

SUMMARY
TP53 missense mutations significantly influence the development and progression of various human cancers via their gain of new functions (GOF) through different mechanisms. Here we report a unique mechanism underlying the GOF of p53-R249S (p53-RS), a p53 mutant frequently detected in human hepatocellular carcinoma (HCC) that is highly related to hepatitis B infection and aflatoxin B1. A CDK inhibitor blocks p53-RS’s nuclear translocation in HCC, whereas CDK4 interacts with p53-RS in the G1/S phase of the cells, phosphorylates it, and enhances its nuclear localization. This is coupled with binding of a peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1) to p53-RS, but not the p53 form with mutations of 4 serines previously shown to be crucial for PIN1-binding. As a result, p53-RS interacts with c-Myc and enhances c-Myc-dependent rDNA transcription key for ribosomal biogenesis. These results unveil a CDK4-PIN1-p53-RS-c-Myc pathway as a novel mechanism for the GOF of p53-RS in HCC.
Keywords: Mutant p53, p53R249S, CDK4, PIN1, c-Myc, gain of function, transcription, Phosphorylation, cell cycle, ribosomal biogenesis, cell proliferation
Graphical Abstract
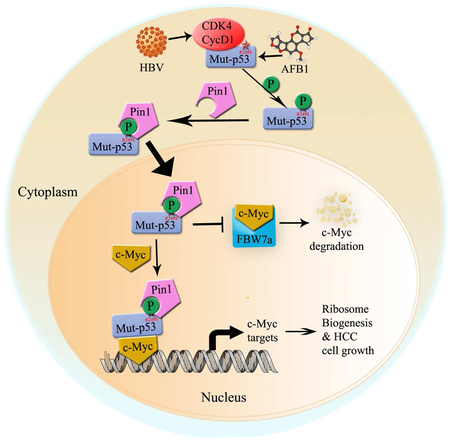
In Brief
The study by Liao et al unveils a unique molecular pathway that conveys selective mutation of the tumor suppressor p53 at a hot spot site solely found in HBV- and aflatoxin B1-associated human liver cancers and renders the mutated p53 more oncogenic in promoting liver cancer cell growth.
INTRODUCTION
The tumor suppressor p53 plays a prominent role in human cancer prevention, as ~50% of human tumors harbor mutated TP53 (Brosh and Rotter, 2009). Among the p53 mutations, more than 80% are missense mutations that mostly occur in p53’s central DNA sequence-specific binding domain. There are six hotspot mutations of p53 at R175, G245, R248, R249, R273 and R282, respectively, identified in various primary and metastatic human cancers. Remarkably, these p53 mutants, besides losing their wild type (wt) functions and exerting their dominant-negative effects (DN) on wt p53’s activity because p53 acts as a homotetrameric transcriptional factor (Kern et al., 1992), can also possess GOFs distinct from their wt counterpart’s activity, significantly influencing the development and progression of cancers (Brosh and Rotter, 2009).
One of the fairly-studied hotspot mutants is p53-R249S (p53-RS) (Hsu et al., 1991; Ozturk, 1991). Interestingly, p53-RS is highly associated with HCC, which is often diagnosed in patients with high exposure to aflatoxin B1 (AFB1) and/or infected with Hepatitis Virus B (HBV). To date, p53-RS is the only hotspot mutant that has been identified among 30% of HCC that harbor p53 mutations (Hsu et al., 1991; Hussain et al., 2007; Qi et al., 2015; Staib et al., 2003). Similar to other hotspot p53 mutants, p53-RS displays both loss of function and DN effects crucial for HCC cell proliferation (Goh et al., 2011; Lee et al., 2012). Yet, it still remains elusive if p53-RS possesses GOF activity critical for development and progression of HCC (Junk et al., 2008; Lee et al., 2012; Yin et al., 1998), and if so, what would be the underlying molecular mechanism.
In addressing these questions, we link a cell cycle-regulated kinase CDK4 (Lim and Kaldis, 2013), PIN1 (Lu and Zhou, 2007), and c-Myc with the GOF activity of p53-RS by performing a set of biochemical, molecular and cellular studies. CDK4 plays a key role in the cell cycle through G1/S phase by forming a complex with cyclin D or A family (Johnson and Walker, 1999), acts as an oncoprotein key for cancer cell proliferation (Sherr, 1996), and is highly expressed in various human cancers (Malumbres and Barbacid, 2009). PIN1 is also highly expressed in human cancers (Yeh and Means, 2007) and plays an oncogenic role by converting inactive oncoproteins into active ones, such as p53 mutants (Girardini et al., 2011) or c-Myc (Farrell et al., 2013). PIN1 binds to phosphorylated target proteins and modifies their confirmation via its peptidyl-prolyl cis-trans isomerase activity (Lu and Zhou, 2007). Often, this modification can mediate subcellular re-localization of target proteins (Ryo et al., 2001). PIN1 can bind HBx to enhance hepatocarcinogenesis in HBV-associated hepatocytes (Datta et al., 2007). c-Myc is a nuclear transcriptional factor essential for proliferation and renewal of stem cells (Bouchard et al., 1998; Takahashi and Yamanaka, 2006) and for survival of various cancer or cancer stem cells (Gordan et al., 2007; Kim et al., 2010) partially by activating gene expression crucial for ribosomal biogenesis(Grandori et al., 2005), and highly expressed in ~80% of human cancers (Dang, 2012). Thus, these oncoproteins are critical for cell proliferation and tumorigenesis.
Our studies as detailed below unveil a unique mechanism for p53-RS’s GOF, i.e., CDK4 specifically binds to and phosphorylates p53-RS, and this phosphorylation facilitates PIN1 binding and subsequent nuclear fractions of this mutant p53. In the nucleus, p53-RS binds to and stabilizes c-Myc by blocking FBW7-mediated degradation, consequently leading to c-Myc activation of rDNA and tRNA transcription. Through these actions, p53-RS executes its GOFs activity crucial for HCC cell proliferation and survival.
RESULTS
CDK4 binds to and phosphorylates p53-RS at G1/S phase.
