

Abstract
Tuberculosis now ranks as the leading cause of death in the world due to a single infectious agent. Current standard of care treatment can achieve very high cure rates for drug-sensitive disease but requires a 6-month duration of chemotherapy. Drug-resistant disease requires significantly longer treatment durations with drugs associated with a higher risk of adverse events. Thus, there is a pressing need for a drug regimen that is safer, shorter in duration and superior to current front-line chemotherapy in terms of efficacy. The TB drug pipeline contains several candidates that address one or more of the required attributes of chemotherapeutic regimens that may redefine the standard of care of this disease. Several new drugs have been reported and novel targets have been identified allowing regimens containing new compounds to trickle into clinical studies. Furthermore, a recent paradigm-shift in understanding the pharmacokinetics of antitubercular drugs is revolutionizing the way we select compounds for clinical progression.
Introduction
Mycobacterium tuberculosis (Mtb) is arguably the most successful pathogen on earth, infecting a third of the world’s population and killing more than a million people each year [1]. The drugs that are used for front-line chemotherapy of drug sensitive disease were developed more than half a century ago with the clinical studies that defined their optimal combination and duration largely completed in the 1970s [2]. While the standard of care is safe and well tolerated, the long duration of chemotherapy even for drug-sensitive disease is the major driver of patient non-adherence which in turn contributes to the emergence of drug resistance. The difficulty in developing clinically efficacious therapeutics that act more quickly is a result of multiple factors dominated by the complex biology of the pathogen and the extraordinary pathology of the disease. Herein, we discuss recent advances in all stages of TB drug development, starting from hit discovery and target validation to late-stage clinical studies. We then review advances in the pharmacokinetics of drugs in TB granulomas and how they relate to clinically observed treatment failures and successes.
Compounds in the Discovery Phase of the Pipeline
Out of the hundreds of compounds used clinically to treat bacterial infections, only 18 are used to treat TB – the majority of which target macromolecular synthesis. Not only is there a need for new drugs, but also an indisputable lack of novel targets. Recently, however, several molecules with novel targets have been identified (Table 1). Several of these hits (broadly defined as a compound with a desirable biological activity within the mid-to low-micromolar range, a clogP < 4, a molecular weight less than 400, and whose activity has been confirmed upon retesting) has undergone chemical optimization to turn them into lead molecules (which are more potent analogs that possess desirable pharmacokinetic properties that would allow their efficacy to be tested in an in vivo model including high aqueous solubility, intermediate microsomal stability, and no Hep G2 hepatotoxicity at 50X of the IC50). These compounds can be grouped into four large categories based on the pathways in which the enzyme they inhibit lie on, and the downstream effect, their inhibition has on the physiology of the bacterium.
Table 1.
Lead compounds with known targets against M. tuberculosis.
| Structure | Chemical Class | Target | Mode of Inhibition | Method of Identification | Activity Profilea |
|---|---|---|---|---|---|
| Inhibitors of enzymes in classically targeted pathways | |||||
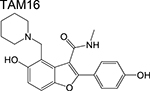 |
Benzofuran [3] | Polyketide synthase 13 (Pks13) | Blocks active site | Whole-cell based high- throughput screen against aerobically grown Mtb H37Rv | IC50 = 0.19 μM MIC = 0.09 μM In vivo active (mouse) |
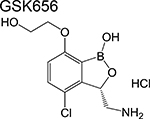 |
Benzoxaboroles [4] | Leucyl-tRNA synthetase (LeuRS) | Forms a covalent adduct with the cis diol of A76 | Structure- guided design of analogs | IC50 = 0.20 μM MIC = 0.08 μM In vivo active (mouse) |
| Inhibitors of the carbon assimilation pathways V | |||||
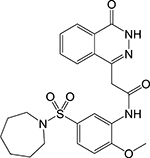 |
Benzopyridazino ne [7] | Fumarate hydratase (Fum) | Binds to an allosteric regulatory site blocking substrate binding | Target based high- throughput screen coupled to diaphorase reduction of resazurin | IC50 = 2.50 μM MIC = 65% inhibition at 250 μM |
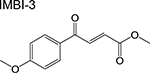 |
Diketoester [8] | Isocitrate lyase (Icl1) | Binds to the catalytic active site | Target- based high- throughput screen monitored via reaction of enzymatic product with phenylhydra zine | IC50 = 31 μM MIC = 0.25–1 μg/mL |
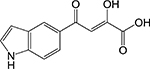 |
Indole diketo acids [9] | Malate synthase (GlcB) | Binds to the catalytic active site | Fragment-based screening coupled with structure-guided design | IC50 = 0.02 μM |
 |
Triazolopyrimidi none [12] Thiadiazole [12] | Flavin- dependent hydroxylas e (HsaAB) | Intramacrop hage screen coupled with counterscreening in cholesterol containing media. | IC50 = 11 μM IC50 = 5 μM | |
| Inhibitors of de novo biosynthesis of macromolecular building blocks | |||||
 |
Azetidine [17] | Tryptophan synthase (TrpAB) | Binds to an allosteric site and stabilizes the closed, active state of the β- subunit | Whole-cell based high- throughput screen | α IC50 = 0.071 μM β IC50 = 0.023 μM MIC = 3 μM In vivo active (zebrafish) |
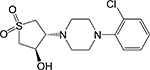 |
Sulfolane [16] | Tryptophan synthase (TrpAB) | Binds to an allosteric site (subunit interface) and may prevent diffusion of indole ring | Whole-cell based high- throughput screen | MIC = 0.76 μM In vivo active (mouse) |
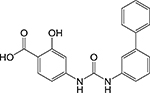 |
Diphenylurea [18] | Cysteine synthase (CysM) | Binds to the active site loop | Target- based high- throughput screen of active site binders | Kd = 4.5 μM MIC = 2.2 μM |
 |
Chromone [19] Diarylurea [19] | Ornithine acetyltrans ferase (ArgJ) | Binds to a shallow allosteric siteb | Target- based medium- throughput in silico screen of FDA- approved drugs followed by in vitro validation | Ki = 139 μM MIC = 5.2 μg/mL Ki = 244 μM MIC = 10 μg/mL |
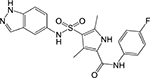 |
Indazolesulfona mide [21] | Inosine monophos phate dehydroge nase (IMPDH, GuaB2) | Binds to the NAD binding pocket and makes extensive contact with the substrate IMP | Whole-cell based high- throughput screen | IC50 = 0.38 μM MIC = 0.09 μM |
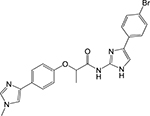 |
Phenylimidazole [22] | Inosine monophos phate dehydroge nase (IMPDH, GuaB2) | Binds to the NAD binding pocket and makes extensive contact with the substrate IMP | Fragment- based screening coupled with structure- guided design of analogs | IC50 = 0.52 μM MIC90 ≥ 50 μM |
| Inhibitors of energy production | |||||
DG70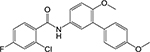
|
Biphenylbenzam ide [23] | Demethylm enaquinon e methyltrassferase (MenG) | Binds to the SAM binding site or substrate binding site* | Whole-cell based pathway- specific screen of compounds known to inhibit Mtb growth | MIC = 4.8 μg/mL |
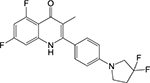 |
Quinolone [24] | NADH:men aquinone oxidpreduc tase (Ndh) | Target-based screen using ligand chemoinfor matic principles; confirmed by wholecell based screen | MIC50 = 0.52 μM (replicating Mtb) MIC50 = 0.076 μM (Wayne model) | |
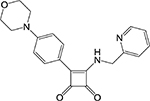 |
Squaramide [25] | ATP synthase (F0) | Binds at the interface of two subunit-c chains and subunit-a likely preventing the rotation of the F0 particle* | Membrane- based biochemical assay measuring oxidative phorphorylat ion ATP output | IC50 = 0.03 μM MIC = 0.50 μM In vivo active (mouse) |
indicated IC50 values refer to inhibitory activity against purified enzymes, and MICs are inhibitory concentrations against whole cells.
enzyme-inhibitor complex was not crystallized but rather, the binding site and binding interactions were deduced based on computational methods.
