

Abstract
The x-ray structure of the monotopic membrane protein (S)-mandelate dehydrogenase (MDH) from Pseudomonas putida reveals an inherent flexibility of its membrane binding segment that might be important for its biological activity. The surface of MDH exhibits a concentration of the positive charges on one side and the negative charges on the other side. The putative membrane binding surface of MDH has a concentric circular ridge, formed by positively charged residues, which projects away from the protein surface by ~4Å; this is unique structural feature and not observed in other monotopic membrane proteins to our knowledge. There are three α-helixes in the membrane binding region. Based on the structure of MDH, it is possible to propose that the interaction of MDH with the membrane is stabilized by coplanar electrostatic interactions, between the positively charged concentric circular ridge and the negatively charged head-groups of the phospholipid bilayer, along with three α-helixes that provide additional stability by inserting into the membrane. The structure reveals the possible orientation of these helixes along with possible role for the individual residues which form those helixes. These α-helixes may play a role in the enzyme’s mobility. The detergent, N-Dodecyl-β-maltoside, is inserted between the membrane binding region and rest of the molecule and may provide structural stability to intra-protein regions by forming hydrogen bonds and close contacts. From the average B-factor of the MDH structure, it is likely that MDH is highly mobile, which might be essential for its intersection in membrane and non-membrane environments, as its substrate (S)-mandelate, is from the cytoplasm, while its electron acceptor is a component of the membrane electron transport chain.
Keywords: x-ray, monotopic, membrane protein, DDM, FMN, α-hydroxy acid enzyme
Graphic abstract
1. Introduction
(S)-Mandelate dehydrogenase (MDH) from Pseudomonas putida is an enzyme present in the mandelate pathway [1]. It catalyzes the oxidation of (S)-mandelate to phenylglycolate as the second of four steps in the mandelate pathway that converts (R) - mandelate to benzoate (Fig.1). The other enzymes are mandelate racemase, which is the first enzyme in the pathway, and benzoylformate decarboxylase followed by benzaldehyde dehydrogenase, which catalyze the third and fourth reactions in the pathway. This pathway helps the organism to grow on mandelic acid as a sole source of carbon and energy.
Fig.1.

(S)-mandelate dehydrogenase catalyzes the oxidation of (S)-mandelate to benzoylformate
MDH is a member of the FMN-containing α-hydroxy acid oxidizing family of enzymes [2]. There is high sequence identity (~30 to 45%) between the members of this family. The members of the family are present in a wide range of organisms including archaebacteria, pseudomonads, yeasts, plants and mammals. Their locations in the cell varies widely from the cytoplasm to the mitochondrial intermembrane space to peroxisomes of the eukaryotic cells [2]. In Pseudomonas putida, MDH is tightly bound to the cytoplasmic membrane and can be released only in the presence of detergents. Like other members of the α-hydroxy acid enzyme family, MDH follows a ping-pong mechanism in which the α-hydroxy acid substrate is first oxidized by the enzyme to form the product and the flavin is reduced to the hydroquinone (the reductive half-reaction). In the second step (the oxidative half-reaction), the reduced flavin is re-oxidized by an electron acceptor by transferring the electrons to it. The mechanism of the reductive half-reaction appears to be similar among the members of the α-hydroxy acid oxidizing family, involving either substrate α-proton abstraction to form carbanion intermediate or the direct transfer of a hydride from the substrate α-proton to the flavin ring[3]. However, the enzyme oxidative half-reaction differs with respect to the electron acceptors among the members. In the case of MDH, the electron acceptor is an organic molecule, a component of the electron transfer chain within the bacterial membrane, possibly a quinone [2]. MDH also differs from other members of the α-hydroxyacid oxidizing family with respect to its physiological substrate (S)-mandelate, which is bulky and a sterically constrained organic molecule compared to small aliphatic substrates utilized by most members of the family[4]. It also prefers substrates with an unsaturated group at the β-carbon position.
MDH belongs to the monotopic class of integral membrane proteins[5] that are inserted into only one side of the phospholipid bilayer. Several attempts have been made for quite a long time to crystallize MDH without success. However, a chimeric construct of MDH was designed in which a 39 residue internal membrane-binding segment of MDH was replaced with a 20 residue segment derived from one of its soluble homologues, Glycolate Oxidase (GOX; Fig.2)[6]. This construct, named MDH-GOX2, retained full catalytic activity of wild-type MDH but was no longer bound to the membrane. MDH-GOX2 yielded well diffracting crystals which enabled the structure to be solved at 2.15 Å resolution [2]. Subsequently, high-resolution structures of MDH-GOX2 both of the oxidized and reduced states at 1.35 Å resolution were obtained [7]. The study of the catalytic mechanism of the chimera was extended by determining structures of the G81A mutant of MDH-GOX2 and in complex with its slow substrates 3-indolelactate and 2-hydroxyoctanoate at 1.8 Å, 2.2 Å and 2.5 Å resolutions respectively [8].
Fig.2.

