

Abstract
Objective –
Unreliable antibodies often hinder the accurate detection of an endogenous protein, and this is particularly true for the cardiac and smooth muscle cofactor, Myocardin (MYOCD). Accordingly, the mouse Myocd locus was targeted with two independent epitope tags for the unambiguous expression, localization, and activity of MYOCD protein.
Approach and Results –
3-component CRISPR was used to engineer a carboxyl-terminal 3xFLAG or 3xHA epitope tag in mouse embryos. Western blotting with antibodies to each tag revealed a protein product of ~150 kDa, a size considerably larger than that reported in virtually all publications. MYOCD protein was most abundant in some adult smooth muscle-containing tissues with surprisingly low-level expression in heart. Both alleles of Myocd are active in aorta since a two-fold increase in protein was seen in mice homozygous versus heterozygous for FLAG-tagged Myocd. ChIP-qPCR studies provide proof-of-principle data demonstrating the utility of this mouse line in conducting genome-wide ChIP-seq studies to ascertain the full complement of MYOCD-dependent target genes in vivo. Although FLAG-tagged MYOCD protein was undetectable in sections of adult mouse tissues, low-passaged VSMC exhibited expected nuclear localization.
Conclusion –
This report validates new mouse models for analyzing MYOCD protein expression, localization, and binding activity in vivo and highlights the need for rigorous authentication of antibodies in biomedical research.
Keywords: CRISPR, Myocardin, smooth muscle, antibody
Introduction
The trustworthy detection, localization, and interactive association of a protein are essential to understanding cellular processes under normal and pathological conditions. Yet, biology is plagued by fallacious antibodies,1 particularly those directed against transcription factors.2 Accordingly, antibodies to a variety of epitope tags have been developed to circumvent the difficulties often encountered with monoclonal or polyclonal antibodies.3 Epitope tagging is commonplace in labs using plasmids to study ectopic protein expression. Far fewer examples exist with the tagging of endogenous protein-coding gene loci in mice,4–6 likely due to the labor-intensive approach of traditional gene targeting. However, the CRISPR-Cas9 genome editing system7–9 has revolutionized the production of novel mouse strains, including those carrying epitope tags.10,11 Here, we present the accurate expression, localization, and binding activity of epitope tagged Myocardin (MYOCD), a serum response factor (SRF) cofactor12 that powerfully drives the program of vascular smooth muscle cell (VSMC) differentiation.13–16
Material and Methods
3-Component CRISPR editing of the Myocd locus
For the Myocd3xFLAG mouse, sgRNA of sequence ACAGCAGUGGUAAACACCCG (synthesized in vitro as described17) was tested for activity in a split reporter assay18 (see Supplemental Figure IIB) and injected (50 ng/μl) with Cas9 mRNA (100 ng/μl, TriLink Biotechnologies) and a symmetric, single-strand oligonucleotide containing the 3xFLAG sequence (100 ng/μl, Ultrapure PAGE-purified, Integrated DNA Technologies) into the pronucleus of C57BL/6J zygotes. The single-strand oligonucleotide was of the same strand as the sgRNA so as to prevent hybridization of the two in the embryo. For the Myocd3xHA mouse, synthetic sgRNA (same as above) and CAS9 protein (both from Synthego) were validated for DNA cleavage in an in vitro tube assay as described by the manufacturer. CAS9 protein and sgRNA were mixed (3 pmol each) as a ribonucleoprotein complex and then combined with a 249 single-strand Megamer oligonucleotide (100 ng/μl, Integrated DNA Technologies) prior to cytoplasmic injection. Viable two-cell stage embryos were transferred to pseudo-pregnant C57BL/6J females.
Local Institutional Animal Care and Use Committees approved all mouse studies.
Mouse pup genotyping, off-targeting, and breeding
One-week old founder pups were toe-clipped or ear-punched and tissue was digested overnight at 55°C in lysis buffer (50 mM KCl, 10 mM Tris-HCL [pH 9], 0.1% Triton X-100, and 0.2 mg/ml proteinase K) on an interval mixer. The next morning, samples were heated at 95°C for 10 minutes to inactivate the proteinase K and spun down at 15,000xg for 10 minutes. Each sample of DNA (1μl) was mixed with 12 μl nuclease-free water, 10 μl AccuStart II Supermix (Quantabio), and 1 μl each of forward and reverse primers (see Table I in the online-only Data Supplement) flanking the double-strand break (see Figure II in the online-only Data Supplement). PCR conditions were 1 cycle of 95°C for 3 minutes followed by 30 cycles of 95°C (30 sec), 58°C (45 sec), and 72°C (1 minute) and a final 7-minute cycle at 72°C.
