007/s00134-007-0926-2. [DOI] [PubMed] [Google Scholar]
- 45.Mihai C, Jubelt B. Post-infectious encephalomyelitis. Curr Neurol Neurosci Rep. 2005;5:440–445. doi: 10.1007/s11910-005-0031-2. [DOI] [PubMed] [Google Scholar]
- 46.de Silva HJ, Hoang P, Dalton H, de Silva NR, Jewell DP, Peiris JB. Immune activation during cerebellar dysfunction following Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg. 1992;86:129–131. doi: 10.1016/0035-9203(92)90536-L. [DOI] [PubMed] [Google Scholar]
- 47.Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, et al. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999;155:395–410. doi: 10.1016/S0002-9440(10)65136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anstey NM, Douglas NM, Poespoprodjo JR, Price RN. Plasmodium vivax: clinical spectrum, risk factors and pathogenesis. Adv Parasitol. 2012;80:151–201. doi: 10.1016/B978-0-12-397900-1.00003-7. [DOI] [PubMed] [Google Scholar]
- 49.Carvalho BO, Lopes SCP, Nogueira PA, Orlandi PP, Bargieri DY, Blanco YC, et al. On the cytoadhesion of Plasmodium vivax—infected erythrocytes. J Infect Dis. 2010;202:638–647. doi: 10.1086/654815. [DOI] [PubMed] [Google Scholar]
- 50.Sahuguet J, Poulet A, Bou Ali H, Parola P, Kaphan E. Postmalaria neurologic syndrome—autoimmune encephalitis with anti-voltage-gated potassium-channel antibodies. Ann Intern Med. 2017;167:70. doi: 10.7326/L16-0651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Please contact author for data requests.