More compounds in pre-clinical development can be found at www.newtbdrugs.org
First, compounds that inhibit distinct enzymes in pathways engaged by current antitubercular drugs in macromolecular biosynthesis. The clinical utility of isoniazid demonstrates the vulnerability of mycolic acid biosynthesis in Mtb at least under certain in vivo environments, and as such, drugs that target enzymes in this pathway have been sought after. Similarly, compounds that inhibit protein synthesis at a level independent of the ribosome are equally interesting because they offer a means to target strains harboring ribosome mutations. Two prominent examples are the benzofuran TAM16 which inhibits the polyketide synthase Pks13 [3], an enzyme involved in mycolic acid synthesis, and the benzoxaboroles which inhibit the leucyl-tRNA synthetase (LeuRS) [4,5]. Both compounds were shown to have activity against Mtb in vitro and in mouse models of infection and offer orthogonal strategies to pathways considered to be old favorites by medicinal chemists.
Second, inhibitors of the TCA cycle and the glyoxylate shunt which is a bypass pathway activated in the absence of carbohydrates allowing bacteria to utilize fatty acids as carbon source [6]. A target-based screen identified a benzopyridazinone that selectively inhibits mycobacterial fumarate hydratase by binding to a unique allosteric site [7]. While promising, this compound had modest potency against Mtb H37Rv likely arising from its low cell permeability. The diketoester IMBI-3 which inhibits isocitrate lyase [8] and the indole diketoacids inhibiting malate synthase [9] were identified by high-throughput screening and structure-guided design. Although only indirect evidence was presented, inhibitors of the central carbon metabolism are likely also active against non-replicating Mtb since carbon assimilation pathways remain active during persistence. While the firstgeneration malate synthase inhibitors demonstrated in vivo efficacy [10], care must be taken when designing competitive inhibitors of the enzymes in the central carbon metabolism due to the similarity between the bacterial and human homologs. In addition to fatty acids, Mtb can also utilize cholesterol as carbon source in the intracellular environment, hence, compounds that inhibit these catabolic processes can aggravate the nutritional limitation imposed by macrophages [11].
Two compounds – a triazolopyrimidinone and a thiadiazole – identified through an intramacrophage high-throughput screen coupled with counter-screening in cholesterolcontaining media were found to inhibit cholesterol catabolism. Measurements of liberated 14CO2 coupled with in vitro biochemical assays pinpointed the target to be the flavindependent hydroxylase HsaAB, an enzyme involved in the degradation of the A/B rings of cholesterol [12]. Intramacrophage screens, which were originally developed to identify inhibitors of Mtb growth in macrophages [13], are particularly useful to identify inhibitors of host processes that Mtb exploits for its survival. It should be noted that such screens have since been extended to an in vitro granuloma model to capture elements of the granuloma biogenesis and thereby identify modulators of these processes [14]. Herein, partially purified protein derivative (PPD) from Mtb is used to coat sepharose beads prior to coincubation with peripheral blood mononuclear cells (PBMC). Alternatively, PBMCs can be co-cultured with exponentially growing BCG at a very low multiplicity of infection (MOI, 1 BCG to 10 PBMC). Cells will naturally be recruited around the beads or around BCG cells and eventually form a granulomatous lesion that recapitulates the in vivo pathology including cellular differentiation into giant cells and epithelioid cells and recruitment of lymphocytes [15]. Modelling the granuloma in vitro presents strategies to study the interactions of bacteria with host cells and how it relates to drug distribution (vide infra) and ultimately clinical efficacy. However, because granulomas are soft materials formed by an assortment of immune cells, robust reproducibility between granulomas grown in different batches could be an issue.
Third, inhibitors of the de novo biosynthesis of macromolecular building blocks. Allosteric inhibitors of tryptophan synthase (TrpAB) [16,17], cysteine synthase (CysM) [18] and ornithine acetyltransferase (ArgJ) [19] were reported, showing rescue of bacterial growth when tryptophan, cysteine, or arginine were exogenously added to the culture medium. TrpAB inhibitors demonstrated efficacy in vivo [17], likely exacerbating the tryptophan starvation imposed by CD-4 T cell-mediated immune response [20]. Concurrently, the in vivo efficacy of the ArgJ inhibitor is likely a combined effect of arginine reduction in Mtb and downregulation of the 5-lipoxygenase pathway in macrophages which further reduces bacterial survival [19]. Targeting inosine monophosphate dehydrogenase (IMPDH), the rate-limiting step in guanosine biosynthesis, by an indazole sulfonamide [21] or a phenyl imidazole [22], on the other hand, proved to be effective in vitro but had no in vivo efficacy which was ascribed to the high levels of guanine in lung tissue which can overcome enzyme inhibition. The discovery and design of inhibitors belonging to the second and third classes mentioned above should therefore be intimately linked with quantitative measurements of bioavailable metabolites and building blocks from the host to be able to predict in vivo and clinical efficacy.
Fourth, inhibitors of energy production. The discovery of bedaquiline (an ATP synthase inhibitor) put forth the essentiality of ATP synthesis and boosted interest in the search for inhibitors of oxidative phosphorylation. The latest addition to the portfolio of energy production inhibitors include a biphenyl benzamide inhibitor of demethylmenaquinone methyltransferase (MenG) [23], the terminal enzyme of menaquinone (MK) biosynthesis; a quinolone scaffold that inhibits the NADH:menaquinone oxidoreductase (Ndh) [24]; and a squaramide that inhibits ATP synthase [25]. Although all three scaffolds were discovered from screening efforts biased towards a specific respiratory enzyme, only two had a validated target. The biphenyl benzamides were demonstrated to engage MenG and the squaramide resistant-conferring mutations mapped to the a and c subunits of ATP synthase.
All these recent studies further shift the paradigm of conventional drug design that have, until recently, banked on targets required only during bacterial replication. The expansion of target space will certainly enable the design of sterilizing drug regimens and hopefully curtail emergence of resistance.