Off-targeting was assessed initially with CCTop,19 Cas-OFFinder,20 and the MIT CRISPR website21 and later with the CRISPOR tool.22 No predicted off-targets were found within two megabases of the on-target double-strand break. We selected the top nine distal off-target sites based on an aggregate analysis of each prediction program. PCR primers flanking each predicted off-target site were synthesized (Integrated DNA Technologies) and used to amplify genomic DNA from founder mice as above only the number of PCR cycles was limited to 28. Each primer pair contained linker sequences positioned at the 5’ end of the template-specific primer sequence necessary for next generation sequencing; the forward primers contained the linker sequence, ACACTGACGACATGGTTCTACA and the reverse primers contained the linker sequence, TACGGTAGCAGAGACTTGGTCT. Following the initial 28-cycle PCR reaction, the samples were verified on an agarose gel and approximately equal amounts were subjected to a second PCR reaction (8 cycles only) using primers containing Illumina adaptor sequence, 10 bp sample-specific barcodes, and the original linker sequences. Amplified sequences were then submitted to the Genomics Core for next generation sequencing on an Illumina HiSeq 2500 platform. This setup allowed for all nine amplicons to be sequenced in one lane of the sequencer. Raw sequence reads were analyzed for indels with CRISPResso.23
Positive founder mice derived from CRISPR-Cas9 genome editing are mosaic.11, 24 We therefore backcrossed each positive founder to C57BL/6J mice for Sanger sequencing to confirm germline transmission of each epitope tag. Sanger sequencing of F1 mice confirmed germline transmission of the correct allele as well as sequence fidelity in and around the edited region. Myocd3xFLAG was successfully bred through the germline (in two founders) and data were similar in both lines. We confirmed one mosaic founder carrying the Myocd3xHA tag, but the line was lost because of dystocia of the pregnant founder. We nevertheless isolated tissues for Western (Figure 1B) and limited immunofluorescence microscopy of adult tissues (data not shown). Each of the CRISPR-edited mouse lines is listed in the Major Resources Table in the Supplemental Material.
Figure 1. In vivo MYOCD protein expression and binding activity.
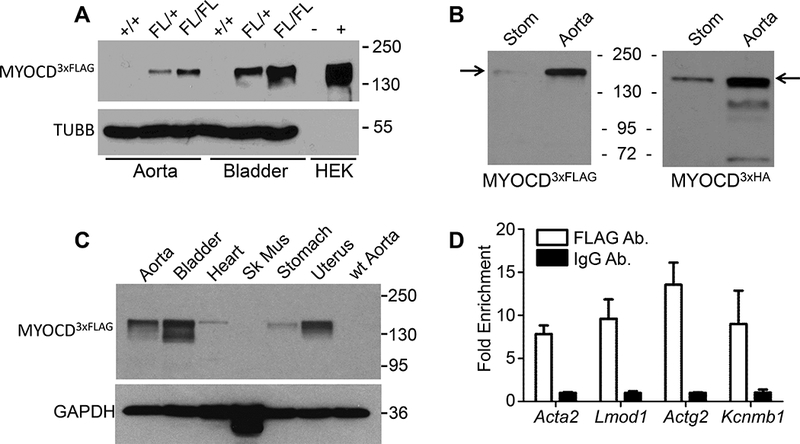
(A) Western blot of MYOCD3xFLAG in wildtype (+/+) versus heterozygous and homozygous FLAG(FL)-tagged MYOCD in indicated male mouse tissues. HEK-293 cells untransfected (−) or transfected (+) with 3xFLAG-tagged v1 isoform of MYOCD served as a control. (B) Parallel Western blots of MYOCD3xFLAG or MYOCD3xHA tagged tissues. (C) Tissue profile of MYOCD3xFLAG expression in indicated female tissues. Similar results were seen in several independent experiments over the course of two years. (D) In vivo ChIP-qPCR in homozygous Myocd3xFLAG aortae for the indicated target genes using FLAG or IgG control. Data are expressed as fold enrichment of FLAG sequence over IgG control. All results were confirmed in at least one independent experiment. Data passed test for normality and paired t-test showed significantly increased enrichment of PCR products with the anti-FLAG antibody, p < 0.01.