As mentioned above, R249S mutation (Figure 1A, S1A) is of high frequency in HCC and closely related to dietary AFB1 and HBV infection. Since serine is frequently modified with phosphorylation, and p53-RS is mostly present in the cytoplasm, but perhaps executes its GOF in the nucleus, we speculated that Ser249 of p53-RS could be phosphorylated, and the phosphorylation might affect its subcellular distribution (Gouas et al., 2010; Xu et al., 2011). To test this idea, we first treated HEK293 cells that stably expressed exogenous Flag-p53-RS with a pan inhibitor of CDK family kinases [JNJ-7706621 (JNJ)], or an inhibitor of MEK1/2 (U0126). Interestingly, JNJ, but not U0126, reduced the nuclear fractions of ectopic p53-RS in the cells in a dose dependent fashion (Figure 1B), suggesting that one of the CDK kinases might be responsible for this nuclear transport of p53-RS. Remarkably, co-immunoprecipitation (IP) assays using synchronized HEK293 cells with expressed Flag-p53-RS showed that CDK4 and CDK7 are co-pulled down with Flag-p53-RS, respectively (Figure 1C). Intriguingly, CKD4 appeared to form more complexes with p53-RS in the G1/S phase, while CDK7 appeared to do so in the G2/M phase. Since CDK7 was previously shown to phosphorylate wt p53 (Lu et al., 1997), CDK4 only bound to p53-RS, but not wt p53 (Figure S1B), and among different point mutants of p53, p53-RS formed more complexes with CDK4 (Figure S1C), we decided to further explore the regulation of p53-RS by this kinase here. First, we performed IP-Western blot (WB) assays using different HCC cells, such as PLC/PRF/5 with p53-RS, Huh7 with p53-C220, and HepG2 with wt p53. Remarkably, anti-CDK4 antibodies pulled down much more endogenous p53-RS than p53-C220, but none of wt p53, with CDK4 (Figure S1E), suggesting that CDK4 might prefer to binding to p53-RS and phosphorylate it. Indeed, CDK4 phosphorylated p53-RS, but not wt p53, in an in vitro 32P-transfer kinase assay (Figure 1D) with Rb as a positive control (Zarkowska and Mittnacht, 1997) (Figure S1G), which was also validated with an antibody generated specifically against the phosphorylated p53-RS at S249 (Figures 1E, S1D and S1E). The S249P250 motif of p53-RS was critical for CDK4 phosphorylation of p53-RS, as the substitution of P250 with alanine diminished its phosphorylation by the CDK4/Cyclin D1 complex (Figure S1F). Consistent with this result, CDK4/Cyclin D1 was the only kinase for p53-RS phosphorylation as CDK2/Cyclin A1, CDK6/Cyclin D1, and CDK4/Cyclin D3 did not phosphorylate p53-RS in vitro (Figures S1H and S1I). Correlated with the result in Figure 1C, p53-RS phosphorylation was detected in the G1/S phase of PLC/PRF/5 cells after synchronization (Figure 1F), and knockdown of Cyclin D1, but not Cyclin D3, in PLC/PRF/5 cells markedly reduced this phosphorylation (Figure S1J), but the phosphorylation were not detected in the human fibroblast-like fetal lung cell line (Figure S1N). In line with these results, more phosphorylated p53-RS proteins were detected in the nucleus than in the cytoplasm of synchronized PLC/PRF/5 cells in G1/S phase (Figures 1G and S1K), and more phosphorylation mimic mutant p53-RDs (p53-R249D) than p53-RS were detected in the nuclear fraction (Figure S1L), whereas treatment of PLC/PRF/5 cells with a specific CDK4/6 inhibitor, PD-0332991 (PD) (Rivadeneira et al., 2010), markedly reduced the nuclear p53-RS level (Figure 1H). Together, these results demonstrate that CDK4/Cyclin D1 can phosphorylate p53-RS at Ser249 in the G1/S phase, consequently leading to its nuclear localization.
Figure 1. CDK4 phosphorylates p53-RS at Ser249 in G1/S phase and mediates its nuclear localization.

A) The sequence comparison of p53WT and p53RS.
B) JNJ-7706621 (JNJ) inhibitor (Pan-CDK inhibitor) blocks p53-RS’s nuclear translocation. Cell fractions of flag-vector- or p53-RS-expressing HEK293 stable cell were treated by JNJ (2 μM or 10 μM) for 16 hours or U0126 (10 μM) for 4 h and then harvested for WB with indicated antibodies.
C) p53-RS interacts with CDK4 or CDK7. The same stable cells as above were synchronized with Nocodazole at 100ng/ml for 16 h and harvested at different time points (different phases) for IP with the anti-Flag antibody followed by WB with indicated antibodies.
D, E) CDK4 phosphorylates p53-RS in vitro. His-p53WT or His-p53RS was purified from E.coli for in vitro kinase reactions using Cyclin D1/CDK4 complexes commercially purchased. Phosphorylated proteins were detected by autoradiography or the anti-phosphor-S249 antibody.
F) p53-RS is phosphorylated at G1/S phase PLC/PRF/5 cells. PLC/PRF/5 cells were synchronized with Nocodazole, and harvested at G1/S phase for IP with IgG or p53 antibody followed by WB with indicated antibodies.
G) The majority of phosphorylated p53-RS in the nucleus. Synchronized PLC/PRF/5 cells were harvested at G1/S phase for subcellular fractionation followed by IP with IgG or the anti-p53 antibody followed by WB with indicated antibodies.
H) A CDK4 inhibitor PD033291 (PD) blocks the nuclear localization of endogenous p53-RS. PLC/PRF/5 cells were treated with or without PD (500 nM) for 16 h, and harvested for analysis of subcellular fractions of endogenous p53-RS by WB with indicated antibodies.
PIN1 interacts with Ser249-phosphorylated p53-RS dependently of CDK4.
Bioinformatic analysis of p53-RS’s amino acid sequence revealed its Ser249/Pro250 motif as a potential PIN1 binding site. To test this possibility, we conducted a co-IP assay using p53-null H1299 cells by comparing ectopic p53-RS with wt p53 as wt p53 was shown to bind to PIN1 before (Zacchi et al., 2002). Considerably more p53-RSs than wt p53s were pulled down with Flag-PIN1 (Figure 2A), suggesting that PIN1 prefers to binding to the mutant p53 over wt p53. Consistently, much more endogenous p53-RS-PIN1 complexes in PLC/PRF/5 cells than wt p53-PIN1 complexes in HepG2 were also pulled down with anti-PIN1 antibodies, and again, S249-phosphorylated p53 was detected in the p53-PIN1 complex from PLC/PRF/5, but not HepG2, cells (Figure 2B). Since there are four known PIN1-binding sites in p53 (Girardini et al., 2011), we compared p53-R280K-4M mutant that harbor mutations at four known PIN1-targeted residues with p53-R280K-4M-RS whose Arg249 is replaced with Ser (Figure 2C) in co-IP assays. Remarkably, mutation of Arg249 to Ser enabled this PIN1-binding defective mutant p53 to bind to PIN1 efficiently (Figure 2D), indicating that PIN1 binds to the Ser249-Pro250 site. We also generated a p53-4M-RS with replacements of 4 known PIN1-targeted residues by Ala in p53-RS, similar to that in Figure 2C, and tested if this mutant can still bind to PIN1. Indeed, this mutant p53-RS still bound to PIN1, and this p53-4M-RS-PIN1 interaction was enhanced by treatment with okadaic acid (OA), a non-specific phosphatase inhibitor (Figure 2E), but eliminated by the treatment of H1299 cell lysates with calf intestine phosphatase (CIP) (Figure 2F). These results demonstrate that PIN1 can specifically bind to the Ser249-Pro250 site of p53-RS highly expressed in HCC cells, and this binding is Ser249 phosphorylation-dependent.
Figure 2. Pin1 interacts with Ser249-phosphorylated p53-RS.

A) Pin1 binds to mutant better than wt p53s when overexpressed in cells. Wt p53 and p53-RS plasmids, respectively, were co-introduced with Flag-PIN 1 into H1299 cells. Protein complexes were pulled down and detected by using co-IP-WB or straight WB with indicated antibodies.
B) Endogenous p53-RS and Pin1 interaction. PLC/PRF/5 or HepG2 cells were used for co-IP with the anti-Pin1 antibody followed by WB with indicated antibodies. * indicates the light chain.
C) The schematic of the functional domains and mutated residues of p53K280-4M and p53K280-4MRS..
D) p53K280-4MRS, but not p53K280-4M, interacts with Pin1. p53K280-4M or p53K280-4MRS was co-introduced with Flag-PIN1 to H1299 cells. Protein complexes were pulled down and detected by using co-IP-WB or straight WB with indicated antibodies.
E) Inhibition of phosphatases with Okadaic acid (OA) leads to elevated p53-4MRS-Pin1 association in cells. p53-4MRS was co-introduced with Flag-PIN1 into H1299 cells. Cells were treated by 1 μM or 5 μM OA for 1 h before harvesting and then used for co-IP-WB or straight WB with indicated antibodies.
F) Dephosphorylation of p53-4MRS by CIP reduces its interaction with Pin1. p53-4MRS were co-introduced with Flag-PIN1 into H1299 cells. After harvesting, cells lysates were treated by CIP for 1 h at 37°C, and then used for co-IP-WB or WB with indicated antibodies.
