

Abstract
Background
Neonatal sepsis represents a unique therapeutic challenge owing to an immature immune system. Necroptosis is a form of programmed cell death that has been identified as an important mechanism of inflammation-induced cell death. Receptor-interacting protein kinase 1 (RIPK1) plays a key role in mediating this process. We hypothesized that pharmacologic blockade of RIPK1 activity would be protective in neonatal sepsis.
Methods
Sepsis was induced in C57BL/6 mouse pups (5-7 days old) by intraperitoneal injection of adult cecal slurry (CS). At 1 h after CS injection, the RIPK1 inhibitor necrostatin-1 (Nec-1, 10 (ig/g body weight) or vehicle (5% DMSO in PBS) was administered via retroorbital injection. At 20 h after CS injection, blood and lung tissues were collected for various analyses.
Results
At 20 h after sepsis induction, vehicle-treated pups showed a marked increase in serum levels of IL-6, IL-β, and IL-18 compared to sham. With Nec-1 treatment, serum levels of IL-6, IL-β, and IL-18 were decreased by 77%, 81%, and 63%, respectively, compared to vehicle. In the lungs, sepsis induction resulted in a 232-, 10-, and 2.8-fold increase in IL-6, IL-β, and IL-18 mRNA levels compared to sham, while Nec-1 treatment decreased these levels to 40-, 4-, and 8-fold, respectively. Expressions of the neutrophil chemokines KC and MIP-2 were also increased in the lungs in sepsis, while Nec-1 treatment dectreased these levels by 81% and 61%, respectively, compared to vehicle. In addition, Nec-1 treatment significantly improved the lung histologic injury score and decreased lung apoptosis in septic pups. Finally, treatment with Nec-1 increased the 7-day survival rate from 0% in the vehicle-treated septic pups to 29% (P = 0.109).
Conclusions
Inhibition of RIPK1 by Nec-1 decreases systemic and pulmonary inflammation, decreases lung injury, and increases survival in neonatal mice with sepsis. Targeting the necroptosis pathway might represent a new therapeutic strategy for neonatal sepsis.
INTRODUCTION
Globally, 5.9 million children under the age of five died in 2015.1 More than half of these children died of severe infection leading to sepsis.2 Moreover, 44% of those deaths occurred during the neonatal period defined as days of life 0-28.2 Sepsis is the third leading cause of neonatal death.3 Hyper-inflammation is one of the characteristic phenomena in sepsis and contributes to multiple organ damage.4 Currently, treatment of sepsis is largely supportive with antibiotics, source control, and hemodynamic and respiratory support. Neonatal sepsis represents a unique therapeutic challenge owing to an immature immune system. A better understanding of factors contributing to organ deterioration during neonatal sepsis will provide an advanced strategy to manage this life-threatening medical condition.
Cell death is an important factor leading to organ dysfunction in sepsis.5, 6 Two predominate forms of cell death are apoptosis and necrosis. Apoptosis is a form of active cell death that occurs both in normal development and in response to cell death stimuli, such as cell damage or stress. Apoptosis is mediated by a caspase-dependent pathway characterized by increased mitochondrial membrane permeability, chromatin condensation, DNA fragmentation, and cell shrinkage.7 In contrast, necrosis is passive, accidental cell death. Necrosis begins with cell swelling, disruption of the plasma and organelle membranes, and finally the cell lyses to release its intracellular components.7 But another programmed cell death, termed necroptosis, has also been identified.8 Necroptosis is a form of caspase-independent and organized necrosis mediated by cellular stress, inflammation, and infection mediated through stimulation of death receptors.9
The signaling pathway of necroptosis is a multi-stage process culminating in a complex set of regulatory proteins.10-12 Briefly, when TNF-α binds to its receptors, TNFR1 or TNFR2, several proteins, including TNFR-associated DEATH domain protein (TRADD) and receptor interaction protein kinase 1 (RIPK1), are recruited to form a transmembrane complex I. Later, RIPKI is deubiquinated by cylindromatosis (CYLD) and dissociates from complex I to form complex II with RIPK3, caspase-8, and Fas-associated protein with death domain (FADD) in the cytosol. The formation of complex II leads to necroptosis.10-12
In times of tissue injury, necroptosis occurs and leads to cell membrane disruption and the release of damage-associated molecular patterns (DAMPs). DAMPs activate the immune system and trigger inflammation.9 Sharma et al demonstrated that inhibition of necroptosis by knocking down RIPK3 not only decreased systemic inflammation but attenuated organ damage in septic animals.13 By screening of a chemical library, necrostatin-1 (Nec-1) was identified as an allosteric inhibitor of RIPK1 that inhibits the necroptosis pathway specifically.14 Several recent papers have demonstrated that necrostatins are able to protect against or attenuate damage to solid organs in animal models of severe inflammation, spinal cord injury, ischemia/reperfusion, and hemorrhagic shock.15-18
In this study, we hypothesized that inhibition of necroptosis via RIPK1 would be protective in neonatal sepsis. To test this hypothesis, we used an animal model of sepsis induced by intraperitoneal injection of adult cecal slurry (CS) to 5- to 7-day old mouse pups. We then examined the effect of administering Nec-1 on the extent of inflammation and severity of lung injury in the septic neonates. Finally, we conducted a 7-day survival study to evaluate the long-term effect of Nec-1 treatment in neonatal sepsis.