RNA isolation, reverse transcription and real-time qPCR
Total RNAs were isolated using RNeasy Mini Kit (Qiagen, #74104) according to the manufacturer’s instructions. Reverse transcription was performed using Bio-Rad iScript cDNA synthesis kit (Bio-Rad, #1708891) after RNA quantitation and DNase I treatment. Real-time qPCR was performed using iTaq™ Universal SYBR® Green Supermix (Bio-Rad, #1725121) with customized primers (see Table I in the online-only Data Supplement).
Western blot
Tissues (from male and female mice, aged 6–10 weeks) were isolated and homogenized in 200μl of cell lysis buffer (Cell Signaling, #9803). After brief centrifugation, supernatants were transferred to fresh 1.5ml microfuge tubes for protein quantitation with DC protein assay kit (Bio-Rad, #5000111). Samples were boiled in LDS sampling buffer (Invitrogen, #NP0007) and subjected to electrophoresis in SDS-PAGE gel. Proteins were transferred to a membrane (PVDF) for 2 hours in cold room at 80V using a Trans-blot machine (Bio-Rad). Membranes were then blocked with 5% non-fat milk in TBS-T for 1 hour and incubated with primary antibody for 1 hour at room temperature. After washing, secondary antibodies were incubated for 1 hour at room temperature followed by immersion in chemiluminescence substrate (Thermo-Fisher, #34080) and exposed to X-ray film. A list of antibodies used is provided in the Major Resource Table in the online-only Data Supplement.
Chromatin immunoprecipitation
At least 5 mouse aortae were isolated and fixed in 10ml of 4% paraformaldehyde at room temperature for 10 minutes. One ml of 1.25M glycine was added to stop crosslinking. Aortae were washed twice with 1xPBS and 1ml of ChIP lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% deoxycholate, 1% Triton X-100 and protease inhibitor cocktail) was added. Tissues were then pulse-sonicated (intervals of 10 second pulse over 2 minutes) and centrifuged at 12,000xg for 10 minutes at 4°C. Supernatants were collected and transferred to fresh 1.5ml microfuge tubes. Mouse anti-FLAG antibody (Sigma-Aldrich, #F3165) and mouse IgG were added to lysate (~500ul each) respectively and incubated for 2 hours at 4°C. 100μl Dynabeads protein G (Thermo-Fisher, #10003D) were washed twice with 1xPBS and resuspended in the original volume of ChIP lysis buffer for each ChIP reaction. Magnetic beads were mixed with antibody-lysate and incubated for 2 hours at 4°C. After five washes with ChIP lysis buffer, samples were treated with proteinase K (10mg/ml) at 55°C on a thermomixer for 1 hour. DNA was then recovered and purified by boiling with Chelex-100 resin (Bio-Rad, #1422822) for downstream assays, (eg, real-time qPCR).
Isolation and culture of mouse aortic smooth muscle cells
We followed a previously published protocol for isolating mouse aortic SMC.25 Briefly, for each independent experiment, three mouse aortae were isolated and placed into ice cold HBSS in a 15-ml conical tubes. Aortae were digested with 3ml of Buffer A (10mg collagenase II, 10mg soybean trypsin inhibitor in 10ml HBSS) at 37°C for 20 minutes. Aortae were then transferred into clean culture dishes loaded with 10ml Buffer B (DMEM/F12 medium with 20%FBS and 1% Pen-Strep, pre-warmed to 37°C). The adventitial layer was peeled off using fine forceps and aortae were transferred into another clean culture dish with 10ml Buffer B. Each aorta was then cut longitudinally and the intimal layer was scraped with angled blunt forceps to remove endothelial cells. Aortae were cut with fine scissors into 1–2mm pieces and transferred to one well of a 6-well culture plate with 3ml Buffer C (3ml Buffer B plus 67ul elastase [Worthington, #LS002279] at 24.8mg/ml). Tissues were incubated 15 minutes at 37°C followed by repeat pipetting with cut 1ml tips to loosen cells. Cells were incubated another 15 minutes at 37°C and dissociated cells were repeat pipetted with uncut 1ml tip 5–6 times. The digestion mixture was transferred to a clean 15ml canonical tube and 3ml Buffer A was added to stop digestion. Samples were centrifuged at 800× g for 5 minutes at 4°C to pellet cells. Cells were resuspended in 5ml Buffer A and plated in a well of 6-well plate. Isolated mouse smooth muscle cells were studied upon reaching 50–70% confluency.