Top: Sequence of (S) Mandelate Dehydrogenase (MDH) from P.putida. Down: Partial sequences of MDH and MDH-GOX2 covering the membrane binding region (residues 170240). The residues replaced with the GOX sequence in MDH-GOX2 is shown in bold letters. The three α-helices of membrane binding segment of MDH are marked as α1 ,α2 and α3 while the only α-helix present in the corresponding segment in MDH-GOX2 marked as α.
All the structures involving the chimera MDH-GOX2 revealed several distinct features which can influence the catalytic activity of the enzyme. The high resolution oxidized and reduced MDH-GOX2 structures show the rearrangement of the hydrogen bonding pattern within the active site that results from binding of a proton to the N-5 position of the anionic hydroquinone form of the reduced flavin prosthetic group[7]. The G81A mutant of MDH-GOX2 shows that reduction in electrophilicity of the flavin ring account for the ~100 fold lower reactivity with the substrate and a modestly higher reactivity with molecular oxygen [8]. The structure of G81A in complex with two of its slow substrates showed that 2-hydroxy octanoate binds in an enzymatically productive mode while the other substrate 3-indolelactate, binds in an unproductive mode [8]. However, how the enzyme interacts with the membrane in the oxidative half reaction and how the electron acceptor substrate reaches the active site from the membrane remained elusive. In this study, we present the 2.2Å structure of the oxidized native form of wild type (S)-mandelate dehydrogenase with the membrane binding segment intact. This structure exhibits inherent flexibility of its membrane binding segment, which may be important for its possible mode of interaction with the membrane as well as for its mobility.
2. Materials and Methods:
2.1. MDH Purification:
Carboxyl terminal histidyl-tagged MDH was expressed by following the published protocol [1, 6], but modifying the enzyme extraction and purification steps. At the end of the expression, the harvested cells were stored at −80°C instead of −20°C in the original protocol. About 20g of frozen cells were thawed and re-suspended in 180ml of 50mM phosphate buffer, pH 7.5. The cells were broken in a Microfludizer and the lysate centrifuged at 5000g for 20min. After removal of the supernatant, the pellet was washed twice with the phosphate buffer. The pellet was then re-suspended in 60ml of 20mM phosphate buffer along with PMSF and 0.36% DDM (rather than 0.1% Triton X-100 as in the published protocol). The suspension was stirred overnight and then centrifuged at 165,000g for 90min and the supernatant retained. The DDM-extract was then loaded on a Ni2+ resin column equibrated with buffer A (50ml phosphate pH7.5, 50ml ethylene glycol 100% and 0.014% DDM). The column was washed extensively with buffer A containing in addition 100mM imidazole. The pure protein was eluted with buffer A containing 400mM imidazole. The protein was passed through a G-25 column equibrated with buffer A and concentrated in a 30K Centricon. The final protein was stored at −80°C in the buffer with 20% ethylene glycol.
2.2. MDH crystallization:
Several detergents were tested and crystals were obtained from six of them (Triton X100, C12E8, 1-S-Octyl-Beta-D-thioglucoside, n-decyl-β-D-maltoside(DM), Cymal6 and n-n-dodecyl-β-D-maltoside(DDM) (with 100mM sodium sulfate, 50mM sodium citrate pH 5.5, 5% PEG400 (v/v) and 0.1mM FMN in the reservoir solution). The detergents were purchased from Anatrace (Maumee, OH, USA), Hampton Research (Aliso Viejo, CA, USA) and Sigma-Aldrich (St Louis, MO, USA). The LCP crystallization trials were also attempted but were not successful. Crystals from the first four detergents diffracted poorly (lower than 5Å resolution) and the diffraction data were highly anisotropic, high mosaicity and spots were elongated.
The data obtained initially with DDM and Cymal6 could be processed (section 2.3 below) to a final resolution of 3.0Å and 3.15Å respectively. Though the non-membrane regions were clearly defined in these maps, regions later attributable to the membrane binding segment was very difficult to model completely which is essential for this study.
The best crystals of MDH were then obtained by the sitting drop method using DDM as the primary detergent. The reservoir solution contained 15%w/v polyethylene glycol monoethyl ether 2000, 20% glycerol, 100mM tri-sodium citrate pH 5.6, 5% polyethylene glycol 200 and 1.3mM FMN. The droplet was prepared with 3μl of protein (concentrated to 20mg/ml) and 1μl of reservoir solution.
2.3. Data collection:
Data for the MDH were collected at the NECAT 24IDE beam line, Advanced Photon Source (APS) equipped with a Microdiffractometer-MD2 and Dectris’s EIGER hybrid pixel detector (Eiger16M) which has a two dimensional array of pin-diodes processed in high resistivity silicon, connected to an array of readout channels designed in CMOS technology. Data were collected at 100K temperature using an x-ray wavelength of 0.98Å. The data were processed using DENZO and SCALEPACK, as a part of HKL2000 package[9].
The structure of MDH was solved by the molecular replacement method with PHASER[10] of PHENIX [11] using coordinates of the high resolution oxidized form of MDH-GOX2 (PDB id 1P4C) with residues 171–230 (covering the membrane binding segment plus a few extra residues on either side) removed (Fig. 2). There are two molecules in the asymmetric unit. Based on a NCS averaged difference Fourier electron density map using COOT [12], all the residues (171–230) were rebuilt in both molecules. A random subset of all reflections (5%,1992 reflections) was set aside for free-R calculation to monitor the refinement and to avoid over-fitting [13]. The refinement of MDH was carried out using PHENIX[11] by subjecting the model to alternative positional and B-factor refinement. During refinement, tight restraints between non-crystallographic symmetry related monomers were maintained.
A simulated annealing step was performed at the beginning of the refinement. A total of 260 water molecules were added to the MDH model along with cations, anions and DDM molecules. The solvent and ion molecules were modelled and verified using the “Motifs and Sites” server of PDB of Europe (www.ebi.ac.uk/pdbe-site/pdbmotif) and the “checkMyMetal” server [14]. The final R and Free-R of the model are 19.7 and 24.0 respectively. Both molecules A and B have reasonably good electron density in the membrane binding segment. In molecule B, DDM inserted itself between the membrane binding segment and the rest of the molecule. The polder density map of DDM is shown in Fig.3. During the refinement of the structure, the Fo-Fc density map showed a very strong density peak about 3.5 Å from the isoalloxazine ring of FMN. The peak was modelled as glycerol. The polder density map of FMN and glycerol is shown in Fig. 4. Based on the analysis using the “Motifs and Sites” server of PDB of Europe, glycerol prefers an environment of charged resides and the FMN environment provided an ideal environment to model it accordingly. However, portions of the side chain for several residues could not be fitted in the weak density and these atoms were assigned zero occupancy. Molecule A contains 69 atoms while the molecule B has 37 atoms with zero occupancy. The first two and last eighteen residues of the cloned sequence, including an engineered hexahistidyl tag at the C-terminus are absent. As molecule B has better defined electron density than molecule A with fewer zero occupancy atoms, most of the analyses were carried with molecule B of MDH. However, a comparison of molecules A and B was made with respect to the FMN environment and the membrane binding segment, to highlight their similarity as well as difference at these very important regions. The final stereochemistry of the model was checked against the Ramachandran map in PROCHECK [15] and MOLPROBITY[16]. The data collection and refinement details are summarized in Table 1.
Fig.3.