MATERIALS AND METHODS
Experimental animals
Pregnant female C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). They were housed in a standard light and temperature controlled room and fed standard rodent diet. Mice were monitored closely to determine accurately the date of birth of their litters. Neonatal mice were used for experiments on days of life 5 to 7. Pups were kept with their mothers throughout the experiment and breast fed ad libitum. The study was approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research. Experimental procedures were performed in agreement with the National Institutes of Health Guidelines on the Use of Laboratory Animals.
Animal model of neonatal sepsis
Neonatal sepsis was induced in C57BL/6 mice at the age of 5 to 7 days by a CS method adapted from Wynn et al19 with some modifications. At this age, sex of the neonate cannot be determined reliably by external characteristics. To prepare the CS, 3 male and 3 female C57BL/6 mice (11 to 13 weeks) were euthanized by CO2 inhalation, and their cecal contents were collected by laparotomy and cecotomy. The cecal contents were pooled, weighed, and then suspended with 5% dextrose in normal saline to a concentration of 90 mg/ml. The CS was filtered through a 70-μm filter to remove large particles the CS was then aliquoted and immediately frozen in liquid nitrogen and stored at -80°C for later use. A fresh aliquot was used for each experiment within 2 h after thawing. For sepsis induction, neonates were removed as a group from their mothers and placed on a 37°C heating pad. Mice were anesthetized by inhalation with 2.5% isoflurane after which CS [0.9 mg/g body weight (BW)] was delivered by intraperitoneal (IP) injection. After recovery from anesthesia, all neonates were returned to the cage with their mothers as a group. Lung and blood samples were collected 20 h after CS injection and stored at -80°C until analysis. For the survival study, the pups were received a diluted dose of CS (0.175 mg/g BW) and monitored for 7 days after CS injection. Sham mice in the study underwent a procedure similar to the experimental mice but received IP injection of normal saline instead of CS.
Administration of Nec-1
Septic neonates were assigned randomly to treatment or vehicle groups. The treatment group received a retroorbital injection of 10 μg/g BW Nec-1 stable (EMD Millipore, Billerica, MA) in a volume of 8 μl/g BW of 5% DMSO/PBS at 1 h after CS injection. The vehicle group received an equivalent volume of 5% DMSO/PBS retroorbitally. In the survival study, due to the reduction in the amount of CS injection, pups received a 6 μg/g BW dose of Nec-1 or equivalent volume vehicle (6 μl/g BW) retroorbitally. The dose of Nec-1 was based on previous publications.20, 21
Enzyme-linked immunosorbent assay (ELISA)
Serum was analyzed by ELISA kits specific for interleukin (IL)-6, IL-β (BD Biosciences, San Jose, CA), and IL-18 (MBL International, Woburn, MA) according to the manufacturer’s instructions.
Real-time polymerase chain reaction (qPCR)
Total RNA was extracted from lung tissues using TRIzol reagent (Invitrogren, Carlsbad, CA). Total RNA (2 μg) underwent reverse transcription using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA). The qPCR reaction was carried out in a 20 μl final volume containing 0.25 μl each of forward and reverse primers, 2 μl cDNA, 7.5 μl DEPC-treated water, and 10 μl Power SYBR Green PCR Master Mix (Applied Biosystems). An Applied Biosystems StepOnePlus real-time PCR machine was used for amplification under the thermal cycling profile of 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Mouse β-actin mRNA levels were used for normalization. Relative expression of mRNA was calculated by the 2-ΔΔCt method, and results are expressed as fold change in comparison to the sham group. The sequence of primers for this study is listed as follow: IL-6 (NM_031168), forward (CCGGAGAGGAGACTTCACAG) and reverse (CAGAATTGCCATTGCACAAC); IL-1β (NM_008361), forward (CAGGATGAGGACATGAGCACC) and reverse (CTCTGCAGACTCAAACTCCAC); IL-18 (NM_008360), forward (GCCTCAAACCTTCCAAATCA) and reverse (TACAGTGAAGTCGGCCAAAG); KC (NM_008176), forward (GCTGGGATTCACCTCAAGAA) and reverse (ACAGGTGCCATCAGAGCAGT); MIP-2 (NM_009140), forward (CCCTGGTTCAGAAAATCATCCA) and reverse (GCTCCTCCTTTCCAGGTCAGT); and β-actin (NM_007393), forward (CGTGAAAAGATGACCCAGATCA) and reverse (TGGTACGACCAGAGGCATACAG).
Histologic evaluation
Lung tissue was placed in 10% formalin prior to embedding in paraffin. Tissue was cut into 5-μm sections and stained with hematoxylin and eosin (H&E). Light microscopy was used to evaluate the degree of lung injury in a blinded fashion. The severity of injury was scored using a system for the assessment of acute lung injury in experimental animals outlined by the American Thoracic Society.22 The weighted score ranged from zero to one and took into account neutrophil infiltration in the alveolar and interstitial spaces, the presence of hyaline membranes, the presence of proteinaceous debris in the airspaces, and the degree of septal thickening. The average of the scores per field was calculated as the final lung injury score in each group.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Lung tissue sections were dewaxed, rehydrated, and immersed in 20 mg/ml of proteinase K at room temperature for 20 min. Tissue samples were then stained with a TUNEL kit (Roche Diagnostics, Indianapolis, IN) and counterstained with DAPI. Apoptotic cells appeared green under a fluorescence microscope and were counted at 200× magnification in 5 visual fields/section. The average of the number of apoptotic cells/field was calculated.
