

I. Introduction
Since the start of the pandemic in the early 1980’s, HIV infection has been a major public health crisis across the world. Currently, around 37 million people are infected with HIV, with 1.8 million of them infected in 2016 alone. Although HIV-infection affects every region of the globe, the majority of infections are found in in sub-Saharan Africa and southeast Asia (UNAIDS 2017). Initially a terminal diagnosis, the advent of combinatorial antiretroviral therapy (cART or HAART) in 1996 dramatically improved the prognosis for HIV, enabling infection to be managed as a chronic condition rather than a terminal illness [1-3]. This therapeutic strategy treated patients with multiple (generally 3) antiretroviral drugs simultaneously, preventing escape mutations within the virus and thereby significantly improving the suppression of HIV replication. Successful cART has lengthened the lifespan and improved the quality of life for infected individuals. However, effects of cART have also created new health issues, as chronic infection and long-term exposure to antiretrovirals have created a suite of new metabolic, cardiovascular and neurologic disorders in infected individuals.
Even when HIV replication is fully suppressed with cART, around 50% of infected individuals still display a variety of neuropathological and neurocognitive sequelae known as NeuroHIV, or when referring specifically to the neurocognitive effects, as HIV-associated neurocognitive disorders (HAND) [1, 4]. While severe forms of neurocognitive impairment are rare in the cART era, the prevalence of HAND is increasing, and deficits in executive functioning, working memory, and psychomotor fluency are still frequently observed in HIV patients [2, 5, 6]. These can significantly impair therapeutic adherence [7] and accelerate the development of peripheral disease [8]. The development of these disorders is initiated by HIV infection of the central nervous system (CNS), which occurs in nearly all infected individuals shortly after initial infection [9, 10]. Within the CNS, the primary targets for HIV are myeloid lineage cells, such as microglia and perivascular macrophages. Infection of these cells is central to HIV neuropathogenesis, as infected myeloid cells generate new virions to further spread infection, and both infected and uninfected myeloid cells produce inflammatory mediators in response to infection [11-16]. The persistence of neurocognitive impairment in virally suppressed individuals suggests subclinical alterations in neurotransmission, neuronal and immune function may underlie the earliest manifestations of NeuroHIV in the cART era [17-23].
Dysregulation of catecholaminergic neurotransmission, particularly the dopaminergic system, has long been correlated with the development of neuroinflammation and HAND. Studies specifically examining role of catecholamines in HIV pathogenesis, particularly focused on NeuroHIV, are relatively few. However, over the course of the epidemic, researchers found that infected individuals show a number of catecholaminergic changes. These include increased damage in dopaminergic regions of the CNS, altered autonomic nerve activity, changes in catecholamine metabolism, stress induced changes in infection and response to cART, and altered viral replication and dysregulated immune responses resulting from changes to catecholaminergic tone [19, 24-48]. Further, data show that catecholamines, particularly dopamine and norepinephrine, are important mediators for neuroimmune crosstalk [47, 49-53]. As the dysregulated immune response is central to the etiology of NeuroHIV, these data suggest that disruptions in catecholaminergic tone in response to HIV infection, drug abuse, stress or specific therapeutic drugs, could exacerbate HIV neuropathogenesis. The dearth of studies in this area, particularly those dissecting the mechanisms by which catecholamines mediate their effects on NeuroHIV, has hindered our ability to understand and effectively treat these components of HIV neuropathogenesis. Therefore, this review will discuss what is known about the role of catecholamines in the development of NeuroHIV. We briefly discuss the state of CNS infection in the cART era, touching on the specific effects in dopaminergic and adrenergic systems, and then discuss catecholamine biology and what is known about the catecholaminergic systems in immune cells. We will then focus on catecholaminergic modulation of HIV infection and neuroinflammatory processes, and how these effects might enhance the development of NeuroHIV. The neurologic complications of HIV are a growing problem within the infected population, making the development of new neuroprotective strategies a pressing medical need. This review will contribute to a better understanding of the bidirectional interactions between catecholamines and HIV infection of the CNS, leading to novel therapeutic targets for the treatment of NeuroHIV.
II. HIV neuropathogenesis in the cART era
Prior to the widespread use of cART, HIV infection of the CNS commonly resulted in significant neuropathology including HIV encephalitis (HIVE), meningitis, microglial nodules, multinucleated giant cells, reactive gliosis, and neuronal injury and death [12, 34, 54, 55]. Severe neurocognitive impairment was also common, with HIV-associated dementia (HAD) found in 15-20% of infected individuals. Significant neuropathology was often coincident with neurocognitive impairment in many infected individuals, but the relationship between neurological damage and cognitive impairment is not entirely causal. Studies demonstrated that HIVE did not explain all HIV-associated dementia, and the best correlate for the development of HIV-associated neuropathology was myeloid cell activation in the CNS [16, 34, 56]. With cART, HIVE and HAD are almost nonexistent, and severe forms of neurocognitive impairment are rare, but the prevalence of mild and asymptomatic forms of neurocognitive impairment have increased, and HAND is still found in 40 – 70% of HIV infected individuals [1, 4, 57-59]. Despite suppression of HIV replication below the limit of detection, fully suppressed individuals still show signs of NeuroHIV, and data show no correlation between neuropathology, neurocognitive impairment, and CNS viral load [23, 58-63]. This indicates that the development of HIV-associated neurological disease does not derive solely from damage associated with actively replicating virus [64-66].
NeuroHIV is initiated by HIV crossing the blood-brain barrier (BBB) to enter the CNS. This is thought to occur primarily via the transmigration of infected CD14+CD16+ monocytes [67], using the monocytes as “Trojan Horses” [68], although some studies suggest alternate routes of entry [69]. These mature CD14+CD16+ monocytes, are enriched in people with HIV and are more susceptible to HIV infection [67, 70]. The enrichment of this population is maintained in the presence of cART treatment [71], and is important to the development of HAND due to the disproportionate transmigration of CD14+CD16+ cells into the CNS [67, 72, 73]. Within the CNS, infected monocytes differentiate into perivascular macrophages and shed new virions, which infect additional macrophages and microglia. In addition to being the primary target for HIV infection in the brain [11-15], myeloid cells are the primary reservoir for HIV in this compartment [74, 75]. Astrocytes are infected at low levels [79, 80], and pericytes may also be infected [81], but the role of these cells in neuropathogenesis is unclear.
Although T-cells are the primary focus of HIV in the periphery, in the CNS the primary cells involved in neuropathogenesis are myeloid cells. This is because relatively few T-cells are present in the CNS, and these mostly surveil brain regions outside the parenchyma [76-78]. Increased T-cells, particularly CD8+ T-cells, are found in the brains of HIV-infected drug abusers and those with CNS-immune reconstitution inflammatory syndrome[47]. Polymorphonuclear neutrophils (PMNs) are the most abundant immune cells in the blood and can contribute to chronic immune activation during HIV infection [82]. However, PMNs do not seem to be infected by HIV [83] and their role in HIV-neuropathogenesis is not clear. Because the cells primarily targeted by HIV are T-cells and macrophages, these cells will be the focus of the remainder of this review.
In healthy individuals, CNS macrophages and microglia act as the primary immune response within the CNS, communicating with neurons by releasing a variety of cytokines and neurotrophic factors important to neuronal health and function [84-87]. Infection of these cells dysregulates production and release of inflammatory cytokines and neurotoxic viral proteins, activating neighboring uninfected cells and inducing further production of factors that perpetuate the neuroinflammatory environment. Infected or activated myeloid populations are thought to be primary drivers of neuroinflammation, and prior to cART, development of dementia correlated with myeloid cell activation [14, 15, 56]. Even with cART, immune activation is a critical component in the development of neurologic disease [88]. Infected brain tissue from cART-treated individuals shows upregulated CD68+ expression, as well as CD16 and CD163 positivity, indicative of significant macrophage and microglia accumulation and activation [66]. Soluble factors of monocyte activation, CD14 and CD163, are increased in the plasma and cerebrospinal fluid (CSF) of HIV+ individuals with neurocognitive impairment [89-92]. Activated monocytes and macrophages produce a number of inflammatory factors including cytokines (IL-6, IL-1β, TNF-α, interferons), chemokines (CCL2, CXCL8, CXCL9, CXCL10), hydrolytic enzymes (matrix-metalloproteinases), and oxidative mediators (nitric oxide) [93, 94]. Production of these and other inflammatory and cytotoxic factors is thought to be central to the neuropathology of HIV infection in cART treated individuals [1, 14, 16, 34, 95].
Overall, while cART has significantly prolonged life and ameliorated HIV-associated disease, HIV infection of the CNS still induces a number of cognitive, behavioral and motor symptoms, along with substantial neuropathology. Data show that the inflammatory processes driving neurological disease persist in the individuals using fully suppressive cART [66, 88, 95-98], indicating that these processes are mediated by interactions distinct from viral replication. Over time, the damage from this chronic inflammation accumulates, interfering with neurotransmission and dysregulating neuroimmune communication. Many of these issues overlap with neurological issues associated with aging, and the synergy between these problems is growing [58] as the age of the HIV-infected population increases. Further, the ability to suppress but not eliminate HIV has resulted in an increased focus on the insults initiated by chronic infection and neuroinflammation, and the resultant changes in neurotransmission and CNS homeostasis.
IIa. The CNS viral reservoir
Reservoirs, are cell populations or anatomical sites that enable HIV to avoid the immune response, and are the primary impediment to eradication of HIV within an infected individual [99]. The importance of reservoirs is shown by the fact that HIV viremia recurs rapidly following interruption of suppressive cART [100, 101], and HAND occurs even in individuals on uninterrupted, suppressive cART therapy [2, 59, 61]. This suggests the CNS reservoir established prior to cART treatment may play an important role and is supported by studies identifying the lowest measured CD4 cell count, CD4 nadir, as one of the best predictors of neurocognitive impairment [102, 103]. As CD4 nadir generally corresponds to a period of unchecked viral replication, during which CNS invasion and establishment of the viral reservoir occur, the importance of CD4 nadir could indicate that some portion of CNS viral insults may be permanent. This is further supported by studies demonstrating lower CD4 nadir is associated with greater long-term decreases in subcortical gray matter and total white matter volume [104-107]. Although the most commonly studied reservoirs are long-lived CD4+ memory T-cells, many studies show that the CNS, which is an immune privileged site, is also often considered to be a reservoir [99, 108]. Post-mortem brain tissue from pre-symptomatic HIV-infected individuals showed HIV-1 gag DNA in parenchymal macrophages and microglia despite the absence of HIV p24 expression [109]. Another study found the amount of HIV DNA in peripheral blood mononuclear cells (PBMC) correlates with the severity of HAND [110], and the follow-up showed patients with HAND maintained high levels of HIV DNA in activated monocytes throughout a five-year course of cART [111]. While these and other studies do not clarify whether the CNS reservoir is latent or active [75], there is substantial evidence showing that the CNS reservoir exists in long term infected perivascular macrophages and microglia [99, 108, 112], which can support HIV-infection for long periods without cytotoxic effects [113-115].
IIb. Dopamine in HIV Neuropathogenesis
The involvement of the dopaminergic system in HIV neuropathogenesis has been hypothesized since very early in the epidemic. In the pre-cART era, elevated neuropathology was found in dopaminergic brain areas including the caudate and putamen, the substantia nigra and the prefrontal cortex (PFC) [12, 24-26, 30, 56, 58, 116-118]. In brain tissue from HIV-infected individuals, dopamine-rich brain regions such as the basal ganglia show the greatest degree of HIV-associated neuroinflammation [18, 95]. These regions also showed increased numbers of infected cells, and greater amounts of viral RNA [12, 28, 117, 118]. These effects were corroborated in simian immunodeficiency virus (SIV) infected macaques treated with L-DOPA and Selegiline to increase CNS dopamine levels. The increased dopamine in these animals significantly increased CNS viral load and exacerbated neuropathology [31, 32]. HIV infection and exposure to viral proteins also interferes with dopamine transporter expression and function [38, 119, 120], and dopaminergic neurons are particularly vulnerable to HIV-induced neurotoxicity [35, 121, 122]. Treatment with cART has shifted regional neuropathology, but patients still show substantial damage to dopamine-rich regions, including striatal dysfunction, increased inflammation, neuronal damage, and diminished resting state functional connectivity [123-128]. PET scans of cART treated HIV-patients with no history of illicit drug use found increased microglial activation in the basal ganglia [129]. High expression of HIV is still seen in these regions [28, 130], as is accumulation of HIV-infected microglia [131]. Additional studies which are discussed below also show specific effects of dopamine HIV infection and myeloid-mediated neuroinflammatory functions.
Polymorphisms in dopamine related genes, and changes in dopaminergic gene expression are also seen during HIV infection. Autopsy data from HIV+ patients have revealed correlations between dopaminergic gene expression and inflammation, suggesting that inflammatory mediators might be linked to dopamine signaling in HIV [19]. Several studies have connected genetic alterations in dopamine receptors (DRD) to neurocognitive impairment. Failure to downregulate the DRD2L gene is associated with unfavorable neuropsychologic and neuropathologic outcomes [19]. The rs6280TC single nucleotide polymorphism (SNP) in DRD3 is correlated with increased rates of cognitive impairment in HIV+ methamphetamine addicts [132], and several SNPs in DRD1 and DRD2 correlated with cognitive function in HIV+ drug abusers [133]. A strong correlation between a specific allele of the dopamine transporter (DAT), DAT 10/10, and HIV infection was found in German and African cohorts of HIV-positive and negative individuals. Individuals with the DAT10/10 allele, whether or not they were infected, showed higher baseline levels of CSF dopamine, suggesting that elevated dopamine is involved in HIV infection [134]. They also showed these changes in CSF dopamine and HIV infection were not correlated with genetic polymorphisms in catechol-O-methyltransferase (COMT, Val158Met), an enzyme involved in catecholamine metabolism, and dopamine receptors DRD2, DRD3, and DRD4 [135], although the specific DRD polymorphisms investigated were not the same as those mentioned above. Interestingly, the Val158Met polymorphism in COMT, while not associated with increased HIV infection, was found by separate groups to be associated with deficits in executive function and prefrontal activity in HIV infected individuals [136, 137].
Infection with HIV may also interfere with dopamine metabolism, thereby altering dopamine concentrations in the CNS. Infected individuals and SIV-infected macaques show decreases in CSF dopamine [27], increased DOPAC, and decreased homovanillic acid (HVA) in the CSF [43, 138]. Studies show decreased CSF dopamine levels and neuronal degeneration in dopaminergic brain regions of HIV patients [42, 139, 140]. This is supported by experiments in SIV-infected macaques showing decreased dopamine and tyrosine hydroxylase (TH)-positive neurons, as well as increased DOPAC in putamen [141]. However, more recent studies from this group found increased CSF dopamine in HIV patients during asymptomatic infection prior to cART initiation [142, 143]. In these studies, CSF dopamine was inversely correlated with CD4 cell counts, suggesting that elevated dopamine may be associated with disease progression [142, 143]. Using behavioral testing, these dopaminergic changes can be detected as dysfunction of frontostriatal circuits that reflect HIV-associated dopaminergic abnormalities [17, 126, 144]. Examination of brain tissue from HIV-infected individuals shows abnormal expression of dopaminergic synaptic proteins in striatum [33] and PFC [19], perhaps reflecting an adaptive shift in response to elevated dopaminergic tone. Together, these data suggest that HIV infection is still facilitated in a dopamine-rich environment. Further, the increase in infection damages dopamine neurons and neuronal structures, interfering with dopaminergic neurotransmission, even in individuals on effective cART. This interference could alter dopaminergic tone in different regions of the CNS, exacerbating the effects of dopamine on HIV infection in these regions and may play a significant role in the development of NeuroHIV.
IIc. Impact of Drug Induced Dopamine in HIV Neuropathogenesis
Unlike the interactions of catecholamines and HIV, the effects of drug-abuse on HIV neuropathogenesis are well studied [145-150], but it is important to mention them as almost all abused substances increase CNS dopamine [151-159]. Thus, the effects of dopamine on HIV infection are particularly important for HIV-infected drug abusers, who constitute 10 – 20% of the HIV-infected population worldwide [160-165]. While there is disagreement in the literature about the direction and specific impacts of drug abuse on HIV infection, it is generally agreed that drugs of abuse can synergistically disrupt dopaminergic transmission [24, 122], and can to alter the development of HAND [166-168]. This suggests HIV-infected drug abusers would be particularly susceptible to dopamine-mediated changes in neuropathogenesis, as the dopaminergic brain regions specifically affected by drugs of abuse, particularly the basal ganglia, are the regions in which exacerbated neuropathology has been found in the presence of dopamine. Indeed, HIV+ individuals with a history of illicit drug use show increased neuropathology and encephalitis at autopsy [170-174], and in the cART era, drug-abuse history remains one of the best predictors of neurocognitive decline [168, 173, 175-180]. Thus, use of illicit drugs, or of therapeutics that disrupt the dopaminergic system, could exacerbate the development of NeuroHIV in drug using HIV+ populations. A major caveat regarding many studies which clearly show an impact of drug abuse on distinct stages of HIV infection and HIV infected cells, is that these experiments are not necessarily examining the impact of dopamine on HIV infection. One issue with these types of studies is that many of them, in both animal models and humans, examine peripheral pathogenesis [181-184]. As addictive drugs exert the majority of their dopaminergic effects within the CNS and dopamine cannot cross the BBB, the mechanisms involved are not likely mediated by elevated dopamine. Similarly, there are many studies which show potentiation of HIV infection by addictive drugs in vitro, in various immune cell types [185-189], particularly human macrophages [190-193]. These studies have provided a tremendous amount of useful information, but a significant caveat for this type of research is that it is generally performed in monoculture systems. It is not known whether treatment of macrophages or T-cells in vitro with drugs such as morphine or cocaine elicits a dopamine response, and it is not likely that the response would be the same as that generated by in vivo exposure of dopamine neurons within the central nervous system. Despite these caveats, the data still demonstrate that use of illicit drugs, or of dopaminergic therapeutics, could exacerbate the development of NeuroHIV by disrupting the dopaminergic neurotransmission in the CNS. Therefore, developing an improved understanding of the reciprocal interactions by which dopaminergic dysfunction and HIV-associated neuropathogenesis exacerbate each other will be critical to future therapeutics required in the developing HIV epidemic.
IId. Adrenergic Catecholamines in HIV Neuropathogenesis
The interaction of the adrenergic system with HIV neuropathogenesis has been studied far less than the interactions of dopamine and HIV. Studies show significant neuroinflammation and atrophy occurs in HIV-infected brain regions encompassing the CNS adrenergic system, such as the brainstem and pons [194-197], with elevated HIV RNA in the hippocampus proper [118]. However, HIV associated-neuropathology in specific adrenergic brain regions such as the locus coeruleus remains undetermined. Early in the epidemic, prior to cART, Glasgow and colleagues found significant adrenal pathology in HIV-infected individuals [198]. In the cART-era, the hippocampus remains significantly impacted, with increased myeloid cell activation [95, 199] and decreased hippocampal volumes [200], and no studies have specifically examined adrenergic brain regions in HIV-infected individuals on cART.
Most of the studies that have examined the adrenergic impact on HIV infection focus on the interactions with sympathetic nervous system (SNS) or the hypothalamus-pituitary-adrenal (HPA) axis. These studies show that the SNS and/or HPA axis are dysregulated in HIV-infected individuals, with decreases in norepinephrine during stress response [39], and hypo-reactivity of the autonomic system and HPA axis [41, 201]. There is a direct correlation between constitutive autonomic nervous system activity and HIV plasma viral load [45], and elevated stress levels, autonomic nervous system activity and norepinephrine are correlated with a poorer response to cART therapy [202, 203]. These studies suggest that the release of the adrenergic catecholamines, particularly norepinephrine, could enhance HIV infection, providing a pathway by which stress or other adrenergic stimulators might impact systemic HIV pathogenesis [40, 46, 204]. This is corroborated by a more recent study which also found that higher levels of peripheral norepinephrine correlate with accelerated disease progression, represented by increased plasma viral load and decreased CD4 counts, over a 4-year period [205]. These studies indicate that disruptions in adrenergic signaling may be important in the development of systemic infection, but that much more work needs to be done in this area.