G) CDK4 knockdown reduces p53-4MRS interaction with Pin1. H1299 cells transfected with CDK4 or control SiRNA for 24 h were transfected with combination of plasmids encoding p53-4MRS, Flag-PIN1, harvested for co-IP-WB or straight WB analysis with antibodies as indicated.
H) CDK4 inhibitor PD reduces p53-4MRS interaction with Pin1. H1299 cells were transfected with combinations of plasmids encoding p53-4MRS, Flag-PIN1 treated with PD inhibitor 24 h before harvested for co-IP-WB or straight WB with indicated antibodies.
I) CDK4 increases p53-4MRS interaction with Pin1. H1299 cells were transfected with combination of plasmids encoding p53-4MRS, Flag-PIN1 or CDK4-HA for co-IP-WB or straight WB with indicated antibodies.
Next, we tested if CDK4 affects PIN1 binding to p53-RS by conducting co-IP-WB assays either after knockdown of CDK4 or treatment of H1299 cells with the CDK4 inhibitor PD after transfection. Indeed, either knockdown of CDK4 or inhibition of CDK4 activity by PD led to the reduction of the PIN1-p53-RS complex level (Figures 2G and 2H). Conversely, overexpression of CDK4 markedly increased the PIN1-p53-RS complex level (Figure 2I) without binding to PIN1 (Figure S1M). These results indicate that CDK4 can enhance PIN1 binding to p53-RS.
PIN1 enhances p53-RS nuclear translocation.
Our findings as shown in (Figures 1-2) and S1) suggest that CDK4 and PIN1 play a role in mediating the nuclear translocation of this phosphorylated p53-RS. Indeed, analysis of subcellular localization of GFP-p53-RS stably expressed in H1299 cells after treatment with or without the CDK4 inhibitor PD or PIN1 inhibitor ATRA (Wei et al., 2015) using time-lapse fluorescence showed that GFP-p53-RS is dynamically oscillated during the cell cycle (Figure 3), as it was localized to the nucleus in G1 phase cells (120-150 min) (Figure 3A), while evenly distributed in mitotic cells (0-60 min) (Figure 3A). Treatment with either PD (Figure 3B) or ATRA (Figure 3C) markedly reduced or delayed the nuclear translocation of GFP-p53-RS and correspondingly increased the percentage of the cells with cytoplasmic GFP-p53-RS (Figure S2A and S2B). Consistently, more p53-RS molecules were detected in the nuclear fraction in the presence of PIN1 by WB analysis (Figure S2C). Also, from this nuclear fraction, p53-RS was co-pulled down with PIN1 by co-IP analysis (Figure S2D), which was confirmed by analysis of these cells in presence or absence of PIN1 using ImageStream Imaging Flow Cytometer (Figures S2E-G). Conversely, PIN1 knockdown reduced the nuclear level, but not the cytoplasmic level of p53-RS, while knockdown of endogenous p53-RS decreased both of its own nuclear and cytoplasmic levels (Figure 3D). Consistently, treating the transfected H1299 cells with an importin inhibitor Importazole (Soderholm et al., 2011) retained most of the PIN1-phosphorylated p53-RS complex in the cytoplasm, but this complex was almost exclusively detected in the nucleus of non-treated cells (Figure 3E). These results indicate that PIN1 is required for the nuclear transport of this CDK4-phosphorylated mutant p53.
Figure 3. Nuclear localization of phosphorylated p53-RS is dependent on Pin1.

A) Time-lapse analysis of GFP-p53 in H1299. GFP-p53-RS was stably expressed in H1299 cells. Frames show the GFP-p53-RS subcellular localization; Arrows indicate the one cell division in mitosis to two daughter cells in G1 phase. For each sequence, t=0 min is the first frame marked by the arrow were observed. Scale bar, 10um. The same for panels B and C below.
B) Time-lapse analysis of GFP-p53 after treatment of 500 nM CDK4 inhibitor (CDK4i) in H1299 cells.
C) Time-lapse analysis of GFP-p53 after treatment with 5 μM Pin1 inhibitor (Pin1i) in H1299 cells.
D) Knockdown of Pin1 decreases nuclear p53-RS level. PLC/PRF/5 cells were transfected with siRNA for Pin1 (SiPin1) or p53-RS (Sip53), and used for subcellular fractionation 72 h after transfection, followed by WB with indicated antibodies.
E) Nuclear translocation of phosphorylated p53-RS is blocked by a transport inhibitor Importazole. H1299 cells were transfected with combination of plasmids encoding p53-4MRS, and/or Flag-PIN1 and treated with Importazole overnight before harvesting for analysis of subcellular fractions by co-IP with IgG or a p53 antibody followed by WB with indicated antibodies.
p53-RS interacts with c-Myc at G1/S phase and regulates its stability.
To determine if p53-RS affects global gene expression in HCC cells once being in the nucleus, we performed a CHIP-on-ChIP analysis in PLC/PRF/5 cells that were transfected with scramble or p53 siRNA. Interestingly, p53-RS knockdown in the cells led to global decrease of expression of rDNA genes and genes that encode ribosomal proteins (Figure S3A). Next, we checked if p53-RS might regulate the expression of the genes for ribosomal biogenesis by interacting with c-Myc. Indeed, we detected the p53-RS-c-Myc complex by co-IP-IB analysis in Flag-p53-RS expressing HEK293 cells at the G1/S phase (Figure 4A). Consistently and interestingly, endogenous c-Myc interacted only with p53-RS in G1/S phase in PLC/PRF/5 cells (Figure 4B), but not with wt p53 in HepG2 cells (Figure 4C), and the same was true to exogenous proteins (Figure S3B). The p53-RS-c-Myc interaction was enhanced by overexpression of CDK4 (Figure 4D), but reduced by knocking down CDK4 (Figure S3C). Also, endogenous c-Myc bound to phosphorylated p53-RS in G1/S phase in PLC/PRF/5 cells (Figure 4E). Interestingly, knockdown of endogenous p53-RS in PLC/PRF/5 cells or BT-549 cells decreased c-Myc protein level, which was rescued by a protease inhibitor MG132 (Figures 4F and S3E), suggesting that p53-RS might regulate c-Myc’s protein level. Indeed, ectopic p53-RS increased the half-life of endogenous c-Myc in p53-null Hep3B cells (Figure 4G). Ectopic p53-RS blocked c-Myc ubiquitination by FBW7a (Figure S3D), a major E3 ligase for degrading c-Myc (Yada et al., 2004), by reducing the binding of c-Myc to FBW7a in H1299 cells (Figure 4H). Finally, the enhanced p53-RS-c-Myc complex was not due to the increased steady state level of p53-RS by CDK4, as the CDK4 inhibitor did not affect the stability of p53-RS, but reduced the half-life of wt p53 to a certain degree (Figure S3F). Also, the phosphorylation mimic p53-RD formed more complexes with c-Myc than did p53-RS (Figure S3G). Altogether, these results indicate that p53-RS binds to and stabilizes c-Myc in the G1/S phase by preventing its FBW7a-mediated ubiquitination and degradation.
Figure 4. p53-RS interacts with c-Myc at G1/S phase and regulates its stability.

A) p53-RS binds to c-Myc at G1/S phase. Flag-vector or Flag-p53RS stably expressed HEK239 cells were synchronized as described in Figure 1C and harvested at G1/S phase for co-IP-WB or straight WB with indicated antibodies.
B) Endogenous p53-RS binds to c-Myc at G1/S phase. PLC/PRF/5 cells were synchronized and harvested at G/S phase for co-IP-WB or straight WB with indicated antibodies.
C) Endogenous wt p53 doesn’t bind to c-Myc. HepG2 cells were synchronized and harvested at G1/S phase co-IP-WB or straight WB with indicated antibodies.