Confocal Microscopy
Mouse aortic smooth muscle cells (MASMC) were cultured on 22×22mm coverslips in 6-well plates. Cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature. Coverslips were rinsed in 1xPBS twice and cells permeabilized with 1× PBS containing 0.1% Triton X-100 on an ice bath. Cells were blocked with 3% BSA for 20 minutes at room temperature and anti-FLAG primary antibody (Sigma-Aldrich, #F1804; 1:100 dilution) was applied at room temperature for 40 minutes. Coverslips were rinsed twice with 1xPBS and cells incubated with fluorescence-conjugated secondary antibodies (1:200) for 40 minutes at room temperature. Cells were then mounted with ProLong™ Gold anti-fade reagent with DAPI (Thermo-Fisher, #P36935) and examined with an Olympus IX81 confocal microscope using uniform settings for all conditions.
Flow cytometry
Aortae derived from the mTmG/Myh11CreERT2 cross were digested as above and freshly dissociated MASMC were resuspended in PBS and submitted to the URMC Flow Cytometry Core. Cells were sorted according to the Tomato (610nm) or GFP (510nm) signal and quantitated as a percentage of total cells stained for GFP or Tomato.
Transfection assays
Rat aortic smooth muscle cells (A7r5, ATCC® CRL-1444™) were transfected with either Myocd siRNA (si-Myocd, Cat# SASI_Rn01_00118674, Sigma-Aldrich) or MISSION® siRNA Universal Negative Control (si-Scramble, Cat# SIC001, Sigma-Aldrich). Cells were mixed with DharmaFECT1 transfection reagent (Lipofectamine 2000, cat# T2001–02, Dharmacon) in a 1:1 ratio followed by incubation at room temperature for 20 minutes. Cell mixtures were then added to the wells of cell plates followed by incubation for 48 hours at 37°C. After incubation, cells were washed twice with ice-cold PBS, scraped, and collected in either NP-40 lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% Nonidet P-40) containing EDTA-free complete protease and phosphatase inhibitor (Sigma-Aldrich/Roche) for protein estimation or Qiazol buffer for mRNA measurement. For protein measurement, 20μg samples were run on SDS-PAGE gels, transferred to nitrocellulose membrane, blocked with 4% non-fat milk, and subsequently incubated with anti-α-tubulin (TUBA1A; cat# sc5286, Santa Cruz), or Myocardin (MYOCD; cat# ab22073, Abcam) followed by incubation with secondary antibodies and detection using SuperSignal™ West Pico Chemiluminescent Substrate (cat# 34080, ThermoFisher Scientific) on autoradiography film. For Myocd mRNA levels total RNA was extracted from aorta and bladder using a Qiagen miRNeasy kit (Cat # 217004). Input RNA quality and concentration were measured using a Synergy HT Take3 Microplate Reader (BioTek, Winooski, VT) and cDNA was prepared using SuperScript IV VILO Master Mix (Cat # 11756500, Invitrogen). QPCR was performed in triplicate using TaqMan™ Fast Advanced Master Mix (Cat# 44–445-57) in an Mx3000p Real-Time PCR System (Stratagene, Santa Clara, CA). The primers for Myocd (Assay ID # Rn01786178), were purchased from ThermoFisher Scientific/TaqMan. Results of mRNA expression were normalized to internal control Gapdh, and relative mRNA expression was determined using the ΔΔCt method.
NanoLC-MS/MS
Rat aorta, urinary bladder, and A7r5 cells were homogenized and lysed in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% Nonidet P-40) containing EDTA-free complete protease and phosphatase inhibitor (Sigma-Aldrich/Roche). Protein levels were measured using Bradford assays (Bio-Rad), after which 30 μg samples were run on SDS-PAGE gels followed by fixing the gel (50% methanol and 10% acetic acid) for 40 minutes at room temperature and washing for 1 hour. Bands corresponding to 100 kDa were cut and subjected to mass spectrometry analysis. The proteins were eluted and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS; Applied Biomics, Hayward, CA). NanoLC fractionation and matrix-assisted laser desorption ionization-time of flight/time of flight (MALDI-TOF/TOF) were followed by a standard search of the National Center for Biotechnology Information and SwissProt databases using MASCOT. The ~150 kDa FLAG-tagged MYOCD band from mouse bladder was similarly processed and analyzed.
