

Introduction:
In 1990, a landmark study established the link between a genetic mutation in the sarcomeric β-myosin heavy chain (MHC) gene and hypertrophic cardiomyopathy [2]. This seminal discovery was the first time a mutation in a sarcomeric gene was causally linked to disease, thus beginning the “Genetic Era” of cardiomyopathies. Since then, over 450 mutations in sarcomere-associated genes encoding thick filament associated proteins, thin filament (TF) proteins, and titin have been implicated in the development of hypertrophic (HCM) and dilated cardiomyopathy (DCM) [3–5]. The recognition of the genetic basis of cardiomyopathic remodeling was key to furthering our understanding of the complex heterogeneity of disease presentation.
While the genetic basis of cardiomyopathies is widely recognized, the link between genotype and phenotype and our understanding of the precise mechanisms underlying these diseases remains unclear. To add to this disconnect, by the time patients become symptomatic, pathology has often progressed past the initial phase (where treatment would be more effective) to advanced end-stage pathology. Strikingly, despite the persistence of heart disease as a leading cause of death, over the last three decades there has been a marked decline in the innovation of cardiovascular pharmaceuticals [6]. Similarly, the only treatments currently available for HCM are indirect, used primarily for symptom palliation. Further complicating this is the oft-noted finding that, similar mutations, clustered in “hot spots” in sarcomeric proteins often lead to disparate phenotypes. We and others have recently shown that to address this disconnect it is paramount to understand the primary biophysical derangements associated with individual mutations.
Given the complexity of genetic cardiomyopathies, it is perhaps more useful to characterize cardiomyopathy-causing mutations based on their primary biophysical insult rather than their late-stage pathology. Linking the biophysical insult to the earliest molecular dysregulation(s) observed, or “binning”, could lead to targeted therapies for the pre-clinical cohort, that alter the natural progression of the disease (Figure 1). The current review will focus on how the molecular changes, elicited by mutations in myofilament proteins (enzymatic thick filament, myosin; and the regulatory TF, Tm, cTnC, cTnI, cTnT), trigger specific pathways that lead to cardiomyopathies. Of note, as titin mutations are largely linked to DCM and represent a particular complex array of mutations, it will not be covered here. Its role in genetic cardiomyopathies is well reviewed elsewhere [7–9].
Figure 1:
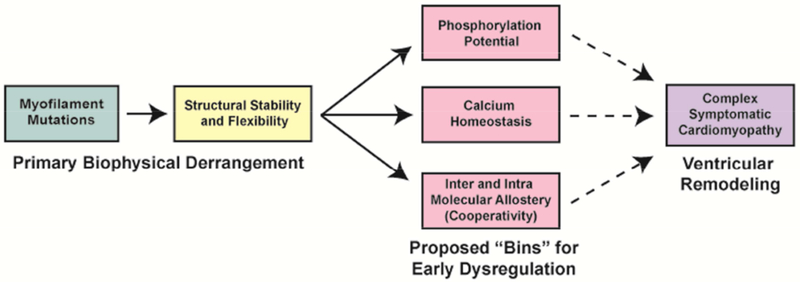
Summary of cardiomyopathic disease progression and proposed “binning”. Dashed line represents the unknown time-course of disease progression between early dysregulation and ventricular remodeling.
We will discuss three proposed “bins” that provide well-studied, mechanistic insight for a wide range of cardiomyopathies in the early and compensatory phase of the disease. These “bins” include:
Phosphorylation Potential: Specifically, how mutations in myofilament proteins alter the ability to respond to physiological β-adrenergic stimulation.
Calcium Homeostasis: Specifically, how mutations in myofilament proteins lead to downstream changes in calcium handling in the myocyte.
Structural Stability and Flexibility: Specifically, how mutations in myofilament proteins alter intra and inter-molecular allostery.
Phosphorylation Potential:
The basic function of the cardiovascular system is to match cardiac output to the hemodynamic demands of the body. The ability of the heart to couple input to output and respond to systemic needs on a beat-to-beat basis is tightly regulated by the autonomic nervous system [10]. At the level of the cardiac myocyte, β-adrenergic (β-AD) stimulation elicits a signaling cascade mediated by PKA-phosphorylation. This culminates in increased lusitropy (relaxation) and inotropy (contractility) allowing the heart to fill more efficiently and beat more rapidly. Downstream targets of PKA phosphorylation in the cardiomyocyte include the myofilament (cTnI), sarcolemma (Na-Ca exchanger, NCX; L-type calcium channel, LTCC) and sarcoplasmic reticulum (SR-bound Ca2+-ATPase, SERCA; phospholamban, PLB; ryanodine receptor, RyR). These targets work synergistically to fine tune the calcium-dependent mechanical response of the myocyte.
The normal structural response to phosphorylation is small yet the functional impact is significant. A robust example of this structure-function relationship is the phosphorylation of cTnI, the inhibitory subunit of cTn, at Ser22/23 (Ser23/24 in mice). Upon β-AD stimulation, PKA-mediated phosphorylation of cTnI elicits an allosteric rearrangement of cTn such that the calcium sensitivity of force development is decreased and ventricular relaxation can occur more rapidly [11]. Phosphorylation of cTnI and calcium sensitivity are intrinsically linked as the role of cTn is two-fold; it regulates the response of the sarcomere to increased demand via phosphorylation, and it is the calcium switch that initiates contraction (discussed below). A critical interface exists between the N-terminal extension of cTnI (cNTnI) and the C-lobe of cTnC that has a reduced affinity in response to PKA-phosphorylation, (Figure 2) [12–14]. This reduction in affinity of cTnI for cTnC mediates the rapid release of calcium from the N-lobe of cTnC (cNTnC) via electrostatic repulsion of cNTnI, and sub-optimal repositioning of cNTnC, thus affecting calcium sensitivity [15, 16].