Studies specifically examining norepinephrine in the context of NeuroHIV are rare, although Dever and colleagues recently found an increase in β-adrenergic receptor gene expression in frontal lobe white matter and/or frontal cortex from HIV-positive individuals with combined neurocognitive impairment and HIV encephalitis [206]. In SIV-infected macaques, enhanced stress reduced survival [207, 208], and SIV replication was enhanced in close proximity to elevated catecholamine levels present in catecholaminergic varicosities [48]. In rodents, the HIV envelope protein gp120 interferes with the β-adrenergic receptor mediated function of microglia and astrocytes [209]. Thus, in the CNS, elevated norepinephrine could exacerbate HIV infection activation of myeloid β-adrenergic receptors, which are increased in the infected brain. As both norepinephrine and epinephrine are increased with stress, a common issue in HIV-infected individuals, these changes may exacerbate both peripheral and CNS infection in a significant proportion of the HIV-infected population. Unfortunately, the data on this subject are quite sparse, and the full effects of adrenergic catecholamines on both the HIV infection process and the associated inflammatory effects remain relatively undefined. To better treat both long term HIV infection and the chronic diseases associated with it, it is important to further define the role of adrenergic catecholamines in HIV infection.
III. Catecholamines
Catecholamines are monoamines, organic compounds containing a catechol ring (ortho-dihydroxybenzene) linked to an amino side-group. Dopamine, norepinephrine or noradrenaline, and epinephrine or adrenaline are catecholamine neurotransmitters derived from the amino acid tyrosine. Catecholamines are synthesized in their cognate neurons, both in cell bodies and in the nerve terminals. From there they are rapidly transported and stored in the endoplasmic reticulum or in vesicles along dendrites and at synaptic terminals [210, 211]. The release and reuptake of these molecules is highly-regulated through complex mechanisms, which act independently or in conjunction with other regulatory mechanisms. These molecules play vital roles in the modulation of behavior, metabolism, autonomic function, and immunity, acting in both the periphery and the CNS [212]. In the CNS, dopamine, norepinephrine, and to a much lesser extent epinephrine enable interneuronal communications that modulate neuronal activity and influence behavior. While the majority of studies on the actions of catecholamines have been performed in neurons, recent studies show that all catecholamines also have immunomodulatory actions. These occur in a number of different immune cell types, including T-cells, myeloid cells and neutrophils, and has been widely reviewed [47, 213-218]. Although these and many other studies clearly demonstrate an immunoregulatory effect of catecholamine interaction with immune cells, it is important to note that many of them used pharmacologic levels (10−6M and higher) of catecholamines or catecholamine receptor agonists/antagonists. Therefore, it is not entirely clear how human immune cells in the CNS, where catecholaminergic concentrations remain unclear, respond to catecholaminergic stimulation in vivo. Further, during HIV infection, these effects may be changed by HIV-associated disruption in catecholaminergic tone.
IIIa. Catecholamine Signaling and Metabolism
Catecholamine metabolism is a critical step in the regulation of activity, as the amount of catecholamine to which a receptor is exposed is controlled by a) reuptake through DAT or the norepinephrine (NET) transporter, and b) synthesis and degradation of the catecholamine. All catecholamines are derived from tyrosine, and are synthesized through a series of enzymatic reactions, beginning with the rate-limiting step, the conversion of tyrosine to L-DOPA by TH (Figure 1). Dopamine is then synthesized from L-DOPA by the action of DOPA decarboxylase, and then either released or hydroxylated by dopamine β-hydroxylase (DBH) to form norepinephrine. The final step in catecholamine biosynthesis is the conversion of norepinephrine to epinephrine by phenylethanolamine-N-methyltransferase (PNMT). There are also a number of pathways by which catecholamines can be catabolized, which are primarily mediated by COMT and monoamine oxidase (MAO), although additional enzymes are active in several stages of the catabolic cycle (Figure 1).
Figure 1.
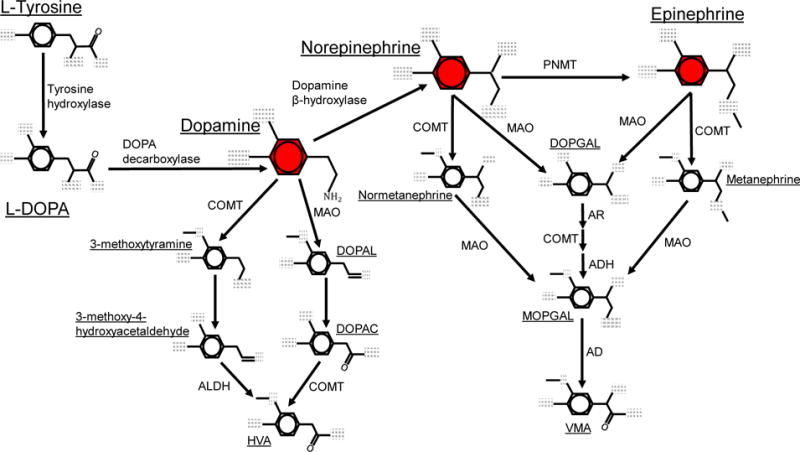
Metabolic pathways for catecholamine biosynthesis and degradation. Metabolic pathways for the formation and degradation of dopamine, norepinephrine and epinephrine (shown in red) are described. Catecholamine synthesis is initiated with the hydroxylation of the amino acid tyrosine is by tyrosine hydroxylase (TH), generating L-DOPA. L-DOPA is then converted to dopamine by DOPA decarboxylase (DDC, also known as aromatic L-amino acid decarboxylase, AADC). Dopamine is hydroxylated by dopamine β-hydroxylase (DBH) to form norepinephrine, which is then converted to epinephrine by phenylethanolamine-N-methyltransferase (PNMT). The catecholamines are primarily metabolized by two enzymatic pathways with catechol-O-methyltransferase (COMT) and monoamine oxidase (MAO). COMT converts dopamine to 3-methoxytyramine, norepinephrine to normetanephrine, and epinephrine to metanephrine via meta-O-methylation. MAO converts dopamine to 3,4-Dihydroxyphenylacetaldehyde (DOPAL), and norepinephrine or epinephrine to 3,4-didydroxyphenylclycoaldehyde (DOPGAL) by oxidative deamination. MAO also converts 3-methoxytyramine to 3-methoxy-4-hydroxyacetaldehyde, and the metanephrines to an unstable aldehyde monohydroxyphenylglycol aldehyde (MOPGAL). MOPGAL is ultimately converted to the final product vanillyl mandelic acid (VMA) by aldehyde reductase. In the final steps of dopamine metabolism, COMT and aldehyde dehydrogenase (ALDH) convert DOPAL and 3-methoxy-4-hydroxyacetaldehyde to the final product homovanilic acid (HVA).
The actions of all catecholamines are mediated through their cognate receptors, dopamine or adrenergic receptors, which are members of the G-protein coupled receptor superfamily [219]. Dopamine receptors are divided into two groups, D1-like (D1 and D5), which couple to Gαs/olf, and D2-like (D2, D3, D4) which are coupled to Gαi/o [219, 220]. Canonically, D1-like receptors activate adenylate cyclase, increasing intracellular cAMP, while D2-like receptors inhibit adenylate cyclase, blocking intracellular cAMP production [219-221]. These receptors can also signal through alternative pathways, the most prominent being mobilization of intracellular Ca2+ and activation of protein kinase C (PKC) through activation of phospholipase C (PLC). These pathways are thought to be mediated by either the Gq/11 protein coupling to D5 or D1:D2 heteromers, or by D2-like dopamine receptors signal through Gβγ, although the specific mechanisms remain controversial [222-224]. Another pathway exclusively activated by D2-like receptors acts through β-arrestin 2 to regulate the activity of Akt and glycogen synthase kinase 3-α/β (GSK3β) [219, 225]. Adrenergic receptors are also divided into two subgroups, α and β, and can be further subdivided into α1, α2, β1, β2, and β3 receptors [226]. Each of the α receptors has distinct pharmacology, with the α 1-receptors coupled to Gαq, and stimulating PLC, while α2-receptors are coupled to Gαi, inhibiting the production of cAMP [227]. All β-adrenergic receptors couple to Gαs, meaning that activation stimulates the production of cAMP [228, 229]. Interestingly, data also show that in some circumstances, β2-receptors may also couple to Gαi, inhibiting cAMP production and potentially interfering with the effects of other β-adrenergic receptors [230].
IIIb. Catecholaminergic System in Immune Cells
Over the past two decades, the data has found that many types of immune cells, including both myeloid cells and T-lymphocytes, express all five dopamine receptors, as well as all the α and β adrenergic receptors. Human monocytes, monocyte-derived macrophages (MDMs) [52, 231, 232] and microglia express mRNA for all five dopamine-receptor subtypes, with the exception of D5 not being found in microglia [233]. We have shown D1, D2, D3 and D4 expression in the MDM plasma membrane, and other studies have also identified dopamine receptors on monocytes by flow cytometry [231, 232, 234-237]. Analysis of mRNA expression and surface proteins also confirm expression of all five dopamine-receptor subtypes in T-cells, although D1-like receptor expression is low and not consistent between studies [234, 238-241].
Expression of adrenergic-receptors on immune cells has been more widely studied than dopamine-receptors. Immunoblotting and pharmacologic assays show human monocytes and macrophages express functional α1, β1, and β2 receptors [242-249]. These cells respond functionally to stimulation by α2 specific agonists [244, 250], however expression of α2 receptors has not been established. The presence of functional adrenergic receptors in human microglia has not been confirmed, but mRNA and pharmacologic data indicate that α1, α2, β1, and β2 receptors are present in rat microglia [251, 252]. T-lymphocytes also express mRNA and protein for β1, β2 and β3 receptors [241, 253]. Pharmacological studies and ligand-binding experiments also indicate that T-cells express α2 receptors [254, 255], but expression of α1 receptors has not been established.
In addition to expression of dopamine and adrenergic receptors, many immune cells express TH, DAT, and the vesicular monoamine transporter 2 (VMAT2) [236, 241, 256, 257]. Although expression of NET is undefined in human cells, NET and MAO-A expression was shown in rodents in a subset of macrophages called sympathetic neuron–associated macrophages (SAMs) [258]. Our data also show that MDM express both the genes (2A) and proteins (2B) for the majority of the enzymes involved in the metabolism of catecholamines, including MAO-A and B, COMT and very small amounts of DBH (Figure 2). Gene expression of PNMT was not found (data not shown). The purpose of immune catecholaminergic systems is not clear, but in T-cells, dopamine acts in an autocrine fashion to regulate both proliferation and TGF-β production [241, 259], and dopamine, but not norepinephrine or epinephrine, blocked a PKC-mediated induction of TH mRNA expression [257]. Elderly human microglia were found to be able to take up and release dopamine after K+ stimulation [233], and catecholamine uptake and release was shown in human PBMC [49, 241, 257, 259, 260]. These data indicate that immune cells can produce, release and metabolize of catecholamines, supporting the concept that these processes are immunoregulatory.
Figure 2. Human macrophages express enzymes necessary for the synthesis of norepinephrine and degradation of catecholamines.
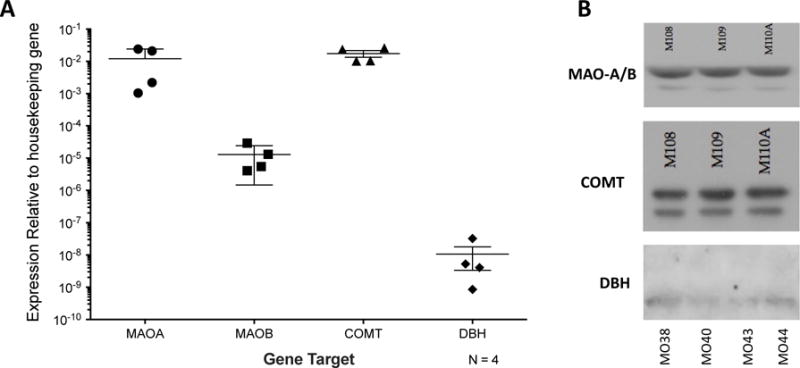
Human macrophages were generated from peripheral blood mononuclear cells that were isolated from whole blood by ficoll density centrifugation, then matured into monocyte-derived macrophages (MDM) by adherence and maturation for 6 days in 10 ng/mL M-CSF. (A) Expression of mRNA for MAO-A and MAO-B, COMT and DBH is seen in mRNA derived from MDM from 4 donors. (B) Analysis of protein lysates from MDM derived from 3 or 4 additional donors shows expression of MAO (antibody did not distinguish A and B), COMT, and DBH protein.
IIIc. Distribution of Catecholamines in the Central Nervous System
The catecholamines found in the central nervous system are primarily dopamine and norepinephrine, while the majority of epinephrine is found in the periphery. Epinephrine has been reported in a small number of brainstem neurons with projections to the thalamus and spinal cord, but very little is known about these neurons [261, 262]. Both dopamine and norepinephrine mediate their effects through volume transmission, acting on distinct brain regions to regulate a number of critical CNS functions. The pathways by which these catecholamines interact with different brain regions are described in Figure 3. As dopamine and norepinephrine are the predominant catecholamines present in the CNS, they are the primary catecholamines influencing the development of NeuroHIV and will be the focus for the remainder of this review.
Figure 3. Anatomical location of catecholaminergic brain regions and projections.
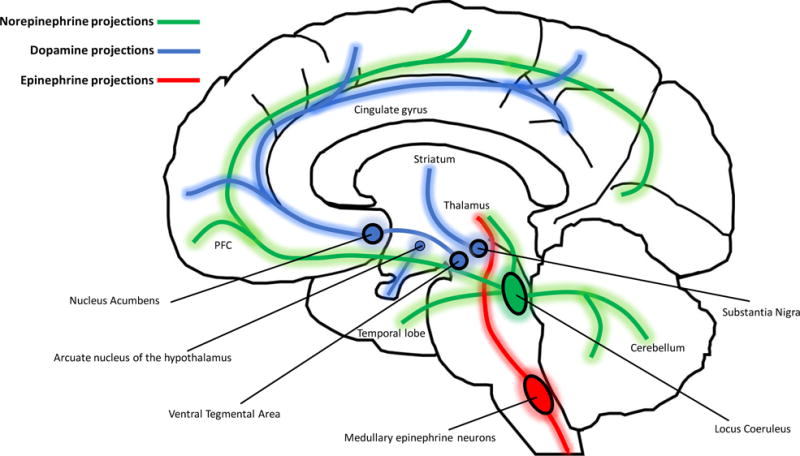
Dopamine is predominantly synthesized in midbrain nuclei that give rise to three major dopaminergic projections. The mesocortical pathway arises from the ventral tegmental area (VTA), and ascends via the median forebrain bundle to innervate prefrontal and cingulate cortices to modulate cognition, motivation, and emotional response. The VTA is also the origin of the mesolimbic pathway that projects to the nucleus acumbens, medial PFC, amygdala, hippocampus to mediate reward-based habits, emotions, and cognition. The nigrostriatal pathway ascends from the midbrain substantia nigra pars compacta (SNpc) to striatal nuclei (caudate and putamen) in the forebrain to modulate production and coordination of movement. There is an additional small group of dopamine neurons in the arcuate nucleus of the hypothalamus that project to the median eminence (via the tuberoinfundibular pathway) where dopamine is released into the hypophyseal portal system and transported to the anterior pituitary to inhibit the release of prolactin. Norepinephrine neurons from the locus coeruleus in the brainstem broadly innervate cortical and midbrain regions including the PFC, temporal lobe (hippocampus and amygdala), thalamus, hypothalamus, and cerebellum. Outside the brain, norepinephrine is released as a transmitter from postganglionic sympathetic nerve terminals near the spinal cord, and released as a hormone from the adrenal medulla into the blood stream. The role of norepinephrine is to modulate alertness and arousal (via cortical projections), and autonomic functions (via symphathetic nerves and adrenal medulla) in order to mobilize the body for action. Unlike norepinephrine and dopamine, the majority of epinephrine is synthesized in the periphery, with more than 90% of circulating epinephrine produced by chromaffin cells in the adrenal medulla. Some epinephrine may be synthesized in medullary epinephrine neurons that project to the thalamus and spinal cord, although very little is known about the function of these neurons.
In the CNS, dopamine is synthesized in dopaminergic neurons of the ventral tegmental area (VTA) (mesocortical and mesolimbic projections), substantia nigra (nigrostriatal projections), and arcuate nucleus of the hypothalamus (tuberoinfundibular pathway) [263]. Dopaminergic projections from the VTA ascend via the median forebrain bundle to innervate cortical and limbic regions. The mesocortical pathway projects to the prefrontal and cingulate cortices and plays major role in cognition, motivation, and emotional response. The mesolimbic pathway projects to the nucleus acumbens, medial PFC, amygdala, and hippocampus to mediate reward-based habits, emotions, and cognition. Dysregulation of this pathway by dopaminergic stimuli plays a major role in the development of various addictions. The nigrostriatal pathway ascends from the midbrain substantia nigra pars compacta (SNpc) to striatal nuclei (caudate and putamen) in the forebrain to modulate production and coordination of movement. Dopaminergic neurons of the tuberoinfundibular pathway project from the arcuate nucleus to the median eminence and release dopamine into the hypophyseal portal system, regulating the release of prolactin from the anterior pituitary. Norepinephrine is predominantly synthesized in the locus coeruleus, as well as the lateral tegmental area of the brainstem. Neurons from the loci coerulei innervate a number of different CNS regions, releasing norepinephrine by volume transmission to modulate alertness and arousal. As mentioned, epinephrine is mostly peripheral, and is synthesized by chromaffin cells in the adrenal medulla. Norepinephrine and epinephrine both act on the autonomic nervous system, regulating fight-or-flight responses, as well as a number of other physiologic functions on tissues throughout the body.
In contrast to fast-acting, synaptic neurotransmitters like glutamate and GABA, dopamine and norepinephrine are more diffusely released along their axonal network in a process known as volume transmission [264, 265]. On midbrain and cortex neurons, most DAT and many dopamine receptors are located extrasynaptically [266-269], so neuronally released dopamine must diffuse through the extracellular space to bind to its receptors and to be taken back up into cells. Synaptic dopamine receptors are also present (particularly in the striatum) and are activated by precise synaptic dopamine release [267, 270]. However, following quantal dopamine release, diffusion occurs too quickly for extra-synaptic DAT uptake, resulting in extra-synaptic spillover and exposure of extra-synaptic cells to dopamine [271, 272]. The size of the region exposed to dopamine depends on complex interactions between the volume and concentration of dopamine released, the volume of extracellular fluid volume, barriers to diffusion, uptake dynamics of DAT, and the sensitivity of regional dopamine receptors [273]. HIV associated damage to dopaminergic neurons can disrupt these interactions and dysregulate dopaminergic tone [36, 119], and all drugs of abuse and many therapeutics interfere with dopamine synthesis, release, uptake, or metabolism, potentially disrupting volume transmission [274, 275]. Thus, HIV infection, particularly when combined with drugs of abuse or dopaminergic therapeutics, exposing other CNS cell types including macrophages and microglia to altered levels of dopamine.