Statistical analyses
Primary data were assessed for normality with a Shapiro-Wilk test or for equal variance (in case of one- or two-way ANOVA) and all datasets displayed normal distributions and variances. Accordingly, we used parametric paired t-test for comparisons between experimental and control conditions or one- and two-way ANOVA for multiple group comparisons. All data analysis was performed in GraphPad Prism 7. Results are expressed as mean ± standard deviation. A value of p<0.05 was considered statistically significant. For Mendelian inheritance of different alleles we used a Chi-square test.
Results
In vivo Myocardin Protein Expression and Binding Activity
There are at least four MYOCD isoforms with predicted molecular weights of 106 kDa (v1, 983 amino acids), 101 kDa (v2, 935 amino acids), 97 kDa (v3, 904 amino acids), and 92 kDa (v4, 856 amino acid).26, 27 Most commercial antibodies report endogenous MYOCD protein of ~100 kDa, consistent with the predicted molecular weights of each isoform. However, upon knockdown of Myocd mRNA there was no corresponding change in a widely assumed MYOCD protein band in A7r5 SMC, mouse and rat aorta and bladder, mouse aortic SMC, and human coronary artery SMC (Figure IB in the online-only Data Supplement and data not shown). Similar results were found using a different siRNA to Myocd in MASMC (Figure IC in the online-only Data Supplement). Mass spectrometry of the excised 100 kDa bands from rat aorta and bladder as well as the A7r5 SMC line revealed no evidence of MYOCD peptides (Table II in the online-only Data Supplement). Thus, while many commercial antibodies are able to reveal ectopic MYOCD expression (not shown), there is question as to the accuracy of detecting the endogenous MYOCD protein.
Accordingly, 3-component CRISPR11 was used to target the last coding exon of Myocd with either 3xFLAG or 3xHA epitope tags (Figure IIA in the online-only Data Supplement). Correct targeting was validated by Sanger sequencing in 3xFLAG (4/35) and 3xHA (1/6) founders with no predicted off-targeting in linkage disequilibrium with the on-target site; sequence analysis of the top nine predicted distal off-target sites showed no mutations, consistent with the high specificity of the sgRNA (data not shown). The Myocd3xFLAG allele was bred through the germline and sibling-matings revealed an expected Mendelian ratio (Figure IID in the online-only Data Supplement). Western blotting supported biallelic expression of the Myocd gene (Figure 1A). The estimated molecular weight of FLAG-tagged MYOCD was ~150 kDa, a size considerably larger than that reported in the literature; Myocd3xHA mice showed a similar sized band (Figure 1B). An independently generated Myocdmyc-HA mouse also revealed MYOCD to have a molecular mass of ~150 kDa (Figure IIIA in the online-only Data Supplement). Adult tissue profiling showed highest expression of MYOCD in bladder and uterus with moderate levels in aorta; surprisingly lower levels of MYOCD protein were consistently seen in the adult heart (Figure 1C and Figure IIIB in the online-only Data Supplement). Importantly, mass spectrometry analysis of the 150 kDa band revealed small quantities of MYOCD protein (Table III in the online-only Data Supplement). ChIP-qPCR of Myocd3xFLAG aortic tissue showed significant enrichment of SRF-binding CArG sequences around several known targets of the SRF-MYOCD transcriptional switch (Figure 1D). Despite repeated efforts under numerous experimental conditions, no consistent MYOCD signal was obtained on tissue sections with an array of antibodies to FLAG (data not shown). These in vivo findings reveal a higher molecular weight of MYOCD than previously reported and define, for the first time, reliable detection of endogenous MYOCD protein expression and binding activity in adult mouse tissues.