D) CDK4 enhances the interaction of p53-RS with c-Myc. H1299 cells were transfected with combinations of plasmids encoding HA-c-Myc, Flag-p53-RS or CDK4-HA for co-IP-WB or straight WB with indicated antibodies.
E) c-Myc interacts with phosphorylated p53-RS in G1/S phase in PLC/PRF/5 cells. PLC/PRF/5 cells were synchronized and harvested at G1/S phase for co-IP-WB or straight WB with indicated antibodies.
F) Knockdown of p53-RS decreases the c-Myc protein level. PLC/PRF/5 cells transfected with p53 or control SiRNA for 72 h were treated with or without MG132 and harvested for WB analysis with indicated antibodies.
G) p53-RS prolongs c-Myc protein half-life. The vector or p53-RS was introduced into Hep3B cell, and the cells were treated by CHX (100 mg/ml) and harvested at the time points as indicated for analysis of endogenous c-Myc proteins by WB with indicated antibodies.
H) p53-RS impairs the binding of c-Myc with FBW7a. H1299 cells were transfected with combinations of plasmids encoding HA-c-Myc, p53-RS or 4xFlag-Fbw7a for co-IP-WB or straight WB with indicated antibodies.
p53-RS increases rDNA transcription.
Next, we decided to validate some of the CHIP on ChIP data by Q-PCR analysis of RNAs isolated from HCC cells with either overexpressed or knocked down p53-RS by siRNA. Indeed, the expression of pre-rRNA, rRNA and tRNA was elevated by overexpression of p53-RS in Hep3B cells (Figures 5A, 5B, S4A and S4B), but downregulated by knockdown of p53-RS or c-Myc in PLC/PRF/5 cells (Figures 5C and S4C). In contrast, knockdown of wt p53 in HepG2 cells increased the expression of pre-rRNA and rRNA, opposite to that after knockdown of c-Myc in the same cells (Figures 5D and S4D). Also, p53-RS co-resided with c-Myc at c-Myc target promoters, such as rDNA or tRNA-leu promoters, as measured by ChIP analysis (Figure 5E), and the CDK4 inhibitor PD dramatically repressed the p53-RS activity (Figure 5F). Knockdown of p53-RS decreased the expression of RPs-encoding genes at RNA levels in HCC cells (Figure 5G), whereas overexpression of p53-RS increased their global, though moderate, expression in HCC cells (Figure 5H). These moderate alterations are not unexpected, as these changes are similar to the physiological modulation of gene expression by c-Myc (Adhikary and Eilers, 2005; Chang et al., 2008). These results indicate that p53-RS enhances c-Myc transcriptional activity and boosts up c-Myc-driven ribosomal biogenesis.
Figure 5. p53-RS increases c-Myc activity on ribosomal biogenesis.

A) p53-RS dramatically increases rRNA expression. Vector or p53-RS plasmid was introduced into Hep 3B cells. RNA levels were analyzed using RT-PCR and Q-PCR.
B) p53-RS increases 5sRNA and tRNA expression. Vector or p53-RS plasmid was introduced into Hep 3B cells. RNA levels were analyzed using RT-PCR and Q-PCR.
C) Knockdown of p53-RS reduces rRNA expression. Sip53 or SiMyc was introduced into PLC/PRF/5 cells that were harvested for RNA analysis by RT-PCR and Q-PCR.
D) Knockdown of wt p53 increases rRNA expression. Sip53 or SiMyc were introduced into HepG2 cells that were harvested for analysis of RNA levels by RT-PCR and Q-PCR.
E) p53-RS binds to c-Myc’s target promoters. Vector or p53-RS plasmid was introduced into Hep3B cells that were harvested for ChIP assay using anti-p53 antibodies, and p53-RS-bound DNAs were analyzed by Q-PCR.
F) CDK4 inhibitor represses rRNA expression. Vector or p53RS were transfected into Hep 3B cells. Cells were treated with 500nM PD033291 for 24 h before harvesting for RNA analysis by RT-PCR and Q-PCR.
G) Knockdown of p53-RS represses the expression of mRNAs for ribosomal proteins. SiP53 was transfected into PLC/PRF/5 cells that were harvested for mRNA analysis by RT-PCR and Q-PCR.
H) p53-RS increases the expression of mRNAs for ribosomal proteins. Vector or the p53-RS plasmid was introduced into Hep 3B cells that were harvested for mRNA analysis by RT-PCR and Q-PCR.
p53-RS renders HCC cells more sensitive to a CDK4 inhibitor.
To test if p53-RS plays a role in regulation of HCC cell proliferation in response to the CDK4 inhibitor PD (Rivadeneira et al., 2010), we introduced ectopic p53-RS into p53-null Hep3B cells and then treated them with different doses of PD. As a result, PD treatment reduced c-Myc levels in a dose-dependent manner in the presence of p53-RS (Figures 6A and 6B), and also the Hep3B cells with ectopic p53-RS were more sensitive to PD than were their parental p53-null cells (Figures 6C and 6D), as the IC50 value for cell growth inhibition decreased by ~2.5-fold in the presence of p53-RS (Figure 6D). Conversely, knockdown of p53-RS conferred resistance of PLC/PRF/5 cells to the CDK4 inhibitor (Figures 6E-6G). Yet, knockdown of wt p53 increased proliferation of HepG2 cells without affecting the sensitivity of the cells to a CDK4 inhibitor (Figure S5C). These results suggest that p53-RS is a downstream player of the CDK4 signaling in HCC cells, rendering the cells more sensitive to the inhibitor of this kinase.
Figure 6. CDK4 inhibitor alleviates p53-RS-dependent cell proliferation.

A) and B) p53-RS can sensitize the reduction of c-Myc protein level by a CDK4 inhibitor, PD033291 (PD). The control or p53-RS plasmid was introduced into p53-null Hep3B HCC cells. Cells were treated by PD for 24 h and harvested for WB with indicated antibodies (A), and the quantification of c-Myc protein level is shown in the graph (B). c-Myc level was quantified against the level of a-tubulin. AU=arbitrary unit.
C) and D) p53-RS makes Hep3B HCC cells more sensitive to the CDK4 inhibitor PD. The control or p53-RS plasmid was introduced into Hep3B cells. Cells were treated by different concentrations of PD for Colony formation assay (C), or cell survival analysis by a WST cell growth kit (D). IC50 values are represented as mean ± SD (n = 3) (D).
E) and F) Knockdown of p53-RS makes PLC/PRF/5 HCC cells less sensitive to the CDK4 inhibitor PD. SiNC or Sip53 was introduced into PLC/PRF/5 cells, and then treated by different concentrations of PD for colony formation assay (E). The quantification of colonies is shown in the graph (F). P values were calculated for the data between columns 1 and 3 (p = 0.0015) and between columns 2 and 4 (p = 0.5), respectively.
G) Cell survival analysis was also conducted in the same set of experiments with different doses of PD by using a WST cell growth kit. Cell survival rates are represented as means ± SD (n = 3) for each time point (D = day, such as 5D = 5 days). P values were calculated as shown on top of the columns.
p53-RS phosphorylation is correlated with HBV infection and high levels of CDK4 and c-Myc in primary HCCs.