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM) and compared by one way analysis of variance (ANOVA and the Student-Newman-Keuls (SNK) test. Survival rate was determined by the Kaplan-Meier estimator and compared by a log-rank test. Differences in values were considered significant if P < 0.05.
RESULTS
Nec-1 decreased systemic inflammation in neonatal sepsis
The extent of systemic inflammation reflects the severity of sepsis.23 We measured serum levels of proinflammatory cytokines IL-6, IL-1β, and IL-18 in the neonates. At 20 h after CS injection, IL-6 levels were increased from 0.32 ng/ml in the sham to 50.62 ng/ml, while Nec-1 treatment decreased the levels of Il-6 by 77% compared to the vehicle (Fig 1, A). Similarly, IL-1β levels in the vehicle group were increased by 251-fold from 1.6 pg/ml in the sham to 396.3 pg/ml at 20 h after CS injection, while Nec-1 treatment decreased IL-β levels by 81% compared to the vehicle (Fig 1, B). IL-18 levels were increased to 1,545 pg/ml after CS injection, whereas its levels were non- detectable in the sham (Fig. 1, C). Nec-1 treatment decreased IL-18 levels by 63% compared to the vehicle (Fig. 1, C).
Fig. 1.
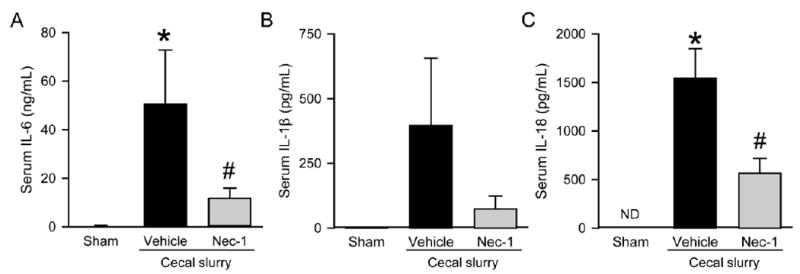
Effect of Nec-1 on serum cytokine levels in septic neonates. C57BL/6 mouse pups were subjected to sepsis by intraperitoneal injection of cecal slurry (CS) and treated with vehicle (5% DMSO/PBS) or Nec-1 (10 (μg/gin vehicle) 1 h thereafter. Serum samples were collected at 20 h after CS injection and measured for (A) IL-6, (B) IL-1β, and (C) IL-18 by ELISA. Data shown as mean ± SEM (n=5-7/group) and compared by ANOVA and SNK test. ND, non-detectable; *P <0.05 vs. sham; #P < 0.05 vs. vehicle.
Nec-1 decreased inflammatory cytokine gene expression in the lungs of septic neonates
Because lung injury is responsible for a substantial percentage of morbidity and mortality in sepsis,24 we then examined inflammation at the pulmonary level. At 20 h after CS injection, there were a significant 232-, 10-, and 2.8-fold increase in the mRNA expressions of IL-6, IL-1β, and IL-18, respectively, in vehicle-treated pups as compared to sham (Fig 2, A-C). Their expression levels were decreased in the Nec-1-treated group by 83%, 60%, and 71% , respectively, compared to the vehicle group (Fig 2, A-C).
Fig. 2.
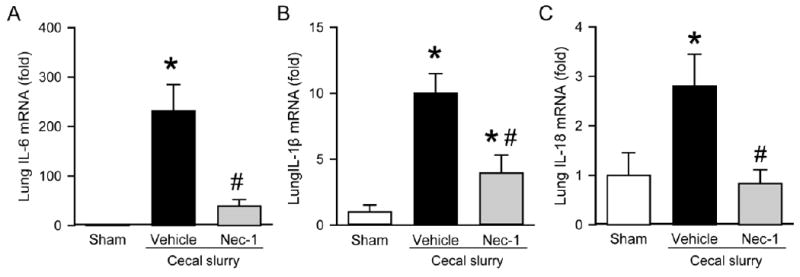
Effect of Nec-1 on inflammatory cytokine expression of mRNA in the lungs of septicneonates. Sepsis was induced as in Figure 1. Lung tissues were collected at 20 h after CS injection. The mRNA levels of (A) IL-6, (B) IL-1β, and (C) IL-18 were measured by qPCR. Each gene expression level was normalized to β-actin. The value in the sham group is designated as 1 for comparison. Data shown as mean ± SEM (n=5-7/group) and compared by ANOVA and SNK test. *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
Nec-1 attenuated lung injury in neonatal sepsis
Lung architecture was examined histologically at 20 h after CS injection. Lung injury was apparent in the vehicle-treated group, as judged by histologic analysis showing marked thickening of the alveolar septal wall, neutrophil infiltration, and congestion compared to the sham (Fig 3, A). In contrast, Nec-1- treated pups demonstrated improved lung morphology compared to the vehicle group (Fig 3, A). Overall, the lung histologic injury score was increased 2.4-fold in the vehicle group compared to sham, whereas the score was decreased by 38% in the Nec-1-treated group as compared to vehicle (Fig 3, B).