Drugs and Drug Regimens under Clinical Development
In contrast to the compounds discussed above which are in the pre-clinical stage, several other candidates are in various phases of clinical development headlined by the recent approval of bedaquiline and delamanid (a nitroimidazole that poisons cells by liberating reactive nitrogen species and blocks cell wall synthesis). As one might suspect, drugs in the earlier stages of the clinical pipeline exhibit greater target diversity relative to those in later stages – a reflection of the recently renewed interest in TB drug development. Across the clinical pipeline, however, inhibition of cell wall synthesis and protein translation is well represented. Therefore, compounds in the various stages of the drug development pipeline with an orthogonal mechanism of action would avoid cross-resistance with drugs currently in clinical use or under clinical evaluation. Methods to filter out inhibitors of common protein targets exist [26,27] and their implementation early in the drug discovery phase would avoid redundancy in target identification and, more importantly, allow development of treatment regimens that can ideally address both drug-sensitive as well as drug-resistant disease.
An investigational new drug can enter the clinical development phase following target identification, lead optimization and demonstration of desirable pharmacokinetic (PK) parameters in various animal models. Ongoing clinical trials for new chemotherapeutic agents or regimens are described in Table 2. While often skipped, Phase 0 (pre-phase 1) assesses the safety of a new drug in fewer than 20 healthy volunteers, with the aim of recapitulating pharmacological profiles observed in various animal models during the preclinical stage. Some notable pre-phase 1 compounds include the bedaquiline analogue TBAJ-587 [28,29] and the semi-synthetic spectinamide 1810 which inhibits the ribosome [30]. TBAJ-587 maintains the bactericidal activity of bedaquiline along with decreased off-target effects although lipophilicity remains an issue for this series, as SAR studies showed that analogs with a lower clogP were only equally as potent as bedaquiline [29]. Spectinamide 1810 has been optimized to maintain activity against drug-resistant Mtb while simultaneously evading intrinsic efflux by Mtb [30] and host-metabolism [31], while effective in vivo, spectinamide efficacy is limited by poor gut permability.
Table 2.
Drugs under Clinical Development.
| Drug | Chemical Class | Phase | Mechanism of Action |
|---|---|---|---|
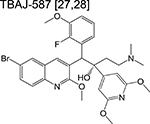 |
diarylquinoline | Pre-phase 1 | Inhibits ATP synthase and inhibits respiration |
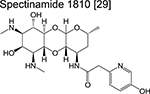 |
spectinamide | Pre-phase 1 | Binds to the ribosome and inhibits protein synthesis |
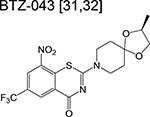 |
benzothiazinone | 1 | Forms a covalent adduct with DprE1 and inhibits arabinogalactan synthesis |
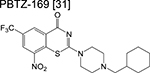 |
benzothiazinone | 1 | Forms a covalent adduct with DprE1 and inhibits arabinogalactan synthesis |
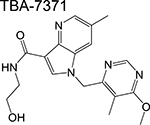 |
azaindole | 1 | Binds to DprE1 and inhibits arabinogalactan synthesis |
| OPC-167832* | dihydrocarbostyril | 1 | Binds to DprE1 and inhibits |
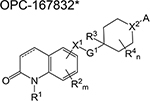 |
arabinogalactan synthesis | ||
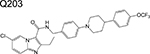 |
imidazopyridine | 1/2 | Binds to the QcrB subunit of cytochrome bc1 and inhibits respiration |
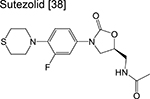 |
oxazolidinone | 1/2 | Binds to the ribosome and inhibits protein synthesis |
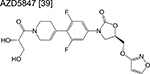 |
oxazolidinone | 2 | Binds to the ribosome and inhibits protein synthesis |
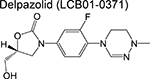 |
oxazolidinone | 2 | Binds to the ribosome and inhibits protein synthesis |
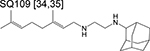 |
ethylenediamine | 2 | Binds to MmpL3 and inhibits cell wall synthesis |
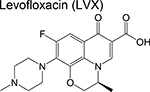 |
fluoroquinolone | 2 | Binds to DNA gyrase and inhibits DNA replication |
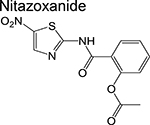 |
nitrothiazole | 2 | Disrupts the membrane potential and pH homeostasis |
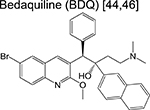 |
diarylquinoline | 3 | Binds to ATP synthase and inhibits respiration |
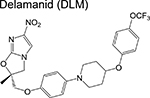 |
nitroimidazole | 3 | Blocks synthesis of mycolic acids and forms NO |
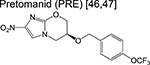 |
nitroimidazole | 3 | Blocks synthesis of mycolic acids and forms NO. |
 |
riminophenazine | 3 | Reduced by NADH dehydrogenase II and subsequently forms reactive oxygen species |
The general structure of OPC-167832 shown here was taken from the US patent No. US2017/0253576 A1.
More compounds in clinical development can be found at www.newtbdrugs.org
Phase 1 trials aim to determine safety and tolerability in humans, alongside studying the pharmacokinetics and pharmacodynamics of new drugs. Because healthy volunteers are usually monitored for a defined period, the optimum dose and formulation can be determined – information that will be used for subsequent phases. Drugs targeting the decaprenylphosphoryl-β-D-ribose-2’-epimerase (DprE1) – an enzyme necessary for the synthesis of D-arabinofuranose – dominate phase 1 of the clinical pipeline. The benzothiazinones BTZ-043 and PBTZ-169 (macozinone) inhibit DprE1 via formation of a covalent adduct with an essential cysteine residue [32,33]. The risk of idiosyncratic drugrelated toxicity due to formation of chemically reactive drug metabolites may be a potential risk factor in development of these benzothiazinones although complete target inactivation could translate to lower doses and thus mitigate clinical toxicities. A trial on tolerability and pharmacokinetics of PBTZ-169 via single and multiple oral administration with an escalating dose was recently concluded. The large discrepancies in plasma drug concentrations of the benzothiazinones in repeated pharmacokinetic studies were found to be due to an in vivo dearomatization (via the enzymatic reduction of the nitrobenzene moiety) resulting in the formation of an air-sensitive Meisenheimer complex [34].
Therefore, results of the phase 1 trial on PBTZ-169, should it be consistent with the reported in vivo reduction, will be essential in determining optimum doses in subsequent phases. Other DprE1 inhibitors in phase 1 development include the azaindole TBA-7371 and the dihydrocarbostyril OPC-167832, with only the former having an active ongoing trial. TBA-7371 inhibits DprE1 non-covalently and may overcome potential toxicities or immune-mediated hypersensitivities of the covalent DprE1 inhibitors.
Other notable drugs in phase 1 development include the QcrB (subunit of cytochrome bc1) inhibitor, Q203 [35]; and the LeuRS inhibitor GSK656 [4]. The high cLogP of the former would be predicted to pose considerable challenges in designing a formulation that could be widely administered to the target population of tuberculosis patients. Both trials are randomized, placebo-controlled, single and multiple ascending dose studies.