The precise amount of catecholamines to which CNS cells are exposed depends on the catecholamine concentrations in the human CNS. These are not well defined in the human brain, but in primates and rodents, basal dopamine levels are generally reported to be between 5 – 40 nM depending on the brain region [276-278]. These can be elevated into the low micromolar range by drugs of abuse [152, 155, 279-283], which is likely to result in significant spillover into the surrounding tissue, exposing immune cells and glia to elevated dopamine. Methamphetamine can also release norepinephrine via reversal of VMAT and DAT [284], increasing levels of norepinephrine in the CNS. The basal levels of norepinephrine in the locus coeruleus and the PFC have been reported between 1-5 nM, and may be elevated by 300% during phasic stimulation [285, 286]. Amphetamine increased basal levels of norepinephrine in the PFC and locus coeruleus up to 50 nM [287], although the significance of this finding in compared to other natural stimuli is not known. The concentration of epinephrine in the hypothalamus has been reported to be lower than 100 picomolar, and is been below the limit of detection in other brain regions [288, 289].
IV. Effects of Dopamine on HIV infection
Dopamine has long been associated with retroviral infection, as studies almost four decades ago showed dopamine-receptor antagonists such as chlorpromazine and haloperidol have a lytic effect on retroviruses and inhibit reverse transcriptase [290, 291]. More recently, Rohr and colleagues found pharmacologic dopamine increases HIV replication in Jurkat T-cells and PBMCs by promoting viral transcription through NF-κB [292, 293], and dopamine also increased HIV production in chronically infected ACH-2 T-cells [294]. Increasing dopamine availability in the CNS of SIV-infected macaques using the MAO-inhibitor selegiline and L-DOPA significantly enhanced CNS viral replication, increased microglial activation, induced vacuole formation and disrupted dendritic architecture [31, 32]. These studies indicated that dopamine, a neurotransmitter, might contribute to the progression of NeuroHIV by directly increasing infection in immune cells.
This connection is supported by studies in the SIV macaque model of HIV, where treatment with methamphetamine, which greatly enhances CNS dopamine levels [283], increased brain viral load. Methamphetamine also increased expression of the HIV co-receptor CCR5 in CNS macrophages, thereby enhancing the susceptibility of these cells to infection [295-297]. Ongoing studies in our lab have also examined the effects of dopamine exposure on HIV infection in vitro using primary human macrophages. These studies use dopamine concentrations (10−10 M – 10−5 M) present in the CNS during drug abuse, homeostatic and pathologic conditions [155, 276, 277, 279-282]. They show that exposing macrophages to elevated dopamine increases their susceptibility to infection. The effect on macrophages is critical to the development of NeuroHIV, as myeloid cells are thought to be the primary drivers of HIV neuropathogenesis. Primary MDM were infected with HIVYU2 or HIVBaL, HIV strains that use the CCR5 co-receptor, which is the main co-receptor involved in the infection of macrophages. Infections in the presence of dopamine, or specific agonists for D1-like (SKF38393) and D2-like (Quinpirole) dopamine receptors show that activation of all dopamine receptor subtypes increases HIV entry into macrophages. Pretreatment with the pan-dopamine receptor antagonist flupenthixol, or the CCR5 antagonist TAK779, blocked the effects of dopamine on HIV entry, indicating that the mechanism is dependent on both CCR5 and dopamine-receptors [231, 298].
The involvement of both D1- and D2-like dopamine receptors suggests activation of a signaling pathway common to both subtypes, although canonically, different dopamine receptor subtypes mediate opposing effects. However, both subtypes of dopamine receptors can also induce Ca2+ release from the endoplasmic reticulum [223], which has been connected with HIV infection in a number of experiments. Interaction of HIV-gp120 with CCR5 induces Ca2+ mobilization via Gαq [299-302], and Ca2+ flux is an essential step for in HIV entry in an astrocyte model of infection [303]. Our own studies in DR1 and DR2-transfected HEK293 cells show that dopamine receptor activation potentiates Ca2+ mobilization via initiated by Gαq [298]. Together, this indicates that Ca2+ release may be the common mechanism by which dopamine receptor activation enhances entry and suggests that PLC-mediated Ca2+ mobilization may be a novel therapeutic target in the prevention of HIV infection.
Although our data point to a Ca2+ mediated mechanism, dopamine-mediated potentiation of HIV infection act through other mechanisms, such as the NF-kB mediated effects shown in T-cells by Rohr and colleagues [292, 293]. Enhanced oxidative stress is another possible mechanism, as dopamine oxidizes to form free radicals and reactive dopamine quinones [304], and studies show HIV LTR driven reporter expression and reactivation of latent HIV in T-cells can be mediated by the interaction of reactive oxygen species (ROS) and NF-kB activation [305, 306]. The effects of dopamine in ACH-2 cells resulted from treatment with pharmacologic levels of dopamine (6 – 10 × 10−5 M), which increased HIV production via an oxidative mechanism. This effect was prevented by the addition of the antioxidant glutathione, indicating that the reactivation of HIV by dopamine was due to oxidative stress [294]. All these data come from T-cells, suggesting this cell type may be more vulnerable to virologic effects induced by oxidative stress, perhaps because macrophages require exposure to ROS for proper polarization and effector function [307, 308]. While our studies show 24-hour exposure to dopamine is not cytotoxic in macrophages (Gaskill et. al., unpublished results), other studies show murine bone-marrow derived macrophages (BMDM) exposed to pharmacologic dopamine (5 × 10−6M) for 24 hours did increase expression of oxidative stress markers [309]. Taken together, these data suggest that dopamine may enhance HIV infection through several different effector pathways, and that these might differ with distinct cell types.
V. Effects of Norepinephrine on HIV infection
The few in vitro studies specifically examining the effects of norepinephrine on HIV infection suggest that this catecholamine can influence the HIV replication process, although the precise mechanism is unclear. Cole and colleagues found concentrations of norepinephrine (10−8 to 10−5M) dose-dependently increased HIV infection of human PBMC stimulated with CD3/CD28 after 6 days of infection. The increase was abrogated by the β-adrenergic receptor antagonists Sotalol and Propranolol, but not the α-adrenergic-receptor antagonist Phentolamine, indicating it was mediated specifically by β-adrenergic receptors [310]. The mechanisms underlying this increase in infection were a reduction in the production of IL-10 [310] and an increase in the surface expression of CCR5 and CXCR4 [45, 50], both of which have been shown to increase HIV infection [311]. The norepinephrine mediated increases in HIV infection occur irrespective of the concentration of the infecting virus and were seen in response infection with both X4 or R5 tropic viral strains [45]. However, another study showed HIV infection of CD8-depleted, CD3/CD28 stimulated PBMC and MDM was significantly decreased by norepinephrine (10−8 – 10−6 M). The decrease in HIV replication was only seen from days 9 to 18 post-infection, and unlike the experiments by Cole et. al., no impact on replication was observed during the first 8 days post-infection. Another distinction between the studies was that the mechanism by which norepinephrine decreased HIV infection was a downregulation of the HIV-1 LTR through inhibition of NF-kB [312].
To clarify these opposing findings, we examined HIV infection of MDM in the presence of the β-adrenergic receptor agonist isoproterenol, as these receptors mediated the effects on HIV infection in the majority of similar experiments. Primary MDM were inoculated with HIV in the presence the isoproterenol. In these experiments, MDM were generated from PBMC, inoculated with HIVYU2 or HIVBaL and infection was assessed using a viral replication assay or viral entry assay as previously described [231, 298]. These experiments showed isoproterenol (10−9 to 10−6 M) did not significantly increase HIV replication at 2 – 6 days post infection (Figure 4A), nor did Isoproterenol (10−8 or 10−5 M) alter the viral entry into these cells (Figure 4B). These results indicate activation of β-adrenergic receptor does not change the entry process of the virus in macrophages, agreeing with the data from Moriuchi and colleagues. These data suggest that norepinephrine does not have a significant impact on HIV infection of MDM infection, while the data are conflicted regarding the impact on infection of CD3/CD28 stimulated PBMC, which is primarily T-cell infection. This could indicate that norepinephrine has different effects on HIV infection in distinct cell types, potentially indicating distinct signaling mechanisms by which norepinephrine mediated its effects. It is also possible that the disparity regarding the infection of activated PBMC is explained by the differences in experimental design, specifically the length of the experiments and of the norepinephrine treatments.
Figure 4.
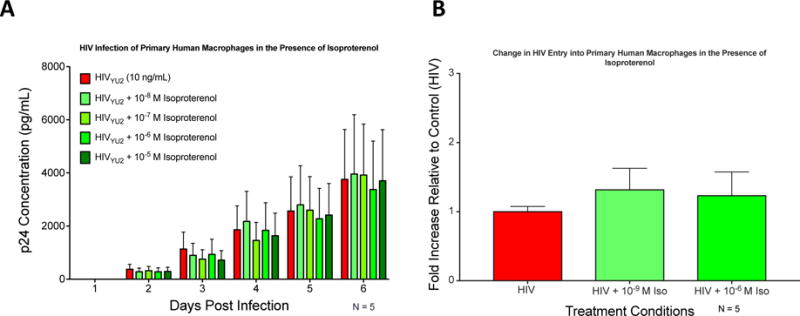
Activation of β-adrenergic receptors does not increase HIV infection in human macrophages. Human macrophages were generated from peripheral blood mononuclear cells that were isolated from whole blood by ficoll density centrifugation, then matured into monocyte-derived macrophages (MDM) by adherence and maturation for 6 days in 10 ng/mL M-CSF. (A) MDM from 5 donors were infected with 10 ng•p24/mL of the brain-derived R5 virus HIVYU2 for 24 hours in the presence (greens) or absence (red) of the isoproterenol (β-adrenergic receptor agonist). Cells were washed after 24 hours and supernatant was collected every 24 hours for 6 days as assayed for p24•Gag as a measure of viral replication. (B) MDM from 5 donors were infected for 2.5 hours with a modified HIVBaL, a lung derived R5 virus, containing an active β-lactamase enzyme, enabling visualization of entry within 9 hours of inoculation. Infections were performed in the absence (red) or presence (greens) of isoproterenol. After 2.5 hours cells were washed to remove excess virus, and incubated in CCF2-AM (ThermoFisher) for 6 hours. After 6 hours, cells were imaged and the percentage of infected cells was enumerated by fluorescent microscopy. Amount of infection displayed as fold change relative to infection with HIV alone, which was set to 1. Statistical analysis for both experiments performed using one-way repeated measures ANOVA.
The data suggest that norepinephrine does not significantly impact the spread of HIV infection in myeloid cells in the CNS, although this is complicated by the study in SIV-infected macaque suggesting that CNS catecholamines can increase SIV infection [48]. The picture is still less clear in regard to peripheral infection, particularly because the in vivo studies suggest elevated norepinephrine exacerbates HIV pathogenesis [40, 45, 46, 204]. Irrespective of these differences, these findings demonstrate that further investigation of the impact of norepinephrine on HIV infection is needed. In vitro models should examine the mechanisms by which norepinephrine could enhance HIV infection in different cell types. Patient studies should investigate the correlation between norepinephrine levels, endogenous immune cell expression of adrenergic receptors and both plasma and CSF viral load. Without this research, it will be difficult to understand and effectively counteract the effects of adrenergic activation of HIV target cells on disease progression.
VI. Role of Catecholamines in HIV-associated Neuroinflammation
While it is clear that CNS catecholamines can act as immunomodulators, their effects on neuroimmunity are not well studied. Further, it remains unclear whether and how disruptions in catecholaminergic neurotransmission contribute to HIV-associated inflammation, particularly in the case of norepinephrine. However, many of the immune functions altered by activation of dopaminergic or adrenergic receptors on immune cells, such as cytokine secretion, chemotaxis and nitric oxide production, are central to the development of NeuroHIV. As myeloid cells are the primary targets for and responders to HIV infection in the CNS, the catecholaminergic effects on the inflammatory mechanisms driven by these cells are most likely to be central to HIV-associated neuropathogenesis. Thus, the interactions between the CNS catecholaminergic systems and myeloid inflammatory processes constitute a bidirectional feedback loop that could enhance the development of NeuroHIV. Therefore, this section will focus mainly on the mechanisms by which dopamine impacts various myeloid cell functions that promote NeuroHIV.
VIa. Production of HIV-associated inflammatory mediators
Many cytokines that promote inflammation, such as IL-1β, IL-6, IL-10, IL-12, IL-18 and TNF-α, are important inflammatory regulators associated with HIV infection [1, 14, 16, 34, 313]. In particular, IL-6 and IL-1β are play an important role in HIV neuropathogenesis through immune mobilization, the activation of inflammatory cascades, and by increasing BBB permeability [313-315]. In the pre-cART era, elevated CSF IL-6 and IL-1β were often reported in individuals with HIV-dementia [316, 317], but have not been linked to cognitive impairment in the cART era [96]. Similarly, the chemokines affected by dopamine, such as CCL2, are important chemoattractants that enhance inflammation by recruiting additional immune cells [318, 319]. Elevated levels of the chemokines CCL2 and CXCL8 are found in the CSF of HIV+ individuals with neurocognitive impairment [96, 320], and CCL2 is increased soon after infection in the CNS of SIV-infected macaques [321]. The release of cytokines and chemokines is critical to the development and persistence of HIV-associated neuroinflammation, and changes in cytokine production have also been shown to alter HIV infection directly [311].
i. Dopamine
Although studies directly examining the effects of dopamine on inflammation in HIV-infected cells are rare, there are a growing number of studies demonstrating that dopamine increases the production of a number of cytokines and chemokines in myeloid cells (Table 1). Further, long term exposure of dopamine neurons to CCL2 increases extracellular dopamine release in rat striatum [322], and inflammatory cytokines have been shown to disrupt basal ganglial function [323]. And postmortem analysis of brains from patients with Parkinsons’ Disease show increased levels of cytokines, including IL-1β, TNF-α and IL-6, in striatal regions [324, 325]. These data suggest that inflammation and damage in dopaminergic brain regions, such as that which occurs in HIV infection, could create a bi-directional feedback loop in which both inflammatory mediators and dopamine levels are increased over time.
Table 1. Effect of dopamine, and dopamine agonists or antagonists on cytokine & NO production.
Effect of dopamine agonists/antagonists on cytokine production in myeloid cells A summary of in-vitro and in-vivo studies that have examined the effect of dopamine receptor agonists and antagonists on cytokine production in immune-cells. Haloperidol and chlorpromazine are non-specific dopamine-receptor antagonists. L-750,667 is a D4 receptor antagonist. Salsolinol and 1 BnTIQ are dopamine metabolites. Pramipexole is a D2/D3 receptor agonist.
| Effect on inflammation | Dopamine agonist/antagonist | cytokine | Experimental model | citation |
|---|---|---|---|---|
| Macrophages | ||||
| ↑ | Dopamine | ↑ IL-6, CCL2 | Human macrophages | [236] |
| ↑↓ | Dopamine | ↑ IL-8, IL-10. ↓ TNF-α | LPS-primed human macrophages | [236] |
| ↑ | Haloperidol, L750.667 (D2-like antagonist) | ↓ IL-6, IL-1β, IL-12 | RAW 264 murine macrophages | [330] |
| ↓ | Dopamine (effect blocked by D1 knockdown) | ↓ IL-1β, IL-18 | Nigericin-stimulated, LPS-primed Mouse BMDM | [326] |
| ↑ | Dopamine | ↑ NO | LPS-primed RAW 264 murine macrophages | [381] |
| ↑ | Salsolinol, 1BnTIQ (dopamine metabolites) | ↑ NO | RAW 264 murine macrophages | [384] |
| ↑ | Dopamine | ↑ iNOS and CXCL9. ↓ IL-10, CCL22 | Tumor-associated macrophages from rat glioma model | [385] |
| ↓ | dopamine (effect blocked by β antagonist) | ↓ IL-12 | LPS-primed mouse macrophages | [335] |
| ↓ | Dopamine (increased by treatment with reserpine) | ↓ TNF-α | LPS- or PMA- treated human U937 monocytoid cells | [327] |
| ↑↓ | Dopamine (through adrenergic receptors) | ↑ IL-10, ↓ IL-12p40 | LPS-treated mouse peritoneal macrophages | [335] |
| ↓ | Fenoldopam (D1-like agonist) | ↓ TNF-α, MIP-2 | LPS-treated mouse peritoneal macrophages | [386] |
| ↑ | Chlorpromazine (Pan dopamine receptor antagonist) | ↑ IL-10 | staphylococcal enterotoxin-stimulated mice | [331] |
| ↑ | Selegiline/L-DOPA | ↑ TNF-α mRNA | SIV infected Macaque Brain | [32] |
| Microglia | ||||
| ↑ | dopamine | ↑ IL-1β, IL-6 | N9 murine microglia | [329] |
| ↑ | Pramipexole (D2/D3 agonist) | ↑ NO | LPS or IFN-γ primed murine microglia | [382] |
| ↑ | Dopamine (effect blocked by D1 and D2 antagonists) | ↓ NO | LPS-primed murine microglia | [51] |
| ↓ | Dopamine (effect blocked by N-acetylcysteine) | ↓ NO | LPS-primed BV-2 murine microglia | [383] |
Data from our laboratory support this hypothesis, showing treatment with physiologic dopamine (10−8 – 10−5M) significantly increased production of CCL2, IL-6, CXCL8 and IL-10, and decreased TNF-α, in unstimulated or LPS-stimulated primary human monocyte derived macrophages [236]. Treatment of murine BMDMs and microglia with pharmacologic dopamine concentrations (1.5 – 2.5 × 10−4M) negatively regulated NLRP3 inflammasome activity, blocking nigericin-induced secretion of IL-1β and IL-18 in a D1-like dopamine receptor dependent manner. Production of IL-1β could also be inhibited in a more physiologically relevant manner through repetitive treatment with 1.5 μM of dopamine every 5 minutes for 2.5 hours [326]. Indirect increases in dopamine also modulate cytokine generation, as blocking VMAT with reserpine, thereby preventing dopamine uptake, also decreased TNF-α in the U937 myeloid cell line [327]. However, selegiline treatment, which increases CNS dopamine, increased TNF-α mRNA expression in SIV-infected macaques [32]. Disruption of striatal dopaminergic neurotransmission by expression of HIV-1 Nef in transgenic mice, potentially altering dopaminergic tone in striatum, increased CCL2 expression in microglia [328]. However, pharmacologic dopamine treatment (10−5 M) of murine microglial N9 cells with increased MAO activity, which should decrease dopamine levels, also increased secretion of IL-6 and IL-1β [329]. And the D2-antagonist haloperidol inhibits secretion of IL-6, IL-1β, and IL-12 in LPS-activated RAW264.7 murine macrophages, while the non-specific dopamine receptor antagonist chlorpromazine enhanced IL-10 production in mice [330, 331]. In many of these studies, inflammatory effects were driven by activity of D1-like dopamine receptors, while anti-inflammatory activities were linked to D2-like dopamine receptors, suggesting that the dopamine receptor subtypes play distinct roles in the regulation of inflammation.
ii. Norepinephrine
Numerous studies examining the effects of adrenergic receptor activation in myeloid cells (Table 2) broadly agree that both norepinephrine and epinephrine mediate predominantly anti-inflammatory effect via β-adrenergic receptors in cells stimulated with immune-activators such as LPS, Aβ or IL-18 [51, 244-246, 250, 251, 332-350]. Stimulation of α-adrenergic receptors in LPS-activated macrophages, microglia and monocytes had the opposite effect, increasing inflammatory cytokine production [249, 250, 345]. A similar pattern was found in resting or unstimulated cells treated with β-adrenergic receptor activation, which increased production of the inflammatory cytokines IL-6 [351-353], IL-1β [351, 353, 354], TNF-α [346, 351], IL-12 [346], IL-18 [355]. Interestingly, in rodent macrophages, β-adrenergic receptors activated by dopamine were also shown to decrease production of IL-12p40 and increase production of IL-10 [356]. Together, these data indicate that both α- and β-adrenergic receptor expression levels and the activation state of the cell are important to determining the effects of adrenergic neurotransmitters on cytokine production.
Table 2.