Figure 2:
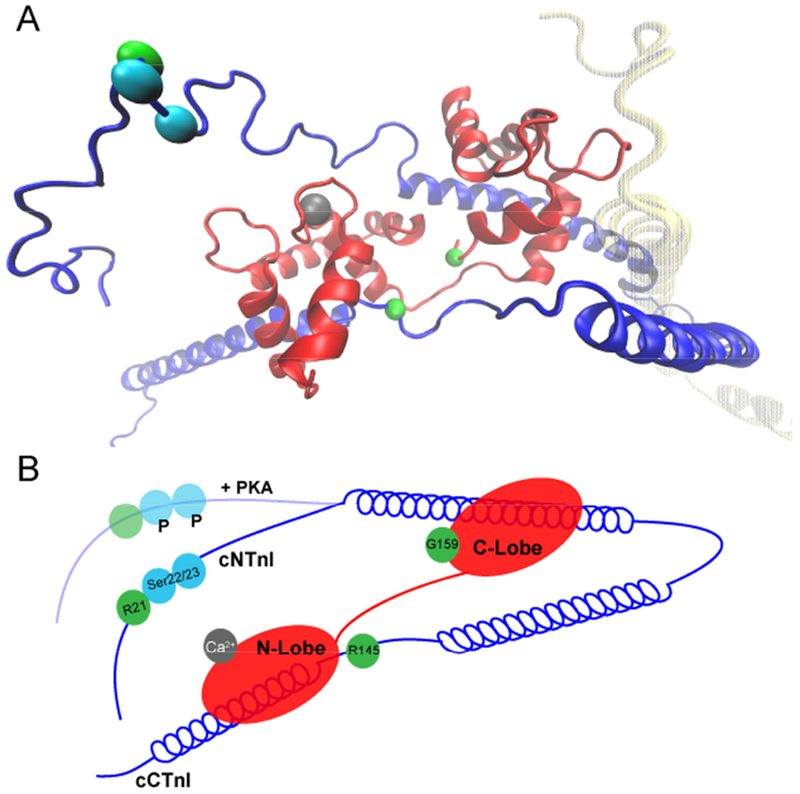
A - Atomistic Model of the cTnI-cTnC Interface generated from the publicly available average structure by JR Exequiel Pineda [1]. cTnI (dark blue), cTnC (red), cTnT (yellow). Dark vs light areas of the protein indicate spatial depth ie: near and far, respectively. cTnT is intentionally faded for clarity. Mutation sites discussed (cTnI R21 and R145, and cTnC G159) are marked with green beads, cTnI Ser22/23 are highlighted with cyan beads, and the active calcium binding pocket is marked with a grey bead. B – 2D representation of the interface, including the structural rearrangement upon PKA phosphorylation.
Impaired response to the β-AD signaling cascade is a commonly reported finding in heart failure [17]. However, in HCM and DCM this desensitization is observed in the absence of changes in the concentration of plasma catecholamines and prior to the onset of heart failure [18–20]. Notably, this failure to augment contractility is often out of proportion to the degree of remodeling, where diastolic dysfunction may be present with or without hypertrophy [21]. These observations beg the question of how a single amino acid substitution within the myofilament could trigger these downstream events leading to a change in β-AD responsiveness. Due to the inherent lack of structure in “intrinsically disordered” segments of cTn, the N-terminal extension of both cTnI and cTnT among others, have been notoriously difficult to study [22, 23]. Recently, molecular dynamics simulations (MDS), a powerful computational tool for monitoring the rapid fluctuation of these flexible segments over an ensemble of conformations, have enhanced the ability to study the dynamic nature of cTn [14, 24]. Thus, mutations in TF proteins that alter this critical interface could represent an early trigger, responsible for the blunted response that is out-of-proportion to the degree of remodeling. In fact, it was postulated as early as 1995 that targeting this interaction may be a promising approach in the treatment of heart failure [25]. While clinical trials using levosimendan (a calcium sensitizer that binds the cTnI-cTnC interface) have demonstrated mixed results, it is not approved for use in the United States since no improvement in short or long-term outcome beyond traditional inotropes (ie: dobutamine) was reported [25–29]. Due to its inherent complexity, an improved understanding of the cTnI-cTnC interface is necessary to properly target interventions that directly ameliorate disruptions caused by myofilament mutations.
Clinically relevant cardiomyopathy-associated mutations have been reported in the proteins of the cTnI-cTnC interface, including cTnT which has known effects on modulating the cTn core [30]. An illustrative example is cTnC G159D (DCM), which was shown to alter the cTnI-cTnC interface by Biesiadecki et al. [31]. Specifically, a blunting of the expected calcium desensitization upon PKA-treatment was observed. Of note, pseudo-phosphorylation, whereby the PKA-targeted sites were mutated to mimic the negatively charged phosphate ions was also found to be insufficient to induce a decrease in calcium sensitivity [31]. These data suggest that these cTnI-PKA sites are inaccessible in the presence of cTnC-G159D. This structural rearrangement was hypothesized to lead to an increased affinity of the C-lobe of cTnC for the cNTnI, the opposite of the physiological effect of phosphorylation of cTnI, thereby hindering the release of calcium from the functional cNTnC. This further suggests that alterations in the cTnI-cTnC interface, rather than changes in the calcium binding pocket on cTnC, govern calcium affinity. While illustrative, the prevalence of mutations in cTnC is very low, suggesting low tolerance and that alterations at the interface are more commonly due to allosteric effects of mutations in other myofilament proteins.
Cardiomyopathy-associated mutations in cTnI and cTnT are more common, with mutations in “hot-spot” regions of both proteins shown to alter β-AD signaling and some proposed to alter the cTnI-cTnC interface [32–37]. Strikingly, these mutations have similar effects on the interface as reported for cTnC-G159D despite being primarily causative for HCM. The HCM-linked cTnI-R145G mutation (R146G in rodents) has been shown to have an increased calcium binding affinity at baseline [34]. This increase was linked to an increased cTnI-cTnC affinity that was not reduced by introduction of Ser23D/24D pseudo-phosphorylation [34]. These results suggest that, as previously noted, PKA is unable to decrease the affinity of cTnI for cTnC in the presence of the mutation. Similarly, cTnI-R21C has been shown to lead to a blunted response to β-AD signaling in vivo, and has demonstrated an increased cTnI-cTnC interface affinity [34]. Notably, the location of these cTnI mutations, discretely located within the N-terminus (R21C) and C-terminus (R145G), highlight the potential for allosteric propagation of structure-induced functional effects within this highly organized system. This is further highlighted in the cTnT-R92L mutation in which we have measured a myofilament specific decrease in PKA-mediated cTnI phosphorylation in the absence of severe downstream calcium handling abnormalities (discussed in depth below) [37]. This suggests that cTnT-R92L is altering the structure of the cTn core and cTnI-cTnC interface, despite its relative distance, blocking PKA from accessing its binding site. MDS suggests that in the presence of cTnT-R92L, cNTnI is closer to cNTnC likely resulting in PKA’s inability to phosphorylate Ser22/23 [1].
While the predominant effects of mutations in this critical interface are to decrease the accessibility of Ser22/23 for PKA, there are a few exceptions. The restrictive cardiomyopathy (RCM) associated cTnI-R145W mutation that has been shown to reduce the interaction frequency between the C-lobe of cTnC and cTnI, yet causes an increase in myofilament calcium sensitivity [33]. Interestingly, this increased sensitivity is opposite to what would be predicted in the face of a decreased affinity of the cTnI-cTnC interface. However, MDS predictions suggest that the significant structural alterations will ultimately negate the functional impact of Ser23/24 phosphorylation [33]. These complex examples of the functional deficits induced by structural alterations underscore the need for continued research into this highly relevant interface within the cTn core.
Although regionally and pathogenically disparate, these mutations have similar effects on the phosphorylation potential of cTnI and calcium-dependent relaxation. This suggests a critical structural mechanism exists whereby cTnI-cTnC interaction regulates the response to β-AD stimulation, a regulation that can become uncoupled by pathogenic myofilament mutations. Thus, pharmacological targeting of the interface may represent a promising area of investigation in treating the progression to heart failure in cardiomyopathies. Promising work recently demonstrated that Epigallocatechin-3-gallate (EGCG) binds the cTnI-cTnC interface and effectively decreases calcium sensitivity maintaining the coupling of cTnI phosphorylation-dependent calcium sensitivity [38]. However, continued research is necessary to translate this therapy into the clinical realm.