In vitro Myocardin Protein Expression
The mTmG reporter mouse28 was crossed with the Myh11CreERT2 mouse29 to indelibly mark VSMC of the aortic wall with GFP. Most aortic cells from passage numbers 1–4 are GFP+; however, beginning at passage number five a notable decrease in GFP+ VSMC was seen (Figure IV in the online-only Data Supplement). Thus, in vitro studies were restricted to cultures of mouse VSMC at passage 1–3. Homozygous Myocd3xFLAG VSMC exhibited similar mRNA levels of SRF/MYOCD target genes as in wild type VSMC, suggesting the FLAG epitope tag did not adversely affect MYOCD activity (Figure VA in the online-only Data Supplement). Western blotting of passage two VSMC showed MYOCD protein comparable in size to that seen in tissues, indicating the molecular weight of MYOCD did not change appreciably upon culture (Figure VB in the online-only Data Supplement). Immunofluorescence microscopy disclosed nuclear-localized MYOCD in cultured VSMC (Figure 2A). Knockdown of Myocd mRNA (Figure 2B) resulted in reduced VSMC FLAG-tagged MYOCD protein expression (Figure 2C and Figure VB-VC in the online-only Data Supplement); we estimate a reduction of at least 10-fold over si-control treated VSMC (Figure 2D). Collectively, these results authenticate a new mouse model for accurate detection of MYOCD protein in both adult tissues and cultured VSMC.
Figure 2. In vitro MYOCD protein expression.

(A) Immunofluorescence microscopy of MYOCD3xFLAG in low-passaged mouse aortic SMC (MASMC). Note abundant nuclear staining in Myocd3xFLAG MASMC versus wild-type controls. (B) Quantitative RT-PCR following siRNA knockdown of Myocd mRNA in cultured MASMC. (C) Immunofluor-escence microscopy of MYOCD3xFLAG in MYH11 positive MASMC treated with si scrambled control (siCtrl, top) or siMyocd (bottom). Scale bar here and in panel A is 10 μm. (D) Quantitative analysis (by two independent, blinded investigators) of number of MYOCD-FLAG positive cells co-staining for MYH11. See also Figure VC in the online-only Data Supplement.
Discussion
We introduce new Myocardin mouse models wherein epitope tags (3xFLAG and 3xHA) were used to independently target the last coding exon of the Myocd locus. The motivation for generating these mice stems from our long-standing difficulty in assessing endogenous levels of MYOCD protein expression in vivo or in vitro in mouse, human, and rat.30 The initial characterization of the epitope tagged mice reported here reveals several notable findings. First, the two-fold increase in MYOCD protein expression of homozygous versus heterozygous Myocd3xFLAG mice indicates biallelic gene transcription. Second, multiple epitope tags demonstrate the molecular weight of the endogenous MYOCD protein to be much higher (~150 kDa) than formerly reported. Indeed, with rare exception,31 the molecular weight of MYOCD protein, when reported, has been between 95–105 kDa. We show in data from two independent laboratories, using distinct reagents and cell types, that a putative MYOCD protein of ~100 kDa does not change with siRNA knockdown of Myocd mRNA. We therefore caution investigators using commercial antibodies to MYOCD and urge rigorous analysis to authenticate the specificity of bands so as to avoid further confusion in the field. The higher molecular weight of MYOCD over its predicted size of ~100 kDa likely reflects a number of post-translational modifications shown previously to occur in MYOCD, at least in cultured model systems.30, 32 Precision CRISPR editing of key amino acids undergoing post-translational modification in the Myocd3xFLAG mouse may reveal more nuanced phenotypes than those reported in conditional Myocd knockouts of the heart and vessel wall.33, 34
Lower molecular weight bands were seen with FLAG antibodies, particularly in aorta, bladder, and uterus (Figure 1 and Supplemental Figures III and V). The identity of these bands is unknown at this time, but they could represent different MYOCD isoforms, differential post-translational modifications of a specific MYOCD isoform, or proteolytic cleavage products of MYOCD. The targeting of the C-terminus of MYOCD with epitope tags, as reported here, should allow for the detection and functional assessment, via precision CRISPR editing, of all previously described MYOCD isoforms.26, 27
Another noteworthy result was the low-level MYOCD protein in adult heart. This finding was seen in every experiment conducted and cannot be attributed to lower mRNA levels since we originally reported Myocd mRNA to be equivalent in heart and aorta (13), a finding we have repeated here (Supplemental Figure VI). The lower levels of MYOCD protein in adult heart as compared to bladder and aorta could reflect a known mechanism of microRNA-mediated repression during cardiac development. Such repression was shown to promote cardiomyocyte maturation while repressing the VSMC gene program.35, 36 Interestingly, Myocd mRNA is elevated in heart failure,37 a condition characterized by the re-emergence of the fetal heart gene program, which includes an array of VSMC genes. Thus, it appears that levels of MYOCD protein are under exquisite control in the normal adult heart for optimal cardiac function.