Effect of adrenergic agonists and antagonists on cytokine production in myeloid cells A summary of in-vitro and in-vivo studies that have examined the effect of adrenergic receptor agonists and antagonists on cytokine production in monocytes, macrophages, and microglia. Phenylephrine is a selective α1 receptor agonist, Norepinephrine (NE) and epinephrine are non-selective α and β receptor agonists. Isoproterenol and isoprenaline are non-selective β receptor agonists. Terbutaline, salmeterol, and formoterol are selective β2 receptor agonists. Clonidine is a centrally acting α2 receptor agonist. Dobutamine is predominantly a β1 receptor agonist with weak effects on β2 and α1 receptors. Butoxamine is a selective β2 receptor antagonist. Phentolamine is a non-selective α receptor antagonist. Yohimbine is predominantly a α2 antagonist that also has moderate affinity for α1 as well as D2 and serotonin receptors. Propranolol is a nonselective β receptor antagonist.
Effect of Adrenergic agonists/antagonists on cytokine production
| Effect on inflammation | Agonist/antagonist | cytokine | Experimental model | citation |
|---|---|---|---|---|
| Monocytes | ||||
| ↑ | Phenylephrine | ↑ IL-1β | LPS-primed human monocytes | [249] |
| ↑ | NE, E, isoproterenol (effect blocked by β2 antagonist) | ↑ IL-18, TNF-α, IFN-γ | Human PBMC | [355] |
| ↑ | NE, E, isoproterenol (effect blocked by β2 antagonist) | ↑ IL-18 | Human monocytes | [355] |
| ↑ | Terbutaline (effect blocked by β antagonist) | ↑ IL-8 | LPS primed human monocytes | [336] |
| ↑ | Isoproterenol | ↑ IL-12, TNF-α, NO | PMA-primed human monocytes | [346] |
| ↑ | NE (effect blocked by β antagonist) | ↓ IL-10 | HIV-infected human monocytes | [310] |
| ↓ | NE, clonidine | ↓ IL-6, TNF-α | LPS-primed whole blood | [244] |
| ↓ | NE (effect blocked by β antagonist) | ↓ IL-6, TNF-α, IL-1β, IL-2, IL-4, IFN-γ | HIV-infected human monocytes | [310] |
| ↓ | Terbutaline, salmeterol, formoterol | ↓ IL-1β | LPS-primed human monocytes | [245] |
| ↓ | NE, E (effect blocked by β2 antagonist) | ↓ TNF-α | LPS-primed human monocytes | [334] |
| ↓ | E, isoproterenol (effect blocked by β2 but not α antagonist) | ↓ MIP-1 | LPS-primed human monocytes | [349] |
| ↓ | Dobutamine, salbutamol | ↓ MIP-1, IL-8 | LPS-primed human monocytes | [350] |
| ↓ | Dobutamine | ↓ CCL2 | LPS-primed human monocytes | [337] |
| ↓ | Salbutamol, butoxamine, terbutaline | ↓ IL-18, IL-12 | LPS-primed human monocytes | [339] |
| ↓ | NE, E, isoproterenol, salbutamol, terbutaline | ↓ TNF-α, IL-12, IFN-γ | IL-18-primed human monocytes | [341] |
| ↓ | Isoproterenol | ↓ TNF-α, IL-12, NO | LPS-primed human monocytes | [346] |
| ↓ | NE, E, terbutaline | ↑ IL-10, IL-4. ↓ IFN-γ | tetanus-primed human monocytes | [332] |
| ↓ | Terbutaline | ↑ IL-10, ↓ TNF-α | LPS primed U937 human monocytes | [336] |
| Macrophages | ||||
| ↑ | NE | ↑ TNF-α | LPS-primed mouse macrophages | [359] |
| ↑ | Phenylephrine | ↑ IL-1β | LPS-primed human macrophages | [249] |
| ↑ | Phentolamine, yohimbine | ↓ IL-1β | LPS-primed mouse macrophages | [250] |
| ↑ | NE (effect blocked by β antagonist) | ↑ IL-6 | U937 human macrophages | [352] |
| ↑ | NE, E | ↑ NO | LPS-primed RAW 264 murine macrophages | [381] |
| ↑ | Salbutamol | ↑ IL-1β, IL-6 | RAW 264 murine macrophages | [353] |
| ↑ | Isoproterenol | ↑ TNF-α, IL-12, NO | PMA-primed mouse macrophages | [346] |
| ↓ | E, isoprenaline, dobutamine, salbutamol | ↓ NO | LPS-primed mouse macrophages | [344] |
| ↓ | Propranolol | ↑ IL-1β | LPS-primed mouse macrophages | [250] |
| ↓ | Isoproterenol (effect blocked by β antagonist) | ↓ TNF-α | LPS-primed mouse macrophages | [345] |
| ↓ | NE, E, formoterol, salbutamol | ↓ IL-27 | LPS-primed mouse macrophages | [340] |
| ↓ | NE, E, formoterol (effect blocked by β2 antagonist) | ↓ TNF-α, IL-8. ↑ IL-10 | LPS-primed porcine macrophages | [342] |
| ↓ | Dopamine (effect blocked by β antagonist) | ↓ IL-12 | LPS-primed mouse macrophages | [335] |
| ↓ | Clenbuterol | ↓ TNF-α, IL-6. ↑ IL-10 | LPS-primed PMA-differentiated U937 human macrophages | [246] |
| ↓ | Isoproterenol | ↓ TNF-α, IL-12, NO | LPS-primed mouse macrophages | [346] |
| Microglia | ||||
| ↑ | Isoproterenol (effect blocked by β antagonists) | ↑ IL-1β | Rat microglia | [354] |
| ↑ | Isoproterenol | ↑ IL-6, IL-1β, ↓TNF-α | Rat microglia | [351] |
| ↑ | Propranolol | ↓ IL-6, IL-1β, ↓TNF-α | Rat microglia isolated after surgical trauma | [351] |
| ↓ | NE, isoproterenol | ↓ TNF-α, CCL2 | Aβ stimulated mouse microglia | [348] |
| ↓ | NE, isoproterenol | ↓ NO | LPS-primed rat microglia | [347] |
| ↓ | NE, epi, isoproterenol, phenylephrine | ↓ NO | LPS-primed N9 murine microglia | [333] |
| ↓ | NE | ↓ TNF-α, IL-6 | LPS-primed mouse microglia | [51] |
| ↓ | NE, phenylephrine, dobutamine, terbutaline | ↓ TNF-α, NO | LPS-primed rat microglia | [251] |
| ↓ | dobutamine, terbutaline | ↓ TNF-α, CCL2, IL-6 | LPS-primed microglia in mouse hippocampal slices | [338] |
| ↑ | Isoproterenol (effect blocked by β antagonists) | ↑ IL-1β | Rat microglia | [354] |
| ↑ | Isoproterenol | ↑ IL-6, IL-1β, TNF-α | Rat microglia | [351] |
There is little research specifically investigating HIV-infected cells, although in HIV-infected PBMC, norepinephrine (10−5 M) significantly decreased production of IL-1β, IL-6, TNF-α, IL-10 and IFN-γ, with the effects on IL-10 and IFN-γ mediated through modulation of cAMP production [310]. And in HIV infected individuals with autonomic system hypo-reactivity, there was an increase in baseline TNF-α [41]. These data suggest that norepinephrine mediated activation of adrenergic receptors, particularly β-adrenergic receptors, may play an important role in the regulation of HIV-associated inflammation. This is supported by studies showing elevated stress in HIV-infected individuals, as stress increases expression of inflammatory cytokines in both the plasma and CNS via activation of β-adrenergic receptors [357]. Thus, physiologically-relevant increases in norepinephrine could exacerbate neuroinflammation during HIV, however more research is needed to determine which cells and cytokines are involved.
VIb. Changes in Chemotaxis and Transmigration
The transport of HIV into the CNS predominantly involves the transmigration of mature CD14+ CD16+ monocytes across the BBB. These mature monocytes are enriched in the blood of HIV infected individuals, and this enrichment of CD14+CD16+ cells is maintained in the presence of cART treatment [71]. These mature monocytes, particularly once they are infected, preferentially transmigrate across the blood brain barrier in response to chemoattractants such as CCL2 [67, 319]. This is a critical process in the development of HIV-associated neurologic disease. These cells not only bring HIV into the CNS, they also contribute to neuroinflammation and differentiate into infected macrophages, which further contribute to NeuroHIV [16, 66, 67, 72]. Thus, changes in the processes by which monocytes transmigrate into the CNS, or move throughout the brain once transmigration are complete, could have a significant impact on neuropathogenesis.
i. Dopamine
Studies in the Berman lab have shown that dopamine receptor expression on monocytes changes with maturation, with mature monocytes expressing increased amounts of D1-like dopamine receptors. Dopamine increased chemokinesis in these cells, and enhanced the CCL2-mediated transmigration of these cells across a BBB model in response to dopamine and the D1-like agonist SKF38393 (10−6 – 5 × 10−5 M) [232, 237]. Notably, the enhancement of transmigration only applied to mature CD14+CD16+ monocytes, with transmigration of immature Cd14lowCD16+ monocytes and T-cell remaining unaffected [237]. This indicates that increased extracellular CNS dopamine could increase the accumulation of these monocytes, and the virus they are transporting, in the CNS, specifically in dopaminergic brain regions. While the effects of dopamine on macrophage chemotaxis are not clear, dopamine has been shown to stimulate microglial chemotaxis. Physiologic dopamine (10−7M) significantly increases migration of human microglia relative to untreated and CCL2 stimulated chemotaxis, an effect mediated by D2-like dopamine receptors, as it was blocked by treatment with the D2-antagonist Spiperone [233]. Similar findings were reported in rodent microglia, significantly increasing chemotaxis in response to physiologic dopamine (10−8 – 10−5 M) or the D1 and D2 agonists dihydrexidine and quinpirole [51]. Dopamine may affect macrophage chemotaxis similarly, but additional research is necessary to confirm this. These studies suggest increased chemotaxis could be an important mechanism by which elevated CNS dopamine exacerbates inflammation and neurocognitive impairment in HIV-infected individuals with altered dopaminergic tone.
ii. Norepinephrine
As with cytokine production, the effects of norepinephrine on myeloid cell chemotaxis seem to be highly context and receptor-dependent. Short-term exposure to low levels of norepinephrine (10−10 M, 2 hrs), or isoproterenol stimulates chemotaxis of human macrophages and monocytes in a β-adrenergic receptor and cAMP dependent manner [358]. However, long term exposure of mouse BMDM to norepinephrine (10−6M, 7 days) inhibited cell proliferation and migration toward CCL2 via downregulation of MHC II and CCR2, although increasing MHC II and CCR2 expression with a lower concentration of norepinephrine (10−8 M), and had no effect of macrophage migration [359]. In murine peritoneal macrophages, varying concentrations of norepinephrine (10−12 M to 10−3 M) induced both adherence and chemotaxis, but the effects varied with the age of the mice from which the cells were derived [360]. A second study from this group confirmed that low-level norepinephrine increased murine macrophage chemotaxis, and suggested this effect was mediated by β-adrenergic receptor activation. They also found that higher levels of norepinephrine enhanced phagocytosis [361]. In microglia, norepinephrine (10−7 M, 2 hrs) and isoproterenol also increase microglial migration in response to Aβ [348]. However, bath application of norepinephrine (3 × 10−5M) to mouse cortical brain slices resulted in significant retraction of microglial processes in both resting and LPS-activated cells, with the specific adrenergic receptor subtypes involved dependent on the activation state of the cells [362]. This suggests the presence of other cells and inflammatory factors could modulate microglial response to norepinephrine. For example, norepinephrine increases release of CCL2 from astrocytes, but inhibited CCL2 expression in microglia [363]. Astrocyte and microglia density vary in different brain regions [364, 365], so norepinephrine modulation of CCL2 expression in different cells may cause region-specific effects on chemotaxis. While the effect of norepinephrine on the chemotaxis or motility of myeloid cells has not been investigated in the context of HIV, these studies suggest that changes in the concentration of this neurotransmitter could distinctly alter myeloid migration within the HIV infected CNS, depending on the microenvironments present in specific brain regions.
VIc. Effects on Nitric Oxide Production
Nitric oxide (NO) is a key molecule in immunity and inflammation. In pathologic conditions, NO can both support or inhibit inflammation, acting in either a cytoprotective or cytotoxic capacity. Nitric oxide is produced by inducible nitric oxide synthase (iNOS or NOS2), generally after induction with immunologic or inflammatory stimuli. A variety of immune cells, particularly innate immune cells such as macrophages, both produce and respond to NO [366]. In the CNS, excessive NO production can contribute to neuronal damage and death via induction of oxidative stress [367-370]. Increases in NO production occurs during HIV infection [371, 372], with both pro- [373, 374] and anti-viral [375-377] effects. In human PBMC, HIV replication is inhibited by NO in acutely infected cells, but stimulated by NO in chronically-infected cells [378]. In the HIV-infected brain, modulation of NO the NO-arginase network in microglia reduces neuroinflammation [379], but also enhances neuroinflammation through up-regulation in response to the HIV proteins Tat and g120 [380]. The opposing effects of NO make it difficult to determine how alterations NO production would affect NeuroHIV [368].
i. Dopamine
A number of studies show dopamine-receptor activation modulates NO production (Table 1). The effects of dopamine human myeloid cells remain undetermined, but in RAW264.7 murine macrophages, pharmacologic dopamine (5 × 10−5 M) enhanced LPS-induced production of NO [381], and the D2/D3 specific agonist pramipexole and increased LPS/IFN-γ-induced secretion of NO from primary murine microglia [382]. Contrary to these findings, stimulation of primary rat microglia with pharmacologic dopamine (10−6M – 10−5M) for 24 hrs significantly reduced LPS mediate NO production [51], and pretreatment with higher dopamine concentrations (3 × 10−6 – 10−4M) significantly reduced LPS-induced NO in BV-2 murine microglial cells [383]. The effects of dopamine were dopamine receptor dependent in the rat microglia, but not the BV-2 cells, in which they were induced by the formation of dopamine quinones. These contradictory findings suggest that dopamine impacts NO production through multiple pathways and receptors, and the specific effects on NO may reflect a concentration-dependent activation of a predominant pathway in each specific context. These data also emphasize the importance of cell origin, culture conditions, and treatment paradigm when interpreting data from various studies. Thus, further studies are needed to determine whether and how the dopaminergic changes occurring in NeuroHIV alter NO production in human myeloid cells.
ii. Norepinephrine
Adrenergic receptor activation also modulates NO production in myeloid cells (Table 2). Szelenyi and colleagues found that isoproterenol increased NO production in PMA-primed human monocytes, but decreased NO production in LPS-primed monocytes. They also found that isoproterenol decreased NO production in LPS-primed murine macrophages [346]. The β-receptor agonists isoprenaline, dobutamine, and salbutamol, as well as epinephrine, inhibited NO production in LPS-primed murine macrophages at a concentration of 10−7M [344]. On the other hand, Chi and colleagues found that norepinephrine and epinephrine increased NO production in murine LPS-primed macrophages, although this study was performed in the RAW264 cell-line using a high concentration (5 × 10−6M) of catecholamine [381]. Adrenergic receptor activation also inhibited NO production in microglia. In LPS-primed rat microglia, norepinephrine, isoproterenol, phenylephrine, dobutamine, and terbutaline reduce NO at multiple concentrations (10−7 – 10−5 M) [251, 347]. In addition, norepinephrine (5 × 10−6M), isoproterenol (2.5 × 10−7M), and phenylephrine (7.8 × 10−6M) all reduced NO produced in LPS-primed murine N9 microglia. These studies suggest that activation of all subtypes of adrenergic receptors can have a suppressive effect on LPS-induced NO production in most myeloid cells. However, there is still little known about how adrenergic receptor activation would impact NO production under basal conditions or during HIV infection. Further studies are needed, particularly in human macrophages to elucidate the mechanism and impact of altered NO production by norepinephrine or epinephrine.
VII. Concluding remarks
The ability to control HIV replication has greatly improved the lifespan and quality of life for infected individuals, but that transformation to a chronic disease has created a number of new issues. Among these effects is the dysregulation of catecholaminergic neurotransmission due to neuroinflammation and neurologic damage, which can result in the exposure of CNS myeloid populations to aberrant levels of catecholamines. Additionally, the effects of cART drugs on catecholaminergic systems remains undetermined, so the potential effects of these therapeutics also require further study. These alterations in catecholaminergic tone can be further exacerbated by the use of illicit drugs or legal therapeutics that modulate dopaminergic and noradrenergic neurotransmission. The effect of brain catecholamines on myeloid cell functions is highly relevant in the cART era, as the persistence of HIV within CNS monocytes and macrophages is a major obstacle to HIV-eradication and the treatment/prevention of HIV-associated neuroinflammation. This review has attempted to disentangle the bidirectional interactions between catecholamines and immune-processes implicated in HIV-infection of the CNS. The data described here suggest that elevated dopaminergic tone in the CNS may increase CNS viral load and enhance neuroinflammation through dopamine-mediated changes in myeloid inflammatory processes and susceptibility to viral infection. Elevated CNS norepinephrine may also alter HIV-infection, particularly in regard to modulation of neuroinflammation, but the direction and magnitude of these effect remain unclear due to the paucity of studies investigating the role of the adrenergic system in HIV infection. Thus, dysregulation of catecholaminergic systems through direct or indirect viral effects, elevated stress, use of illicit drugs or catecholaminergic therapeutics, or a combination of these factors, is likely to exacerbate NeuroHIV.
However, more research is needed to determine the precise mechanisms responsible for the immunomodulatory effects of catecholamines, including the involvement of specific receptors and signaling pathways in immune cells. This is particularly true regarding the immunomodulatory effects of the adrenergic system, which remains poorly understood in the context of HIV infection. Research in this area is complicated by contradictory findings that are likely due to variations in culture conditions, treatment paradigms, and specificity of receptor agonists and antagonists. Our summary of catecholaminergic modulation of inflammatory mediators (Tables 1 and 2) reflects these contradictions. Further, they suggest that the disparate dopamine and adrenergic receptor subtypes play distinct and potentially opposing roles in inflammation. Continuing research that utilizes receptor-specific pharmacologic assays and gene-level knockdown may reveal these specific roles. Because of the differences in neurotransmission among mammals, it is important to use both in vitro systems involving human cells and animal models to interrogate the precise molecular mechanisms and pathways involved in this type of neuroimmune communication. Further, new technologies enabling investigation of human CNS cells, using technologies such as iPSC co-culture or multi-cell type brain organoids, provide novel platforms in which to expand these studies. Determining the effects of catecholamines on myeloid cells during HIV infection is critical to developing treatments for chronically infected individuals and may reveal important contraindications for the use of catecholaminergic therapeutics in HIV infection. Further, the proliferation of psychiatric drugs in todays’ medical settings suggests that the poorly defined interactions we have described here between catecholamines and immune cells likely have larger implications for other neuroinflammatory diseases and psychiatric disorders. Therefore, understanding the interactions between catecholamine neurotransmission and immune function, particularly in myeloid cells, is critical to the creation of strategies that will ameliorate the long-term effects of catecholaminergic dysregulation in HIV-infected individuals.
Highlights.