Calcium Homeostasis:
In addition to blunted response to β-AD stimulation and subsequent calcium-dependent relaxation in the heart, an extensively studied mechanism of genetic cardiomyopathy-driven disease pathogenesis is altered calcium homeostasis. This critical signaling molecule is intimately involved in excitation-contraction coupling (ECC), reviewed in [39]. In brief, depolarization of the sarcolemma leads to the opening of voltage gated LTCC, allowing for an influx of calcium into the myocyte. LTCC are organized in close proximity to the SR (the main intracellular calcium store) via T-tubules. RyR located on the membrane of the SR bind free calcium, thereby opening and raising intracellular calcium via calcium-induced calcium-release (CICR). Free calcium binds to cTnC leading to cross bridge formation and force-generation. Upon release of calcium from cTnC, free calcium is removed from the myocyte primarily via SERCA2a, lowering free calcium to resting levels. Thus, given the role of ECC in dynamic regulation of systolic and diastolic function, and its well-described dysregulation in sudden cardiac death, the components of this system are potential targets for precise therapeutic interventions.
Many studies have described common alterations in myocellular calcium handling in HCM and DCM including prolonged calcium transients, increased resting calcium levels, and reduced SR calcium content [40–43]. However, to link these downstream changes to primary insults at the myofilament level, we must define alterations in the buffering capacity of the myofilament. An extensively studied aspect of this myofilament-calcium axis is the calcium sensitivity of force generation in myofibrils and/or reconstituted proteins. Studies have reported disease-specific alterations in force generation whereby HCM-associated mutations are described as calcium-sensitized while DCM-associated mutations are described as calcium-desensitized [44–46]. Of note, while the dysregulation in calcium-sensitivity is proposed to be a secondary mechanism in disease progression, the subsequent downstream alterations in myocellular calcium handling are posited to have differential roles in pathogenesis. Specifically, the increased calcium transients and peak amplitude of calcium seen in multiple models of DCM suggest a maintained ECC pathway in which the downstream alterations are initiated to re-sensitize the myofilament and restore systolic function [47–49]. Alternatively, HCM-associated mutations are typically characterized by increased calcium levels and reduced, prolonged calcium transients, despite progressive diastolic dysfunction with normal, or enhanced systolic function [37, 50–52]. Thus, unlike DCM, these downstream disruptions suggest an uncoupling of the excitation-contraction pathway in HCM that potentiates disease pathogenesis (Figure 3).
Figure 3:
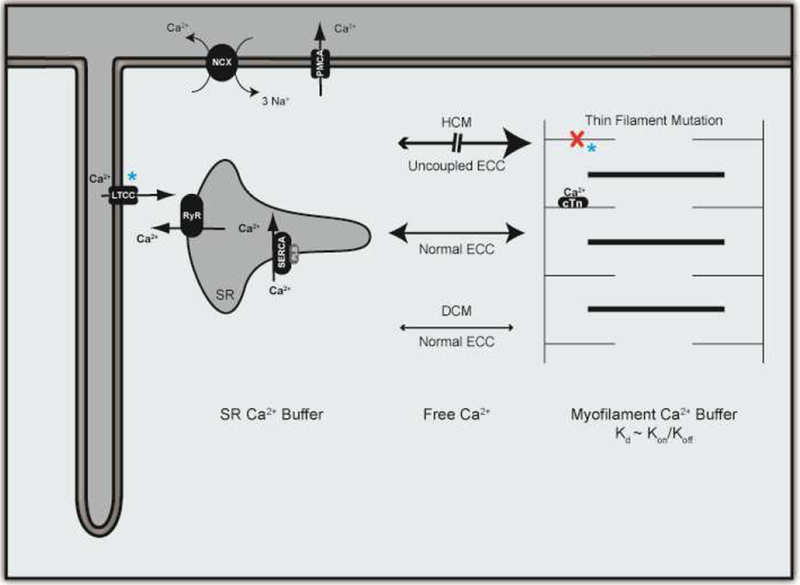
Calcium Homeostasis and Dysregulation. L-type calcium channel (LTCC), Ryanodine Receptor (RyR), Sarcoplasmic Reticulum (SR), SR-bound calcium ATP-ase (SERCA), plasma membrane calcium ATP-ase (PMCA). Blue stars indicate current and proposed sites of therapeutic intervention for modulation of calcium homeostasis.
Many therapies implemented in the clinical management of heart failure have been targeted to restoring normal calcium homeostasis in an effort to improve diastolic and/or systolic function [53, 54]. In a pilot trial utilizing diltiazem, an LTCC blocker, in a pre-clinical cohort of sarcomeric HCM, Ho et.al described a reduction in progressive diastolic dysfunction and cardiac remodeling with treatment. Furthermore, after treatment cessation, progressive cardiac remodeling occurred, implicating diltiazem as an early intervention for HCM patients [53]. Of note, the trial demonstrated early intervention for patients carrying MYBPC3 mutations, while MYH7 mutation carriers exhibited progressive cardiac remodeling and continued diastolic dysfunction throughout treatment [53, 55]. The authors speculated a gene-specific response to diltiazem treatment, highlighting the necessity of defining the natural history of disease pathogenesis to implement efficacious interventions.
Therapeutics targeted to restoring calcium sensitivity of the myofilament have been proposed to be a more direct intervention in calcium homeostasis. To date, while small molecule calcium sensitizers have been successful in clinical trials at restoring cardiac function, the complexity of adverse effects make these therapies less than desirable for patients [56]. To improve these therapies, studies are on-going to understand the precise mechanisms by which cTnC and the TF modulate calcium sensitivity. These advancements include additional small molecules (i.e.: EMD 57033) and the design of engineered cTnC variants, which Davis et al. have shown to be a potential gene therapy option for modulating calcium sensitization [57–59]. While the paradigm of calcium sensitivity offers insight into alterations in myofilament calcium handling, it is a steady-state measurement, thereby lacking resolution of the dynamic processes that govern affinity. Thus, it is imperative to investigate the kinetics of myofilament calcium handling, specifically the rates of calcium association (kon) and dissociation (koff), to understand the dynamic alterations mediating this divergent calcium affinity.
Myofilament calcium kinetics have long been over-looked due to the rapid rate of calcium exchange with cTnC as compared to the rate of relaxation [60, 61]. Recent studies, however, have suggested that these rates may link the dynamic, myofilament-driven insults that initiate the downstream calcium dysregulation to the differential presentation of disease seen in patients [1, 60, 62–65]. For example, three HCM-causative mutations all found in cTnT (R92L, R92W, and R92Q) are known to have varying degrees of cardiac remodeling and risk of sudden cardiac death [66–69]. Myofibers independently expressing these mutations were shown to have a nearly identical increases in calcium sensitivity of force generation [46, 70] but differential downstream calcium dysregulation [37, 71]. Investigation into the dissociation kinetics revealed no change in the koff for cTnT-R92Q TFs [60], while cTnT-R92L and cTnT-R92W caused a significant decrease and increase in koff, respectively [1]. These data suggest that the comparable increase in calcium affinity is likely the result of mutation-specific alterations in dynamic myofilament calcium handling.