Polder electron density map for DDM at +2.5σ level. DDM is shown as ball and stick.
Fig.4.

Polder electron density map for FMN and Glycerol at +3.5σ level. FMN and Glycerol are shown as ball and stick and in red and blue colors respectively.
Table 1.
Data Collection, Structure Determination and Refinementa
| Crystal | MDH |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.98 |
| Space group | I222 |
| Unit cell dimensions | |
| a,b,c (Å) | 123.4 129.9 143.3 |
| Res. limit (Å) | 50–2.2 (2.28–2.2) |
| I/Sigma(I) | 9.7 (1.2) |
| Rmerge (%) | 16.7 (93.3) |
| Rpim (%) | 8.0 (55.4) |
| CC1/2 (%)- highest shell resolution | 50.7 |
| Completeness (%) | 93.7 (69.0) |
| Redundancy | 4.9(3.3) |
| Refinement | |
| Resolution range (Å) | 41.5–2.2 (2.25–2.2) |
| R-work (%) | 19.7 (27.4) |
| Rfree (%) | 24.0 (29.3) |
| R (working + test) (%) | 19.8 |
| No. of Reflections | 54963 |
| Model | |
| No. of amino acids | 746 |
| No. of water molecules | 260 |
| Residues in generously allowed region | 2 |
| Residues in disallowed regions | 2 (Glu32 in both molecules) |
| No. of residues with alternate conformation | |
| Stereo-chemical ideality | |
| Bonds (Å) | 0.01 |
| Angles (deg) | 1.17 |
| Dihedral angles (deg) | 14.6 |
| Planarity (Å) | 0.01 |
Values in parentheses are for the outer shell
The rms deviation and structure analysis were calculated using the programs CCP4[17], COOT[12] and CCP4MG [18]. The hydrogen bonding parameters were defined as d(X….A) ≤ 3.6Å, d(H…..A) < 3.0Å and <(X-H….A) > 90° where d is the distance, X is the hydrogen bond donor and A is the hydrogen bond acceptor[19]. The hydrogen bonds were calculated using contact program of CCP4[17] and CCP4MG[18]. All the software used in this project was curated by SBGRID[20].
3. Results:
3.1. Structure analysis
The structure of the membrane associated wild type MDH has been determined at 2.2Å resolution (Fig. 5). MDH has a subunit molecular weight of ~43kD and crystallized in the orthorhombic space group I222. There are two molecules in the asymmetric unit. The subunit folds as a β8α8 TIM barrel and is similar in conformation to MDH-GOX2 and GOX. The polypeptide chain could be traced from residues Gln3 to Glu375 in both molecules using the NCS averaged (2fo-fc) and (fo-fc) electron density maps and refined as described in Materials and Methods. The Ramachandran map prepared using PROCHECK[15] showed that Glu32 in both molecules lies in a disallowed region, a feature observed in two other members of this family of proteins (flavocytochrome B2 and GOX) as well as in MDH-GOX2, the G81A mutant of MDH-GOX2 and in its complex structures[2, 7, 8, 21, 22]. The MOLPROBITY[16] showed Ser304 as an outlier both in molecules A and B. However, the electron density of Ser304 in the 2fofc map is excellent at the 1.5σ level.
Fig.5.