In order to examine the clinical relevance of our findings as shown in Figures 1-6, we collected primary HCC samples from the First Affiliated Hospital of Nanchang University in China. Our DNA sequencing analysis (Figure 7C) identified eight of them with R249S (Figure 7A). Consistent with previous studies ((Figure S1A), HCCs harboring p53-RS were all positive with HBV, whereas some of selected wt p53-containing HCCs were HBV-negative (Figure S6). Also, p53 protein levels were drastically higher in most of the eight p53-RS HCC specimens than that in wt p53-containing HCC samples by WB analysis (Figure 7A). In order to detect S249 phosphorylation, we enriched p53 proteins from selected HCC samples by IP (using more proteins in wild type p53-containing HCC samples than that in p53-RS-harboring HCC samples) followed by WB with anti-phosphor-S249 antibodies. Indeed, we detected phosphorylated S249 in four out of six p53-RS-containing samples with high levels of p53 protein, but not in the wt p53-containing samples, even though the p53 protein input was equivalent (Figure 7B). In line with the results in Figures 1-5, the CDK4 and c-Myc levels were also higher in the four HCC samples (#64, #180, #74, and #79) with S249-phosphorylated p53-RS than in those HCC samples with wt or non-S249 phosphorylated p53 (Figure 7B), consistent with database (Figures 5SA and 5SB). Also, PIN1 levels in p53-RS-containing HCCs were moderately higher than that in wt p53-containing HCCs (Figure 7A). We also detected the p53-RS-CDK4-c-Myc complex in the aforementioned two pairs of p53-RS-containing HCC tumors, but not in non-p53-RS-containing HCC samples, by co-IP-WB analysis (Figure 7D). In sum, these results in Figures 7A-7D and S6, together with the results in Figures 1-6 and S1-S5, demonstrate that CDK4 mediates the activation of p53-RS by phosphorylating its Ser249 and enhancing its association with PIN1 and nuclear localization, consequently boosting c-Myc-dependent ribosomal biogenesis, cell proliferation (Figure 7E), and in doing so, p53-RS makes HCC cells more sensitive to the inhibition of CDK4.
Figure 7. Phosphorylation of p53-R249S is well correlated with high levels of CDK4, Pin1 and c-Myc in primary human HCCs.

A) Concurrence of high expression of p53-R249S, c-Myc and CDK4 in HCCs. Equal amounts of HCC samples with or without p53-R249S were analyzed by WB with indicated antibodies.
B) p53-R249S is phosphorylated in HCCs. HCC samples from Panel A were subjected to IP using the anti-p53 DO-1 followed by WB with the anti-phosphor-S249 antibody and the rabbit monoclonal anti-p53 EPR17343 for total p53 protein level, respectively (of note, equal amounts of total p53 protein inputs were used for IP-WB for all HCC tissues used here by increasing 5-6 fold more total proteins in wt p53 containing HCCs than in p53-RS-containing HCCs).
C) R249S mutations were identified in HCC specimens by TP53-Exon 7 sequencing.
D) p53-R249S binds to c-Myc and CDK4 in primary HCCs. HCC samples with or without p53-R249S mutation were subjected to co-IP-WB assays with the anti-p53 antibody, and bound proteins were detected by WB with indicated antibodies. The input proteins were presented in the lower panels, and arrowheads point to target proteins.
E) A model for the CDK4-p53-R249S-PIN1-c-Myc signaling pathway in HCC.
DISCUSSION
The p53 R249S (p53-RS) is the sole hotspot mutant in HCC(Bressac et al., 1991; Gouas et al., 2009) and also possess GOF in promoting HCC cell proliferation, growth, survival and metastasis(Junk et al., 2008; Yin et al., 1998). However, it has remained completely unclear how this mutant p53 executes these oncogenic GOF activities. Our study as presented here shows a surprising finding that the substitution of Arg249 with Ser in p53 converts the p53-RS into a substrate of CDK4/Cyclin D1 in the G1/S phase of the cell cycle (Figure 1). Many tumorigenic events, including liver carcinogenesis, ultimately drive proliferation by impinging on CDK4 complexes in the G1 phase of the cell cycle (Asghar et al., 2015). Also, other studies suggest that the HBV X protein (HBx) has a role in the development of HBV-associated HCC (Kremsdorf et al., 2006) by increasing CDK4 kinase activity (Gearhart and Bouchard, 2010), suggesting that CDK4 plays an important role of facilitating the development of HBV-positive HCC. Consistent with these studies, our analysis of the TCGA genomic database also showed that the p53-RS is highly related to amplification of CCND1, CCND2, MYC or CDK4 (Figure S5A), and mRNA of CDK4 is increased in HCC than in normal liver in ONCOMINE (Figure S5B). Interestingly, p53-RS becomes a novel substrate of CDK4/Cyclin D1, but not CDK2/Cyclin A1, CDK4/Cyclin D3, or CDK6/Cyclin D1 (Figures 1 and S1), both in vitro and in HCC cells. It is this CDK4/Cyclin D1-mediated phosphorylation at Ser249 that enables p53-RS to acquire a new GOF via interaction with PIN1 and c-Myc as described below.
Remarkably, Ser249 phosphorylation of p53-RS by CDK4 enhances its binding to PIN1 and facilitates its PIN1-dependent nuclear localization (Figures 2-3 and S2). Although PIN1 was previously shown to interact with wt p53 via its Ser46-Pro47 motif or other hot spot mutant p53s via their PIN1-binding motifs (Girardini et al., 2011; Zacchi et al., 2002), we unveiled the Ser249-Pro250 motif as a novel binding site of PIN1, which is dependent on CDK4 phosphorylation (Figures 2G-2I). This was further verified by using a mutant p53 devoid of all four of the previously identified PIN1-binding sites, as this mutant with the substitution of Arg249 with Ser was still able to bind to PIN1 (Figures 2D and S2A). Also, this binding facilitates the nuclear localization of p53-RS, as overexpression of ectopic PIN1 increased the nuclear fraction of p53-RS, which was blocked by an importin inhibitor, whereas knockdown of PIN1 reduced the nuclear p53-RS level (Figure 3). Consistently, more phosphorylation mimic mutant p53-RDs than p53-RSs were detected in nuclear fractions when overexpressed in cells (Figure S1L). Hence, our findings demonstrate that PIN1 can bind to CDK4 phosphorylated p53-RS at the Ser249-Pro-250 motif and mediate its nuclear transport in HCC cells.
More remarkably, four lines of evidence demonstrate that nuclear p53-RS, but not wt p53, interacts with c-Myc in G1/S phase cells: 1) Ectopic p53-RS and c-Myc were co-immunoprecipitated in the G1/S phase fraction of synchronized cells (Figure 4A); 2) Endogenous and phosphorylated p53-RS in PCL/PRF/5 cells, but not endogenous wt p53 in HepG2 cells, formed a complex with c-Myc in G1/S phase (Figures 4B, 4C and 4E); 3) Overexpression of CDK4 enhanced the formation of the p53-RS-c-Myc complex (Figure 4D); 4) p53-RS co-resided with c-Myc at several c-Myc target promoters (Figure 5E).
Interestingly, the p53-RS-binding makes c-Myc more stable, as knockdown of p53-RS in PLC/PRF/5 cells reduced c-Myc protein level, which was rescued by a 26S proteasome inhibitor, while overexpression of p53-RS extended the half-life of c-Myc (Figures 4F and 4G). Consistently, p53-RS reduced the binding of c-Myc to its E3 ligase FBW7a (Figure 4H; Figure S3D). These results indicate that by binding to c-Myc, p53-RS protects the former from FBW7-mediated proteolysis, leading to the increase of c-Myc levels, and suggest that p53-RS might enhance c-Myc transcriptional activity by stabilizing this protein, while p53-RS could also associate with c-Myc at the latter’s target promoters (Figure 5E) and enhancing its transcriptional activity. Indeed, p53-RS promotes c-Myc-mediated expression of rRNA, tRNAs, and ribosomal proteins-encoding genes globally, and consequently support c-Myc-driven HCC cell proliferation and survival (Figure 5). As a result, p53-RS can sensitize HCC cells to a CDK4 inhibitor, but knockdown of endogenous p53-RS makes the HCC cells less sensitive to this inhibitor (Figure 6). Thus, our studies demonstrate a unique cell-cycle regulated signaling pathway that mediates the execution of the GOF of p53-RS by promoting c-Myc activity once located in the nucleus via CDK4-PIN1-involved mechanisms (Figure 7E).