Fig. 3.
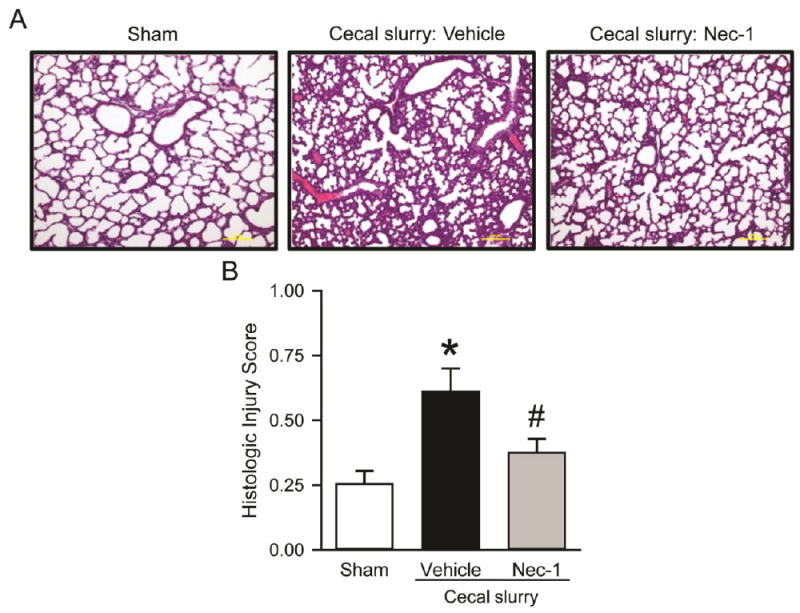
Effect of Nec-1 on lung morphology after neonatal sepsis. Sepsis was induced as in Figure 1. Lung tissues were collected at 20 h after CS injection and subjected to histologic analysis. (A) Representative images of H&E-stained lung tissue sections under light microscope at 100× magnification. (B) Histologic injury scores of the lung in each group were graded as described in Methods. Data shown as mean ± SEM (n=5-7/group) and compared by ANOVA and SNK test. *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
Nec-1 decreased gene expression of inflammatory chemokines in the lungs of septic neonates
Excessive neutrophil infiltration into tissues is another factor that contributes to organ damage.25 The motility of neutrophils is in part regulated by various chemokines.26 We next evaluated neutrophil infiltration into the lungs by measuring the expression of the neutrophil chemoattractants KC and MIP-2 at 20 h after CS injection. There was a significant increase in mRNA levels of both KC and MIP-2 in vehicle treated pups by 654- and 313-fold as compared to the sham, while their levels were decreased by 81% and 61%(, respectively, with Nec-1 treatment as compared to vehicle. (Fig 4, A and B).
Fig. 4.
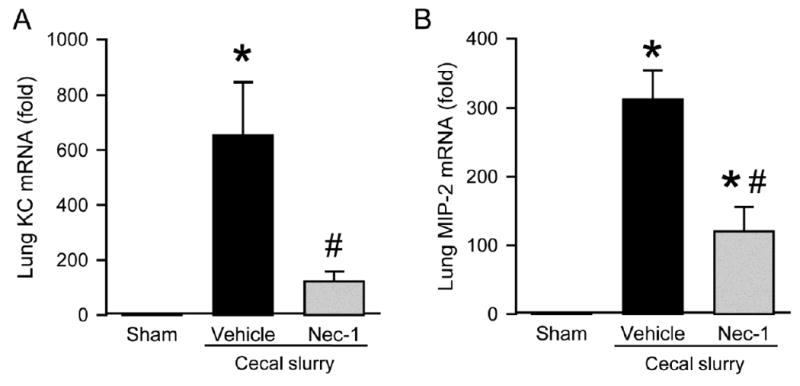
Effect of Nec-1 on inflammatory chemokine expression of mRNA in the lungs of septic neonates. Sepsis was induced as in Figure 1 \. Lung tissues were collected at 20 h after CS injection. The mRNA levels of (A) KC and (B) MIP-2 were measured by qPCR. Each gene expression level was normalized to β-actin. The value in the sham group is designated as 1 for comparison. Data shown as mean ± SEM (n=5-7/group) and compared by ANOVA and SNK test. *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
Nec-1 decreased apoptosis in the lungs of septic neonates
Apoptosis in the lungs can be triggered by inflammation and is an important component of tissue damage and immune dysfunction in sepsis.27 We used the TUNEL assay to detect apoptotic cells in the lungs of the neonates. As shown in Fig 5, A, apoptotic cells were barely detected in the sham, while many TUNEL-positive cells were seen in the vehicle-treated pups. With Nec-1 treatment, the number of the TUNEL-positive cells was decreased by 60% from 13 cells/HPF in the vehicle group to 5.2 cells/HPF (Fig 5, B).
Fig. 5.