In contrast to phase 1 of the pipeline, there is significant target diversity and a good mix of new experimental drugs and FDA-approved drugs for other indications under phase 2 clinical development (Table 2). Phase 2 of clinical trials involve testing the efficacy of a new drug in disease patients and determining whether it causes side effects in these cohorts. Most drug efficacy in this phase is evaluated through an Early Bactericidal Activity (EBA) study, wherein bacterial burden in patient sputum is measured before the start of treatment and monitored every 2 days for the first two weeks of treatment while on monotherapy with the drug of interest [36]. Ethical concerns about the risk of development of acquired drug resistance remain a concern despite limited evidence for drug resistance emerging in this short time frame. A new drug is considered to have an EBA if there is a significant drop in the bacterial count/mL of sputum/day as measured by standard CFU enumeration. While commonly used in early clinical monitoring of chemotherapeutic efficacy, EBA itself, has no correlation in clinical experience in achieving durable cure [37].
Among the novel compounds in phase 2 is the ethylenediamine SQ109, an inhibitor of MmpL3 function – a transporter of trehalose monomycolate [38]. SQ109 was not active alone in smear-positive pulmonary TB patients and neither did it increase the efficacy of 10 mg/kg of rifampicin (even at doses as high as 300 mg) in an EBA study [39]. A more recent trial of SQ109 in various combinations with high-dose rifampicin (R), moxifloxacin (M), isoniazid (H) and pyrazinamide (Z) (compared to the standard TB regimen: HRZE, E = ethambutol) also did not observe significant potentiation by SQ109 as time to culture conversion on solid media were similar in all treatment arms of the trial [40]. SQ109 exhibited synergistic interactions with rifampicin and isoniazid in a mouse model of chronic TB [41], therefore, the factors that prevent this synergy in human patients should be studied prior to advancing it to later stages of the pipeline. SQ109 does not directly bind to MmpL3 [42] and is expected to affect processes due to its ability to dissipate the transmembrane proton gradient [43] and, as a result, while the scaffold itself may have little promise for TB treatment, specific inhibitors of MmpL3 currently in the drug development pipeline may yield more promising clinical results.
Three linezolid analogs (which target translation), sutezolid, LCB01–0371, and AZD5847 are also in phase 2 of the pipeline. High-doses of sutezolid alone exhibited a log reduction in sputum colony forming units (CFU) on the first 14 days of administration indicating good efficacy in smear-positive TB patients, despite exhibiting an inferior prognosis relative to HRZE [44]. A more recent trial examining the extended sputum EBA using various doses of LCB01–0371 for 15 days (compared to linezolid) is ongoing and currently recruiting patients. Finally, AZD5847 showed modest EBA when dosed at 500 mg and 800 mg twice daily although adverse side effects were apparent at higher doses [45].
Trials to shorten the duration of drug-sensitive tuberculosis to 4 months by substituting a fluoroquinolone for isoniazid or ethambutol have all failed [46–48]. The previously approved fluoroquinolones, levofloxacin and moxifloxacin, are now being evaluated in phase 2 trials designed at improving existing regimens for drug resistant disease. Three studies (MDR-END, OptiQ, and NEXT) containing levofloxacin (in combination with other drugs) in one or more of the treatment arms are active and recruiting patients with MDRTB [49]. All of these will be evaluating treatment success following 9–24 months of treatment with anticipated completion in 2019. One treatment arm in another phase 2 trial (NC-005) contains moxifloxacin (M) in combination with bedaquiline (B), pretomanid (Pa), and pyrazinamide [50]. Congruently, the simpliciTB (NC-008) trial will be comparing a 4month BPaMZ regimen to the standard 6-month HRZE/HR regimen and is anticipated to commence mid-2018. The resulting BPaMZ combination is targeted towards MDR-TB patients and has the potential to shorten treatment duration. Finally, the FDA-approved antiparasitic drug nitazoxanide [51] is being evaluated for its EBA in drug-susceptible TB patients compared to the standard HRZE regimen in patients in Haiti.
Phase 3 of the pipeline (Table 3) is represented by drug regimens containing bedaquiline, linezolid (L) and the nitroimidazoles, pretomanid and delamanid. In murine models of TB, drug combinations containing bedaquiline and/or pretomanid exhibited sterilizing activity [52], perhaps rationalizing the overrepresentation of these drugs in phase 3 regimens. The STAND trial (Shortening Treatment by Advancing Novel Drugs, previously NC-001 during its phase 2 development) using a PaMZ combination, was the first regimen to be tested in the clinic that contained a novel drug [50]. Promising results returned in 2016, in accordance with the superior EBA of PaMZ during phase 2 studies [53,54] with the caveat that EBA has no utility in predicting sterilizing cure as mentioned above. The TB Drug Alliance has since given precedence to advancing the BPaMZ (NC-005) regimen further into clinical development. The Nix-TB trial (which later transitioned into the ZeNix-TB or NC-007 in November 2017) evaluates the efficacy of BPaL in patients with MDR-TB and XDR-TB. Initial results showed that of the 20 patients who have completed the 6- to 9month regimen and have been followed to the primary endpoint (6 months post-treatment), only 1 exhibited microbiological relapse (current standard of care is associated with a 5% relapse), suggesting potential for this treatment regimen.
Table 3.
Representative Clinical Trials in Advanced Phases of the Pipeline.
| Trial Name (NCT number) | Included Interventions | Phase | Start – End Dates |
|---|---|---|---|
| NC-005 (NCT02193776) | BDQ, PRE, MOX, PZA | 2 | Nov 2014 – Mar 2018 |
| MDR-END (NCT02619994) | LZD, DLM, LVX, PZA | 2 | Jan 2016 – Dec 2019 |
| Opti-Q (NCT01918397) | LVX, Optimized background regimen | 2 | Jan 2015 – Mar 2019 |
| NEXT (NCT02454205) | LZD, BDQ, LVX, PZA + (ETH, INH, TRZ) | 2/3 | Oct 2015 – Jan 2019 |
| NC-008, SimpliciTB (NCT03338621) | PRE, BDQ, MOX, PZA | 2 | Aug 2018 – Mar 2022 |
| RIPENACTB (NCT03281226) | RIF, INH, PZA, EMB, NAC | 2 | Dec 2016 – Dec 2019 |
| STAND (NCT023442886) | MOX, PRE, PZA | 3 | Feb 2015 – May 2018 |
| NC-007, ZeNix-TB (NCT03086486) | PRE, LZD, BDQ | 3 | Nov 2017 – Jan 2022 |
| endTB (NCT02754765) | BDQ, DLM, CFZ, LVX, MOX, LZD, PZA | 3 | Dec 2016 – Apr 2021 |
| TB-PRACTECAL (NCT02589782) | BDQ, PRE, MOX, LZD, CFZ | 2/3 | Jan 2017 – Mar 2021 |
| STREAM (NCT02409290) | MOX, CFZ, EMB, PZA, INH, PTH, KAN, LVX, BDQ | 3 | Apr 2016 – Dec 2021 |
| (NCT02333799) | BDQ, PRE, LZD | 3 | Mar 2015 – Oct 2021 |
| RIFASHORT (NCT02581527) | RIF, INH, EMB, PZA | 3 | Feb 2017 – Dec 2020 |
| WHIP3TB (NCT02980016) | RPT, INH | 3 | Nov 2016 – Sept 2019 |
*BDQ = bedaquiline, PRE = pretomanid, MOX = moxifloxacin, PZA = pyrazinamide, LZD = linezolid, DLM = delamanid, LVX = levofloxacin, ETH = ethionamide, INH = isoniazid, TRZ = terizidone, NAC = N-acetylcysteine, CFZ = clofazimine, EMB = ethambutol, PTH = prothionamide, RPT = rifapentine.