Further investigation regarding the structural effects via MDS revealed mutation-specific repositioning of the N-terminus of cTnI, with respect to the calcium binding pocket that, in part, governed these changes in dissociation rate [1]. In parallel, a series of studies coupling in silico, in vitro, and in vivo approaches suggested that changes in calcium kinetics at the level of the TF and thus calcium homeostasis may also be regulated by an altered affinity of cTnI for actin and/or cTnC [62, 63, 72]. While these studies were primarily focused on integrating the structural and functional responses of numerous phosphorylation events of cTnI, cardiomyopathy-associated mutations are known to propagate their effects to the critical cTnI-cTnC interface, as discussed above. It follows that baseline structural insults that result in altered myofilament calcium sensitivity are likely intrinsically linked to the phosphorylation potential of the cTn core. Thus, these primary structural alterations may represent a therapeutic target that simultaneously ameliorates alterations in calcium handling and phosphorylation potential.
While it is implausible to design a small molecule specific to each mutation that changes myofilament calcium kinetics, a subset of therapies targeted to restoration of the structural insults caused by these mutations (i.e.: repositioning of cTnI for restitution of the cTnI-cTnC and/or cTnI-actin equilibrium) may prove to be an efficient intervention. Furthermore, these therapies may also be applied to patients that are shown to have an intact ECC (i.e.: DCM) by stabilizing these molecular interactions that play a primary role in calcium sensitivity and thus regulation of contraction and relaxation. Continued research investigating atomic level alterations coupled to whole animal function will further our knowledge of how structure informs function, making the end goal of rationally designed, targeted small molecule therapeutics attainable.
Structural Stability and Flexibility:
It has become increasingly clear that binary classifications of disease mechanism in cardiomyopathies (such as hyper-contractile vs hypo-contractile and calcium sensitized vs desensitized discussed above) may not be sufficient to describe the heterogeneity of disease presentation necessary for efficacious treatment. To address this, investigation into linking the primary structural insult, caused by single amino acid substitutions, to changes in the stability of the myofilament protein and/or its ability to interact with neighboring proteins within the cardiac sarcomere is ongoing. Indeed, the function of the myofilament, depends on its innate ability to transmit seemingly small structural changes to relatively distant portions of the complex via allosteric activation [14]. In this case, perturbations caused by mutations could alter the native stability of individual proteins or protein-protein interfaces at sites spatially distinct from the primary biophysical insult. The consequences of these “effects-at-a-distance” lead to initiation of pathogenic downstream signaling and ultimately the complexity observed in sarcomeric cardiomyopathies.
Mutations in the motor domain of myosin have long been postulated to disrupt the formation of strong cross-bridges [73]. Recent improvements in the ability to purify functional human β-myosin have led to significant advances in determining the structural insults caused by mutations [74]. For instance, the β-myosin mutations M531R (left ventricular noncompaction) and S532P (DCM) located in an actin-binding interface, differentially perturb the local structure [73]. A second study showed that R453C (HCM) decreases the structural stability of the motor head throughout the crossbridge cycle, suggesting an alteration in ATP binding and/or hydrolysis [75]. Thus, it is possible that via changes in structural stability of the motor domain, these mutations could be disrupting a communication pathway for actin binding-sites [76]. These examples highlight the importance of myofilament protein and protein-protein interface stability for the function of this highly evolved multi-subunit machine.
Another example of these crucial structural stabilities and protein-protein interfaces is the complex association of the proteins of the regulatory TF, responsible for transmitting the calcium status of the myocyte to the sarcomere and muscle contraction. For a more comprehensive review of the mechanism of contraction refer to [77, 78]. Briefly, at the onset of contraction CICR leads to cTnC binding calcium (described above). Through a series of allosteric rearrangements in cTn, Tm shifts into the actin groove exposing the myosin binding sites on actin and allowing for strong cross-bridge formation and force generation [79]. Central to TF function is the “gatekeeper”‘ Tm whose position is a dynamic equilibrium, moving over the surface of actin between three states: Blocked, Closed, and Open [78–80]. While Tm’s movement between these three states is tightly regulated, it must remain semi-flexible in order to rapidly couple myocellular calcium status to force generation [81, 82].
Governing the interface between actin and Tm are many weak electrostatic interactions that are central to Tm’s function in regulating cross-bridge formation [80, 83, 84]. Tm, an α-helical coiled-coil dimer, has a surface charge that is largely negative with pockets of mostly acidic residues contributing to its binding to actin [85, 86], it spans actin forming a continuous, flexible filament that strengthens its interaction with actin via interdigitating N- and C-termini (Figure 4). The head-to-tail association of subsequent dimers is crucial for the polymerization of Tm, its affinity for actin, and its cooperativity [87–90]. Further stabilizing this head-to-tail overlap is the α-helical component of the cTnT N-terminal domain resulting in a five-helix bundle at the overlap [1, 30, 81, 91–94]. This arrangement allows for the flexibility and stability required to support the dynamic range of motion along actin necessary for the regulation of cross-bridge cycling.
Figure 4:
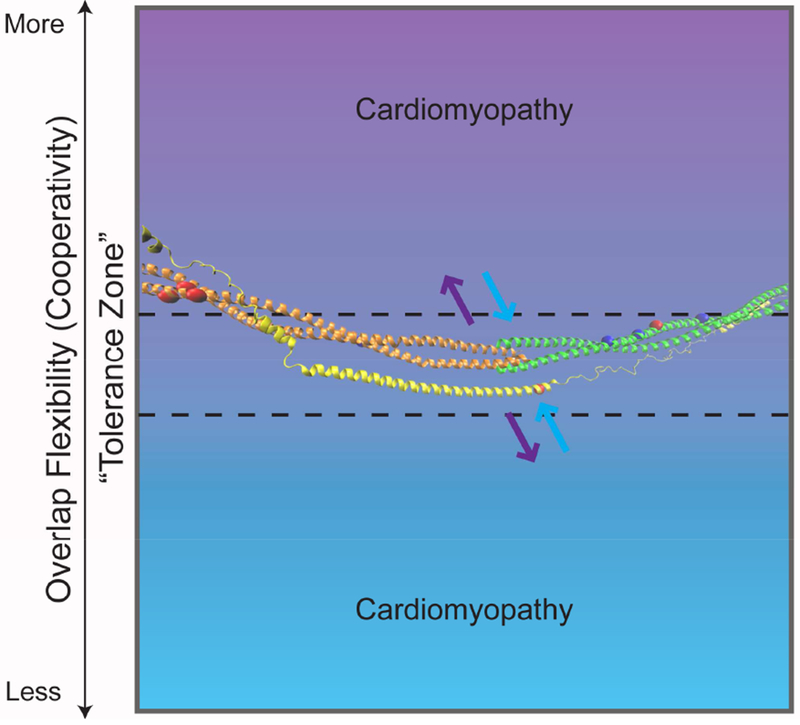
Tm-Overlap Flexibility including the proposed “tolerance zone”. The semi-flexible Tm-overlap can vary within this zone (dotted line), becoming more (purple) or less (blue) flexible, outside of which is associated with disease. Overlaid on the gradient is the atomistic model generated from the publicly available average structure, by JR Exequiel Pineda, of the Tm N-terminus (orange), Tm C-terminus (green), and the N-terminal extension of cTnT (yellow) [1]. The arrows represent decreased (purple) or increased (blue) interaction of the five-helix bundle that comprises the Tm-overlap. Included on the Tm and cTnT model are the sites of the discussed mutations (Tm-D175, E180, L185, E62, E40, E54, D84, D230; cTnT R92) with HCM in red and DCM in dark blue.