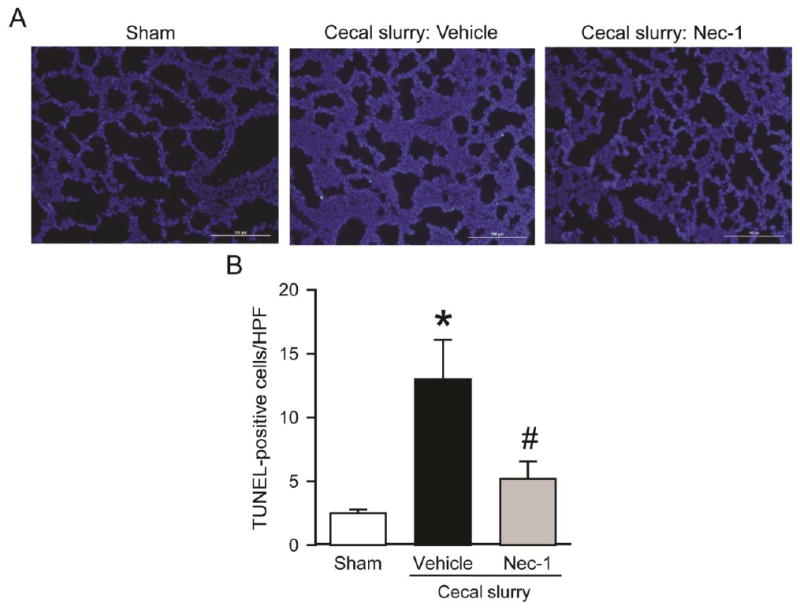
Effect of Nec-1 on lung apoptosis after neonatal sepsis. Sepsis was induced as in Figure 1. Lung tissues were collected at 20 h after CS injection. (A) Representative images of lung tissue sections stained with TUNEL (green fluorescence) for apoptosis and DAPI (blue fluorescence) for nuclei at 200× magnification. (B) The graph represents the number of TUNEL-positive cells averaged over 10 high power fields (HPF)/section. Data shown as mean ± SEM (n=5-7/group) and compared by ANOVA and SNK test. *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
Nec-1 improved survival in neonatal sepsis
To further determine the overall long-term effect of Nec-1 treatment, we performed a survival study in the septic neonates. Pups were injected with a diluted concentration of CS followed by Nec-1 or vehicle treatment one hour later. These neonates were monitored daily for seven days. All pups in the vehicle group were dead within 3 days, while there was a 29% survival in the Nec-1-treated group after 7 days (Fig. 6).
Fig. 6.
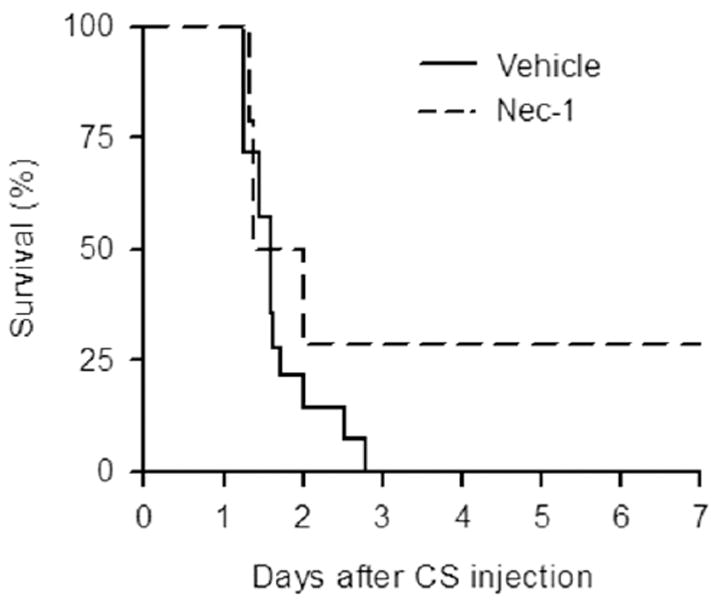
Effect of Nec-1 on the survival of septic neonates. C57BL/6 mouse pups were subjected to sepsis as in Figure 1. treated with vehicle (6% DMSO/PBS) or Nec-1 (6 (μg/g) 1 h after. Pups were monitored for survival for seven days. Survival rates were analyzed by the Kaplan-Meier estimator using a log-rank test. There was no survival in the vehicle group compared to 29% survival in the Nec-1 group (n=14/group; P = 0.109).
DISCUSSION
Childhood sepsis, including neonatal sepsis, is a global problem. Worldwide, sepsis remains one of the leading causes of pediatric death. There are an estimated 30 million cases of pediatric sepsis annually with approximately 8 million mortalities.28 Neonatal sepsis is a particularly challenging condition due to differences in the immune system of neonates and older children/adults.29
Sepsis has been defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.30 The neonatal immune response to pathogens is bimodal. The first phase appears to be a largely unregulated, disproportionate release of proinflammatory cytokines. This is sometimes referred to as a “cytokine storm” and can quickly lead to organ dysfunction and death. The second phase is the secretion of anti-inflammatory cytokines. All together, this can result in relative immune suppression, another factor that contributes to the morbidity and mortality of neonatal sepsis.31
In this study, using an established model of murine polymicrobal intra-abdominal neonatal sepsis, we demonstrated that treatment with Nec-1, an inhibitor of RIPK1, after induction of sepsis decreased systemic and pulmonary inflammation, attenuated lung injury, and decreased neutrophil chemoattractant expression and the number of apoptotic cells in the lungs. We also demonstrated that Nec-1 treatment improved a 7-day survival rate in septic neonates. These results suggests that necroptosis plays an important role on the severity and outcomes in neonatal sepsis.