Worm/Ribbon diagram of MDH. The membrane binding region is shown as a ribbon and colored based on hydrophobicity. Hydrophobic residues are in pale brown and nonhydrophobic residues are in blue color. The three α-helixes of the membrane binding region are marked as α1, α2 and α3. The rest of the molecule is shown as worms. FMN is shown as ball and stick in black color.
3.1.1. DDM binding site in MDH:
DDM is inserted in-between the membrane binding segment and the rest of the MDH molecule (Fig. 6 & 7). DDM forms several hydrogen bonds with residues just preceding the membrane binding segment and several close contacts with residues of the membrane binding segment (Fig.7). In addition to DDM, a polyethylene glycol (PEG) and a phosphate anion along with five water molecules interact with the membrane binding region of molecule B by forming hydrogen bonds with them. In molecule A, several ions, solute and solvent molecules interact with the membrane binding segment through hydrogen bonds (Fig.8).
Fig.6.

DDM provides stability to the membrane binding segment of MDH by inserting between it and the rest of the molecule. FMN and DDM are shown as ball and stick and in red and blue colors respectively. The membrane binding segment is shown as a ribbon in black color. The three α-helixes of the membrane binding region are marked as α1, α2 and α3. The rest of the molecule is shown as a yellow surface.
Fig.7.

Stereo view of the binding site of DDM in MDH. DDM is shown as ball and stick and in gold color. Cα- trace of the membrane binding segment is shown as thin bonds in red. Residues which form hydrogen bonds are shown in full. The hydrogen bonds are shown as dotted lines in black color. Residues which are within 4Å from DDM of MDH’s molecule B are shown as thick bonds in black. Water molecules are shown as green sphere. PEG and PO4 are shown as thick bonds in blue and lawn green colors respectively.
Fig.8.

Stereo view of the interactions of solvent and ion molecules with the membrane binding segment of molecule A of MDH (Residues 177–229). Cα- trace of the membrane binding segment is shown as a thin bond in red color. Residues which form hydrogen bonds are shown in full. The hydrogen bonds are shown as dotted lines in black color. Residues which are within 4A from DDM of MDH’s molecule B are shown as thick bonds in black color. Water molecules are shown as green spheres. Glycerol, SO4, Ethylene glycol and citric acid are shown as thick bonds in blue, lawn green, gold and magenta colors respectively.
3.2. Membrane binding segment of MDH
The membrane binding segment consist of about 53 residues (177–229) and forms three α-helices and random coils (Fig.2, 5 & 9). The three helices comprise residues 181–188, 191200 and 216–226 respectively (Fig. 9). This contrasts to an earlier prediction based on sequence that the first ~20 residues (177–196) form an amphipathic β-sheet[6]. However, the presence of the third α-helix (216–226) was predicted correctly.
Fig.9.

Ribbon and Ball/stick diagram of helixes (α1, α2 and α3) of the membrane binding segment. In the transparent surface diagram, hydrophobic, basic and acidic residues are shown as green, blue, red colors respectively. The rest of the amino acids are in pink colors.
The crystal packing of the membrane binding segments of both the crystallographically independent molecules of the asymmetric unit is shown in Fig. 10. The segments are stacked one over the other in an extended fashion along the a-axis with molecule A aggregating in the form of a tilted square. The membrane binding segments of the independent molecules are in close proximity with each other thereby forming a hydrophobic region with helix-a3 being the closest and is packed in an anti-parallel fashion at a distance of ~8Å. The van der Waals interactions between them plays a role in stabilizing the packing of the molecules.
Fig.10.

Packing of the membrane binding segment of crystallographically independent molecules (A and B are shown in red and green colors respectively) viewed along the b-axis. The axes are also marked in figure. The molecular four-fold axis is passing along the c-axis.
3.3. Comparison of MDH with MDH-GOX2 and GOX:
MDH crystallized in an I-centered orthorhombic space group with two molecules in the asymmetric unit (a.u.) while the MDH-GOX2 was crystallized in an I-centered tetragonal space group with one molecule per a.u. [2]. However, similar to MDH-GOX2, MDH forms a stable tetramer. The PISA [23] confirms that the biological assembly of MDH is a tetramer. The structure of MDH closely resembles the oxidized form of MDH-GOX2 in the regions 3176 and 230–376 (Fig. 11). However, it adopts a different conformation between the residues 177–229. When the monomer of MDH and MDH-GOX2 are aligned, the root mean square (rms) deviation of Ca atoms is 0.48 Å for 329 equivalent atoms. This similarity extends to their quaternary structures. Comparing subunit of MDH with GOX indicates an rms deviation of 1.21 Å for 337 equivalent Ca atoms (Fig.11). Unlike GOX which is packed in octamers, the quaternary structure of MDH does not extend beyond the tetramer like in MDH-GOX2[2].
Fig.11.