Catecholamines significantly alter the development of HIV neuropathogenesis
Catecholamines alter HIV-infection and HIV-associated immune functions
Activation of b-adrenergic receptors does not alter HIV infection in macrophages
Catecholamine immunomodulation could be a therapeutic target for treatment of HAND
Acknowledgments
We would like to thank Dr. Patrick Osei-Owusu for his critical review of this manuscript and insightful advice. We would also like to thank Kaitlyn Runner and Emily Nickoloff in the Gaskill lab for their support and suggestions. These studies were funded by grants from the National Institutes of Drug Abuse, R01 DA039005 (PJG). The authors were also funded in part by the Public Health Service, National Institutes of Health, under the Ruth L. Kirschstein National Research Service Award NIMH T32 MH079785 (PI, Jay Rappaport; with Brian Wigdahl serving as the PI of the Drexel University College of Medicine component and Olimpia Meucci as Co-Director). The contents of the paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Abbreviations
- BBB
blood brain barrier
- BMDM
bone marrow derived macrophages
- cART
combination anti-retroviral therapy
- CNS
central nervous system
- COMT
catechol-O-methyltransferase
- CSF
cerebrospinal fluid
- DAT
dopamine transporter
- DBH
dopamine beta hydroxylase
- DRD
dopamine receptor
- HAD
HIV-associated dementia
- HAND
HIV-associated neurocognitive disorder
- HIVE
HIV-associated encephalitis
- HPA
hypothalamic-pituitary axis
- HVA
homovanillic acid
- iNOS
inducible nitric oxide synthase
- MAO
monoamine oxidase
- MDM
monocyte derived macrophages
- NET
norepinephrine transporter
- NO
nitric oxide
- PBMC
peripheral blood mononuclear cells
- PFC
prefrontal cortex
- PKC
protein kinase C
- PLC
phospholipase C
- PMN
Polymorphonuclear neutrophil
- PNMT
phenylethanolamine-N-methyltransferase
- ROS
reactive oxygen species
- SAM
sympathetic neuron–associated macrophages
- SIV
simian immunodeficiency virus
- SNP
single nucleotide polymorphism
- SNpc
substantia nigra pars compacta
- SNS
sympathetic nervous system
- TH
tyrosine hydroxylase
- VMAT2
vesicular monoamine transporter 2
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saylor D, et al. HIV-associated neurocognitive disorder–pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(4):234–48. doi: 10.1038/nrneurol.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heaton RK, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rappaport J, Volsky DJ. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J Neurovirol. 2015;21(3):235–41. doi: 10.1007/s13365-015-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heaton RK, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75(23):2087–96. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woods SP, et al. HIV-associated deficits in action (verb) generation may reflect astrocytosis. J Clin Exp Neuropsychol. 2010;32(5):522–7. doi: 10.1080/13803390903264130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melrose RJ, et al. Compromised fronto-striatal functioning in HIV: an fMRI investigation of semantic event sequencing. Behav Brain Res. 2008;188(2):337–47. doi: 10.1016/j.bbr.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 7.Hinkin CH, et al. Medication adherence among HIV+ adults: effects of cognitive dysfunction and regimen complexity. Neurology. 2002;59(12):1944–50. doi: 10.1212/01.wnl.0000038347.48137.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant I, et al. Asymptomatic HIV-associated neurocognitive impairment increases risk for symptomatic decline. Neurology. 2014;82(23):2055–62. doi: 10.1212/WNL.0000000000000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis LE, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42(9):1736–9. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- 10.Valcour V, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82. doi: 10.1093/infdis/jis326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koenig S, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–93. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 12.Navia BA, et al. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–35. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 13.Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–62. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- 14.Kraft-Terry SD, et al. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64(1):133–45. doi: 10.1016/j.neuron.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rappaport J, Volsky DJ. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J Neurovirol. 2015 doi: 10.1007/s13365-015-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdo TH, Lackner A, Williams KC. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol Rev. 2013;254(1):102–13. doi: 10.1111/imr.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ipser JC, et al. HIV infection is associated with attenuated frontostriatal intrinsic connectivity: a preliminary study. J Int Neuropsychol Soc. 2015;21(3):203–13. doi: 10.1017/S1355617715000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harezlak J, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS. 2011;25(5):625–33. doi: 10.1097/QAD.0b013e3283427da7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelman BB, et al. Prefrontal dopaminergic and enkephalinergic synaptic accommodation in HIV-associated neurocognitive disorders and encephalitis. J Neuroimmune Pharmacol. 2012;7(3):686–700. doi: 10.1007/s11481-012-9345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ernst T, et al. Abnormal brain activation on functional MRI in cognitively asymptomatic HIV patients. Neurology. 2002;59(9):1343–9. doi: 10.1212/01.wnl.0000031811.45569.b0. [DOI] [PubMed] [Google Scholar]
- 21.Anderson AM, et al. CSF biomarkers of monocyte activation and chemotaxis correlate with magnetic resonance spectroscopy metabolites during chronic HIV disease. J Neurovirol. 2015;21(5):559–67. doi: 10.1007/s13365-015-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen AB, et al. Cerebral FDG-PET scanning abnormalities in optimally treated HIV patients. J Neuroinflammation. 2010;7:13. doi: 10.1186/1742-2094-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gelman BB, et al. Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. J Acquir Immune Defic Syndr. 2013;62(5):487–95. doi: 10.1097/QAI.0b013e31827f1bdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes MG, et al. Nigral degeneration in acquired immune deficiency syndrome (AIDS) Acta Neuropathol. 1991;82(1):39–44. doi: 10.1007/BF00310921. [DOI] [PubMed] [Google Scholar]
- 25.Aylward EH, et al. Reduced basal ganglia volume in HIV-1-associated dementia: results from quantitative neuroimaging. Neurology. 1993;43(10):2099–104. doi: 10.1212/wnl.43.10.2099. [DOI] [PubMed] [Google Scholar]
- 26.Hestad K, et al. Regional brain atrophy in HIV-1 infection: association with specific neuropsychological test performance. Acta Neurol Scand. 1993;88(2):112–8. doi: 10.1111/j.1600-0404.1993.tb04201.x. [DOI] [PubMed] [Google Scholar]
- 27.Berger JR, et al. Cerebrospinal fluid dopamine in HIV-1 infection. AIDS. 1994;8(1):67–71. doi: 10.1097/00002030-199401000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Fujimura RK, et al. HIV-1 proviral DNA load across neuroanatomic regions of individuals with evidence for HIV-1-associated dementia. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16(3):146–52. doi: 10.1097/00042560-199711010-00002. [DOI] [PubMed] [Google Scholar]
- 29.Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol. 2000;14(3):214–21. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- 30.Itoh K, Mehraein P, Weis S. Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol. 2000;99(4):376–84. doi: 10.1007/s004010051139. [DOI] [PubMed] [Google Scholar]
- 31.Czub S, et al. Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol. 2001;101(2):85–91. doi: 10.1007/s004010000313. [DOI] [PubMed] [Google Scholar]
- 32.Czub S, et al. Modulation of simian immunodeficiency virus neuropathology by dopaminergic drugs. Acta Neuropathol. 2004;107(3):216–26. doi: 10.1007/s00401-003-0801-3. [DOI] [PubMed] [Google Scholar]
- 33.Gelman BB, et al. Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J Neuroimmune Pharmacol. 2006;1(4):410–20. doi: 10.1007/s11481-006-9030-6. [DOI] [PubMed] [Google Scholar]
- 34.Anthony IC, Bell JE. The Neuropathology of HIV/AIDS. Int Rev Psychiatry. 2008;20(1):15–24. doi: 10.1080/09540260701862037. [DOI] [PubMed] [Google Scholar]
- 35.Agrawal L, et al. Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res. 2010;1306:116–30. doi: 10.1016/j.brainres.2009.09.113. [DOI] [PubMed] [Google Scholar]
- 36.Ferris MJ, et al. Hyperdopaminergic tone in HIV-1 protein treated rats and cocaine sensitization. J Neurochem. 2010;115(4):885–96. doi: 10.1111/j.1471-4159.2010.06968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitting S, Booze RM, Mactutus CF. HIV-1 Proteins, Tat and gp120, Target the Developing Dopamine System. Curr HIV Res. 2015;13(1):21–42. doi: 10.2174/1570162x13666150121110731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaskill PJ, et al. HIV, Tat and dopamine transmission. Neurobiol Dis. 2017;105:51–73. doi: 10.1016/j.nbd.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar M, et al. Norepinephrine response in early HIV infection. J Acquir Immune Defic Syndr. 1991;4(8):782–6. [PubMed] [Google Scholar]
- 40.Kumar M, et al. The HPA axis in HIV-1 infection. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S89–93. doi: 10.1097/00126334-200210012-00010. [DOI] [PubMed] [Google Scholar]
- 41.Kumar M, et al. HIV-1 infection and its impact on the HPA axis, cytokines, and cognition. Stress. 2003;6(3):167–72. doi: 10.1080/10253890310001605376. [DOI] [PubMed] [Google Scholar]
- 42.Kumar AM, et al. Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol. 2009;15(3):257–74. doi: 10.1080/13550280902973952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koutsilieri E, et al. Monoamine metabolite levels in CSF of SIV-infected rhesus monkeys (Macaca mulatta) Neuroreport. 1997;8(17):3833–6. doi: 10.1097/00001756-199712010-00034. [DOI] [PubMed] [Google Scholar]
- 44.Koutsilieri E, et al. Involvement of dopamine in the progression of AIDS Dementia Complex. J Neural Transm. 2002;109(3):399–410. doi: 10.1007/s007020200032. [DOI] [PubMed] [Google Scholar]
- 45.Cole SW, et al. Impaired response to HAART in HIV-infected individuals with high autonomic nervous system activity. Proc Natl Acad Sci U S A. 2001;98(22):12695–700. doi: 10.1073/pnas.221134198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole SW. Psychosocial influences on HIV-1 disease progression: neural, endocrine, and virologic mechanisms. Psychosom Med. 2008;70(5):562–8. doi: 10.1097/PSY.0b013e3181773bbd. [DOI] [PubMed] [Google Scholar]
- 47.Gaskill PJ, et al. Drug Induced Increases in CNS Dopamine Alter Monocyte, Macrophage and T Cell Functions: Implications for HAND. J Neuroimmune Pharmacol. 2013 doi: 10.1007/s11481-013-9443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloan EK, et al. Enhanced replication of simian immunodeficiency virus adjacent to catecholaminergic varicosities in primate lymph nodes. J Virol. 2006;80(9):4326–35. doi: 10.1128/JVI.80.9.4326-4335.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flierl MA, et al. Catecholamines-crafty weapons in the inflammatory arsenal of immune/inflammatory cells or opening pandora's box? Mol Med. 2008;14(3–4):195–204. doi: 10.2119/2007-00105.Flierl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cole SW, Jamieson BD, Zack JA. cAMP up-regulates cell surface expression of lymphocyte CXCR4: implications for chemotaxis and HIV-1 infection. J Immunol. 1999;162(3):1392–400. [PubMed] [Google Scholar]
- 51.Farber K, Pannasch U, Kettenmann H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci. 2005;29(1):128–38. doi: 10.1016/j.mcn.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Gaskill PJ, et al. Characterization and function of the human macrophage dopaminergic system: implications for CNS disease and drug abuse. J Neuroinflammation. 2012;9(1):203. doi: 10.1186/1742-2094-9-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarkar C, et al. The immunoregulatory role of dopamine: an update. Brain Behav Immun. 2010;24(4):525–8. doi: 10.1016/j.bbi.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Everall IP, Hansen LA, Masliah E. The shifting patterns of HIV encephalitis neuropathology. Neurotox Res. 2005;8(1–2):51–61. doi: 10.1007/BF03033819. [DOI] [PubMed] [Google Scholar]
- 55.Bell JE. The neuropathology of adult HIV infection. Rev Neurol (Paris) 1998;154(12):816–29. [PubMed] [Google Scholar]
- 56.Glass JD, et al. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38(5):755–62. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 57.Cysique LA, Maruff P, Brew BJ. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J Neurovirol. 2004;10(6):350–7. doi: 10.1080/13550280490521078. [DOI] [PubMed] [Google Scholar]
- 58.Clifford KM, et al. Progressive Brain Atrophy Despite Persistent Viral Suppression in HIV Over Age 60. J Acquir Immune Defic Syndr. 2017 doi: 10.1097/QAI.0000000000001489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van den Dries LWJ, et al. Neurocognitive Impairment in a Chronically Well-Suppressed HIV-Infected Population: The Dutch TREVI Cohort Study. AIDS Patient Care STDS. 2017;31(8):329–334. doi: 10.1089/apc.2017.0038. [DOI] [PubMed] [Google Scholar]
- 60.McArthur JC, et al. Attenuated central nervous system infection in advanced HIV/AIDS with combination antiretroviral therapy. Arch Neurol. 2004;61(11):1687–96. doi: 10.1001/archneur.61.11.1687. [DOI] [PubMed] [Google Scholar]
- 61.Sanford R, et al. Association of Brain Structure Changes and Cognitive Function With Combination Antiretroviral Therapy in HIV-Positive Individuals. JAMA Neurol. 2017 doi: 10.1001/jamaneurol.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker LM, et al. The Effect of Central Nervous System Penetration Effectiveness of Highly Active Antiretroviral Therapy on Neuropsychological Performance and Neuroimaging in HIV Infected Individuals. J Neuroimmune Pharmacol. 2015;10(3):487–92. doi: 10.1007/s11481-015-9610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Underwood J, et al. Grey and white matter abnormalities in treated HIV-disease and their relationship to cognitive function. Clin Infect Dis. 2017 doi: 10.1093/cid/cix301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sevigny JJ, et al. Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology. 2004;63(11):2084–90. doi: 10.1212/01.wnl.0000145763.68284.15. [DOI] [PubMed] [Google Scholar]
- 65.McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157(1–2):3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 66.Tavazzi E, et al. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr HIV Res. 2014;12(2):97–110. doi: 10.2174/1570162x12666140526114956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams DW, et al. Monocytes Mediate HIV Neuropathogenesis: Mechanisms that Contribute to HIV Associated Neurocognitive Disorders. Curr HIV Res. 2014 doi: 10.2174/1570162x12666140526114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peluso R, et al. A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology. 1985;147(1):231–6. doi: 10.1016/0042-6822(85)90246-6. [DOI] [PubMed] [Google Scholar]
- 69.Dohgu S, et al. Human immunodeficiency virus-1 uses the mannose-6-phosphate receptor to cross the blood-brain barrier. PLoS One. 2012;7(6):e39565. doi: 10.1371/journal.pone.0039565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ellery PJ, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007;178(10):6581–9. doi: 10.4049/jimmunol.178.10.6581. [DOI] [PubMed] [Google Scholar]
- 71.Castley A, et al. Elevated plasma soluble CD14 and skewed CD16+ monocyte distribution persist despite normalisation of soluble CD163 and CXCL10 by effective HIV therapy: a changing paradigm for routine HIV laboratory monitoring? PLoS One. 2014;9(12):e115226. doi: 10.1371/journal.pone.0115226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer-Smith T, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. 2001;7(6):528–41. doi: 10.1080/135502801753248114. [DOI] [PubMed] [Google Scholar]
- 73.Buckner CM, et al. Characterization of monocyte maturation/differentiation that facilitates their transmigration across the blood-brain barrier and infection by HIV: implications for NeuroAIDS. Cell Immunol. 2011;267(2):109–23. doi: 10.1016/j.cellimm.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aquaro S, et al. Long-term survival and virus production in human primary macrophages infected by human immunodeficiency virus. J Med Virol. 2002;68(4):479–88. doi: 10.1002/jmv.10245. [DOI] [PubMed] [Google Scholar]
- 75.Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS. 2013;8(3):165–9. doi: 10.1097/COH.0b013e32835fc601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wekerle H, et al. Cellular immune reactivity within the CNS. 1986;9(Supplement C):271–277. [Google Scholar]
- 77.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28(2):254–60. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 78.Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci. 2012;15(8):1096–101. doi: 10.1038/nn.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eugenin EA, Berman JW. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci. 2007;27(47):12844–50. doi: 10.1523/JNEUROSCI.4154-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Churchill MJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66(2):253–8. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- 81.Cho HJ, et al. HIV Alters Gap Junction-Mediated Intercellular Communication in Human Brain Pericytes. Front Mol Neurosci. 2017;10:410. doi: 10.3389/fnmol.2017.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yaseen MM, et al. The role of polymorphonuclear neutrophils during HIV-1 infection. Arch Virol. 2018;163(1):1–21. doi: 10.1007/s00705-017-3569-9. [DOI] [PubMed] [Google Scholar]
- 83.Spear GT, et al. Analysis of lymphocytes, monocytes, and neutrophils from human immunodeficiency virus (HIV)-infected persons for HIV DNA. J Infect Dis. 1990;162(6):1239–44. doi: 10.1093/infdis/162.6.1239. [DOI] [PubMed] [Google Scholar]
- 84.Kennedy RH, Silver R. Neuroimmune Signaling: Cytokines and the CNS. In: Pfaff DW, Volkow ND, editors. Neuroscience in the 21st Century. Springer New York; New York, NY: 2016. pp. 1–41. [Google Scholar]
- 85.Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci. 2013;7:34. doi: 10.3389/fncel.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faraco G, et al. Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berl) 2017;95(11):1143–1152. doi: 10.1007/s00109-017-1573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spudich SS. Immune activation in the central nervous system throughout the course of HIV infection. Curr Opin HIV AIDS. 2016;11(2):226–33. doi: 10.1097/COH.0000000000000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burdo TH, et al. Elevated sCD163 in plasma but not cerebrospinal fluid is a marker of neurocognitive impairment in HIV infection. AIDS. 2013;27(9):1387–95. doi: 10.1097/QAD.0b013e32836010bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kamat A, et al. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr. 2012;60(3):234–43. doi: 10.1097/QAI.0b013e318256f3bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lyons JL, et al. Plasma sCD14 is a biomarker associated with impaired neurocognitive test performance in attention and learning domains in HIV infection. J Acquir Immune Defic Syndr. 2011;57(5):371–9. doi: 10.1097/QAI.0b013e3182237e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamat A, et al. A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PLoS One. 2012;7(2):e30881. doi: 10.1371/journal.pone.0030881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Laskin DL, Pendino KJ. Macrophages and inflammatory mediators in tissue injury. Annu Rev Pharmacol Toxicol. 1995;35:655–77. doi: 10.1146/annurev.pa.35.040195.003255. [DOI] [PubMed] [Google Scholar]