It is well documented that levels of the b pro-inflammatory cytokines TNF-α, IL-β, IL-and IL-8 increase rapidly and substantially during the early phases of neonatal sepsis. In addition, a recent report indicated that serum IL-18 levels were increased in neonatal sepsis and suggested that disruption of the IL-18/IL-1R1/IL-17A axis represents a novel therapeutic approach for human neonates with sepsis. 32 Given this, cytokine levels have been investigated as a diagnostic tool for early identification of sepsis in neonates, a patient population in whom initial signs of sepsis are frequently nonspecific.33 Both the severity and the prognosis of neonatal sepsis are thought to be related to the discordance between proinflammatory and anti-inflammatory cytokine levels.34 As expected, we detected that serum levels of IL-6, IL-β, and IL-18 are markedly increased at 20 hours after CS injection. Treatment with Nec-1 can effectively decreased their levels in the circulation, supporting the potential of developing Nec-1 against neonatal sepsis in clinical studies.
In addition to generalized systemic effects, neonatal sepsis frequently results in acute lung injury or worsening of chronic lung conditions. Furthermore, infants with sepsis with respiratory compromise have increased morbidity and mortality compared to infants without respiratory compromise.35 For this reason, we examined lung tissue closely in this study. In this neonatal sepsis model, we observed increased local inflammation in the lungs, as evidenced by the increase in expression of IL-6, IL-β, and IL-18 mRNA at 20 hours after CS injection for sepsis induction. This inflammation was significantly attenuated by treatment with Nec-1. Neutrophil infiltration is another factor that contributes to lung injury; infiltration is initiated by the increase in neutrophil chemoattractants. We also detected increased expression of KC and MIP-2 mRNA in the lungs of septic neonates. Again, their expressions were significantly decreased in the septic neonates treated with Nec-1.
Moreover, histologic examination revealed marked morphologic changes with edema and parenchymal damage in the lungs of the neonates at 20 hours after CS injection. In addition we detected the apoptotic cells indicated by TUNEL-positive staining after sepsis induction. With Nec-1 treatment, lung damage was mitigated, and lung apoptosis was inhibited in septic neonates. Taken together, we have demonstrated that inhibition of necroptosis can limit inflammation and lung injury in this model of neonatal sepsis in the mouse.
Inflammation can be induced by both infectious and non-infectious etiologies. In times of infection, pathogen-associated molecular patterns (PAMPs) stimulate inflammation by binding to their receptors and triggering the expression of pro-inflammatory cytokines and chemokines. In sterile injury, stimuli of cell death can lead to necrosis and the release of DAMPs which in turn activate an inflammatory response.36, 37 In sepsis, DAMPs and PAMPs can work alone or in combination to stimulate inflammation. There is an exaggerated and prolonged proinflammatory cytokine response to PAMPs from the invading organisms, and DAMPs are released from cells dying in the circulation.38
The protective effects of necrostatin in adult small animal models of both sterile and infectious modes of inflammation have been documented in recent years. Necrostatin has been shown to protect myocardial tissue in rats with acute myocardial infarction,39 liver tissues in murine models of ischemia/reperfusion injury,40 against acute respiratory distress syndrome in rats,41 and neural tissue in brain ischemia/reperfusion studies in both rats and mice.42, 43 Additionally, inhibition of necroptosis (via RIPK3 gene deletion) has been shown to be protective in an adult animal model of polymicrobial sepsis.13, 21 Moreover, we recently reported that the severity of organ injury and inflammation are attenuated in the septic neonates from RIPK3-deficiency mice in comparison with the wild-type mouse neonates.44 It is difficult to identify the formation of complex II for necroptosis in the tissue samples; however, the activity of Nec-1 in inhibiting necroptosis has been well demonstrated. Whether Nec-1 has other effect needs to further investigation. To our knowledge this is the first study demonstrating the protective effects of necrostatin in neonatal sepsis. In the meantime, inhibitors of RIPK1 have progressed to the clinical stage. In 2016, Denali Therapeutics (South San Francisco, CA) announced its intention to start a Phase 1 clinical trial for necrostatin in healthy volunteers to assess its safety, tolerability, and pharmacokinetics (PRNewswire, Aug. 25, 2016). They also planned to treat patients suffering from amyotrophic lateral sclerosis and Alzheimer’s disease. GSK2982772, another RIPK1 inhibitor discovered by GlaxoSmithKline, is currently in Phase 2a clinical studies for psoriasis, rheumatoid arthritis, and ulcerative colitis.45, 46
The development of therapeutic agents for sepsis is quite challenging due to disappointing outcomes from multiple clinical trials.47 Sepsis is composed of both hyperinflammatory and immunosuppresive phases with substantial overlap between the two.48 In addition, over-inhibition of the inflammatory response with a treatment may lead to immunosuppression and increased risk of secondary infection. So far, there is no suitable biomarker that can quickly and accurately identify the stage of sepsis in patients. Therefore, targeted therapies to meet individual patient needs have been difficult to achieve. Another contributing factor to the failure of novel therapies of sepsis is the gap in the translation of animal studies to clinical outcomes. Although the development of a better animal model has advocated testing drugs, current rodent models still can provide a tool to study the pathophysiology of sepsis and to identify a drug-able target at its early development stage. In this study, we demonstrated the beneficial effects of Nec-1 on attenuating inflammation and improving survival in septic mouse neonates, suggesting that necroptosis plays a role in regulating the severity of sepsis, but we acknowledge that other pathways and mechanisms also contribute to the deleterious effects of sepsis. A combined treatment strategy, like often employed in cancer treatment, may be another option for sepsis. Our finding provides a proof- of-concept that RIPK1 can be a target for treating neonatal sepsis. RIPK1 inhibitors are currently in the clinical studies against other inflammatory diseases. Thus, development of RIPK1 inhibitors as therapeutic agents for sepsis is warranted for further investigation.