Stereo view of the superposition of Cα trace of MDH (black), GOX (coral) (pdb id: 1GOX) and MDH-GOX2 (gold) (pdb id:1P4C). The MDH membrane binding segment (residues 177–229) along with the corresponding segment from MDH-GOX2 (residues 177–210) and GOX (residues 176–209) are shown as ribbons in their respective colors. The three α-helixes of the membrane binding region are marked as α1, α2 and α3. The FMN is shown as ball and stick in red color.
The dimer interface of MDH contains both hydrophobic and hydrophilic interactions. There are 26 inter-chain conventional hydrogen bonds (N-H...O and O-H....O) between the two molecules as well as 14 inter-chain C-H...O hydrogen bonds and one N-H....S hydrogen bond at the interface[19, 24]. The solvent-accessible area of MDH at the interface is 1467Å2 which is similar to MDH-GOX2’s 1444Å2[18]. The orientation and conformation of FMN is identical to MDH-GOX2 with N5 of FMN accepting a hydrogen bond from the amide of Gly81[2].
In MDH-GOX2, 39 of the putative 53-residues membrane binding segment of MDH is replaced by 20 residues from its soluble homologue GOX (Fig.2). Residues 195 to 203 of MDH-GOX2 (corresponding to 216–226 of MDH; helix-α3) adopts a α-helix conformation. When superimposed on MDH, this helix is oriented approximately ~100° from helix-α3 (Fig.11). Interestingly, the corresponding segment in the intact GOX structure involving residues 199–205 also adopts an α-helical conformation, but its orientation is similar to helix-α3 of MDH (Fig.11). In other words, this helix-α3 adopts an identical orientation in MDH and GOX (even though the helices differ in sequence) but differs by ~100° in MDH-GOX2.
3.4. Comparison of FMN environment of MDH and MDH-GOX2
The superposition of the FMN and its environment (residues or chemical groups within 4Å of FMN) indicates that FMN adopts a similar orientation in MDH and MDH-GOX2 (Fig. 12). The sulfate ion in MDH-GOX2 is replaced by glycerol in MDH. In this region, there are six water molecules in MDH while MDH-GOX2 has eight water molecules. There are five conserved water molecules between them. All the water molecules which are at the tail region of FMN are conserved between MDH and MDH-GOX2. Unlike in MDH-GOX2 where four water molecules are present near the isoalloxazine ring, there is no water molecule in MDH. Of these four water molecules, three are involved in connecting Tyr26 to Tyr131 in MDH-GOX2 [7]. All these three water molecules along with sulfate ion are replaced by glycerol in MDH.
Fig.12.

Stereo view of the superposition of the FMN environment in MDH and MDH-GOX2. Seven active site residues conserved among α-hydroxy acid oxidizing enzymes along with Gly81 are shown. FMN, Glycerol and SO4 are shown as ball and stick. Water molecules are shown as spheres. MDH and MDH-GOX2 are in red and black colors respectively.
It is interesting to note that glycerol connects Tyr26 and Tyr131 directly through O-H....O and C-H O hydrogen bonds. Both the sulfate ion and glycerol form S-H...O, N-H...O, O-H....O. and C-H....O hydrogen bonds with active site residues as well as FMN in their respective enzymes (Fig. 13). Of the seven conserved active site residues along with Gly81, Tyr26, Tyr131 and Arg165 vary significantly between the MDH and MDH-GOX2 structures (Fig. 12). Tyr26 and Tyr131 have rotated ~9° and 35° and moved about 1 Å and 3Å between MDH and MDH-GOX2. In the case of Arg165, the χ4 shows significant difference between MDH and MDH-GOX2.
Fig. 13.