- 95.Anthony IC, et al. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64(6):529–36. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- 96.Yuan L, et al. Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. J Neurovirol. 2013;19(2):144–9. doi: 10.1007/s13365-013-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yadav A, Collman RG. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol. 2009;4(4):430–47. doi: 10.1007/s11481-009-9174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spudich S, et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J Infect Dis. 2011;204(5):753–60. doi: 10.1093/infdis/jir387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Churchill MJ, et al. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol. 2016;14(1):55–60. doi: 10.1038/nrmicro.2015.5. [DOI] [PubMed] [Google Scholar]
- 100.Pinkevych M, et al. HIV Reactivation from Latency after Treatment Interruption Occurs on Average Every 5-8 Days–Implications for HIV Remission. PLoS Pathog. 2015;11(7):e1005000. doi: 10.1371/journal.ppat.1005000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hill AL, et al. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc Natl Acad Sci U S A. 2014;111(37):13475–80. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ellis RJ, et al. CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS. 2011;25(14):1747–51. doi: 10.1097/QAD.0b013e32834a40cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Munoz-Moreno JA, et al. Nadir CD4 cell count predicts neurocognitive impairment in HIV-infected patients. AIDS Res Hum Retroviruses. 2008;24(10):1301–7. doi: 10.1089/aid.2007.0310. [DOI] [PubMed] [Google Scholar]
- 104.Cohen RA, et al. Effects of nadir CD4 count and duration of human immunodeficiency virus infection on brain volumes in the highly active antiretroviral therapy era. J Neurovirol. 2010;16(1):25–32. doi: 10.3109/13550280903552420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jernigan TL, et al. Clinical factors related to brain structure in HIV: the CHARTER study. J Neurovirol. 2011;17(3):248–57. doi: 10.1007/s13365-011-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tate DF, et al. Regional areas and widths of the midsagittal corpus callosum among HIV-infected patients on stable antiretroviral therapies. J Neurovirol. 2011;17(4):368–79. doi: 10.1007/s13365-011-0033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hua X, et al. Disrupted cerebral metabolite levels and lower nadir CD4 + counts are linked to brain volume deficits in 210 HIV-infected patients on stable treatment. Neuroimage Clin. 2013;3:132–42. doi: 10.1016/j.nicl.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kramer-Hammerle S, et al. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111(2):194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 109.Thompson KA, et al. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. 2011;179(4):1623–9. doi: 10.1016/j.ajpath.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shiramizu B, et al. Amount of HIV DNA in peripheral blood mononuclear cells is proportional to the severity of HIV-1-associated neurocognitive disorders. J Neuropsychiatry Clin Neurosci. 2009;21(1):68–74. doi: 10.1176/appi.neuropsych.21.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kusao I, et al. Cognitive performance related to HIV-1-infected monocytes. J Neuropsychiatry Clin Neurosci. 2012;24(1):71–80. doi: 10.1176/appi.neuropsych.11050109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gray LR, et al. Is the central nervous system a reservoir of HIV-1? Curr Opin HIV AIDS. 2014;9(6):552–8. doi: 10.1097/COH.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ho DD, Rota TR, Hirsch MS. Infection of monocyte/macrophages by human T lymphotropic virus type III. J Clin Invest. 1986;77(5):1712–5. doi: 10.1172/JCI112491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nicholson JK, et al. In vitro infection of human monocytes with human T lymphotropic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) J Immunol. 1986;137(1):323–9. [PubMed] [Google Scholar]
- 115.Busca A, Saxena M, Kumar A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J Biol Chem. 2012;287(18):15118–33. doi: 10.1074/jbc.M111.312660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aylward EH, et al. Magnetic resonance imaging measurement of gray matter volume reductions in HIV dementia. Am J Psychiatry. 1995;152(7):987–94. doi: 10.1176/ajp.152.7.987. [DOI] [PubMed] [Google Scholar]
- 117.Kure K, et al. Cellular localization of an HIV-1 antigen in subacute AIDS encephalitis using an improved double-labeling immunohistochemical method. Am J Pathol. 1990;136(5):1085–92. [PMC free article] [PubMed] [Google Scholar]
- 118.Wiley CA, et al. Distribution of brain HIV load in AIDS. Brain Pathol. 1998;8(2):277–84. doi: 10.1111/j.1750-3639.1998.tb00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang GJ, et al. Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain. 2004;127(Pt 11):2452–8. doi: 10.1093/brain/awh269. [DOI] [PubMed] [Google Scholar]
- 120.Chang L, et al. Decreased brain dopamine transporters are related to cognitive deficits in HIV patients with or without cocaine abuse. Neuroimage. 2008;42(2):869–78. doi: 10.1016/j.neuroimage.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bennett BA, Rusyniak DE, Hollingsworth CK. HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res. 1995;705(1–2):168–76. doi: 10.1016/0006-8993(95)01166-8. [DOI] [PubMed] [Google Scholar]
- 122.Nath A, et al. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol. 2000;14(3):222–7. doi: 10.1177/026988110001400305. [DOI] [PubMed] [Google Scholar]
- 123.du Plessis S, et al. Prefrontal cortical thinning in HIV infection is associated with impaired striatal functioning. J Neural Transm (Vienna) 2016;123(6):643–51. doi: 10.1007/s00702-016-1571-0. [DOI] [PubMed] [Google Scholar]
- 124.Becker JT, et al. Subcortical brain atrophy persists even in HAART-regulated HIV disease. Brain Imaging Behav. 2011;5(2):77–85. doi: 10.1007/s11682-011-9113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gongvatana A, et al. Progressive cerebral injury in the setting of chronic HIV infection and antiretroviral therapy. J Neurovirol. 2013;19(3):209–18. doi: 10.1007/s13365-013-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.du Plessis S, et al. HIV Infection Is Associated with Impaired Striatal Function during Inhibition with Normal Cortical Functioning on Functional MRI. J Int Neuropsychol Soc. 2015;21(9):722–31. doi: 10.1017/S1355617715000971. [DOI] [PubMed] [Google Scholar]
- 127.Bairwa D, et al. Case control study: magnetic resonance spectroscopy of brain in HIV infected patients. BMC Neurol. 2016;16(1):99. doi: 10.1186/s12883-016-0628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ann HW, et al. Characteristics of Resting-State Functional Connectivity in HIV-Associated Neurocognitive Disorder. PLoS One. 2016;11(4):e0153493. doi: 10.1371/journal.pone.0153493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vera JH, et al. Neuroinflammation in treated HIV-positive individuals: A TSPO PET study. Neurology. 2016;86(15):1425–32. doi: 10.1212/WNL.0000000000002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kumar AM, et al. Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: quantification using real-time reverse transcriptase-polymerase chain reaction. J Neurovirol. 2007;13(3):210–24. doi: 10.1080/13550280701327038. [DOI] [PubMed] [Google Scholar]
- 131.Kure K, et al. Morphology and distribution of HIV-1 gp41-positive microglia in subacute AIDS encephalitis. Pattern of involvement resembling a multisystem degeneration. Acta Neuropathol. 1990;80(4):393–400. doi: 10.1007/BF00307693. [DOI] [PubMed] [Google Scholar]
- 132.Gupta S, et al. Dopamine receptor D3 genetic polymorphism (rs6280TC) is associated with rates of cognitive impairment in methamphetamine-dependent men with HIV: preliminary findings. J Neurovirol. 2011;17(3):239–47. doi: 10.1007/s13365-011-0028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jacobs MM, et al. HIV-related cognitive impairment shows bi-directional association with dopamine receptor DRD1 and DRD2 polymorphisms in substance-dependent and substance-independent populations. J Neurovirol. 2013 doi: 10.1007/s13365-013-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Horn A, et al. Increases in CSF dopamine in HIV patients are due to the dopamine transporter 10/10-repeat allele which is more frequent in HIV-infected individuals. J Neural Transm. 2013;120(10):1411–9. doi: 10.1007/s00702-013-1086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Horn A, et al. The dopamine-related polymorphisms BDNF, COMT, DRD2, DRD3, and DRD4 are not linked with changes in CSF dopamine levels and frequency of HIV infection. J Neural Transm (Vienna) 2016 doi: 10.1007/s00702-016-1659-6. [DOI] [PubMed] [Google Scholar]
- 136.Bousman CA, et al. COMT Val158Met Polymorphism, Executive Dysfunction, and Sexual Risk Behavior in the Context of HIV Infection and Methamphetamine Dependence. Interdiscip Perspect Infect Dis. 2010;2010:678648. doi: 10.1155/2010/678648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sundermann EE, et al. Genetic predictor of working memory and prefrontal function in women with HIV. J Neurovirol. 2014 doi: 10.1007/s13365-014-0305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Larsson M, et al. Cerebrospinal fluid catecholamine metabolites in HIV-infected patients. J Neurosci Res. 1991;28(3):406–9. doi: 10.1002/jnr.490280313. [DOI] [PubMed] [Google Scholar]
- 139.Obermann M, et al. Substantia nigra hyperechogenicity and CSF dopamine depletion in HIV. J Neurol. 2009;256(6):948–53. doi: 10.1007/s00415-009-5052-3. [DOI] [PubMed] [Google Scholar]
- 140.Kumar AM, et al. Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol. 2011;17(1):26–40. doi: 10.1007/s13365-010-0003-4. [DOI] [PubMed] [Google Scholar]
- 141.Scheller C, et al. Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkeys due to microglia activation. J Neurochem. 2005;95(2):377–87. doi: 10.1111/j.1471-4159.2005.03373.x. [DOI] [PubMed] [Google Scholar]
- 142.Horn A, et al. Increases in CSF dopamine in HIV patients are due to the dopamine transporter 10/10-repeat allele which is more frequent in HIV-infected individuals. J Neural Transm (Vienna) 2013;120(10):1411–9. doi: 10.1007/s00702-013-1086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Scheller C, et al. Increased dopaminergic neurotransmission in therapy-naive asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. J Neural Transm (Vienna) 2010;117(6):699–705. doi: 10.1007/s00702-010-0415-6. [DOI] [PubMed] [Google Scholar]
- 144.Plessis S, et al. HIV infection results in ventral-striatal reward system hypo-activation during cue processing. AIDS. 2015;29(11):1335–43. doi: 10.1097/QAD.0000000000000680. [DOI] [PubMed] [Google Scholar]
- 145.Nath A, et al. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S62–9. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- 146.Lucas GM, et al. Illicit drug use and HIV-1 disease progression: a longitudinal study in the era of highly active antiretroviral therapy. Am J Epidemiol. 2006;163(5):412–20. doi: 10.1093/aje/kwj059. [DOI] [PubMed] [Google Scholar]
- 147.Byrd DA, et al. Neurocognitive impact of substance use in HIV infection. J Acquir Immune Defic Syndr. 2011;58(2):154–62. doi: 10.1097/QAI.0b013e318229ba41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hauser KF, et al. Opiate drug use and the pathophysiology of neuroAIDS. Curr HIV Res. 2012;10(5):435–52. doi: 10.2174/157016212802138779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gannon BM, Reichard EE, Fantegrossi WE. Psychostimulant Abuse and HIV Infection: cocaine, methamphetamine, and “bath salts” cathinone analogues. Curr Addict Rep. 2014;1(3):237–242. doi: 10.1007/s40429-014-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hidalgo M, V, Atluri S, Nair M. Drugs of Abuse in HIV infection and neurotoxicity. Front Microbiol. 2015;6:217. doi: 10.3389/fmicb.2015.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bhavan KP, V, Kampalath N, Overton ET. The aging of the HIV epidemic. Curr HIV/AIDS Rep. 2008;5(3):150–8. doi: 10.1007/s11904-008-0023-3. [DOI] [PubMed] [Google Scholar]
- 152.Boileau I, et al. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse. 2003;49(4):226–31. doi: 10.1002/syn.10226. [DOI] [PubMed] [Google Scholar]
- 153.D’Aquila PS, et al. The role of dopamine in the mechanism of action of antidepressant drugs. Eur J Pharmacol. 2000;405(1–3):365–73. doi: 10.1016/s0014-2999(00)00566-5. [DOI] [PubMed] [Google Scholar]
- 154.Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–44. doi: 10.1016/j.freeradbiomed.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 155.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85(14):5274–8. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13(5):177–84. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 157.Urban NB, et al. Dopamine release in chronic cannabis users: a [11c]raclopride positron emission tomography study. Biol Psychiatry. 2012;71(8):677–83. doi: 10.1016/j.biopsych.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30(2):215–38. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 159.Volkow ND, et al. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9(6):557–69. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- 160.Beyrer C, et al. Epidemiologic links between drug use and HIV epidemics: an international perspective. J Acquir Immune Defic Syndr. 2010;55(Suppl 1):S10–6. doi: 10.1097/QAI.0b013e3181f9c0c9. [DOI] [PubMed] [Google Scholar]
- 161.Degenhardt L, Hall W. Extent of illicit drug use and dependence, and their contribution to the global burden of disease. The Lancet. 2012;379(9810):55–70. doi: 10.1016/S0140-6736(11)61138-0. [DOI] [PubMed] [Google Scholar]
- 162.El-Bassel N, et al. Drug use as a driver of HIV risks: re-emerging and emerging issues. Curr Opin HIV AIDS. 2014;9(2):150–5. doi: 10.1097/COH.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mathers BM, et al. Global epidemiology of injecting drug use and HIV among people who inject drugs: a systematic review. Lancet. 2008;372(9651):1733–45. doi: 10.1016/S0140-6736(08)61311-2. [DOI] [PubMed] [Google Scholar]
- 164.Kipp AM, Desruisseau AJ, Qian HZ. Non-injection drug use and HIV disease progression in the era of combination antiretroviral therapy. J Subst Abuse Treat. 2011;40(4):386–96. doi: 10.1016/j.jsat.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Strathdee SA, Stockman JK. Epidemiology of HIV among injecting and non-injecting drug users: current trends and implications for interventions. Curr HIV/AIDS Rep. 2010;7(2):99–106. doi: 10.1007/s11904-010-0043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pitcher J, et al. Neuronal ferritin heavy chain and drug abuse affect HIV-associated cognitive dysfunction. J Clin Invest. 2014;124(2):656–69. doi: 10.1172/JCI70090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Basso MR, Bornstein RA. Neurobehavioural consequences of substance abuse and HIV infection. J Psychopharmacol. 2000;14(3):228–37. doi: 10.1177/026988110001400306. [DOI] [PubMed] [Google Scholar]
- 168.Nath A. Human immunodeficiency virus-associated neurocognitive disorder: pathophysiology in relation to drug addiction. Ann N Y Acad Sci. 2010;1187:122–8. doi: 10.1111/j.1749-6632.2009.05277.x. [DOI] [PubMed] [Google Scholar]
- 169.Gosztonyi G, et al. Neuropathologic analysis of postmortal brain samples of HIV-seropositive and -seronegative i.v. drug addicts. Forensic Sci Int. 1993;62(1–2):101–5. doi: 10.1016/0379-0738(93)90052-c. [DOI] [PubMed] [Google Scholar]
- 170.Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1(2):182–91. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- 171.Bouwman FH, et al. Variable progression of HIV-associated dementia. Neurology. 1998;50(6):1814–20. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- 172.Oehmichen M, et al. Neuropathology in non-human immunodeficiency virus-infected drug addicts: hypoxic brain damage after chronic intravenous drug abuse. Acta Neuropathol. 1996;91(6):642–6. doi: 10.1007/s004010050478. [DOI] [PubMed] [Google Scholar]
- 173.Chana G, et al. Cognitive deficits and degeneration of interneurons in HIV+ methamphetamine users. Neurology. 2006;67(8):1486–9. doi: 10.1212/01.wnl.0000240066.02404.e6. [DOI] [PubMed] [Google Scholar]
- 174.Smith DB, Simmonds P, Bell JE. Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J Neurovirol. 2014;20(1):28–38. doi: 10.1007/s13365-013-0225-3. [DOI] [PubMed] [Google Scholar]
- 175.Meade CS, et al. Independent effects of HIV infection and cocaine dependence on neurocognitive impairment in a community sample living in the southern United States. Drug Alcohol Depend. 2015;149:128–35. doi: 10.1016/j.drugalcdep.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Meyer VJ, et al. HIV and recent illicit drug use interact to affect verbal memory in women. J Acquir Immune Defic Syndr. 2013;63(1):67–76. doi: 10.1097/QAI.0b013e318289565c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rippeth JD, et al. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc. 2004;10(1):1–14. doi: 10.1017/S1355617704101021. [DOI] [PubMed] [Google Scholar]
- 178.Green JE, Saveanu RV, Bornstein RA. The effect of previous alcohol abuse on cognitive function in HIV infection. Am J Psychiatry. 2004;161(2):249–54. doi: 10.1176/appi.ajp.161.2.249. [DOI] [PubMed] [Google Scholar]
- 179.Weber E, et al. Substance use is a risk factor for neurocognitive deficits and neuropsychiatric distress in acute and early HIV infection. J Neurovirol. 2013;19(1):65–74. doi: 10.1007/s13365-012-0141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Webber MP, et al. A prospective study of HIV disease progression in female and male drug users. AIDS. 1999;13(2):257–62. doi: 10.1097/00002030-199902040-00014. [DOI] [PubMed] [Google Scholar]
- 181.Kim SG, et al. Cocaine-mediated impact on HIV infection in humanized BLT mice. Sci Rep. 2015;5:10010. doi: 10.1038/srep10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Roth MD, et al. Cocaine and sigma-1 receptors modulate HIV infection, chemokine receptors, and the HPA axis in the huPBL-SCID model. J Leukoc Biol. 2005;78(6):1198–203. doi: 10.1189/jlb.0405219. [DOI] [PubMed] [Google Scholar]
- 183.Moore RD, Keruly JC, Chaisson RE. Differences in HIV disease progression by injecting drug use in HIV-infected persons in care. J Acquir Immune Defic Syndr. 2004;35(1):46–51. doi: 10.1097/00126334-200401010-00006. [DOI] [PubMed] [Google Scholar]
- 184.Cook JA, et al. Crack cocaine, disease progression, and mortality in a multicenter cohort of HIV-1 positive women. AIDS. 2008;22(11):1355–63. doi: 10.1097/QAD.0b013e32830507f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Addai AB, et al. Cocaine modulates HIV-1 integration in primary CD4+ T cells: implications in HIV-1 pathogenesis in drug-abusing patients. J Leukoc Biol. 2015;97(4):779–90. doi: 10.1189/jlb.4A0714-356R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Peterson PK, et al. Morphine promotes the growth of HIV-1 in human peripheral blood mononuclear cell cocultures. AIDS. 1990;4(9):869–73. doi: 10.1097/00002030-199009000-00006. [DOI] [PubMed] [Google Scholar]
- 187.Peterson PK, et al. Cocaine potentiates HIV-1 replication in human peripheral blood mononuclear cell cocultures. Involvement of transforming growth factor-beta. J Immunol. 1991;146(1):81–4. [PubMed] [Google Scholar]
- 188.Napuri J, et al. Cocaine enhances HIV-1 infectivity in monocyte derived dendritic cells by suppressing microRNA-155. PLoS One. 2013;8(12):e83682. doi: 10.1371/journal.pone.0083682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Toussi SS, et al. Short communication: Methamphetamine treatment increases in vitro and in vivo HIV replication. AIDS Res Hum Retroviruses. 2009;25(11):1117–21. doi: 10.1089/aid.2008.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dhillon NK, et al. Cocaine-mediated enhancement of virus replication in macrophages: implications for human immunodeficiency virus-associated dementia. J Neurovirol. 2007;13(6):483–95. doi: 10.1080/13550280701528684. [DOI] [PubMed] [Google Scholar]
- 191.Reynolds JL, et al. Morphine and galectin-1 modulate HIV-1 infection of human monocyte-derived macrophages. J Immunol. 2012;188(8):3757–65. doi: 10.4049/jimmunol.1102276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liang H, et al. Methamphetamine enhances HIV infection of macrophages. Am J Pathol. 2008;172(6):1617–24. doi: 10.2353/ajpath.2008.070971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li Y, et al. Methadone enhances human immunodeficiency virus infection of human immune cells. J Infect Dis. 2002;185(1):118–22. doi: 10.1086/338011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Honeycutt JB, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest. 2016;126(4):1353–66. doi: 10.1172/JCI84456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Budka H, et al. Brain Pathology induced by infection with the human immunodeficiency virus (HIV) A histological, immunocytochemical, and electron microscopical study of 100 autopsy cases. Acta Neuropathol. 1987;75(2):185–98. doi: 10.1007/BF00687080. [DOI] [PubMed] [Google Scholar]