Given the innate complexity of the system, it is not surprising that cardiomyopathy-associated mutations could locally alter the weak electrostatic interactions leading to propagation of structural effects to distant sites in cTn, as discussed above. In 2000 it was first posited that mutations altering the surface charge of Tm could be associated with DCM, whereby a charge reversal alters its electrostatic interactions with actin [95]. Furthermore, a large number of clinically relevant mutations exist near the Tm-overlap region, highlighting its functional significance. We and others have shown that such mutations on both Tm and cTnT propagate their structural effects altering Tm stability, Tm-overlap flexibility (cooperativity), and actin/cTnT affinity in a mutation specific manner [48, 94, 96–101]. Notably, it was first postulated by Heller et al. that alterations in thermal stability of the termini of Tm could translate to changes in Tm flexibility [96]. In a study exploring the stability of Tm dimers containing the DCM causing mutations Tm-E40K and Tm-E54K compared to the HCM causing Tm-E62Q and Tm-L185R it was shown that while the mutations did not affect the α-helical content of the Tm dimer, they did have a significant effect on the thermal denaturation (stability) [97]. Similarly, we have shown that the DCM causative Tm-D230N does not alter the α-helicity of Tm but leads to an increase in thermal stability of Tm-D230N dimers specifically at the C-terminus proximal to the mutation [48]. Strikingly the pathogenic outcome related to each mutation depends on the precise location within the coiled-coil as well as the “magnitude” of the amino acid change (charge gain/loss, size change, etc). For instance, regionally similar mutations Tm-E180G and Tm-D175N cause phenotypically different cardiomyopathies yet both had a similar effect on the thermal stability of Tm and increased local flexibility despite being in a Tn-binding pocket [102–104]. By contrast, the regionally distinct, mutations Tm-D84N and Tm-D230N both result in the same charge loss and increase overall Tm stability and cause phenotypically different cardiomyopathies [48, 105–107]. These changes in the absence of an altered secondary structure (helicity) suggest that the mutational effects could be propagating to the Tm-overlap, a highly sensitive functional domain, affecting its ability to regulate cross-bridge cycling. Furthermore, it suggests that the location of the mutation on Tm (coiled-coil position) and effects on regional stability of the individual proteins alone are not sufficient to predict pathogenicity.
Mutationally-induced changes in Tm dimer inter-digitation at the critical Tm-overlap could affect cooperativity, along its length, with adjacent Tm molecules as well as its ability to interact with actin and cTnT. Studies in reconstituted TFs have demonstrated that for some Tm mutations there is a change in the affinity of Tm for its neighboring proteins (cTnT and/or actin). For instance, with Tm-E180G and Tm-D175N it was shown that the affinity for actin was slightly reduced for Tm-D175N heterodimers (an effect that was much greater for Tm-D175N homodimers) with no apparent effect on the affinity for cTnT for either mutation [101, 103]. Similarly, but in the opposite direction, mutations Tm-D84N and Tm-D230N demonstrated an increased affinity for actin [105]. These changes in actin affinity, while compelling, are likely not sufficient to explain the segregation of HCM and DCM given the complexity of the structural effects. In fact, studies comparing the HCM mutations Tm-E180G and Tm-E180V demonstrated that when bound to actin, Tm-E180G destabilized Tm suggesting a more flexible (less cooperative) Tm-overlap while, in stark contrast, Tm-E180V strongly stabilized Tm suggesting a less flexible Tm-overlap. [108]. Moreover, these affinity studies did not directly address the mutational effects on the complete Tm-overlap (containing actin, cTn, and Tm). To bridge this gap, we have recently demonstrated via in vitro experimentation coupled to computational modeling, that the Tm-D230N mutation decreased the flexibility (increased cooperativity) of the full TF via compaction of the overlap region [94]. Furthermore, when the approach was extended to include the HCM-linked cTnT-R92L due to its proximity to the overlap, a weaker overlap interaction (decreased stability) and more flexible (less cooperative) Tm was reported, in opposition to what was seen with DCM-linked Tm-D230N [94]. Lastly, recent studies on cardiomyopathy-associated mutations in a Tm-binding region of cTnT demonstrated altered binding affinity of cTnT for Tm whereby HCM mutations decreased affinity while DCM mutations increased affinity [100]. Interestingly cTnT-R92L was shown to decrease affinity of cTnT for Tm correlating well with our previous findings. Of note, not all studied mutations fit this defined paradigm, indicating a regional specificity for the mutational effects.
Together these data suggest that mutations that give rise to cardiomyopathies can differentially affect both the stability of the affected protein and the flexibility of the critical overlap region independent of phenotypic outcome, leading to disease via distinct molecular mechanisms (Figure 4). It is likely that a complex interplay between stability and affinity governs pathogenicity dependent on the position of the mutation (cTnT-binding domain, actin-binding domain, etc) and the amino acid change. This complexity underscores our incomplete understanding of how single amino acid substitutions give rise to cardiomyopathies while simultaneously shedding light on the vast heterogeneity of disease presentation.
As with mutations that induce changes in calcium kinetics, it is likely implausible to generate small molecules to address the structural effects of each cardiomyopathy causing mutation. However, understanding how a mutation alters the intra- and inter-molecular allostery of affected proteins, could lead to targeted therapies that correct specific sets of structural alterations. For instance, direct modulators of myosin function are in several phases of clinical trials [109–111]. Omecamtiv mecarbil, a cardiac myosin activator in testing for its efficacy in patients with systolic heart failure has been shown to directly bind myosin S1 near the actin binding domain favoring the strongly bound (open) force-generating state. Alternitavely, mavacamten (MYK-461), has been shown to directly modulate contractility via decreasing enzymatic activity of myosin and the population of strongly bound cross-bridges in HCM [110, 111]. Similarly, small molecules could be designed that target the TF with the goal of optimizing Tm cooperativity (via stabilization or destabilization of the overlap) toward a “tolerance zone” (Figure 4). While work continues on understanding mutationally-induced structural effects of the Tm-overlap, future studies will provide insight into the design of targeted molecules that act to ameliorate altered Tm flexibility at the earliest stages.