Ball and stick diagram of the glycerol interactions with FMN and active site residues of MDH. Carbon and oxygen atoms of glycerol are shown in green and red colors respectively. The FMN and active site residues are in black color. The O-H...O and C-H....O hydrogen bonds are shown in black and red colors respectively.
4. Discussion:
4.1. Structural stability
Based on the difficulty in the model building of the membrane binding segment using the lower resolution Cymal6 and the early DDM data (section 2.2) and by comparing the environment around the membrane binding segment in molecule A and B (Fig.7 & 8) suggested that DDM along with water molecules and ions might play a role in stabilizing the structure of MDH. The DDM might provide more stability to molecule B as the density is better with fewer zero occupancy atoms compared to molecule A. In molecule B, there are only five water molecules that interact with the membrane binding segment compared to ten water molecules in molecule A. By comparing the environments of the membrane binding segment of crystallographically independent molecules, it appears that the presence of DDM eliminates the necessity for other ions and solute molecules to stabilize the structure of the membrane binding segment. By comparing the quality of the data collected using the crystals grown with DDM as a primary detergent in two different crystallization conditions showed the importance of precipitation agents in the crystallization conditions in getting good and high resolution data.
The average B-factor for MDH is 46.8Å2 which indicates a high degree of mobility or flexibility which might help the MDH to facilitate interaction in two different environments. One of its substrates (the reductant, mandelate) is derived from the cytoplasm and the other (the oxidant, possibly a quinone) is derived from the membrane.
Comparison of the FMN environment of MDH and MDH-GOX2 indicates that the sulfate ion is replaced by glycerol in MDH (Fig.12 & 13). The glycerol contains three hydroxyl groups and they form several O-H....O and C-H….O hydrogen bonds with FMN and with some of the active site residues. A detailed comparison of the FMN environment in MDH and MDH-GOX2 shows several significant differences, especially in the conformation of the conserved active site residues Tyr26, Tyr131 and Arg165. Also, the sulfate and water molecules near the isoalloxazine ring are replaced by glycerol. As both MDH-GOX2 and MDH exhibit similar catalytic efficiency (kcat=290 s−1 (MDH) and 205 s−1 (MDH-GOX2))([6], such differences do not appear to influence the catalytic activity of these enzyme.
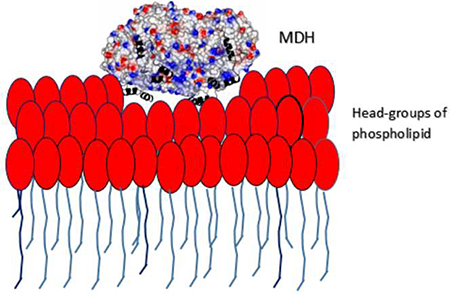
4.2. Proposed Interactions of MDH with membrane
The MDH is crystallized with DDM as the primary detergent and it is likely to maintain the protein close to its native conformation. MDH forms a biological tetramer with circular 4-fold symmetry with a positively charged concentric circular ridge which project away by ~4Å from the protein surface (Fig.14). MDH has a distinctly positive surface on one side while the opposite side is predominantly electronegative. For catalytic efficiency, the MDH tetramer should be oriented perpendicular to the phospholipid head groups so that all four subunit would have equal access to the membrane (Fig. 14). The electropositive surface of the tetramer could provide stable binding with the negatively charged head groups of the phospholipid bilayer of the membrane (Fig.14). The membrane binding segment of MDH which contains three distinct α-helical segments per monomer might provide additional anchoring to the membrane by inserting into it, thereby bringing the enzyme close to its electron acceptor (Fig.5, 9 and 14). Based on the structure, it could be predicted that all the three helixes would be oriented perpendicular to the phospholipid bilayer and the hydrophobic amino acids which form these helixes should have their sidechains directed into the membrane to provide additional stability for binding to the membrane (Fig.9). Based on the structure, it could plausible that Ala181 and Val184 of helix-α1, Pro191,Trp193 and Phe197 of helix-αa2 and Leu216 and Met223 of helix-α3 might have their sidechains inserted into membrane. Based on the Kyte-Doolittle hydrophobicity scale [25], the hydrophobicity of helixes α1, α2 and α3 are 8.7, −8.2 and −0.8 respectively. This indicates that helix- α1 might insert deeply into the membrane compared to the other two helices. It leads us to suggestion that the oxidant which is part of membrane electron transport chain, may utilize helix- α1 to reach the enzyme’s active site. Also, on comparing the crystal structures of MDH, MDH-GOX2 and GOX (Fig.11), it can be speculated that these three helixes could migrate across the membrane surface and attach to an appropriate location so that the reductive/oxidative half reaction could take place (Fig. 14). Both the helixes α1 and α2 project away from the electropositive surface by ~13Å (Fig. 14).
Fig.14.

Stereoview of surface diagram of the MDH tetramer with four fold axis passing through perpendicularly at the middle of the teramer. The positive, negative and neutral surface are colored as blue, red and white. The membrane binding segment is shown as a ribbon in black color. The three α-helixes are marked as α1,α2 and α3.
The 53-residues membrane binding segment of MDH contains 26 hydrophobic, 18 hydrophilic, 6 basic and 4 acidic residues. The average B-factor of the membrane binding segment is 63.0Å2 compared to 43.5Å2 for the rest of the molecule. The three α-helixes in this segment contain both hydrophobic and non-hydrophobic residues (Fig.2, 5&9). There are also several residues in this segment that have adopted a random coil structure. The high flexibility of this segment may allow it to interact both with the membrane and cytoplasmic regions of the organism. It could also possible that when the membrane binding segment is in its natural environment and interacts with the negatively charged phospholipid head groups of the bilayer, the flexibility of this segment might be somewhat reduced.
4.3. Possible additional role of helix-α3:
The third helix (α3) is close to a channel leading to FMN (Fig. 6 & 9). It could be possible that due to its location in the enzyme, this third α-helix (α3) could also act as a gate to provide access of the reduced flavin to the quinone component of the membrane electron transport. Comparison of the crystal structures of MDH with the G81A mutant of MDH-GOX2 in complex with its slow substrate, 2-hydroxyoctonoate (PDB id: 2a85) which binds in an enzymatically productive mode shows the orientation of helix-α3 with respect to the substrate (Fig.15). The α3-helix is ~14Å from the 2-hydroxyoctonoate in the complex structure and is oriented away from the channel while in MDH, it is ~11Å from the substrate and is blocking the channel. It provides an indirect evidence for the flexibility of helix-α3 and its possible role as a gate for the oxidant.
Fig.15.