It is known that sex affects immune response to sepsis. In our study, however, due to the difficulty in identification of the sex of a neonatal mouse, we are unable to classify mice by their exprior to treatment. This is a limitation of using 5- to 7-day neonates for the study. In summary, we have demonstrated that in a murine model of neonatal sepsis, Nec-1 decreased systemic and pulmonary inflammation and decreased the histologic lung injury and apoptosis. Finally, we have shown that treatment of septic pups with Nec-1 improved 7-day survival from non-survival to 29%. Therefore, pharmacologic targeting of the necroptosis pathway may provide a novel strategy for managing neonatal sepsis
Acknowledgments
Supported by the National Institutes of Health grant R35GM118337 to PW.
Abbreviations
- ANOVA
analysis of variance
- CS
cecal slurry
- CYLD
Cylindromatosis
- DAMPs
damage-associated molecular patterns
- DAPI
4’6-diamidino-2-phenylindole
- ELISA
Enzyme-linked immunosorbent assay
- FADD
Fas-associated protein with death domain
- H&E
hematoxylin and eosin staining
- IL-1β
interleukin 1-beta
- IL-6
interleukin 6
- IL-18
interleukin 18
- IP
intraperitoneal
- KC
Keratinocyte chemoattractant
- MIP-2
Macrophage-Inflammatory Protein-2
- Nec-1
Necrostatin-1
- PAMPs
pathogen-associated molecular patterns
- PBS
phosphate buffered saline
- RIPK1
Receptor-interacting protein kinase 1
- RIPK3
Receptor-interacting protein kinase 3
- SEM
Standard error of the mean
- SNK
Student-Newman-Keuls test
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- TNF-α
tumor necrosis factor alpha
- TNFR
tumor necrosis factor alpha receptor
- TRADD
TNFR-associated DEATH domain protein
Footnotes
Published in part as an abstract for the 12th Annual Conference on Academic Surgical Congress, February 7 9, 2017, Las Vegas, NV.
The authors declare no conflict of interest for this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.You D, Hug L, Ejdemyr S, Idele P, Hogan D, Mathers C, et al. Global, regional, and national levels and trends in under-5 mortality between 1990 and 2015, with scenario- based projections to 2030: a systematic analysis by the UN Inter-agency Group for Child Mortality Estimation. Lancet. 2015;386:2275–86. doi: 10.1016/S0140-6736(15)00120-8. [DOI] [PubMed] [Google Scholar]
- 2.Rahman AE, Iqbal A, Hoque DM, Moinuddin M, Zaman SB, Rahman QS, et al. Managing Neonatal and Early Childhood Syndromic Sepsis in Sub-District Hospitals in Resource Poor Settings: Improvement in Quality of Care through Introduction of a Package of Interventions in Rural Bangladesh. PLoS One. 2017;12:e0170267. doi: 10.1371/journal.pone.0170267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qazi SA, Stoll BJ. Neonatal sepsis: a major global public health challenge. Pediatr Infect Dis J. 2009;28:S1–2. doi: 10.1097/INF.0b013e31819587a9. [DOI] [PubMed] [Google Scholar]
- 4.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329–42. doi: 10.1189/jlb.0912437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhan C, Dipankar P, Chakraborty P, Sarangi PP. Role of cellular events in the pathophysiology of sepsis. Inflamm Res. 2016;65:853–68. doi: 10.1007/s00011-016-0970-x. [DOI] [PubMed] [Google Scholar]
- 6.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–59. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Smith CC, Yellon DM. Necroptosis, necrostatins and tissue injury. J Cell Mol Med. 2011;15:1797–806. doi: 10.1111/j.1582-4934.2011.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 10.Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015;6:e1975. doi: 10.1038/cddis.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol. 2012;82:249–58. doi: 10.1016/j.critrevonc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–37. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma A, Matsuo S, Yang WL, Wang Z, Wang P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit Care. 2014;18:R142. doi: 10.1186/cc13970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Wang H, Tao Y, Zhang S, Wang J, Feng X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience. 2014;266:91–101. doi: 10.1016/j.neuroscience.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Su X, Wang H, Kang D, Zhu J, Sun Q, Li T, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem Res. 2015;40:643–50. doi: 10.1007/s11064-014-1510-0. [DOI] [PubMed] [Google Scholar]
- 17.Wen S, Ling Y, Yang W, Shen J, Li C, Deng W, et al. Necroptosis is a key mediator of enterocytes loss in intestinal ischaemia/reperfusion injury. J Cell Mol Med. 2017;21:432– 43. doi: 10.1111/jcmm.12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Dai W, Lin C, Wang F, He L, Shen M, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013;2013:706156. doi: 10.1155/2013/706156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynn JL, Scumpia PO, Delano MJ, O’Malley KA, Ungaro R, Abouhamze A, et al. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28:675–83. doi: 10.1097/SHK.0b013e3180556d09. [DOI] [PubMed] [Google Scholar]