Other ongoing phase 3 trials that contain bedaquiline and delamanid in new regimens include the endTB and TB-PRACTECAL trials. The drug-intensive endTB trial evaluates the efficacy of bedaquiline and/or delamanid in combination with linezolid, pyrazinamide, clofazimine and a fluoroquinolone in patients with fluoroquinolone-sensitive MDR-TB. The TB-PRACTECAL trial, on the other hand, studies the short treatment regimens composed of BPaL in combination with either moxifloxacin or clofazimine for 6 months in MDR-TB patients. Finally, bedaquiline is a component of a six-drug regimen in two of the treatment arms in the STREAM trial for patients with MDR-TB. This study aims to identify a fullyoral 9-month regimen without any adverse effects and microbiological relapse [55].
There are several other ongoing phase 2 and phase 3 trials which cover the whole clinical spectrum of tuberculosis – from TB-HIV co-infections, to latent TB treatment and optimizing drug regimens for TB in children.
The most significant limitation in progressing drugs into the phase 2 clinical studies is predicting their potential at achieving sterilizing cure in patients. Mouse studies have typically been applied retrospectively for predicting sterilizing cure in patients although newer mouse models that recapitulate some of the salient aspects of human disease have been developed and are currently being used in prioritizing drugs and drug regimens [56]. The marmoset model of tuberculosis is a non-human primate model that more closely recapitulates human disease, but more importantly, using this model with current front-line chemotherapy in comparison to an inferior drug regimen accurately mimicked bacillary load reduction and sterilizing efficacy in humans [57]. Although this model remains a low-throughput costly barrier in drug development, it could certainly help prioritize those drugs that have the highest potential in achieving sterilizing cure. Importantly, studies in marmosets have highlighted the utility of PET-CT imaging as a non-invasive tool in monitoring chemotherapeutic efficacy and have helped to launch clinical trial to use PET-CT imaging as a diagnostic tool to monitor which patients are cured more quickly during therapy (ClinicalTrials.gov Identifier: NCT028218). If PET-CT imaging can be used successfully to predict treatment outcomes, it will replace EBA as a measure of early chemotherapeutic efficacy although the ultimate readout of sterilizing cure will remain to be relapse rates within a year after stopping treatment at least within the foreseeable future.
Pharmacokinetics of Drugs in Lung Granulomas
The granuloma, arguably the most distinctive pathological hallmark associated with TB, is a compact and organized conglomeration of macrophages, monocytes and other immune cells designed to “wall-off” Mtb from the surrounding lung tissue. While granuloma formation is a robust immune response that effectively contains the infection, it fails to eradicate the bacterium. At its core is a group of infected macrophages which can either transform into epithelioid cells (thought to be more phagocytic) or to accumulate lipids to become foamy cells [58]. These cellular granulomas provide a hostile, nutrient-poor environment for the bacilli. In contrast, evidence also suggests that foamy macrophages sustain persistent bacteria [59]. Over time, participating cells undergo necrosis to form regions called the caseum, where the bacilli are released to the extracellular environment. Caseous granulomas eventually transform into cavities by erosion into and fusion with nearby airways, promoting dissemination of Mtb to other hosts [58]. Granulomas within the same host exhibit significant heterogeneity [56], generating a very complex pathology with both interlesion and intralesion diversity.
The lesion heterogeneity in TB leads to distinct subpopulations of bacteria that differ in their metabolic states (based on the nutritional capacity of their microenvironment as well as presence of antibacterial metabolites), which subsequently leads to varying phenotypic tolerance to chemotherapies [60]. This notion has been one of the guiding principles in developing drug regimens, that is, effective drug combinations should be able to target multiple subpopulations of the bacilli. Only recently have we realized that lesion heterogeneity also affects drug pharmacokinetics as well.
The complex architecture coupled with the multitude of extracellular biomolecules in granulomas result in varying drug permeabilities which leads to spatiotemporal periods of monotherapy, likely exerting selective pressure for resistance. In fact, a recent study estimated that multidrug resistance can occur in 1% of patients who are completely compliant with their treatment regimen due to variability in drug pharmacokinetics [61]. This novel paradigm came to light with the recent correlation found between the sterilizing activity of rifampicin and pyrazinamide and their drug distribution in lesions of TB patients visualized using imaging mass spectrometry [62]. It was found that rifampicin and pyrazinamide accumulated in both the necrotic foci and the subtending cellular layers (Figure 1), with the former accumulating after multiple doses and the later exhibiting a dose-independent accretion [62,63]. Knowing that the bacilli in caseum are largely nonreplicating [64], and that rifampicin maintains good efficacy against non-replicating Mtb, the observed rifampicin distribution in TB lesions possibly explains its sterilizing activity in the clinic. This is also likely the case for pyrazinamide – a prodrug that at least in vitro, exerts antitubercular activity under low pH conditions – as intramacrophage Mtb are known to reside in acidic compartments although the pH of human caseum within the granuloma is less clearly defined. In support of this, pyrazinamide was inactive in Kramnik mice (C3HeB/FeJ) that had large necrotic lesions due to the neutral pH of the caseum [65]. The two other first-line drugs, isoniazid and ethambutol, were also found to have good lesion penetration and a sustained accumulation in necrotic foci [62,66].
Figure 1. Lesion pharmacokinetics of representative anti-TB drugs.

(A) Differences in the physicochemical properties of drugs, coupled with the complex architecture of TB granulomas result in varying permeability profiles across different lesion types. The result is periods of monotherapy that generate selective pressure for emergence of resistance. (B) Imaging MS (top) and H&E (bottom) stained section of necrotic nodules and cavities (outlined in white) in rabbit lungs showing accumulation of rifampicin 6 hrs after the 7th daily dose. Image in (B) courtesy of Dr. Brendan Prideaux and Dr. Veronique Dartois of New Jersey Medical School, Rutgers, The State University of New Jersey
The power of lesion pharmacokinetic parameters in predicting treatment outcomes is further exemplified by two recent studies. In a comparative study of the responses of BALB/c and C3HeB/FeJ mice to bedaquiline with and without pyrazinamide, two distinct populations were consistently observed in C3HeB/FeJ mice – one which responded well to the treatment and one which responded less favorably [67]. This bimodal response was attributed to the ability of C3HeB/FeJ mice to form both cellular and caseous lesions, in contrast to BALB/c mice which can only form cellular lesions. It was found that while pyrazinamide exhibited similar distribution in lesions between the two mouse strains, bedaquiline preferentially accumulated in cellular lesions [67]. It was therefore likely that bedaquiline failed to target the large reservoir of bacilli in the caseous lesions of C3HeB/FeJ mice (Figure 1) leading to a population of mice that responded less favorably to treatment. Similarly, a recent study of rifapentine distribution in rabbits with cavitary lesions, showed that while rifapentine was able to distribute into cellular and fibrotic cavity walls to the same extent as rifampicin, it was inferior in partitioning into the caseum of cavitary lesions [68]. These results correlated well with a recent phase II trial that found an inverse correlation between lung cavity size and response to high doses of rifapentine [69].