Stereo view of the surface diagram of MDH in light green color. The membrane binding segment of MDH is shown as a ribbon in coral color. The superposed corresponding segment in the G81A mutant of MDH-GOX2 in complex with 2-hydroxyoctanoate is shown as a ribbon in pink color. The three α-helixes of the membrane binding region are marked as α1, α2 and α3. The helix-α3 which is predicted to act as a gate for the oxidant is away from the channel in the G81A complex, while blocking it in MDH. The FMN and 2-hydroxyoctanoate are shown as ball and stick and in black and red colors respectively.
4.4. Comparison of MDH with other monotopic membrane proteins
Structures of several monotopic membrane proteins have been solved, which include yeast NADH dehydrogenase (Ndi 1), prostaglandin H2 synthase (PHS), squalene-hopene cyclase (SHC) and the C2 membrane binding domains of coagulation factors Va and Viiia [26–31].
The Ndi1, PHS and SHC are dimeric proteins and are oriented in such a way that the putative membrane binding region of both monomers are coplanar, and thereby bind to the membrane at the same time. MDH forms a 4-fold symmetric tetramer and the electropositive surfaces of the four monomers are coplanar and bind to the membrane similar to those proteins. In the case of the C-2 coagulation factor domains, there are several hydrophobic residues forming short loops which interact with the membrane along with electrostatic stabilization. The presumed membrane binding surface of Ndi 1 is a hydrophobic ridge surrounded on both left and right sides by concentrated positive charge patches. This ridge, which consists of two α-helixes and two β-sheets per monomer, may be inserted into the membrane and the negatively charged lipid head groups would interact with the positively charged patches.
MDH adopts a strategy similar to the Ndi1 and C2- coagulation factor domains in its hydrophobic membrane insertion along with coplanar electrostatic interaction. Moreover, the membrane binding surface of MDH has an unique concentric circular ridge which projects away from the protein surface by ~4Å (Fig. 14) which is not observed in other monotopic membrane proteins to our knowledge. Also, MDH is unique in its interaction with the membrane with its three distinct α-helixes that act like prongs.
5. Conclusions:
The PDB data base contains less than 1% of unique membrane protein entries and the addition of the structure of the monotopic membrane protein (S)-dehydrogenase to this database is very significant. The structure exhibits its possible mode of interaction with the membrane. The coplanar electrostatic interaction between its elevated positively charged concentric circular ridge and the negatively charged head groups of the phospholipid bilayer forms the main stabilization force. The membrane binding segment of this enzyme consists of three α-helices which can provide additional stability by inserting into the membrane. The structure also shows the possible orientation of the helices during membrane interaction along with the roles of individual amino acids in membrane insertion. Based on the structure, it is possible that helix- α1 might be inserted into the membrane deeper than other two helices, as its hydrophobicity is far higher. This leads to a further speculation that the oxidant which is part of membrane electron transport chain may utilize helix- α1 to reach the enzyme’s active site and helix-α3 could act as a gate thereby allowing only the specific oxidant to reach the active site.
Highlights:
-
(1)
Membrane binding segment contains three α-helixes.
-
(2)
Three helixes provide stability in membrane interaction.
-
(3)
Formation of concentric circular positively charged ridge is unique
-
(4)
Electrostatic interaction is the main force of stabilization in membrane interaction.
5. Acknowledgement:
This work and the beamline NECAT 24IDE used to collect data are supported by award GM103403 from NIGMS of NIH and the NIH grant HL-121718 (WL). The Eiger 16M detector on 24IDE beamline is funded by a NIH-ORIP HEI grant (S10OD021527). We thank Professor Ealick, Cornell University and Dr. Bharat Reddy, University of Chicago for helpful discussions and Professor Perozo, University of Chicago for allowing us to use his lab facilities. Use of APS is supported by the U.S. DOE, office of Science and Contract No. DE-AC02– 06CH11357.
Abbreviations:
- MDH
(S)- Mandelate Dehydrogenase
- MDH-GOX2
Chimera of (S)-Mandelate Dehydrogenase
- GOX
Glycolate Oxidase
- FMN
Flavin mononucleotide
- DDM
n-dodecyl-β-D-maltoside
- rms
root mean square
- au
asymmetric unit
Footnotes
Data deposition: The x-ray coordinates of the (S) Mandelate Dehydrgenase have been deposited in the PDB under code 6BFG.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mitra B, Gerlt JA, Babbitt PC, Koo CW, Kenyon GL, Joseph D, Petsko GA, A novel structural basis for membrane association of a protein: construction of a chimeric soluble mutant of (S)-mandelate dehydrogenase from Pseudomonas putida, Biochemistry, 32 (1993) 12959–12967. [DOI] [PubMed] [Google Scholar]
- [2].Sukumar N, Xu Y, Gatti DL, Mitra B, Mathews FS, Structure of an active soluble mutant of the membrane-associated (S)-mandelate dehydrogenase, Biochemistry, 40 (2001) 9870–9878. [DOI] [PubMed] [Google Scholar]
- [3].Stenberg K, Clausen T, Lindqvist Y, Macheroux P, Involvement of Tyr24 and Trp108 in substrate binding and substrate specificity of glycolate oxidase, European journal of biochemistry / FEBS, 228 (1995) 408–416. [PubMed] [Google Scholar]