- 20.Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62:1847–57. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–38. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinhart K, Bauer M, Riedemann NC, Hartog CS. New approaches to sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev. 2012;25:609–34. doi: 10.1128/CMR.00016-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American- European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–24. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 25.Hoesel LM, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, et al. Harmful and protective roles of neutrophils in sepsis. Shock. 2005;24:40–7. doi: 10.1097/01.shk.0000170353.80318.d5. [DOI] [PubMed] [Google Scholar]
- 26.Alves-Filho JC, de Freitas A, Spiller F, Souto FO, Cunha FQ. The role of neutrophils in severe sepsis. Shock. 2008;30(Suppl 1):3–9. doi: 10.1097/SHK.0b013e3181818466. [DOI] [PubMed] [Google Scholar]
- 27.Shao L, Meng D, Yang F, Song H, Tang D. Irisin-mediated protective effect on LPS- induced acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. 2017;487:194–200. doi: 10.1016/j.bbrc.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 28.Dugani S, Kissoon N. Global advocacy needed for sepsis in children. J Infect. 2017;74(Suppl 1):S61–S5. doi: 10.1016/S0163-4453(17)30193-7. [DOI] [PubMed] [Google Scholar]
- 29.Kumar SK, Bhat BV. Distinct mechanisms of the newborn innate immunity. Immunol Lett. 2016;173:42–54. doi: 10.1016/j.imlet.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–10. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khaertynov KS, Boichuk SV, Khaiboullina SF, Anokhin VA, Andreeva AA, Lombardi VC, et al. Comparative Assessment of Cytokine Pattern in Early and Late Onset of Neonatal Sepsis. J Immunol Res. 2017;2017:8601063. doi: 10.1155/2017/8601063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wynn JL, Wilson CS, Hawiger J, Scumpia PO, Marshall AF, Liu JH, et al. Targeting IL- 17A attenuates neonatal sepsis mortality induced by IL-18. Proc Natl Acad Sci U S A. 2016;113:E2627–35. doi: 10.1073/pnas.1515793113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye Q, Du LZ, Shao WX, Shang SQ. Utility of cytokines to predict neonatal sepsis. Pediatr Res. 2017;81:616–21. doi: 10.1038/pr.2016.267. [DOI] [PubMed] [Google Scholar]
- 34.Machado JR, Soave DF, da Silva MV, de Menezes LB, Etchebehere RM, Monteiro ML, et al. Neonatal sepsis and inflammatory mediators. Mediators Inflamm. 2014;2014:269681. doi: 10.1155/2014/269681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tewabe T, Mohammed S, Tilahun Y, Melaku B, Fenta M, Dagnaw T, et al. Clinical outcome and risk factors of neonatal sepsis among neonates in Felege Hiwot referral Hospital, Bahir Dar, Amhara Regional State, North West Ethiopia 2016: a retrospective chart review. BMC Res Notes. 2017;10:265. doi: 10.1186/s13104-017-2573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem. 2016;85:743–63. doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- 37.Kearney CJ, Martin SJ. An Inflammatory Perspective on Necroptosis. Mol Cell. 2017;65:965–73. doi: 10.1016/j.molcel.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 38.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5:36–44. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu YR, Xu HM. Protective effect of necrostatin-1 on myocardial tissue in rats with acute myocardial infarction. Genet Mol Res. 2016:15. doi: 10.4238/gmr.15027298. [DOI] [PubMed] [Google Scholar]
- 40.Hong JM, Kim SJ, Lee SM. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol Appl Pharmacol. 2016;308:1–10. doi: 10.1016/j.taap.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 41.Pan L, Yao DC, Yu YZ, Li SJ, Chen BJ, Hu GH, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem Biophys Res Commun. 2016;478:1602–8. doi: 10.1016/j.bbrc.2016.08.163. [DOI] [PubMed] [Google Scholar]
- 42.Yang R, Hu K, Chen J, Zhu S, Li L, Lu H, et al. Necrostatin-1 protects hippocampal neurons against ischemia/reperfusion injury via the RIP3/DAXX signaling pathway in rats. Neurosci Lett. 2017;651:207–15. doi: 10.1016/j.neulet.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 43.Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–89. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen LW, Jacob A, Yang WL, Bolognese AC, Prince J, Nicastro JM, et al. Deficiency of receptor-interacting protein kinase 3 (RIPK3) attenuates inflammation and organ injury in neonatal sepsis. J Pediatr Surg. 2017 doi: 10.1016/j.jpedsurg.2017.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris PA, King BW, Bandyopadhyay D, Berger SB, Campobasso N, Capriotti CA, et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J Med Chem. 2016;59:2163–78. doi: 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- 46.Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem. 2017;60:1247–61. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 47.Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 2014;13:741–58. doi: 10.1038/nrd4368. [DOI] [PubMed] [Google Scholar]
- 48.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–74. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]