As previously shown (Hsu et al., 1991; Hussain et al., 2007; Qi et al., 2015; Staib et al., 2003), p53-RS was the only hotspot mutation in HCC. Indeed, our screening ~200 HCC samples identified 8 cases with p53-RS positive from China. Although we need to collect more HCC samples to perform a statistically significant study, this study would take much longer time to complete, as there are some significant differences in terms of p53 mutation incidences in HCCs in different provinces of China (Gouas et al., 2009; Liu et al., 2002; Qi et al., 2015) (Figure S1A). However, we did find out that p53-RS phosphorylation at S249 is well correlated with high levels of CDK4 and c-Myc in p53-RS positive HCC samples (Figure 7), which is in line with our HCC cellular studies (Figures 1-6). These p53-RS positive HCCs were also well correlated with HBV infection (Figure S6). Also, the phosphorylated p53-RS indeed formed a complex with CDK4 and c-Myc in two pairs of p53-RS-containg HCC tissues, but not in non-p53-RS-containing HCC samples (Figure 7D). These results are consistent with our HCC cell-based studies above and previous studies by others, showing that HBV infection is highly associated with CDK4 activation and high levels of c-Myc (Ayub et al., 2013; Hansen et al., 1993; Terradillos et al., 1997). Hence, our study using several clinical HCC samples further verifies the concept that HBV infection might in part help HCCs select the R249S mutation of p53 by activating CDK4 that in turn phosphorylates S249 and enhances the binding of phosphorylated p53-RS to PIN1, facilitating p53-RS nuclear relocation, and once in the nucleus, phosphorylated p53-RS interacts with c-Myc and stabilizes this transcriptional factor by inhibiting FBW7a-mediated c-Myc ubiquitination and degradation, consequently activating c-Myc and c-Myc-driven ribosomal biogenesis and promoting cell survival (Figure 7E).
The identification of the unique CDK4-PIN1-p53-RS-c-Myc pathway in HCC cells has two folds of biological or translational significance. First, our findings can explain why this mutant p53 only displays a dominant-negative function without apparent GOF in a mouse p53R246S/R246S line (human R249S) (Lee et al., 2012). This is probably because in the mice, p53-R246S cannot execute its GOF by associating with c-Myc without highly active CDK4 and/or PIN1 in their liver cells. This speculation is well in line with the fact that the p53-RS is highly associated with AFB1 and HBV-positive HCCs that harbor many other active oncoproteins, such as CDK4, PIN1 or c-Myc (Chisari et al., 1989; Jung et al., 2007; Pang et al., 2007). Also, our findings suggest that co-targeting p53-RS with CDK4, PIN1 or c-Myc might be more effective for anti-HCC therapy, as all these oncoproteins are highly active in and related to HCC.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact, Dr. Hua Lu (hlu2@tulane.edu)
METHODS DETAILS
Plasmids and antibodies
The V5-tagged plasmid pLenti6/V5-p53 and pLenti6/V5-p53-R249S were purchased from Addgene created by Dr. Bernard Futscher. The Flag-p53-R249S expression plasmid was generated by point mutant from Flag/HA-p53 were gifted from Dr. Wei Gu, using the following primers, 5-ATGGGCGGCATGAACCGGAGTCCCATCCTCA-3 and 5-TACCCGCCGTACTTGGCCTCAGGGTAGGAGT-3. Plasmid encoding the His-tagged p53 was generated into the pET30a vector, His-p53-RS (S249S) was mutant based on His-p53. The no-tag plasmid pcDNA3.1-p53, pcDNA3.1-p53-4M, pcDNA3.1-p53K280 and pcDNA3.1-p53K280-4M were obtained from Dr. Giannino Del Sal, no-tag pcDNA3.1-p53-RS (S249R) and pcDNA3.1-p53-4MRS and pcDNA3.1-p53K280-4MRS were generated by point mutant. The Flag-tagged plasmid pcDNA3.1-PIN1 was gifted from Dr. Kunping Lu. The HA-tagged CDK4 plasmid was purchased from Addgene created by Dr. Sander van den Heuvel. The HA-c-Myc and Flag-FBW7a plasmid were gifted from Dr. Ping Wang. Anti-Flag (Sigma-Aldrich, St. Louis, MO, USA), anti-HA (F-7, Santa Cruz Biotechnology), anti-GFP (B-2, Santa Cruz Biotechnology), anti-p53 (DO-1, FL-393 Santa Cruz Biotechnology), anti-p21(CP74, Neomarkers, Fremont, CA, USA), anti-PINl (H-123, Santa Cruz Biotechnology and #3722, Cell Signaling Technology) anti-H3 (#4499, Cell Signaling Technology) anti-GAPDH (#5174, Cell Signaling Technology), anti-α-actin (C4, Santa Cruz Biotechnology), CDK Antibody Sampler Kit (#9868, Cell Signaling Technology) and anti-c-Myc (ab32072, Abcam), were commercially purchased. Anti-phosphor-S249 antibody was generated using a peptide GMNRp(S)PILTI-cys (NeoBioLab Cambridge, Massachusetts, USA), and the non-phosphorylation peptide GMNRSPILTI-cys is used for blocking the non-specific signaling. Okadaic Acid (OA) was purchased from Cell signaling technology, and alkaline phosphatase, Calf Intestinal (CIP), was purchased from New England BioLabs Inc. The pan CDK inhibitor JNJ-7706621 (JNJ), the MEK1/2 inhibitor U0126, the p38 MAPK inhibitor SB203580, the PI3K inhibitor Wortmannin and the CDK4 inhibitor PD0332991 were purchased from Selleckchem. The importin inhibitor Importazole was purchased from Sigma.
Cell culture and transient transfection
Human liver cancer cell lines HepG2, PLC/PRF/5 and Hep3B were gifted from Dr. Tong Wu at Tulane University.
The stable HKE293 cells expressing vector or Flag-p53-RS were established by G418, the stable Hep 3B cells expressing vector or p53-RS were infected by lenti-virus of pLenti6 vector or pLentif6-p53-RS, then established by Blasticdin.
Human cancer cell lines HepG2, PLC/PRF/5 Hep3B and H1299 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 0.1 mg/ml streptomycinn. All cells were maintained at 37 °C in a 5% CO2 humidified atmosphere. Cells seeded on the plate overnight were transfected with plasmids as indicated in figure legends using the TurboFect transfection reagent by following the manufacturer’s protocol (Thermo Scientific). Cells were harvested at 30-48h post-transfection for future experiments.
Cell fractionation
This experiment was performed as described previously (Liao et al., 2014), Briefly, ~106 cells were collected, washed twice with PBS, and resuspended in 1 ml buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT) for 30 min on ice. Phenylmethylsulfonyl fluoride was added to a final concentration of 0.2 mM, and the mixture was then Dounce homogenized until all cytoplasmic membranes were disrupted. For cytosolic isolation, cells were centrifuged at 228 χ g for 5 min at 4 °C to obtain the supernatant as cytoplasm, and pellets were washed by Buffer A twice and stored as nuclear extracts (NE).
Western blotting
Cells were harvested and lysed in lysis buffer consisting of 50 mMTris/HCl (pH7.5), 0.5% Nonidet P-40 (NP-40), 1 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.2 mMphenylmethylsulfonyl fluoride (PMSF), 10 μM pepstatin A and 1mMleupeptin. Equal amounts of clear cell lysate (20–80 μg) were used for Western blotting (WB) analyses as described previously(Wang et al., 2015).