- 196.Elovaara I, et al. Mild brain atrophy in early HIV infection: the lack of association with cognitive deficits and HIV-specific intrathecal immune response. J Neurol Sci. 1990;99(2–3):121–36. doi: 10.1016/0022-510x(90)90149-h. [DOI] [PubMed] [Google Scholar]
- 197.Kallianpur KJ, et al. HIV DNA in CD14+ reservoirs is associated with regional brain atrophy in patients naive to combination antiretroviral therapy. AIDS. 2014;28(11):1619–24. doi: 10.1097/QAD.0000000000000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Glasgow BJ, et al. Adrenal pathology in the acquired immune deficiency syndrome. Am J Clin Pathol. 1985;84(5):594–7. doi: 10.1093/ajcp/84.5.594. [DOI] [PubMed] [Google Scholar]
- 199.Langford D, et al. Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J Acquir Immune Defic Syndr. 2003;34(5):467–74. doi: 10.1097/00126334-200312150-00004. [DOI] [PubMed] [Google Scholar]
- 200.Kallianpur KJ, et al. Oxidative mitochondrial DNA damage in peripheral blood mononuclear cells is associated with reduced volumes of hippocampus and subcortical gray matter in chronically HIV-infected patients. Mitochondrion. 2016;28:8–15. doi: 10.1016/j.mito.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chittiprol S, et al. Progressive dysregulation of autonomic and HPA axis functions in HIV-1 clade C infection in South India. Psychoneuroendocrinology. 2008;33(1):30–40. doi: 10.1016/j.psyneuen.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 202.Cole SW, et al. Psychological risk factors for HIV pathogenesis: mediation by the autonomic nervous system. Biol Psychiatry. 2003;54(12):1444–56. doi: 10.1016/s0006-3223(02)01888-7. [DOI] [PubMed] [Google Scholar]
- 203.Ironson G, et al. Perceived stress and norepinephrine predict the effectiveness of response to protease inhibitors in HIV. Int J Behav Med. 2008;15(3):221–6. doi: 10.1080/10705500802219606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Schneiderman N, Ironson G, Siegel SD. Stress and health: psychological, behavioral, and biological determinants. Annu Rev Clin Psychol. 2005;1:607–28. doi: 10.1146/annurev.clinpsy.1.102803.144141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ironson G, et al. Psychosocial and Neurohormonal Predictors of HIV Disease Progression (CD4 Cells and Viral Load): A 4 Year Prospective Study. AIDS Behav. 2015;19(8):1388–97. doi: 10.1007/s10461-014-0877-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dever SM, Rodriguez M, El-Hage N. beta-Adrenergic receptor gene expression in HIV-associated neurocognitive impairment and encephalitis: implications for MOR-1K subcellular localization. J Neurovirol. 2016;22(6):866–870. doi: 10.1007/s13365-016-0464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sloan EK, et al. Social stress enhances sympathetic innervation of primate lymph nodes: mechanisms and implications for viral pathogenesis. J Neurosci. 2007;27(33):8857–65. doi: 10.1523/JNEUROSCI.1247-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Capitanio JP, Mendoza SP, Lerche NW. Individual differences in peripheral blood immunological and hormonal measures in adult male rhesus macaques (Macaca mulatta): evidence for temporal and situational consistency. Am J Primatol. 1998;44(1):29–41. doi: 10.1002/(SICI)1098-2345(1998)44:1<29::AID-AJP3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 209.Levi G, et al. Human immunodeficiency virus coat protein gp120 inhibits the beta-adrenergic regulation of astroglial and microglial functions. Proc Natl Acad Sci U S A. 1993;90(4):1541–5. doi: 10.1073/pnas.90.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kopin IJ. Catecholamine metabolism: basic aspects and clinical significance. Pharmacol Rev. 1985;37(4):333–64. [PubMed] [Google Scholar]
- 211.Kelly RB. Storage and release of neurotransmitters. Cell. 1993;72(Suppl):43–53. doi: 10.1016/s0092-8674(05)80027-3. [DOI] [PubMed] [Google Scholar]
- 212.Barnes MA, Carson MJ, Nair MG. Non-traditional cytokines: How catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine. 2015;72(2):210–9. doi: 10.1016/j.cyto.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Marino F, Cosentino M. Adrenergic modulation of immune cells: an update. Amino Acids. 2013;45(1):55–71. doi: 10.1007/s00726-011-1186-6. [DOI] [PubMed] [Google Scholar]
- 214.Pinoli M, Marino F, Cosentino M. Dopaminergic Regulation of Innate Immunity: a Review. J Neuroimmune Pharmacol. 2017 doi: 10.1007/s11481-017-9749-2. [DOI] [PubMed] [Google Scholar]
- 215.Barnes MA, Carson MJ, Nair MG. Non-traditional Cytokines: How catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine. 2015 doi: 10.1016/j.cyto.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bellinger DL, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci. 2014;182:15–41. doi: 10.1016/j.autneu.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 217.Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. 2015;6:171. doi: 10.3389/fphar.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Scanzano A, et al. Adrenergic modulation of migration, CD11b and CD18 expression, ROS and interleukin-8 production by human polymorphonuclear leukocytes. Inflamm Res. 2015;64(2):127–35. doi: 10.1007/s00011-014-0791-8. [DOI] [PubMed] [Google Scholar]
- 219.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 220.Kebabian JW, Beaulieu M, Itoh Y. Pharmacological and biochemical evidence for the existence of two categories of dopamine receptor. Can J Neurol Sci. 1984;11(1 Suppl):114–7. doi: 10.1017/s0317167100046254. [DOI] [PubMed] [Google Scholar]
- 221.Missale C, et al. Dopamine receptors: from structure to function. Physiol Rev. 1998;78(1):189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 222.Hernandez-Lopez S, et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20(24):8987–95. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chun LS, et al. D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol. 2013;84(2):190–200. doi: 10.1124/mol.113.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.So CH, et al. Calcium signaling by dopamine D5 receptor and D5-D2 receptor hetero-oligomers occurs by a mechanism distinct from that for dopamine D1-D2 receptor hetero-oligomers. Mol Pharmacol. 2009;75(4):843–54. doi: 10.1124/mol.108.051805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Peterson SM, et al. Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci U S A. 2015;112(22):7097–102. doi: 10.1073/pnas.1502742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32(4):213–8. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hoffman BB, Lefkowitz RJ. Alpha-adrenergic receptor subtypes. N Engl J Med. 1980;302(25):1390–6. doi: 10.1056/NEJM198006193022504. [DOI] [PubMed] [Google Scholar]
- 228.Maguire ME, Ross EM, Gilman AG. beta-Adrenergic receptor: ligand binding properties and the interaction with adenylyl cyclase. Adv Cyclic Nucleotide Res. 1977;8:1–83. [PubMed] [Google Scholar]
- 229.Ross EM, et al. Relationship between the beta-adrenergic receptor and adenylate cyclase. J Biol Chem. 1977;252(16):5761–75. [PubMed] [Google Scholar]
- 230.Fu Q, Wang Q, Xiang YK. Insulin and beta Adrenergic Receptor Signaling: Crosstalk in Heart. Trends Endocrinol Metab. 2017;28(6):416–427. doi: 10.1016/j.tem.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gaskill PJ, et al. Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol. 2009;175(3):1148–59. doi: 10.2353/ajpath.2009.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Coley JS, et al. Dopamine Increases CD14+CD16+ Monocyte Migration and Adhesion in the Context of Substance Abuse and HIV Neuropathogenesis. PLoS One. 2015;10(2):e0117450. doi: 10.1371/journal.pone.0117450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mastroeni D, et al. Microglial responses to dopamine in a cell culture model of Parkinson's disease. Neurobiol Aging. 2009;30(11):1805–17. doi: 10.1016/j.neurobiolaging.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.McKenna F, et al. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol. 2002;132(1–2):34–40. doi: 10.1016/s0165-5728(02)00280-1. [DOI] [PubMed] [Google Scholar]
- 235.<Macrophage Phagocytosis Assay - Zymosan.pdf>.
- 236.Gaskill PJ, et al. Characterization and function of the human macrophage dopaminergic system: implications for CNS disease and drug abuse. J Neuroinflammation. 2012;9:203. doi: 10.1186/1742-2094-9-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Calderon TM, et al. Dopamine Increases CD14(+)CD16(+) Monocyte Transmigration across the Blood Brain Barrier: Implications for Substance Abuse and HIV Neuropathogenesis. J Neuroimmune Pharmacol. 2017;12(2):353–370. doi: 10.1007/s11481-017-9726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Huang Y, et al. Roles of dopamine receptor subtypes in mediating modulation of T lymphocyte function. Neuro Endocrinol Lett. 2010;31(6):782–91. [PubMed] [Google Scholar]
- 239.Cosentino M, et al. Dopaminergic receptors and adrenoceptors in circulating lymphocytes as putative biomarkers for the early onset and progression of multiple sclerosis. J Neuroimmunol. 2016;298:82–9. doi: 10.1016/j.jneuroim.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 240.Besser MJ, Ganor Y, Levite M. Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFalpha or both. J Neuroimmunol. 2005;169(1–2):161–71. doi: 10.1016/j.jneuroim.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 241.Cosentino M, et al. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood. 2007;109(2):632–42. doi: 10.1182/blood-2006-01-028423. [DOI] [PubMed] [Google Scholar]
- 242.Grisanti LA, et al. Pro-inflammatory responses in human monocytes are beta1-adrenergic receptor subtype dependent. Mol Immunol. 2010;47(6):1244–54. doi: 10.1016/j.molimm.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Fragala MS, et al. Leukocyte beta2-adrenergic receptor expression in response to resistance exercise. Med Sci Sports Exerc. 2011;43(8):1422–32. doi: 10.1249/MSS.0b013e31820b88bc. [DOI] [PubMed] [Google Scholar]
- 244.Maes M, et al. The effects of noradrenaline and alpha-2 adrenoceptor agents on the production of monocytic products. Psychiatry Res. 2000;96(3):245–53. doi: 10.1016/s0165-1781(00)00216-x. [DOI] [PubMed] [Google Scholar]
- 245.Linden M. The effects of beta 2-adrenoceptor agonists and a corticosteroid, budesonide, on the secretion of inflammatory mediators from monocytes. Br J Pharmacol. 1992;107(1):156–60. doi: 10.1111/j.1476-5381.1992.tb14479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Izeboud CA, et al. Participation of beta-adrenergic receptors on macrophages in modulation of LPS-induced Cytokine release. J Recept Signal Transduct Res. 1999;19(1–4):191–202. doi: 10.3109/10799899909036645. [DOI] [PubMed] [Google Scholar]
- 247.Rouppe van der Voort C, et al. Neuroendocrine mediators up-regulate alpha1b- and alpha1d-adrenergic receptor subtypes in human monocytes. J Neuroimmunol. 1999;95(1–2):165–73. doi: 10.1016/s0165-5728(99)00011-9. [DOI] [PubMed] [Google Scholar]
- 248.Heijnen CJ, et al. Cytokines regulate alpha(1)-adrenergic receptor mRNA expression in human monocytic cells and endothelial cells. J Neuroimmunol. 2002;125(1–2):66–72. doi: 10.1016/s0165-5728(02)00034-6. [DOI] [PubMed] [Google Scholar]
- 249.Grisanti LA, et al. alpha1-adrenergic receptors positively regulate Toll-like receptor Cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther. 2011;338(2):648–57. doi: 10.1124/jpet.110.178012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Engler KL, et al. Autocrine actions of macrophage-derived catecholamines on interleukin-1 beta. J Neuroimmunol. 2005;160(1–2):87–91. doi: 10.1016/j.jneuroim.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 251.Mori K, et al. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology. 2002;43(6):1026–34. doi: 10.1016/s0028-3908(02)00211-3. [DOI] [PubMed] [Google Scholar]
- 252.Tanaka KF, et al. Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res. 2002;70(2):232–7. doi: 10.1002/jnr.10399. [DOI] [PubMed] [Google Scholar]
- 253.Borger P, et al. Beta-adrenoceptor-mediated inhibition of IFN-gamma, IL-3, and GM-CSF mRNA accumulation in activated human T lymphocytes is solely mediated by the beta2-adrenoceptor subtype. Am J Respir Cell Mol Biol. 1998;19(3):400–7. doi: 10.1165/ajrcmb.19.3.2765. [DOI] [PubMed] [Google Scholar]
- 254.Jetschmann JU, et al. Expression and in-vivo modulation of alpha- and beta-adrenoceptors on human natural killer (CD16+) cells. J Neuroimmunol. 1997;74(1–2):159–64. doi: 10.1016/s0165-5728(96)00221-4. [DOI] [PubMed] [Google Scholar]
- 255.von Dossow V, et al. Clonidine attenuated early proinflammatory response in T-cell subsets after cardiac surgery. Anesth Analg. 2006;103(4):809–14. doi: 10.1213/01.ane.0000237308.28739.d8. [DOI] [PubMed] [Google Scholar]
- 256.Amenta F, et al. Identification of dopamine plasma membrane and vesicular transporters in human peripheral blood lymphocytes. J Neuroimmunol. 2001;117(1–2):133–42. doi: 10.1016/s0165-5728(01)00317-4. [DOI] [PubMed] [Google Scholar]
- 257.Ferrari M, et al. Dopaminergic D1-like receptor-dependent inhibition of tyrosine hydroxylase mRNA expression and catecholamine production in human lymphocytes. Biochem Pharmacol. 2004;67(5):865–73. doi: 10.1016/j.bcp.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 258.Pirzgalska RM, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. 2017;23(11):1309–1318. doi: 10.1038/nm.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bergquist J, et al. Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop. Proc Natl Acad Sci U S A. 1994;91(26):12912–6. doi: 10.1073/pnas.91.26.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Marino F, et al. Endogenous catecholamine synthesis, metabolism storage, and uptake in human peripheral blood mononuclear cells. Exp Hematol. 1999;27(3):489–95. doi: 10.1016/s0301-472x(98)00057-5. [DOI] [PubMed] [Google Scholar]
- 261.Fuller RW. Pharmacology of brain epinephrine neurons. Annu Rev Pharmacol Toxicol. 1982;22:31–55. doi: 10.1146/annurev.pa.22.040182.000335. [DOI] [PubMed] [Google Scholar]
- 262.Mefford IN. Epinephrine in mammalian brain. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12(4):365–88. doi: 10.1016/0278-5846(88)90099-1. [DOI] [PubMed] [Google Scholar]
- 263.Scarr E, et al. Cholinergic connectivity: it's implications for psychiatric disorders. Front Cell Neurosci. 2013;7:55. doi: 10.3389/fncel.2013.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Fuxe K, et al. Volume transmission and its different forms in the central nervous system. Chin J Integr Med. 2013;19(5):323–9. doi: 10.1007/s11655-013-1455-1. [DOI] [PubMed] [Google Scholar]
- 265.Fuxe K, et al. Volume Transmission in Central Dopamine and Noradrenaline Neurons and Its Astroglial Targets. Neurochem Res. 2015 doi: 10.1007/s11064-015-1574-5. [DOI] [PubMed] [Google Scholar]
- 266.Paspalas CD, Goldman-Rakic PS. Microdomains for dopamine volume neurotransmission in primate prefrontal cortex. J Neurosci. 2004;24(23):5292–300. doi: 10.1523/JNEUROSCI.0195-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Jansson A, et al. On the distribution patterns of D1, D2, tyrosine hydroxylase and dopamine transporter immunoreactivities in the ventral striatum of the rat. Neuroscience. 1999;89(2):473–89. doi: 10.1016/s0306-4522(98)00317-0. [DOI] [PubMed] [Google Scholar]
- 268.Nirenberg MJ, et al. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J Neurosci. 1996;16(2):436–47. doi: 10.1523/JNEUROSCI.16-02-00436.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14(1):88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rice ME. Distinct regional differences in dopamine-mediated volume transmission. Prog. Brain Res. 2000;125:277–90. doi: 10.1016/S0079-6123(00)25017-6. [DOI] [PubMed] [Google Scholar]
- 271.Fuxe K, et al. Volume Transmission in Central Dopamine and Noradrenaline Neurons and Its Astroglial Targets. Neurochem Res. 2015;40(12):2600–14. doi: 10.1007/s11064-015-1574-5. [DOI] [PubMed] [Google Scholar]
- 272.Rice ME, Cragg SJ. Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev. 2008;58(2):303–13. doi: 10.1016/j.brainresrev.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cragg SJ, et al. Dopamine-mediated volume transmission in midbrain is regulated by distinct extracellular geometry and uptake. J Neurophysiol. 2001;85(4):1761–71. doi: 10.1152/jn.2001.85.4.1761. [DOI] [PubMed] [Google Scholar]
- 274.Volkow ND, et al. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature. 1997;386(6627):830–3. doi: 10.1038/386830a0. [DOI] [PubMed] [Google Scholar]
- 275.Hudson AL, Lalies MD, Silverstone P. Venlafaxine enhances the effect of bupropion on extracellular dopamine in rat frontal cortex. Can J Physiol Pharmacol. 2012;90(6):803–9. doi: 10.1139/y2012-045. [DOI] [PubMed] [Google Scholar]
- 276.Parsons LH, Justice JB., Jr Extracellular concentration and in vivo recovery of dopamine in the nucleus accumbens using microdialysis. J Neurochem. 1992;58(1):212–8. doi: 10.1111/j.1471-4159.1992.tb09298.x. [DOI] [PubMed] [Google Scholar]
- 277.Venton BJ, et al. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J Neurochem. 2003;87(5):1284–95. doi: 10.1046/j.1471-4159.2003.02109.x. [DOI] [PubMed] [Google Scholar]
- 278.Gonon FG, Buda MJ. Regulation of dopamine release by impulse flow and by autoreceptors as studied by in vivo voltammetry in the rat striatum. Neuroscience. 1985;14(3):765–74. doi: 10.1016/0306-4522(85)90141-1. [DOI] [PubMed] [Google Scholar]
- 279.Carboni E, et al. Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience. 1989;28(3):653–61. doi: 10.1016/0306-4522(89)90012-2. [DOI] [PubMed] [Google Scholar]
- 280.Kimmel HL, Ginsburg BC, Howell LL. Changes in extracellular dopamine during cocaine self-administration in squirrel monkeys. Synapse. 2005;56(3):129–34. doi: 10.1002/syn.20135. [DOI] [PubMed] [Google Scholar]
- 281.Schiffer WK, et al. Selegiline potentiates cocaine-induced increases in rodent nucleus accumbens dopamine. Synapse. 2003;48(1):35–8. doi: 10.1002/syn.10183. [DOI] [PubMed] [Google Scholar]
- 282.Zachek MK, et al. Simultaneous monitoring of dopamine concentration at spatially different brain locations in vivo. Biosens Bioelectron. 2010;25(5):1179–85. doi: 10.1016/j.bios.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Xi ZX, et al. A single high dose of methamphetamine increases cocaine self-administration by depletion of striatal dopamine in rats. Neuroscience. 2009;161(2):392–402. doi: 10.1016/j.neuroscience.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Truong JG, et al. Age-dependent methamphetamine-induced alterations in vesicular monoamine transporter-2 function: implications for neurotoxicity. J Pharmacol Exp Ther. 2005;314(3):1087–92. doi: 10.1124/jpet.105.085951. [DOI] [PubMed] [Google Scholar]
- 285.Mateo Y, Meana JJ. Determination of the somatodendritic alpha2-adrenoceptor subtype located in rat locus coeruleus that modulates cortical noradrenaline release in vivo. Eur J Pharmacol. 1999;379(1):53–7. doi: 10.1016/s0014-2999(99)00488-4. [DOI] [PubMed] [Google Scholar]
- 286.Fernandez-Pastor B, et al. Characterization of noradrenaline release in the locus coeruleus of freely moving awake rats by in vivo microdialysis. Psychopharmacology (Berl) 2005;180(3):570–9. doi: 10.1007/s00213-005-2181-y. [DOI] [PubMed] [Google Scholar]
- 287.Florin SM, Kuczenski R, Segal DS. Regional extracellular norepinephrine responses to amphetamine and cocaine and effects of clonidine pretreatment. Brain Res. 1994;654(1):53–62. doi: 10.1016/0006-8993(94)91570-9. [DOI] [PubMed] [Google Scholar]
- 288.Fuller RW, Perry KW. Lowering of epinephrine concentration in rat brain by 2,3-dichloro-alpha-methyl-benzylamine, an inhibitor of norepinephrine N-methyltransferase. Biochem Pharmacol. 1977;26(21):2087–90. doi: 10.1016/0006-2952(77)90024-7. [DOI] [PubMed] [Google Scholar]