Summary:
In this review we have focused on three “bins” that comprise sets of biophysical changes elicited by cardiomyopathy-linked mutations in the myofilament: altered interaction of the cTnI-cTnC interface, changes in calcium kinetics, and altered protein stability and flexibility. The current binary paradigm of proposed mechanisms has proven to be inadequate to describe the heterogeneity of disease presentation. While current therapies focus on symptom palliation, we and others have proposed that a more nuanced classification could lead to direct interventions based on the earliest dysregulation. Such early, targeted therapies could change the trajectory of disease progression in the preclinical cohort. Notably, the “bins” discussed here are a subset of this complexity and do not address other known avenues including energetics, sarcomere assembly, and protein turnover. Continued research is necessary to address the complexity of cardiomyopathic progression and develop efficacious therapeutics.
KEY POINTS.
Current therapeutics aimed at symptom palliation do not address the complex heterogeneity of disease presentation in genetic cardiomyopathies.
We discuss three proposed “bins” that provide mechanistic insight in the early and compensatory phases of cardiomyopathic progression.
A more refined classification based on primary biophysical derangements could lead to direct interventions that target early dysregulation.
SYNOPSIS.
In this review we focus on three “bins” that comprise sets of biophysical derangements elicited by cardiomyopathy-associated mutations in the myofilament. Current therapies focus on symptom palliation and do not address the disease at its core. We and others have proposed that a more nuanced classification could lead to direct interventions based on early dysregulation changing the trajectory of disease progression in the preclinical cohort. Continued research is necessary to address the complexity of cardiomyopathic progression and develop efficacious therapeutics
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT
Nothing to disclose
References:
- [1].Williams MR, Lehman SJ, Tardiff JC, Schwartz SD. Atomic resolution probe for allostery in the regulatory thin filament. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:3257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. [DOI] [PubMed] [Google Scholar]
- [3].Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nature Clinical Practice Cardiovascular Medicine. 2008;5:158. [DOI] [PubMed] [Google Scholar]
- [4].McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circulation research. 2017;121:731–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Braunwald E. Cardiomyopathies. Circulation research. 2017;121:711. [DOI] [PubMed] [Google Scholar]
- [6].Hwang TJ, Lauffenburger JC, Franklin JM, Kesselheim AS. Temporal Trends and Factors Associated With Cardiovascular Drug Development, 1990 to 2012. JACC: Basic to Translational Science. 2016;1:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MRG, et al. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Frontiers in Cardiovascular Medicine. 2016;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gerull B The Rapidly Evolving Role of Titin in Cardiac Physiology and Cardiomyopathy. Canadian Journal of Cardiology. 2015;31:1351–9. [DOI] [PubMed] [Google Scholar]
- [9].Golbus J, Puckelwartz M, Fahrenbach J, Dellefave-Castillo L, Wolfgeher D, McNally E. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Solaro R, Rarick H. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ Research. 1998;83:471–80. [DOI] [PubMed] [Google Scholar]
- [11].Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circulation research. 1995;76:1028–35. [DOI] [PubMed] [Google Scholar]
- [12].Rao V, Cheng Y, Lindert S, Wang D, Oxenford L, McCulloch AD, et al. PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils. Biophys J. 2014;107:1196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, et al. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circulation research. 2001;88:1059–65. [DOI] [PubMed] [Google Scholar]
- [14].Manning E, Tardiff J, Schwartz S. A model of calcium activation of the cardiac thin filament. Biochemistry. 2011;50:7405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ. Effects of protein kinase A phosphorylation on signaling between cardiac troponin I and the N-terminal domain of cardiac troponin C. Biochemistry. 1997;36:13305–11. [DOI] [PubMed] [Google Scholar]
- [16].Hwang PM, Cai F, Pineda-Sanabria SE, Corson DC, Sykes BD. The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:14412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Najafi A, Sequeira V, Kuster DW, van der Velden J. beta-adrenergic receptor signalling and its functional consequences in the diseased heart. European journal of clinical investigation. 2016;46:362–74. [DOI] [PubMed] [Google Scholar]
- [18].Schumacher C, Becker H, Conrads R, Schotten U, Pott S, Kellinghaus M, et al. Hypertrophic cardiomyopathy: a desensitized cardiac beta-adrenergic system in the presence of normal plasma catecholamine concentrations. Naunyn-Schmiedeberg’s archives of pharmacology. 1995;351:398–407. [DOI] [PubMed] [Google Scholar]
- [19].Choudhury L, Guzzetti S, Lefroy DC, Nihoyannopoulos P, McKenna WJ, Oakley CM, et al. Myocardial beta adrenoceptors and left ventricular function in hypertrophic cardiomyopathy. Heart (British Cardiac Society). 1996;75:50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. The Journal of biological chemistry. 1999;274:22251–6. [DOI] [PubMed] [Google Scholar]
- [21].Spirito P, Maron BJ, Chiarella F, Bellotti P, Tramarin R, Pozzoli M, et al. Diastolic abnormalities in patients with hypertrophic cardiomyopathy: relation to magnitude of left ventricular hypertrophy. Circulation. 1985;72:310–6. [DOI] [PubMed] [Google Scholar]
- [22].Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. [DOI] [PubMed] [Google Scholar]
- [23].Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, et al. Ca2+-regulated structural changes in troponin. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Papadaki M, Marston SB. The Importance of Intrinsically Disordered Segments of Cardiac Troponin in Modulating Function by Phosphorylation and Disease-Causing Mutations. Frontiers in physiology. 2016;7:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, et al. Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circulation research. 1995;77:107–13. [DOI] [PubMed] [Google Scholar]
- [26].Robertson IM, Baryshnikova OK, Li MX, Sykes BD. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochemistry. 2008;47:7485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kasikcioglu HA, Cam N. A review of levosimendan in the treatment of heart failure. Vascular Health and Risk Management. 2006;2:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Abbate A, Van Tassell BW. Levosimendan in advanced heart failure: where do we stand? Journal of cardiovascular pharmacology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mehta RH, Leimberger JD, van Diepen S, Meza J, Wang A, Jankowich R, et al. Levosimendan in Patients with Left Ventricular Dysfunction Undergoing Cardiac Surgery. The New England journal of medicine. 2017;376:2032–42. [DOI] [PubMed] [Google Scholar]
- [30].Palm T, Graboski S, Hitchcock-DeGregori S, Greenfield N. Disease-causing mutations in cardiac troponin T: Identification of a critical tropomyosin-binding region. Biophys J. 2001;81:2827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Biesiadecki BJ, Kobayashi T, Walker JS, Solaro RJ, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circulation research. 2007;100:1486–93. [DOI] [PubMed] [Google Scholar]