The failure of recent clinical trials to shorten treatment of drug-sensitive disease where isoniazid or ethambutol was substituted with a fluoroquinolone could also retrospectively be explained, at least in part, by modeling the plasma and tissue pharmacokinetics and pharmacodynamics with activity against the various sub-populations of bacilli in the lesions. Using simulated exposures in typical patient populations, it was found that none of these fluoroquinolones were predicted to be particularly bactericidal against Mtb within the caseum although moxifloxacin may have had some superior activity against Mtb residing in host cells due to its higher intracellular partitioning [70].
The studies mentioned thus far epitomize the impact of lesion heterogeneity on localized drug response and demonstrate its predictive power in clinical outcomes. Therefore, it’s not surprising that many of these studies advocate for measurements of lesion pharmacokinetic parameters in various animal models prior to advancing drugs into clinical studies. These studies also engrave the notion that serum drug concentrations don’t always correlate with drug concentration at the site of infection – an idea that seems intuitive for TB given the complex histopathology observed in patients’ lungs. However, a big drawback in the widespread application of lesion pharmacokinetic studies is the costly and invasive methodologies used and the requirement for sophisticated instruments to determine such parameters. Nevertheless, steps in the right direction are currently being made to accurately predict favorable lesion penetration of candidate drugs using in vitro models [64,71,72]. For example, profiling 279 compounds for caseum binding, coupled with in silico analysis led to the drafting of empirical rules that predict the extent of caseum binding and hence, drug diffusion into necrotic foci [72]. Because the caseum is entirely acellular drug penetration is dependent more on physicochemical effects rather than biological consequence. Further, only the fraction of free compounds (those not trapped inside macrophages) can passively diffuse into this matrix. As this diffusion proceeds inward, drugs bind to macromolecules in the outer rim of the caseum decreasing bioavailable concentration of drugs for continued inward diffusion. Various lipophilicity parameters including overall solubility, number of aromatic rings, molecular shape and number of sp2 carbons showed correlations with caseum binding. The sum of the hydrophobicity with the number of aromatic rings was found to correlate well with caseum binding. Taken together, these empirical measures could be used as guiding principles in property-based drug design during the early phases of drug discovery. However, while these guidelines can be predictive, the fact that a small number of drugs was used for this analysis indicate that this empirical rule should be taken with caution until a more comprehensive and definitive picture of physicochemical properties that define drug distribution is available.
It is tempting to argue that while validation of new drug targets and discovery of new scaffolds are important, these efforts will ultimately be rendered futile if the eventual drug doesn’t reach the entirety of the Mtb population. Therefore, greater attention must be paid to examine the fundamental factors that dictate drug distribution in TB lesions. Furthermore, the studies mentioned in this section were done in mouse and rabbit models of pulmonary TB which, as one might suspect, have subtle yet crucial differences with the human pathology.
Conclusion and Outlook
Several limitations still exist in developing effective and fast-acting TB treatments, chief among which are the complex and heterogenous nature of the disease pathology. There are several other shortcomings and gaps in knowledge in the field that we did not cover, including understanding the biology of different subpopulations of Mtb and whether these are clinically relevant, and development of a quick and accurate diagnostic method to both detect the bacteria and monitor response to chemotherapy. For now, we conclude by echoing the propositions we mentioned above. First, expanding the target space will be streamlined by the early incorporation of triage methods that filter out inhibitors of the most vulnerable drug targets. This will likely lead to drug combinations that attack the bacteria at multiple points curtailing emergence of resistance and lead to a sterilizing cure. Second, optimization of drug regimens should occur in animal models that accurately recapitulate the pathology of human pulmonary TB. New animal models are available that are still not necessarily widely applied in pre-clinical studies. Finally, pharmacokinetic parameters of active compounds must be established in robust animal models and used as a metric in advancing clinical candidates. This will lead to a better idea of the optimum dose prior to entering the clinical stage and lower the failure rate at the later stages of the pipeline. While there is still a long way to go to develop the optimum treatment course, the flurry of compounds within the pipeline and the diversity in pre-clinical development certainly offers some hope towards the goal of realizing a world rid of TB.
Acknowledgement
This work was funded by the Intramural Research Program of NIAID/ NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Collaborators GBDT: The global burden of tuberculosis: results from the Global Burden of Disease Study 2015. Lancet Infect Dis 2018, 18:261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchison DA: The diagnosis and therapy of tuberculosis during the past 100 years. Am J Respir Crit Care Med 2005, 171:699–706. [DOI] [PubMed] [Google Scholar]
- 3.*.Aggarwal A, Parai MK, Shetty N, Wallis D, Woolhiser L, Hastings C, Dutta NK, Galaviz S, Dhakal RC, Shrestha R, et al. : Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170:249–259 e225. This report reinforces the utility of whole-genome sequencing of resistant mutants for target identification coupled with structure-guided drug design in the transition between lead molecules and drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X, Hernandez V, Rock FL, Choi W, Mak YSL, Mohan M, Mao W, Zhou Y, Easom EE, Plattner JJ, et al. : Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol1(3H)-ol (GSK656). J Med Chem 2017, 60:8011–8026. [DOI] [PubMed] [Google Scholar]
- 5.Palencia A, Li X, Bu W, Choi W, Ding CZ, Easom EE, Feng L, Hernandez V, Houston P, Liu L, et al. : Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob Agents Chemother 2016, 60:6271–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee W, VanderVen BC, Fahey RJ, Russell DG: Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem 2013, 288:6788–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasbekar M, Fischer G, Mott BT, Yasgar A, Hyvonen M, Boshoff HI, Abell C, Barry CE 3rd, Thomas CJ: Selective small molecule inhibitor of the Mycobacterium tuberculosis fumarate hydratase reveals an allosteric regulatory site. Proc Natl Acad Sci U S A 2016, 113:7503–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Zhou S, Deng Q, Li X, Meng J, Guan Y, Li C, Xiao C: Identification of a novel inhibitor of isocitrate lyase as a potent antitubercular agent against both active and non-replicating Mycobacterium tuberculosis. Tuberculosis (Edinb) 2016, 97:38–46. [DOI] [PubMed] [Google Scholar]