Microscopy and Image Analysis
For immunofluorescent staining, cells were fixed, stained, and mounted as described previously (Zhou et al., 2016). For live-cell imaging, GFP-p53RS stable H1299 cells were placed on poly-L-Lysine coated glass bottom culture dishes (MatTek Corporation) for 8–12 hr before imaging. Time-lapse microscopy was performed on an Olympus VivaView FL microscope (Louisiana Cancer Research Center of Shared Resources of Imaging).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay was performed using antibodies as indicated in the figure legends and described previously (Liao and Lu, 2013). The reverse cross-linked immuoprecipitated DNA fragments were purified using GeneJET gel extraction kit (Thermo Scientific) followed by PCR analyses for the c-Myc-responsive DNA elements on the promoters of human rDNA, E2F2 and eIF4E using the following primers, 5’-AACGGTGGTGTGCGTTCCC-3’ and 5’-TCTCGTCTCACTCAAACCGCC-3’ for human rDNA, 5’-TCACCCCTCTGCCATTAAAGG-3’ and 5’-AGCAGTGTATTCCCCAGGCC-3’ for human E2F2, and 5’-AAGCCTCTCGTTACTCACGC-3’ and 5’-AGATTCAAACCGATTGGCC-3’ for eIF4E (Dai et al., 2007; Dai et al., 2010).
In Vitro p53-RS Ser249 Kinase Assay
The p53-RS Ser-249 kinase assay was carried out using a previously described method (Keller et al., 2001) using [γ-32P]-ATP. Substrates included 100 ng of His-p53 and 100 ng of His-p53-RS, and 1 μg of the kinase CDK4/CycD1 complex (ProQinase) was used. Kinase assays were also done using unlabeled ATP (1 mM) followed by SDS-PAGE, and then phosphorylated S249 was detected by WB using the anti-p53-Ser249 antibody.
ChIP-on-CHIP and bioinformatics analysis
ChIPs from the PLC/PRF/5 cell lines samples were performed according to the Agilent protocol version 11.3 (http://www.chem.agilent.com), using anti-mouse IgG (sc-2025, Santa Cruz) and anti-p53 (sc-126 X, Santa Cruz) mAbs. ChIP-on-CHIP analysis was conducted at Haywood Genetics Center of Tulane University School of Medicine. The bioinformatics analysis of ChIP-on-CHIP data were carried out by the Cancer Crusaders Next Generation Sequence Analysis Core of the Tulane Cancer Center. Experiments were triplicate, and genes with over 1.5-fold increase in expression (P<0.05) were shown from the experiments.
Immunoprecipitation
Immunoprecipitation (IP) was conducted using antibodies as indicated in the figure legends and described previously(Wang et al., 2015). Briefly, ~500 to 1000 μg of proteins were incubated with indicated antibodies at 4 °C for 4 h or overnight. Protein A or G beads (Santa Cruz Biotechnology) were then added, and the mixture was left to incubate at 4 °C for additional 1 to 2 h. The beads were washed at least three times with lysis buffer. Bound proteins were detected by IB with antibodies as indicated in the figure legends.
Reverse transcription and quantitative PCR analyses
Total RNA was isolated from cells using Trizol (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Total RNAs of 0.5 to 1μg were used as templates for reverse transcription using poly-(T)20 primers and M-MLV reverse transcriptase (Promega, Madison, WI, USA). Quantitative PCR (Q-PCR) was conducted using SYBR Green Mix according to the manufacturer’s protocol (BioRad, Hercules, CA, USA). The primers for human p53, p21, ribosomal protein, rRNA, tRNA, and GAPDH were used as previously described (Sun et al., 2008).
RNA interference
The siRNAs against PIN1, CDK4, c-Myc and p53 were commercially purchased. 40~60nM of siRNAs were introduced into cells using TurboFect transfection reagent following the manufacturer’s protocol. Cells were harvested ~72 h after transfection for IB or Q-PCR.
Cell viability assay
To assess the long term cell survival, the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Rockville, MD, USA) was used according to the manufacturer’s instructions. Cell suspensions were seeded at 2,000 cells per well in 96-well culture plates at 12 h post-transfection. Cell viability was determined by adding WST-8 at a final concentration of 10% to each well, and the absorbance of the samples was measured at 450 nm using a Microplate Reader (Molecular Device, SpecrtraMax M5e, Sunnyvale, CA, USA) every 24 h for 4 days.
Colony formation assay
Cells were trypsinized and seeded with the same amount on 10-cm plates following siRNA transfection for 12 to 18 h. The medium was changed every 3 days until the colonies were visible. Blasticdin was added in the medium when stable cell lines were used in the experiment. Cells were then fixed by methanol and stained by crystal violet solution at RT for 30 min. ImageJ was used for quantification of colonies.
Human Hepatocellular carcinoma specimens
Hepatocellular carcinoma (HCC) tissue samples were collected and archived at the First Affiliated Hospital of Nanchang University, Jiangxi, China (Zhou et al., 2016). Fresh HCC cancer samples were immediately snap-frozen in liquid nitrogen and stored at −80°C until their use in. All patients provided written informed consent to participate in the study and all primary HCC samples without preoperative radiotherapy were included and confirmed by pathologists.
DNA sequencing for p53-RS in HCC tissues
Genomic DNA was extracted from ground HCC tissue samples. DNA was amplified by PCR to generate 110 bp product encompassing codon 249 located at exon 7 of TP53. The primers used were: P1 5 ’-GTTGGCTCTGACTGTACCAC-3’ and P2 5’-CTGGAGTCTTCCAGTGTGAT-3’. The PCR products were sequenced by GENEWIZ and aligned using NCBI BLAST (Yang et al., 1998).
Analysis of primary HCC specimens
Equal amounts (50 μg) of total proteins from liver cancer tissues were analyzed by WB using antibodies indicated in the figure legends. As the p53-R249S levels were much higher, adjusted amounts of total proteins were used for R249S phosphorylation analysis by IP-WB assays. Briefly, 100~300 μg total proteins from R249S samples and 2000 μg total proteins from non-R249S samples were subjected to IP analysis using the mouse monoclonal anti-p53 DO-1 (Santa Cruz Biotechnology). Phosphorylation of p53-R249S was assessed by WB using the primary anti-phosphor-S249 antibody. The non-phosphorylation peptide GMNRSPILTI-cys was used to block the non-specific signals before the secondary antibody was used. The total p53 proteins were determined using the rabbit monoclonal anti-p53 EPR17343 (abcam).