- [32].Cheng Y, Regnier M. Cardiac troponin structure-function and the influence of hypertrophic cardiomyopathy associated mutations on modulation of contractility. Archives of Biochemistry and Biophysics. 2016;601:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dvornikov AV, Smolin N, Zhang M, Martin JL, Robia SL, de Tombe PP. Restrictive Cardiomyopathy Troponin I R145W Mutation Does Not Perturb Myofilament Length-dependent Activation in Human Cardiac Sarcomeres. The Journal of biological chemistry. 2016;291:21817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cheng Y, Rao V, Tu AY, Lindert S, Wang D, Oxenford L, et al. Troponin I Mutations R146G and R21C Alter Cardiac Troponin Function, Contractile Properties, and Modulation by Protein Kinase A (PKA)-mediated Phosphorylation. The Journal of biological chemistry. 2015;290:27749–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Pinto JR, Solis RS, Dweck D, Liang J, Diaz-Perez Z, et al. Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. The Journal of biological chemistry. 2012;287:2156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dweck D, Sanchez-Gonzalez MA, Chang AN, Dulce RA, Badger CD, Koutnik AP, et al. Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. The Journal of biological chemistry. 2014;289:23097–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guinto P, Haim T, Dowell-Martino C, Sibinga N, Tardiff J. temporal and mutation-specific alterations in ca2+ homeostasis differentially determine the progression of ctnt-related cardiomyopathies in murine models. Am J Physiol Heart Circ Physiol. 2009;297:H614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Papadaki M, Vikhorev PG, Marston SB, Messer AE. Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovascular research. 2015;108:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and Excitation-Contraction Coupling in the Heart. Circulation research. 2017;121:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Research in Cardiology. 1997;92:87–93. [DOI] [PubMed] [Google Scholar]
- [41].Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–84. [DOI] [PubMed] [Google Scholar]
- [42].Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, et al. Genotype-Dependent and -Independent Calcium Signaling Dysregulation in Human Hypertrophic Cardiomyopathy. Circulation. 2016;134:1738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, et al. Late Sodium Current Inhibition Reverses Electromechanical Dysfunction in Human Hypertrophic Cardiomyopathy; Clinical Perspective. Circulation. 2013;127:575. [DOI] [PubMed] [Google Scholar]
- [44].Pan S, Sommese RF, Sallam KI, Nag S, Sutton S, Miller SM, et al. Establishing disease causality for a novel gene variant in familial dilated cardiomyopathy using a functional in-vitro assay of regulated thin filaments and human cardiac myosin. BMC medical genetics. 2015;16:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, Ruppel KM. Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human beta-cardiac myosin. PloS one. 2013;8:e83403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chandra M, Tschirgi M, Tardiff J. Increase in tension-dependent ATP consumption induced by cardiac troponin T mutation. Am J Physiol Heart Circ Physiol. 2005;289:H2112–9. [DOI] [PubMed] [Google Scholar]
- [47].Ramratnam M, Salama G, Sharma RK, Wang DW, Smith SH, Banerjee SK, et al. Gene-Targeted Mice with the Human Troponin T R141W Mutation Develop Dilated Cardiomyopathy with Calcium Desensitization. PloS one. 2016;11:e0167681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lynn ML, Tal Grinspan L, Holeman TA, Jimenez J, Strom J, Tardiff JC. The structural basis of alpha-tropomyosin linked (Asp230Asn) familial dilated cardiomyopathy. Journal of molecular and cellular cardiology. 2017;108:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Du C, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Research. 2007;101:185–94. [DOI] [PubMed] [Google Scholar]
- [50].Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, et al. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circulation research. 2003;92:428–36. [DOI] [PubMed] [Google Scholar]
- [51].Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Flenner F, Friedrich FW, Ungeheuer N, Christ T, Geertz B, Reischmann S, et al. Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovascular research. 2016;109:90–102. [DOI] [PubMed] [Google Scholar]
- [53].Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, et al. Diltiazem treatment for preclinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart failure. 2015;3:180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet (London, England). 2016;387:1178–86. [DOI] [PubMed] [Google Scholar]
- [55].Ho CY, Cirino AL, Lakdawala NK, Groarke J, Valente AM, Semsarian C, et al. Evolution of hypertrophic cardiomyopathy in sarcomere mutation carriers. Heart (British Cardiac Society). 2016 [DOI] [PubMed] [Google Scholar]
- [56].Pollesello P, Papp Z, Papp JG. Calcium sensitizers: What have we learned over the last 25 years? International journal of cardiology. 2016;203:543–8. [DOI] [PubMed] [Google Scholar]
- [57].Liu B, Lee RS, Biesiadecki BJ, Tikunova SB, Davis JP. Engineered troponin C constructs correct disease-related cardiac myofilament calcium sensitivity. The Journal of biological chemistry. 2012;287:20027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shettigar V, Zhang B, Little SC, Salhi HE, Hansen BJ, Li N, et al. Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nature communications. 2016;7:10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].White J, Lee JA, Shah N, Orchard CH. Differential effects of the optical isomers of EMD 53998 on contraction and cytoplasmic Ca2+ in isolated ferret cardiac muscle. Circulation research. 1993;73:61–70. [DOI] [PubMed] [Google Scholar]
- [60].Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PloS one. 2012;7:e38259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Davis JP, Tikunova SB. Ca(2+) exchange with troponin C and cardiac muscle dynamics. Cardiovascular research. 2008;77:619–26. [DOI] [PubMed] [Google Scholar]
- [62].Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PM, et al. Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration. Frontiers in physiology. 2016;7:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Siddiqui JK, Tikunova SB, Walton SD, Liu B, Meyer M, de Tombe PP, et al. Myofilament Calcium Sensitivity: Consequences of the Effective Concentration of Troponin I. Frontiers in physiology. 2016;7:632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, et al. Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. Journal of molecular and cellular cardiology. 2014;76:257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Little SC, Biesiadecki BJ, Kilic A, Higgins RS, Janssen PM, Davis JP. The rates of Ca2+ dissociation and cross-bridge detachment from ventricular myofibrils as reported by a fluorescent cardiac troponin C. The Journal of biological chemistry. 2012;287:27930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, et al. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29:549–55. [DOI] [PubMed] [Google Scholar]
- [67].Forissier JF, Carrier L, Farza H, Bonne G, Bercovici J, Richard P, et al. Codon 102 of the cardiac troponin T gene is a putative hot spot for mutations in familial hypertrophic cardiomyopathy. Circulation. 1996;94:3069–73. [DOI] [PubMed] [Google Scholar]
- [68].Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–12. [DOI] [PubMed] [Google Scholar]
- [69].Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. The New England journal of medicine. 1995;332:1058–64. [DOI] [PubMed] [Google Scholar]
- [70].Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, Solaro RJ. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol Heart Circ Physiol. 2001;280:H705–13. [DOI] [PubMed] [Google Scholar]