- 9.Huang HL, Krieger IV, Parai MK, Gawandi VB, Sacchettini JC: Mycobacterium tuberculosis Malate Synthase Structures with Fragments Reveal a Portal for Substrate/Product Exchange. J Biol Chem 2016, 291:27421–27432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krieger IV, Freundlich JS, Gawandi VB, Roberts JP, Gawandi VB, Sun Q, Owen JL, Fraile MT, Huss SI, Lavandera JL, et al. : Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem Biol 2012, 19:1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey AK, Sassetti CM: Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A 2008, 105:4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, et al. : Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog 2015, 11:e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christophe T, Jackson M, Jeon HK, Fenistein D, Contreras-Dominguez M, Kim J, Genovesio A, Carralot JP, Ewann F, Kim EH, et al. : High content screening identifies decaprenylphosphoribose 2’ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog 2009, 5:e1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva-Miranda M, Ekaza E, Breiman A, Asehnoune K, Barros-Aguirre D, Pethe K, Ewann F, Brodin P, Ballell-Pages L, Altare F: High-content screening technology combined with a human granuloma model as a new approach to evaluate the activities of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother 2015, 59:693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puissegur MP, Botanch C, Duteyrat JL, Delsol G, Caratero C, Altare F: An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cell Microbiol 2004, 6:423–433. [DOI] [PubMed] [Google Scholar]
- 16.Abrahams KA, Cox JAG, Futterer K, Rullas J, Ortega-Muro F, Loman NJ, Moynihan PJ, Perez Herran E, Jimenez E, Esquivias J, et al. : Inhibiting mycobacterial tryptophan synthase by targeting the inter-subunit interface. Sci Rep 2017, 7:9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.**.Wellington S, Nag PP, Michalska K, Johnston SE, Jedrzejczak RP, Kaushik VK, Clatworthy AE, Siddiqi N, McCarren P, Bajrami B, et al. : A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat Chem Biol 2017, 13:943–950. This paper describes the inhibition of a highly dynamic, multi-subunit metabolic enzyme with in vivo essentiality via uncompetetive mechanisms using a small molecule that binds to an allosteric site. This paper opened up a new paradigm in the design of next-generation antimicrobials that don’t necessarily bind to the active site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunner K, Maric S, Reshma RS, Almqvist H, Seashore-Ludlow B, Gustavsson AL, Poyraz O, Yogeeswari P, Lundback T, Vallin M, et al. : Inhibitors of the Cysteine Synthase CysM with Antibacterial Potency against Dormant Mycobacterium tuberculosis. J Med Chem 2016, 59:6848–6859. [DOI] [PubMed] [Google Scholar]
- 19.Mishra A, Mamidi AS, Rajmani RS, Ray A, Roy R, Surolia A: An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a novel combinatorial therapy. EMBO Mol Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, et al. : Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 2013, 155:1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.*.Park Y, Pacitto A, Bayliss T, Cleghorn LA, Wang Z, Hartman T, Arora K, Ioerger TR, Sacchettini J, Rizzi M, et al. : Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect Dis 2017, 3:18–33. This paper argues that while inhibition of essential enzymes should intuitively lead to in vivo efficacy, levels of bioavailable metabolites can rescue bacterial growth and render the drug inactive. Hence, drug discovery efforts should not bias against non-essential targets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapero A, Pacitto A, Singh V, Sabbah M, Coyne AG, Mizrahi V, Blundell TL, Ascher DB, Abell C: Fragment-Based Approach to Targeting Inosine-5’-monophosphate Dehydrogenase (IMPDH) from Mycobacterium tuberculosis. J Med Chem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sukheja P, Kumar P, Mittal N, Li SG, Singleton E, Russo R, Perryman AL, Shrestha R, Awasthi D, Husain S, et al. : A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. MBio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong WD, Gibbons PD, Leung SC, Amewu R, Stocks PA, Stachulski A, Horta P, Cristiano MLS, Shone AE, Moss D, et al. : Rational Design, Synthesis, and Biological Evaluation of Heterocyclic Quinolones Targeting the Respiratory Chain of Mycobacterium tuberculosis. J Med Chem 2017, 60:3703–3726. [DOI] [PubMed] [Google Scholar]
- 25.Tantry SJ, Markad SD, Shinde V, Bhat J, Balakrishnan G, Gupta AK, Ambady A, Raichurkar A, Kedari C, Sharma S, et al. : Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J Med Chem 2017, 60:1379–1399. [DOI] [PubMed] [Google Scholar]
- 26.**.Naran K, Moosa A, Barry CE 3rd, Boshoff HI, Mizrahi V, Warner DF: Bioluminescent Reporters for Rapid Mechanism of Action Assessment in Tuberculosis Drug Discovery. Antimicrob Agents Chemother 2016, 60:6748–6757. This paper reported two M. tuberculosis strains that can be used to rapidly triage hits from whole-cell screening to filter out cell wall biosynthesis inhibitors and DNA damaging agents avoiding the identification of redundant targets downstream of the MOA studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arora K, Ochoa-Montano B, Tsang PS, Blundell TL, Dawes SS, Mizrahi V, Bayliss T, Mackenzie CJ, Cleghorn LA, Ray PC, et al. : Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2014, 58:6962–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi PJ, Sutherland HS, Tong AST, Blaser A, Franzblau SG, Cooper CB, Lotlikar MU, Upton AM, Guillemont J, Motte M, et al. : Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorg Med Chem Lett 2017, 27:5190–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutherland HS, Tong AST, Choi PJ, Conole D, Blaser A, Franzblau SG, Cooper CB, Upton AM, Lotlikar MU, Denny WA, et al. : Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorg Med Chem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Bruhn DF, Lee RB, Zheng Z, Janusic T, Scherbakov D, Scherman MS, Boshoff HI, Das S, Rakesh, et al. : Structure-Activity Relationships of Spectinamide Antituberculosis Agents: A Dissection of Ribosomal Inhibition and Native Efflux Avoidance Contributions. ACS Infect Dis 2017, 3:72–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhura DB, Liu J, Meibohm B, Lee RE: Phase II Metabolic Pathways of Spectinamide Antitubercular Agents: A Comparative Study of the Reactivity of 4-Substituted Pyridines to Glutathione Conjugation. Medchemcomm 2016, 7:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foo CS, Lechartier B, Kolly GS, Boy-Rottger S, Neres J, Rybniker J, Lupien A, Sala C, Piton J, Cole ST: Characterization of DprE1-Mediated Benzothiazinone Resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2016, 60:6451–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makarov V, Manina G, Mikusova K, Mollmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, et al. : Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324:801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kloss F, Krchnak V, Krchnakova A, Schieferdecker S, Dreisbach J, Krone V, Mollmann U, Hoelscher M, Miller MJ: In Vivo Dearomatization of the Potent Antituberculosis Agent BTZ043 via Meisenheimer Complex Formation. Angew Chem Int Ed Engl 2017, 56:2187–2191. [DOI] [PubMed] [Google Scholar]
- 35.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, et al. : Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 2013, 19:1157–1160. [DOI] [PubMed] [Google Scholar]
- 36.Diacon AH, Donald PR: The early bactericidal activity of antituberculosis drugs. Expert Rev Anti Infect Ther 2014, 12:223–237. [DOI] [PubMed] [Google Scholar]
- 37.Jindani A, Dore CJ, Mitchison DA: Bactericidal and sterilizing activities of antituberculosis drugs during the first 14 days. Am J Respir Crit Care Med 2003, 167:1348–1354. [DOI] [PubMed] [Google Scholar]
- 38.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, 3rd, et al. : SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2012, 56:1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinrich N, Dawson R, du Bois J, Narunsky K, Horwith G, Phipps AJ, Nacy CA, Aarnoutse RE, Boeree MJ, Gillespie SH, et al. : Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J Antimicrob Chemother 2015, 70:1558–1566. [DOI] [PubMed] [Google Scholar]