The use of a FLAG antibody in ChIP-qPCR studies provides proof-of-principle data supporting ChIP-seq studies in the vessel wall under normal or stress-induced conditions. In this context, a growing number of SRF-independent target genes of MYOCD have been reported.30 The ability to generate mice in which each Myocd allele carries either a 3xFLAG or 3xHA tag offers a rigorous approach to define novel DNA binding sequences and the corresponding DNA-binding transcription factors associated with MYOCD. The mice reported here will also be useful for the discovery of new MYOCD binding proteins or RNAs, especially the class of long non-coding RNA. These and other studies will further illuminate MYOCD biology, thus providing a more integrative understanding of how this cofactor functions under normal and pathological conditions.
In contrast to adult tissues, nuclear-localized MYOCD was observed consistently in cultured VSMC as demonstrated in siRNA knockdown studies. Previous mouse studies showed FLAG-tagged protein expression in tissue sections38 suggesting there may be some steric hindrance in detecting the MYOCD protein in vivo. It is possible the epitope is masked through interactions of MYOCD with other molecules in the cell.30, 32 On the other hand, we have observed consistent staining in adult mouse tissues carrying an inducible HA-tagged Myocd transgene (unpublished), and preliminary staining of aorta carrying the Myocd3xHA tag suggests that it could be a more discriminating epitope tag than 3xFLAG for immunostaining, though this will require further analysis. Importantly, genetically marked VSMC progressively decrease from culture indicating the need for careful experimental design and circumspect conclusions drawn from in vitro primary VSMC experiments.
The CRISPR revolution has expedited the generation of new mouse models.11 We anticipate similarly tagged mice, as those reported here, will be developed for rigorous protein analyses and accelerated discoveries in vascular biology.
Supplementary Material
Highlights.
Non-discriminating antibodies have hampered the reliable detection of Myocardin protein.
CRISPR-Cas9 genome editing was used to knock-in epitope tags at the C-terminal end of the Myocardin locus.
The molecular mass of Myocardin protein is ~150 kDa, a size much larger than that reported in the vast literature.
Epitope tagging of Myocardin allows for facile detection of nuclear Myocardin in cultured cells as well as associated DNA binding as revealed with ChIP-qPCR.
Epitope tagging of genes with CRISPR affords an unambiguous assessment of protein expression, localization, and associated interactions in the cell.
Acknowledgements
We thank Dr. Lin Gan and Dr. Min Deng of the University of Rochester Genome Editing Resource Facility for their expert RNA and ribonucleoprotein complex microinjections and embryo transfers. We also thank Ms Diane Singer of Albany Medical College for her expert protocol of culturing mouse aortic SMC.
Source of Funding
This work was supported by NIH grants HL117907 to JMM; HL132574 (SG and JMM); and HL122686 (to XL). PH was supported from the Indiana Clinical and translational Sciences Institute funded in part by UL1TR001108.
Abbreviations
- 3cCRISPR
Three-component CRISPR
- CRISPR
Clustered regularly interspaced short palindromic repeats
- Cas9
CRISPR-associated protein 9
- sgRNA
Single guide RNA
- HDR
Homology directed repair
- MASMC
Mouse aortic smooth muscle cell(s)
- MYOCD
Myocardin
- PAM
Protospacer adjacent motif
- RFLP
Restriction fragment length polymorphism
- SRF
Serum response factor
- VSMC
Vascular smooth muscle cell(s)
Footnotes
Disclosures
None.