- [4].Lehoux IE, Mitra B, (S)-Mandelate dehydrogenase from Pseudomonas putida: mechanistic studies with alternate substrates and pH and kinetic isotope effects, Biochemistry, 38 (1999) 5836–5848. [DOI] [PubMed] [Google Scholar]
- [5].Blobel G, Intracellular protein topogenesis, Proceedings of the National Academy of Sciences of the United States of America, 77 (1980) 1496–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xu Y, Mitra B, A highly active, soluble mutant of the membrane-associated (S)-mandelate dehydrogenase from Pseudomonas putida, Biochemistry, 38 (1999) 12367–12376. [DOI] [PubMed] [Google Scholar]
- [7].Sukumar N, Dewanti AR, Mitra B, Mathews FS, High resolution structures of an oxidized and reduced flavoprotein. The water switch in a soluble form of (S)-mandelate dehydrogenase, The Journal of biological chemistry, 279 (2004) 3749–3757. [DOI] [PubMed] [Google Scholar]
- [8].Sukumar N, Dewanti A, Merli A, Rossi GL, Mitra B, Mathews FS, Structures of the G81A mutant form of the active chimera of (S)-mandelate dehydrogenase and its complex with two of its substrates, Acta crystallographica, 65 (2009) 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Otwinowski Z, Minor W, Processing of x-ray diffraction data collected by oscillation methods., Methods Enzymol, 276 (1997) 307–326. [DOI] [PubMed] [Google Scholar]
- [10].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ, Phaser crystallographic software, J. Appl. Cryst, 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adams PD, Grosse-Kunstleve RW, Hung LW, loerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC, PHENIX: building new software for automated crystallographic structure determination, Acta crystallographica, 58 (2002) 1948–1954. [DOI] [PubMed] [Google Scholar]
- [12].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta crystallographica, 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [13].Brunger AT, Free R value: a novel statistical quantity for assessing the accuracy of crystal structures, Nature, 355 (1992) 472–475. [DOI] [PubMed] [Google Scholar]
- [14].Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W, CheckMyMetal: a macromolecular metal-binding validation tool, Acta Crystallogr D Struct Biol, 73 (2017) 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Laskowski R, Thornton J, Moss D, MacArthur M, PROCHECK: a program to check the stereochemical quality of protein structures., J. Appl. Cryst, 26 (1993) 283–291. [Google Scholar]
- [16].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta crystallographica, 66 (2010) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].CCP4, Collaborative Computational Project Number 4, Acta Crystallogr. Sect. D Biol. Crystallogr, 50 (1994) 760–763. [DOI] [PubMed] [Google Scholar]
- [18].Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M, Developments in the CCP4 molecular-graphics project, Acta crystallographica, 60 (2004) 2288–2294. [DOI] [PubMed] [Google Scholar]
- [19].Sukumar N, Crystallographic studies on B12 binding proteins in eukaryotes and prokaryotes, Biochimie, 95 (2013) 976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P, Collaboration gets the most out of software, Elife, 2 (2013) e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xia ZX, Mathews FS, Molecular structure of flavocytochrome b2 at 2.4 A resolution, Journal of molecular biology, 212 (1990) 837–863. [DOI] [PubMed] [Google Scholar]
- [22].Lindqvist Y, Refined structure of spinach glycolate oxidase at 2 A resolution, Journal of molecular biology, 209 (1989) 151–166. [DOI] [PubMed] [Google Scholar]
- [23].Krissinel E, Stock-based detection of protein oligomeric states in jsPISA, Nucleic acids research, 43 (2015) W314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sukumar N, Mathews FS, Langan P, Davidson VL, Role of Protein Dynamics in electron transfer - A joint X-ray and Neutron Study on Amicyanin, Proceedings of the National Academy of Sciences of the United States of America, 107 (2010), 6817–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kyte J, Doolittle RF, A simple method for displaying the hydropathic character of a protein, Journal of molecular biology, 157 (1982) 105–132. [DOI] [PubMed] [Google Scholar]
- [26].Macedo-Ribeiro S, Bode W, Huber R, Quinn-Allen MA, Kim SW, Ortel TL, Bourenkov GP, Bartunik HD, Stubbs MT, Kane WH, Fuentes-Prior P, Crystal structures of the membrane-binding C2 domain of human coagulation factor V, Nature, 402 (1999) 434–439. [DOI] [PubMed] [Google Scholar]
- [27].Wendt KU, Lenhart A, Schulz GE, The structure of the membrane protein squalene-hopene cyclase at 2.0 A resolution, Journal of molecular biology, 286 (1999) 175–187. [DOI] [PubMed] [Google Scholar]
- [28].Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC, Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents, Nature, 384 (1996) 644–648. [DOI] [PubMed] [Google Scholar]
- [29].Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL, Structure of the C2 domain of human factor VIII at 1.5 A resolution, Nature, 402 (1999) 439–442. [DOI] [PubMed] [Google Scholar]
- [30].Picot D, Loll PJ, Garavito RM, The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1, Nature, 367 (1994) 243–249. [DOI] [PubMed] [Google Scholar]
- [31].Iwata M, Lee Y, Yamashita T, Yagi T, Iwata S, Cameron AD, Maher MJ, The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates, Proceedings of the National Academy of Sciences of the United States of America, 109 (2012) 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]