- 40.**.Boeree MJ, Heinrich N, Aarnoutse R, Diacon AH, Dawson R, Rehal S, Kibiki GS, Churchyard G, Sanne I, Ntinginya NE, et al. : High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect Dis 2017, 17:39–49. This study demonstrated that a multi-arm, multi-stage trial can greatly speed-up the development of drug regimens in advanced phases of clinical trials. This concept is particularly useful in multicenter, high-burden tuberculosis study sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikonenko BV, Protopopova M, Samala R, Einck L, Nacy CA: Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob Agents Chemother 2007, 51:1563–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Z, Meshcheryakov VA, Poce G, Chng SS: MmpL3 is the flippase for mycolic acids in mycobacteria. Proc Natl Acad Sci U S A 2017, 114:7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, et al. : Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2014, 58:6413–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallis RS, Dawson R, Friedrich SO, Venter A, Paige D, Zhu T, Silvia A, Gobey J, Ellery C, Zhang Y, et al. : Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. PLoS One 2014, 9:e94462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furin JJ, Du Bois J, van Brakel E, Chheng P, Venter A, Peloquin CA, Alsultan A, Thiel BA, Debanne SM, Boom WH, et al. : Early Bactericidal Activity of AZD5847 in Patients with Pulmonary Tuberculosis. Antimicrob Agents Chemother 2016, 60:6591–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jindani A, Harrison TS, Nunn AJ, Phillips PP, Churchyard GJ, Charalambous S, Hatherill M, Geldenhuys H, McIlleron HM, Zvada SP, et al. : High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med 2014, 371:1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merle CS, Fielding K, Sow OB, Gninafon M, Lo MB, Mthiyane T, Odhiambo J, Amukoye E, Bah B, Kassa F, et al. : A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med 2014, 371:1588–1598. [DOI] [PubMed] [Google Scholar]
- 48.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PP, Nunn AJ, Consortium RE: Four-month moxifloxacin-based regimens for drugsensitive tuberculosis. N Engl J Med 2014, 371:1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bouton TC, Phillips PPJ, Mitnick CD, Peloquin CA, Eisenach K, Patientia RF, Lecca L, Gotuzzo E, Gandhi NR, Butler D, et al. : An optimized background regimen design to evaluate the contribution of levofloxacin to multidrug-resistant tuberculosis treatment regimens: study protocol for a randomized controlled trial. Trials 2017, 18:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murray S, Mendel C, Spigelman M: TB Alliance regimen development for multidrug-resistant tuberculosis. Int J Tuberc Lung Dis 2016, 20:38–41. [DOI] [PubMed] [Google Scholar]
- 51.Shigyo K, Ocheretina O, Merveille YM, Johnson WD, Pape JW, Nathan CF, Fitzgerald DW: Efficacy of nitazoxanide against clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2013, 57:2834–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL: Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 2011, 55:5485–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, van Niekerk C, Everitt D, Winter H, Becker P, et al. : 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 2012, 380:986–993. [DOI] [PubMed] [Google Scholar]
- 54.Dawson R, Diacon AH, Everitt D, van Niekerk C, Donald PR, Burger DA, Schall R, Spigelman M, Conradie A, Eisenach K, et al. : Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: a phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385:1738–1747. [DOI] [PubMed] [Google Scholar]
- 55.Moodley R, Godec TR, Team ST: Short-course treatment for multidrug-resistant tuberculosis: the STREAM trials. Eur Respir Rev 2016, 25:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lenaerts A, Barry CE, 3rd, Dartois V: Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev 2015, 264:288–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Via LE, England K, Weiner DM, Schimel D, Zimmerman MD, Dayao E, Chen RY, Dodd LE, Richardson M, Robbins KK, et al. : A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob Agents Chemother 2015, 59:4181–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramakrishnan L: Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 2012, 12:352–366. [DOI] [PubMed] [Google Scholar]
- 59.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F: Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 2009, 10:943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barry CE 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D: The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 2009, 7:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T: Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 2011, 204:1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, Chen C, Kaya F, Weiner DM, Chen PY, et al. : The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med 2015, 21:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DeMarco VP, Ordonez AA, Klunk M, Prideaux B, Wang H, Zhuo Z, Tonge PJ, Dannals RF, Holt DP, Lee CK, et al. : Determination of [11C]rifampin pharmacokinetics within Mycobacterium tuberculosis-infected mice by using dynamic positron emission tomography bioimaging. Antimicrob Agents Chemother 2015, 59:5768–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.*.Sarathy JP, Via LE, Weiner D, Blanc L, Boshoff H, Eugenin EA, Barry CE 3rd, Dartois VA: Extreme Drug Tolerance of Mycobacterium tuberculosis in Caseum. Antimicrob Agents Chemother 2018, 62 This study describes the development of an ex vivo drug susceptibility testing against M. tuberculosis in their most clinically-relevant setting - the cavity caseum. This paper also demonstratred that bacilli within the caseum are indeed nonreplicating. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanoix JP, Ioerger T, Ormond A, Kaya F, Sacchettini J, Dartois V, Nuermberger E: Selective Inactivity of Pyrazinamide against Tuberculosis in C3HeB/FeJ Mice Is Best Explained by Neutral pH of Caseum. Antimicrob Agents Chemother 2016, 60:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zimmerman M, Lestner J, Prideaux B, O’Brien P, Dias-Freedman I, Chen C, Dietzold J, Daudelin I, Kaya F, Blanc L, et al. : Ethambutol Partitioning in Tuberculous Pulmonary Lesions Explains Its Clinical Efficacy. Antimicrob Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.*.Irwin SM, Prideaux B, Lyon ER, Zimmerman MD, Brooks EJ, Schrupp CA, Chen C, Reichlen MJ, Asay BC, Voskuil MI, et al. : Bedaquiline and Pyrazinamide Treatment Responses Are Affected by Pulmonary Lesion Heterogeneity in Mycobacterium tuberculosis Infected C3HeB/FeJ Mice. ACS Infect Dis 2016, 2:251–267. This study uncovered key pharmacokinetic parameters, including caseum drug distribution, that may impact the course of treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rifat D, Prideaux B, Savic RM, Urbanowski ME, Parsons TL, Luna B, Marzinke MA, Ordonez AA, DeMarco VP, Jain SK, et al. : Pharmacokinetics of rifapentine and rifampin in a rabbit model of tuberculosis and correlation with clinical trial data. Sci Transl Med 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Savic RM, Weiner M, MacKenzie WR, Engle M, Whitworth WC, Johnson JL, Nsubuga P, Nahid P, Nguyen NV, Peloquin CA, et al. : Defining the optimal dose of rifapentine for pulmonary tuberculosis: Exposure-response relations from two phase II clinical trials. Clin Pharmacol Ther 2017, 102:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pienaar E, Sarathy J, Prideaux B, Dietzold J, Dartois V, Kirschner DE, Linderman JJ: Comparing efficacies of moxifloxacin, levofloxacin and gatifloxacin in tuberculosis granulomas using a multi-scale systems pharmacology approach. PLoS Comput Biol 2017, 13:e1005650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarathy JP, Liang HH, Weiner D, Gonzales J, Via LE, Dartois V: An In Vitro Caseum Binding Assay that Predicts Drug Penetration in Tuberculosis Lesions. J Vis Exp 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.**.Sarathy JP, Zuccotto F, Hsinpin H, Sandberg L, Via LE, Marriner GA, Masquelin T, Wyatt P, Ray P, Dartois V: Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect Dis 2016, 2:552–563. This is the first report of semi-empirical rules that predict the caseum distribution potential of a lead molecule based solely on its structure. [DOI] [PMC free article] [PubMed] [Google Scholar]