References
- 1.Kosmidou C, Efstathiou NE, Hoang MV, et al. Issues with the specificity of immunological reagents for NLRP3: Implications for age-related macular degeneration. Sci. Rep 2018;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Partridge EC, Watkins TA, Mendenhall EM. Every transcription factor deserves its map: Scaling up epitope tagging of proteins to bypass antibody problems. Bioessays. 2016;38:801–811. [DOI] [PubMed] [Google Scholar]
- 3.Jarvik JW, Telmer CA. Epitope tagging. Annu Rev Genet. 1998;32:601–618. [DOI] [PubMed] [Google Scholar]
- 4.Chen YI, Maika SD, Stevens SW. Epitope tagging of proteins at the native chromosomal loci of genes in mice and in cultured vertebrate cells. J. Mol. Biol 2006;361:412–419. [DOI] [PubMed] [Google Scholar]
- 5.Lee WJ, Kraus P, Lufkin T. Endogenous tagging of the murine transcription factor Sox5 with hemaglutinin for functional studies. Transgenic Res. 2012;21:293–301. [DOI] [PubMed] [Google Scholar]
- 6.Ferrando RE, Newton K, Chu F, Webster JD, French DM. Immunohistochemical detection of flag-tagged endogenous proteins in knock-in mice. J. Histochem. Cytochem 2015;63:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miano JM, Zhu QM, Lowenstein CJ. A CRISPR path to engineering new genetic mouse models for cardiovascular research. Arterioscler Thromb Vasc Biol. 2016;36:1058–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang DZ, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. [DOI] [PubMed] [Google Scholar]
- 14.Du K, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol. Cell. Biol 2003;23:2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res 2003;92:856–864. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Wang DZ, Pipes GCT, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U. S. A 2003;100:7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Y, Slivano OJ, Christie CK, Cheng AW, Miano JM. CRISPR-Cas9 genome editing of a single regulatory element nearly abolishes target gene expression in mice--brief report. Arterioscler Thromb Vasc Biol. 2015;35:312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, Ikawa M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep 2013;3:3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL. Cctop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One. 2015;10:e0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae S, Park J, Kim JS. Cas-offinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS, Concordet JP. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol. 2016;34:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen ST, Zhang M, Deng JM, Usman SJ, Smith CN, Parker-Thornburg J, Swinton PG, Martin JF, Behringer RR. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol 2014;393:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saddouk FZ, Sun LY, Liu YF, Jiang M, Singer DV, Backs J, Van Riper D, Ginnan R, Schwarz JJ, Singer HA. Ca2+/calmodulin-dependent protein kinase II-gamma (CaMKIIgamma) negatively regulates vascular smooth muscle cell proliferation and vascular remodeling. FASEB J. 2016;30:1051–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Creemers EE, Sutherland LB, Oh J, Barbosa AC, Olson EN. Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol. Cell 2006;23:83–96. [DOI] [PubMed] [Google Scholar]
- 27.Imamura M, Long X, Nanda V, Miano JM. Expression and functional activity of four myocardin isoforms. Gene. 2010;464:1–10. [DOI] [PubMed] [Google Scholar]
- 28.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent cre reporter mouse. Genesis. 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 29.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med 2008;14:64–68. [DOI] [PubMed] [Google Scholar]
- 30.Miano JM. Myocardin in biology and disease. J Biomed Res. 2015;29:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morita T, Hayashi K. Arp5 is a key regulator of myocardin in smooth muscle cells. J. Cell Biol 2014;204:683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng XL. Myocardin and smooth muscle differentiation. Arch. Biochem. Biophys 2014;543:48–56. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Proweller A, Epstein JA, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J. Clin. Invest 2008;118:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang J, Lu MM, Cheng L, Yuan LJ, Zhu X, Stout AL, Chen M, Li J, Parmacek MS. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc. Natl. Acad. Sci. U. S. A 2009;106:18734–18739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wystub K, Besser J, Bachmann A, Boettger T, Braun T. Mir-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013;9:e1003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heidersbach A, Saxby C, Carver-Moore K, Huang Y, Ang YS, de Jong PJ, Ivey KN, Srivastava D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. eLife. 2013;2:e01323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torrado M, Lopez E, Centeno A, Medrano C, Castro-Beiras A, Mikhailov AT. Myocardin mRNA is augmented in the failing myocardium: Expression profiling in the porcine model and human dilated cardiomyopathy. J. Mol. Med 2003;81:566–577. [DOI] [PubMed] [Google Scholar]
- 38.Zheng S, Ghitani N, Blackburn JS, Liu JP, Zeitlin SO. A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: Their application to understanding the effect of increasing the length of normal Huntingtin’s polyglutamine stretch on CAG140 mouse model pathogenesis. Molecular brain. 2012;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.