- [71].Ferrantini C, Coppini R, Pioner JM, Gentile F, Tosi B, Mazzoni L, et al. Pathogenesis of Hypertrophic Cardiomyopathy is Mutation Rather Than Disease Specific: A Comparison of the Cardiac Troponin T E163R and R92Q Mouse Models. Journal of the American Heart Association. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chung JH, Biesiadecki BJ, Ziolo MT, Davis JP, Janssen PM. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Frontiers in physiology. 2016;7:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aksel T, Choe Yu E, Sutton S, Ruppel KM, Spudich JA. Ensemble Force Changes that Result from Human Cardiac Myosin Mutations and a Small-Molecule Effector. Cell Reports. 2015;11:910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Resnicow DI, Deacon JC, Warrick HM, Spudich JA, Leinwand LA. Functional diversity among a family of human skeletal muscle myosin motors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bloemink M, Deacon J, Langer S, Vera C, Combs A, Leinwand L, et al. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac beta-myosin. The Journal of biological chemistry. 2014;289:5158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nikolaeva OP, Orlov VN, Bobkov AA, Levitsky DI. Differential scanning calorimetric study of myosin subfragment 1 with tryptic cleavage at the N-terminal region of the heavy chain. European journal of biochemistry. 2002;269:5678–88. [DOI] [PubMed] [Google Scholar]
- [77].Batters C, Veigel C, Homsher E, Sellers JR. To understand muscle you must take it apart. Frontiers in physiology. 2014;5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological reviews. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- [79].McKillop D, Geeves M. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophysical Journal. 1993;65:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gordon A, Regnier M, Homsher E. Skeletal and cardiac muscle contraction activation: Tropomyosin “rocks and rolls”. News Physiol Sci. 2001;16:49–55. [PubMed] [Google Scholar]
- [81].Li XE, Orzechowski M, Lehman W, Fischer S. Structure and flexibility of the tropomyosin overlap junction. Biochemical and biophysical research communications. 2014;446:304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Li X, Lehman W, Fischer S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. Journal of Structural Biology. 2010;170:313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gordon A, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological reviews. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- [84].Holmes K, Lehman W. Gestalt-binding of tropomyosin to actin filaments. J Muscle Res Cell Motil. 2008;29:213–9. [DOI] [PubMed] [Google Scholar]
- [85].Barua B, Pamula M, Hitchcock-DeGregori S. Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc Natl Acad Sci USA. 2011;108:10150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Barua B, Fagnant PM, Winkelmann DA, Trybus KM, Hitchcock-DeGregori SE. A periodic pattern of evolutionarily conserved basic and acidic residues constitutes the binding interface of actintropomyosin. The Journal of biological chemistry. 2013;288:9602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cho YJ, Liu J, Hitchcock-DeGregori SE. The amino terminus of muscle tropomyosin is a major determinant for function. The Journal of biological chemistry. 1990;265:538–45. [PubMed] [Google Scholar]
- [88].Brown JH, Kim KH, Jun G, Greenfield NJ, Dominguez R, Volkmann N, et al. Deciphering the design of the tropomyosin molecule. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Phillips G, Jr, Fillers J, Cohen C. Tropomyosin crystal structure and muscle regulation. J Mol Biol. 1986;192:111–31. [DOI] [PubMed] [Google Scholar]
- [90].Mak AS, Smillie LB. Non-polymerizable tropomyosin: preparation, some properties and F-actin binding. Biochemical and biophysical research communications. 1981;101:208–14. [DOI] [PubMed] [Google Scholar]
- [91].Greenfield NJ, Huang YJ, Swapna GV, Bhattacharya A, Rapp B, Singh A, et al. Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J Mol Biol. 2006;364:80–96. [DOI] [PubMed] [Google Scholar]
- [92].Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, et al. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc Natl Acad Sci USA. 2008;105:7200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Frye J, Klenchin VA, Rayment I. Structure of the tropomyosin overlap complex from chicken smooth muscle: insight into the diversity of N-terminal recognition. Biochemistry. 2010;49:4908–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].McConnell M, Tal Grinspan L, Williams MR, Lynn ML, Schwartz BA, Fass OZ, et al. Clinically Divergent Mutation Effects on the Structure and Function of the Human Cardiac Tropomyosin Overlap. Biochemistry. 2017;56:3403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. Journal of molecular and cellular cardiology. 2001;33:723–32. [DOI] [PubMed] [Google Scholar]
- [96].Heller M, Nili M, Homsher E, Tobacman L. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivty and tropomyosin N-domain flexibility. The Journal of biological chemistry. 2003;278:41742–8. [DOI] [PubMed] [Google Scholar]
- [97].Chang AN, Greenfield NJ, Singh A, Potter JD, Pinto JR. Structural and protein interaction effects of hypertrophic and dilated cardiomyopathic mutations in alpha-tropomyosin. Frontiers in physiology. 2014;5:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI. Effects of Two Familial Hypertrophic Cardiomyopathy Mutations in α-Tropomyosin, Asp175Asn and Glu180Gly, on the Thermal Unfolding of Actin-Bound Tropomyosin. Biophysical Journal. 2004;87:3922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Loong CKP, Zhou H- X, Chase PB. Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac α-tropomyosin. FEBS Letters. 2012;586:3503–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gangadharan B, Sunitha MS, Mukherjee S, Chowdhury RR, Haque F, Sekar N, et al. Molecular mechanisms and structural features of cardiomyopathy-causing troponin T mutants in the tropomyosin overlap region. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:11115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Janco M, Kalyva A, Scellini B, Piroddi N, Tesi C, Poggesi C, et al. α-Tropomyosin with a D175N or E180G Mutation in Only One Chain Differs from Tropomyosin with Mutations in Both Chains. Biochemistry. 2012;51:9880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves M, Levitsky D. Effects of two familial hypertrophic cardiomyopathy mutations in alpha tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys J. 2004;87:3922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, et al. Effects of Two Familial Hypertrophic Cardiomyopathy-Causing Mutations on α-Tropomyosin Structure and Function. Biochemistry. 1997;36:4637–42. [DOI] [PubMed] [Google Scholar]
- [104].White SP, Cohen C, Phillips GN Jr. Structure of co-crystals of tropomyosin and troponin. Nature. 1987;325:826. [DOI] [PubMed] [Google Scholar]
- [105].Gupte TM, Haque F, Gangadharan B, Sunitha MS, Mukherjee S, Anandhan S, et al. Mechanistic heterogeneity in contractile properties of alpha-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. The Journal of biological chemistry. 2015;290:7003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].van de Meerakker JB, Christiaans I, Barnett P, Lekanne Deprez RH, Ilgun A, Mook OR, et al. A novel alpha-tropomyosin mutation associates with dilated and non-compaction cardiomyopathy and diminishes actin binding. Biochimica et biophysica acta. 2013;1833:833–9. [DOI] [PubMed] [Google Scholar]
- [107].Lakdawala N, Dellefave L, Redwood C, Sparks E, Cirino A, Depalma S, et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Matyushenko AM, Shchepkin DV, Kopylova GV, Popruga KE, Artemova NV, Pivovarova AV, et al. Structural and Functional Effects of Cardiomyopathy-Causing Mutations in the Troponin T-Binding Region of Cardiac Tropomyosin. Biochemistry. 2017;56:250–9. [DOI] [PubMed] [Google Scholar]
- [109].Hwang PM, Sykes BD. Targeting the sarcomere to correct muscle function. Nat Rev Drug Discov. 2015;14:313–28. [DOI] [PubMed] [Google Scholar]
- [110].Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A smallmolecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (New York, NY). 2016;351:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kawas RF, Anderson RL, Ingle SRB, Song Y, Sran AS, Rodriguez HM. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. The Journal of biological chemistry. 2017;292:16571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]