- 289.Hernandez L, Paredes D, Rada P. Feeding behavior as seen through the prism of brain microdialysis. Physiol Behav. 2011;104(1):47–56. doi: 10.1016/j.physbeh.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 290.Wunderlich V, Fey F, Sydow G. Antiviral effect of haloperidol on Rauscher murine leukemia virus. Arch Geschwulstforsch. 1980;50(8):758–62. [PubMed] [Google Scholar]
- 291.Wunderlich V, Sydow G. Lytic action of neurotropic drugs on retroviruses in vitro. Eur J Cancer. 1980;16(9):1127–32. doi: 10.1016/0014-2964(80)90170-x. [DOI] [PubMed] [Google Scholar]
- 292.Rohr O, et al. Dopamine stimulates expression of the human immunodeficiency virus type 1 via NF-kappaB in cells of the immune system. Nucleic Acids Res. 1999;27(16):3291–9. doi: 10.1093/nar/27.16.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Rohr O, et al. CREB and COUP-TF mediate transcriptional activation of the human immunodeficiency virus type 1 genome in Jurkat T cells in response to cyclic AMP and dopamine. J Cell Biochem. 1999;75(3):404–13. [PubMed] [Google Scholar]
- 294.Scheller C, et al. Dopamine activates HIV in chronically infected T lymphoblasts. J Neural Transm (Vienna) 2000;107(12):1483–9. doi: 10.1007/s007020070012. [DOI] [PubMed] [Google Scholar]
- 295.Kumar R, et al. Chronic morphine exposure causes pronounced virus replication in cerebral compartment and accelerated onset of AIDS in SIV/SHIV-infected Indian rhesus macaques. Virology. 2006;354(1):192–206. doi: 10.1016/j.virol.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 296.Marcondes MC, et al. Methamphetamine increases brain viral load and activates natural killer cells in simian immunodeficiency virus-infected monkeys. Am J Pathol. 2010;177(1):355–61. doi: 10.2353/ajpath.2010.090953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Najera JA, et al. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016;17(1):7. doi: 10.1186/s12865-016-0145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Gaskill PJ, et al. Dopamine Receptor Activation Increases HIV Entry into Primary Human Macrophages. PLoS One. 2014;9(9):e108232. doi: 10.1371/journal.pone.0108232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Arthos J, et al. CCR5 signal transduction in macrophages by human immunodeficiency virus and simian immunodeficiency virus envelopes. J Virol. 2000;74(14):6418–24. doi: 10.1128/jvi.74.14.6418-6424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Liu QH, et al. HIV-1 gp120 and chemokines activate ion channels in primary macrophages through CCR5 and CXCR4 stimulation. Proc Natl Acad Sci U S A. 2000;97(9):4832–7. doi: 10.1073/pnas.090521697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Melar M, Ott DE, Hope TJ. Physiological levels of virion-associated human immunodeficiency virus type 1 envelope induce coreceptor-dependent calcium flux. J Virol. 2007;81(4):1773–85. doi: 10.1128/JVI.01316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Arai H, I, Charo F. Differential regulation of G-protein-mediated signaling by chemokine receptors. J Biol Chem. 1996;271(36):21814–9. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- 303.Harmon B, Ratner L. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J Virol. 2008;82(18):9191–205. doi: 10.1128/JVI.00424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19(4):1484–91. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Pyo CW, et al. Reactive oxygen species activate HIV long terminal repeat via post-translational control of NF-kappaB. Biochem Biophys Res Commun. 2008;376(1):180–5. doi: 10.1016/j.bbrc.2008.08.114. [DOI] [PubMed] [Google Scholar]
- 306.Yang HC, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest. 2009;119(11):3473–86. doi: 10.1172/JCI39199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Fazal N. The role of reactive oxygen species (ROS) in the effector mechanisms of human antimycobacterial immunity. Biochem Mol Biol Int. 1997;43(2):399–408. doi: 10.1080/15216549700204191. [DOI] [PubMed] [Google Scholar]
- 308.Tan HY, et al. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid Med Cell Longev. 2016;2016:2795090. doi: 10.1155/2016/2795090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Dichtl S, et al. Dopamine promotes cellular iron accumulation and oxidative stress responses in macrophages. Biochem Pharmacol. 2017 doi: 10.1016/j.bcp.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 310.Cole SW, et al. Norepinephrine accelerates HIV replication via protein kinase A-dependent effects on Cytokine production. J Immunol. 1998;161(2):610–6. [PubMed] [Google Scholar]
- 311.Wang J, et al. Regulation of CC chemokine receptor 5 and CD4 expression and human immunodeficiency virus type 1 replication in human macrophages and microglia by T helper type 2 Cytokines. J Infect Dis. 2002;185(7):885–97. doi: 10.1086/339522. [DOI] [PubMed] [Google Scholar]
- 312.Moriuchi M, et al. Norepinephrine inhibits human immunodeficiency virus type-1 infection through the NF-kappaB inactivation. Virology. 2006;345(1):167–73. doi: 10.1016/j.virol.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 313.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 314.Brabers NA, Nottet HS. Role of the pro-inflammatory Cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006;36(7):447–58. doi: 10.1111/j.1365-2362.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- 315.Benveniste EN. Cytokine circuits in brain. Implications for AIDS dementia complex. Res Publ Assoc Res Nerv Ment Dis. 1994;72:71–88. [PubMed] [Google Scholar]
- 316.Gallo P, et al. Human immunodeficiency virus type 1 (HIV-1) infection of the central nervous system: an evaluation of Cytokines in cerebrospinal fluid. J Neuroimmunol. 1989;23(2):109–16. doi: 10.1016/0165-5728(89)90029-5. [DOI] [PubMed] [Google Scholar]
- 317.Perrella O, et al. Cerebrospinal fluid Cytokines in AIDS dementia complex. J Neurol. 1992;239(7):387–8. doi: 10.1007/BF00812156. [DOI] [PubMed] [Google Scholar]
- 318.Eugenin EA, et al. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26(4):1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Williams DW, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One. 2013;8(7):e69270. doi: 10.1371/journal.pone.0069270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Baggiolini M. Novel aspects of inflammation: interleukin-8 and related chemotactic Cytokines. Clin Investig. 1993;71(10):812–4. doi: 10.1007/BF00190326. [DOI] [PubMed] [Google Scholar]
- 321.Witwer KW, et al. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS One. 2009;4(12):e8129. doi: 10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Guyon A, et al. Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience. 2009;162(4):1072–80. doi: 10.1016/j.neuroscience.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 323.Felger JC, Miller AH. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol. 2012;33(3):315–27. doi: 10.1016/j.yfrne.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Mogi M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147–50. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 325.Mogi M, et al. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165(1–2):208–10. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 326.Yan Y, et al. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell. 2015;160(1–2):62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 327.Capellino S, et al. Catecholamine-producing cells in the synovial tissue during arthritis: modulation of sympathetic neurotransmitters as new therapeutic target. Ann Rheum Dis. 2010;69(10):1853–60. doi: 10.1136/ard.2009.119701. [DOI] [PubMed] [Google Scholar]
- 328.Acharjee S, et al. HIV-1 Nef expression in microglia disrupts dopaminergic and immune functions with associated mania-like behaviors. Brain Behav Immun. 2014;40:74–84. doi: 10.1016/j.bbi.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 329.Trudler D, et al. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem. 2014;129(3):434–47. doi: 10.1111/jnc.12633. [DOI] [PubMed] [Google Scholar]
- 330.Yamamoto S, et al. Haloperidol Suppresses NF-kappaB to Inhibit Lipopolysaccharide-Induced Pro-Inflammatory Response in RAW 264 Cells. Med Sci Monit. 2016;22:367–72. doi: 10.12659/MSM.895739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Tarazona R, et al. Chlorpromazine amplifies macrophage-dependent IL-10 production in vivo. J Immunol. 1995;154(2):861–70. [PubMed] [Google Scholar]
- 332.Agarwal SK, Marshall GD., Jr Beta-adrenergic modulation of human type-1/type-2 Cytokine balance. J Allergy Clin Immunol. 2000;105(1 Pt 1):91–8. doi: 10.1016/s0091-6749(00)90183-0. [DOI] [PubMed] [Google Scholar]
- 333.Chang JY, Liu LZ. Catecholamines inhibit microglial nitric oxide production. Brain Res Bull. 2000;52(6):525–30. doi: 10.1016/s0361-9230(00)00291-4. [DOI] [PubMed] [Google Scholar]
- 334.Guirao X, et al. Catecholamines increase monocyte TNF receptors and inhibit TNF through beta 2-adrenoreceptor activation. Am J Physiol. 1997;273(6 Pt 1):E1203–8. doi: 10.1152/ajpendo.1997.273.6.E1203. [DOI] [PubMed] [Google Scholar]
- 335.Hasko G, et al. Dopamine suppresses IL-12 p40 production by lipopolysaccharide-stimulated macrophages via a beta-adrenoceptor-mediated mechanism. J Neuroimmunol. 2002;122(1–2):34–9. doi: 10.1016/s0165-5728(01)00459-3. [DOI] [PubMed] [Google Scholar]
- 336.Kavelaars A, et al. Beta 2-adrenergic activation enhances interleukin-8 production by human monocytes. J Neuroimmunol. 1997;77(2):211–6. doi: 10.1016/s0165-5728(97)00076-3. [DOI] [PubMed] [Google Scholar]
- 337.Li CY, et al. Dobutamine inhibits monocyte chemoattractant protein-1 production and chemotaxis in human monocytes. Anesth Analg. 2003;97(1):205–9. doi: 10.1213/01.ane.0000066013.34263.54. table of contents. [DOI] [PubMed] [Google Scholar]
- 338.Markus T, et al. beta-Adrenoceptor activation depresses brain inflammation and is neuroprotective in lipopolysaccharide-induced sensitization to oxygen-glucose deprivation in organotypic hippocampal slices. J Neuroinflammation. 2010;7:94. doi: 10.1186/1742-2094-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Mizuno K, et al. Beta2-adrenergic receptor stimulation inhibits LPS-induced IL-18 and IL-12 production in monocytes. Immunol Lett. 2005;101(2):168–72. doi: 10.1016/j.imlet.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 340.Roewe J, et al. Neuroendocrine Modulation of IL-27 in Macrophages. J Immunol. 2017;199(7):2503–2514. doi: 10.4049/jimmunol.1700687. [DOI] [PubMed] [Google Scholar]
- 341.Takahashi HK, et al. Effect of beta 2-adrenergic receptor stimulation on interleukin-18-induced intercellular adhesion molecule-1 expression and Cytokine production. J Pharmacol Exp Ther. 2003;304(2):634–42. doi: 10.1124/jpet.102.042622. [DOI] [PubMed] [Google Scholar]
- 342.Bacou E, et al. beta2-adrenoreceptor stimulation dampens the LPS-induced M1 polarization in pig macrophages. Dev Comp Immunol. 2017;76:169–176. doi: 10.1016/j.dci.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 343.Muthu K, et al. Adrenergic modulation of Cytokine release in bone marrow progenitor-derived macrophage following polymicrobial sepsis. J Neuroimmunol. 2005;158(1–2):50–7. doi: 10.1016/j.jneuroim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 344.Sigola LB, Zinyama RB. Adrenaline inhibits macrophage nitric oxide production through beta1 and beta2 adrenergic receptors. Immunology. 2000;100(3):359–63. doi: 10.1046/j.1365-2567.2000.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Spengler RN, et al. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol. 1994;152(6):3024–31. [PubMed] [Google Scholar]
- 346.Szelenyi J, et al. Dual beta-adrenergic modulation in the immune system: stimulus-dependent effect of isoproterenol on MAPK activation and inflammatory mediator production in macrophages. Neurochem Int. 2006;49(1):94–103. doi: 10.1016/j.neuint.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 347.Dello Russo C, et al. Inhibition of microglial inflammatory responses by norepinephrine: effects on nitric oxide and interleukin-1beta production. J Neuroinflammation. 2004;1(1):9. doi: 10.1186/1742-2094-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Heneka MT, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107(13):6058–63. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Li CY, et al. Adrenaline inhibits lipopolysaccharide-induced macrophage inflammatory protein-1 alpha in human monocytes: the role of beta-adrenergic receptors. Anesth Analg. 2003;96(2):518–23. doi: 10.1097/00000539-200302000-00040. table of contents. [DOI] [PubMed] [Google Scholar]
- 350.Li CY, et al. Dobutamine modulates lipopolysaccharide-induced macrophage inflammatory protein-1alpha and interleukin-8 production in human monocytes. Anesth Analg. 2003;97(1):210–5. doi: 10.1213/01.ane.0000066257.38180.04. table of contents. [DOI] [PubMed] [Google Scholar]
- 351.Wang J, et al. Beta-adrenoceptor mediated surgery-induced production of pro-inflammatory Cytokines in rat microglia cells. J Neuroimmunol. 2010;223(1–2):77–83. doi: 10.1016/j.jneuroim.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 352.Li M, et al. Norepinephrine induces the expression of interleukin-6 via beta-adrenoreceptor-NAD(P)H oxidase system -NF-kappaB dependent signal pathway in U937 macrophages. Biochem Biophys Res Commun. 2015;460(4):1029–34. doi: 10.1016/j.bbrc.2015.02.172. [DOI] [PubMed] [Google Scholar]
- 353.Tan KS, et al. Beta2 adrenergic receptor activation stimulates pro-inflammatory Cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19(2):251–60. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 354.Tomozawa Y, et al. Participation of cAMP and cAMP-dependent protein kinase in beta-adrenoceptor-mediated interleukin-1 beta mRNA induction in cultured microglia. Neurosci Res. 1995;22(4):399–409. doi: 10.1016/0168-0102(95)00922-g. [DOI] [PubMed] [Google Scholar]
- 355.Takahashi HK, et al. Beta 2-adrenergic receptor agonist induces IL-18 production without IL-12 production. J Neuroimmunol. 2004;151(1–2):137–47. doi: 10.1016/j.jneuroim.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 356.Hasko G, et al. Dopamine suppresses IL-12 p40 production bylipopolysaccharide-stimulated macrophages via a beta-adrenoceptor-mediatedmechanism. J Neuroimmunol. 2002;122 doi: 10.1016/s0165-5728(01)00459-3. [DOI] [PubMed] [Google Scholar]
- 357.Johnson JD, et al. Catecholamines mediate stress-induced increases in peripheral and central inflammatory Cytokines. Neuroscience. 2005;135(4):1295–307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- 358.Straub RH, et al. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J Leukoc Biol. 2000;67(4):553–8. doi: 10.1002/jlb.67.4.553. [DOI] [PubMed] [Google Scholar]
- 359.Xiu F, Stanojcic M, Jeschke MG. Norepinephrine inhibits macrophage migration by decreasing CCR2 expression. PLoS One. 2013;8(7):e69167. doi: 10.1371/journal.pone.0069167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ortega E, Garcia JJ, De la Fuente M. Modulation of adherence and chemotaxis of macrophages by norepinephrine. Influence of ageing. Mol Cell Biochem. 2000;203(1–2):113–7. doi: 10.1023/a:1007094614047. [DOI] [PubMed] [Google Scholar]
- 361.Garcia JJ, et al. Regulation of phagocytic process of macrophages by noradrenaline and its end metabolite 4-hydroxy-3-metoxyphenyl-glycol. Role of alpha- and beta-adrenoreceptors. Mol Cell Biochem. 2003;254(1–2):299–304. doi: 10.1023/a:1027345820519. [DOI] [PubMed] [Google Scholar]
- 362.Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288(21):15291–302. doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Madrigal JL, et al. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. J Neurosci. 2009;29(1):263–7. doi: 10.1523/JNEUROSCI.4926-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Houades V, et al. Shapes of astrocyte networks in the juvenile brain. Neuron Glia Biol. 2006;2(1):3–14. doi: 10.1017/S1740925X06000081. [DOI] [PubMed] [Google Scholar]
- 365.Lawson LJ, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 366.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2(10):907–16. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 367.Dawson VL, et al. Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci U S A. 1993;90(8):3256–9. doi: 10.1073/pnas.90.8.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Torre D, Pugliese A, Speranza F. Role of nitric oxide in HIV-1 infection: friend or foe? Lancet Infect Dis. 2002;2(5):273–80. doi: 10.1016/s1473-3099(02)00262-1. [DOI] [PubMed] [Google Scholar]
- 369.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587(2):250–6. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 370.Corasaniti MT, et al. Death of cultured human neuroblastoma cells induced by HIV-1 gp120 is prevented by NMDA receptor antagonists and inhibitors of nitric oxide and cyclooxygenase. Neurodegeneration. 1995;4(3):315–21. doi: 10.1016/1055-8330(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 371.Torre D, et al. Production of nitric oxide from peripheral blood mononuclear cells and polymorphonuclear leukocytes of patients with HIV-1. AIDS. 1995;9(8):979–80. doi: 10.1097/00002030-199508000-00027. [DOI] [PubMed] [Google Scholar]
- 372.Groeneveld PH, et al. Increased production of nitric oxide correlates with viral load and activation of mononuclear phagocytes in HIV-infected patients. Scand J Infect Dis. 1996;28(4):341–5. doi: 10.3109/00365549609037916. [DOI] [PubMed] [Google Scholar]
- 373.Blond D, et al. Nitric oxide synthesis enhances human immunodeficiency virus replication in primary human macrophages. J Virol. 2000;74(19):8904–12. doi: 10.1128/jvi.74.19.8904-8912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Dawson VL, et al. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13(6):2651–61. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Lin YL, et al. Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J Virol. 1997;71(7):5227–35. doi: 10.1128/jvi.71.7.5227-5235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Persichini T, et al. Nitric oxide inhibits HIV-1 replication in human astrocytoma cells. Biochem Biophys Res Commun. 1999;254(1):200–2. doi: 10.1006/bbrc.1998.9880. [DOI] [PubMed] [Google Scholar]
- 377.Persichini T, et al. Nitric oxide inhibits the HIV-1 reverse transcriptase activity. Biochem Biophys Res Commun. 1999;258(3):624–7. doi: 10.1006/bbrc.1999.0581. [DOI] [PubMed] [Google Scholar]
- 378.Mannick JB, et al. Nitric oxide modulates HIV-1 replication. J Acquir Immune Defic Syndr. 1999;22(1):1–9. doi: 10.1097/00042560-199909010-00001. [DOI] [PubMed] [Google Scholar]
- 379.Aalinkeel R, et al. Galectin-1 Reduces Neuroinflammation via Modulation of Nitric Oxide-Arginase Signaling in HIV-1 Transfected Microglia: a Gold Nanoparticle-Galectin-1 “Nanoplex” a Possible Neurotherapeutic? J Neuroimmune Pharmacol. 2017;12(1):133–151. doi: 10.1007/s11481-016-9723-4. [DOI] [PubMed] [Google Scholar]
- 380.Persichini T, et al. Cross-Talk Between NO Synthase Isoforms in Neuro-Inflammation: Possible Implications in HIV-Associated Neurocognitive Disorders. Curr Med Chem. 2016;23(24):2706–2714. doi: 10.2174/0929867323666160809100452. [DOI] [PubMed] [Google Scholar]
- 381.Chi DS, et al. Regulation of nitric oxide production from macrophages by lipopolysaccharide and catecholamines. Nitric Oxide. 2003;8(2):127–32. doi: 10.1016/s1089-8603(02)00148-9. [DOI] [PubMed] [Google Scholar]
- 382.Huck JH, et al. De novo expression of dopamine D2 receptors on microglia after stroke. J Cereb Blood Flow Metab. 2015;35(11):1804–11. doi: 10.1038/jcbfm.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Yoshioka Y, et al. Dopamine inhibits lipopolysaccharide-induced nitric oxide production through the formation of dopamine quinone in murine microglia BV-2 cells. J Pharmacol Sci. 2016;130(2):51–9. doi: 10.1016/j.jphs.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 384.Shavali S, Combs CK, Ebadi M. Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: relevance to Parkinson's disease. Neurochem Res. 2006;31(1):85–94. doi: 10.1007/s11064-005-9233-x. [DOI] [PubMed] [Google Scholar]
- 385.Qin T, et al. Dopamine induces growth inhibition and vascular normalization through reprogramming M2-polarized macrophages in rat C6 glioma. Toxicol Appl Pharmacol. 2015;286(2):112–23. doi: 10.1016/j.taap.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 386.Bone NB, et al. Frontline Science: D1 dopaminergic receptor signaling activates the AMPK-bioenergetic pathway in macrophages and alveolar epithelial cells and reduces endotoxin-induced ALI. J Leukoc Biol. 2016 doi: 10.1189/jlb.3HI0216-068RR. [DOI] [PMC free article] [PubMed] [Google Scholar]