Analysis of interaction of p53-RS, CDK4 and c-Myc in primary HCC specimens
Lysates of liver cancer samples were used for co-IP-WB assays to determine if p53-R249S interacts with CDK4 and c-Myc in vivo. Briefly, about 2-5 mg of total proteins were first precleared with Protein A/G beads (Santa Cruz, USA) for 30 min followed by incubation with the anti-p53 antibody DO-1 for 4 h and Protein A/G beads for 1 h. The immunoprecipitated proteins were analyzed by WB with the antibodies as indicated.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-p53 (DO-1) | Santa Cruz Biotechnology | Cat# sc-126 AB_628082 |
| Mouse monoclonal anti-p53 (DO-1) | Santa Cruz Biotechnology | Cat# sc-126 X |
| Rabbit polyclonal anti-p53 (FL-393) | Santa Cruz Biotechnology | Cat# sc-6243 AB_653753 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025 AB_737182 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 5174 AB_10622025 |
| Rabbit monoclonal anti-Histone H3 | Cell Signaling Technology | Cat# 4499 AB_10544537 |
| Mouse monoclonal anti-cdc2 (CDK1) | Cell Signaling Technology | Cat# 9116P AB_2074795 |
| Rabbit monoclonal anti-CDK2 | Cell Signaling Technology | Cat# 2546P AB_2276129 |
| Rabbit monoclonal anti-CDK4 | Cell Signaling Technology | Cat# 12790P AB_2631166 |
| Rabbit monoclonal anti-CDK6 | Cell Signaling Technology | Cat# 13331P |
| Mouse monoclonal anti-CDK7 | Cell Signaling Technology | Cat# 2916P AB_10827986 |
| Rabbit monoclonal anti-CDK9 | Cell Signaling Technology | Cat# 2316P AB_2291505 |
| Mouse monoclonal anti-CDK4 | Santa Cruz Biotechnology | Cat# sc-56277 AB_1121419 |
| Rabbit polyclonal anti-Phosphorylation of S249 | This paper | N/A |
| Mouse monoclonal anti-Flag M2 | Sigma-Aldrich | Cat# F3165 AB_259529 |
| Mouse monoclonal anti-Cyclin D1 | Santa Cruz Biotechnology | Cat# sc-20044 AB_627346 |
| Mouse monoclonal anti-Cyclin D3 | Cell Signaling Technology | Cat# 2936P AB_10841292 |
| Rabbit polyclonal anti-Pin1 | Cell Signaling Technology | Cat# 3722S AB_10692654 |
| Rabbit polyclonal anti-Pin1 | Santa Cruz Biotechnology | Cat# sc-15340 AB_2237080 |
| Mouse monoclonal anti-Pin1 | Santa Cruz Biotechnology | Cat# sc-46660 AB_628132 |
| Rabbit monoclonal anti-PARP | Cell Signaling Technology | Cat# 9532S AB_10695538 |
| Rabbit monoclonal anti-c-Myc | Abcam | Cat# ab32072 AB_731658 |
| Mouse monoclonal anti-GFP | Santa Cruz Biotechnology | Cat# sc-9996 AB_627695 |
| Mouse monoclonal anti-P21WAF1 | NeoMarkers | Cat#MS-891-P1 |
| Mouse monoclonal anti-beta-Actin | Sigma-Aldrich | Cat# A1978 AB_476692 |
| Mouse monoclonal anti-beta-Actin | Santa Cruz Biotechnology | Cat# sc-47778 AB_626632 |
| Mouse monoclonal anti-alpha-tubulin | Sigma-Aldrich | Cat# 00020911 AB_10013740 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, Light Chain Specific for Western blotting after IP | Jackson ImmunoResearch | Cat# 115-035-174 AB_2338512 |
| Peroxidase IgG Fraction Monoclonal Mouse Anti-Rabbit IgG, Light Chain Specific | Jackson ImmunoResearch | 211-032-171 AB_2339149 |
| Mouse IgG Isotype Control | Thermo Fisher | Cat# 31903 AB_10959891 |
| Rabbit IgG Isotype Control | Thermo Fisher | Cat# 31235 AB_243593 |
| Bacterial and Virus Strains | ||
| DH5a competent cells | NEW ENGLAND BioLabs | Cat# C2987H |
| BL21(DE3) competent cells | NEW ENGLAND BioLabs | Cat# C2527H |
| Stbl3 competent cells | Thermo Fisher | Cat# A10469 |
| Biological Samples | ||
| Hepatocellular carcinoma (HCC) tissue samples | The First Affiliated Hospital of Nanchang University, Jiangxi, China. | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| JNJ-7706621 | Selleckchem | Cat# S1249 |
| U0126-EtOH | Selleckchem | Cat# S1102 |
| PD-0332991 (Palbociclib) | Selleckchem | Cat# S1116 |
| Nocodazole | Millipore | Cat# 487929 |
| Okadaic acid (OA) | Cell Signaling Technology | Cat# 5934 |
| Alkaline Phosphatase, Calf Intestinal (CIP) | NEW ENGLAND BioLabs | Cat# M0290S |
| ATRA Pin1 inhibitor | Kunping Lu | N/A |
| Importazole | Sigma-Aldrich | Cat#SML0341 |
| MG132 | Selleckchem | Cat#S8410 |
| Cycloheximide (CHX) | Sigma-Aldrich | C7698 |
| P53S-249-phos-peptide | This paper | N/A |
| P53S-249-non-phos-peptide | This paper | N/A |
| γ32P-ATP | Perkin-Elmer | NEG002A250UC |
| Human His-p53-WT | This paper | N/A |
| Human His-p53-S249 | This paper | N/A |
| Human His-p53-SA(S249A250) | This paper | N/A |
| GST-Rb(379-928) | This paper | N/A |
| CDK4/Cyclin D1 Active | SignalChem | Cat# C31-10G |
| CDK4/Cyclin D3 Active | SignalChem | Cat# C31-18G |
| CDK2/Cyclin A1 Active | SignalChem | Cat# C29-10BG |
| CDK6/Cyclin D1 Active | SignalChem | Cat# C35-18H |
| iTAq Universal SYBR-green Supermix | Bio-Rad | Cat# 1725125 |
| Critical Commercial Assays | ||
| Cell Counting Kit-8 | Dojindo Molecular Technologies | Cat#CK04 |
| Deposited Data | ||
| Raw and processed data - ChIP-on-CHIP analysis In PLC/PRF/5 cells | This paper | GSE100968 |
| Experimental Models: Cell Lines | ||
| Human: HKE293 cells | ATCC | CRL-1573 |
| Human: HEK293 cells with Flag/HA-vector | This paper | N/A |
| Human: HEK293 cells with Flag/HA-p53-S249 | This paper | N/A |
| Human: PLC/PRF/5 cells | Tong Wu | Tulane University |
| Human: HepG2 cells | Tong Wu | Tulane University |
| Human: Huh7 cells | Tong Wu | Tulane University |
| Human: Hep 3B cells | ATCC | HB-8064 |
| Human: BT-549 cells | ATCC | HTB-122 |
| Human: H1299 cells | ATCC | CRL-5803 |
| Human: H1299 cells with GFP-p53-RS cells | This paper | N/A |
| Human: Hep 3B cells with plenti6/V5-vector cells | This paper | N/A |
| Human: Hep 3B cells with plenti6/V5-p53-RS cells | This paper | N/A |
| Human: WI38 cells | ATCC | CCL-75 |
| Oligonucleotides | ||
| SiRNA: SiCyclin D1-1 | Sigma-Aldrich | SASI_Hs01_002139 09 |
| SiRNA: SiCyclin D1-2 | Sigma-Aldrich | SASI_Hs01_002139 08 |
| SiRNA: SiCyclin D3-1 | Sigma-Aldrich | SASI_Hs01_000501 84 |
| SiRNA: SiCyclin D3-2 | Sigma-Aldrich | SASI_Hs01_000501 85 |
| SiRNA: SiCdk4(h) | Santa Cruz Biotechnology | Cat# sc-29261 |
| SiRNA: SiPin1(h) | Santa Cruz Biotechnology | Cat# sc-36230 |
| SiRNA: Sip53(h) | Thermo Fisher | 106141 |
| SiRNA: Sip53(h) | Sigma-Aldrich | SASI_Hs01_000563 96 |
| SiRNA: SiMyc | Santa Cruz Biotechnology | Cat# sc-29226 |
| RT-PCR primers | This paper | See Table S1 |
| Recombinant DNA | ||
| Plasmids: pIRESneo3-Flag/HA-p53-S249 | This paper | N/A |
| Plasmids: pET-30a-His-p53-WT | This paper | N/A |
| Plasmids: pET-30a-His-p53-S249 | This paper | N/A |
| Plasmids: pCMV-neo-Bam-CDK4-HA | Addgene | Addgene Plasmid #1876 |
| Plasmids: pcDNA3.1-Flag-p53 | This paper | N/A |
| Plasmids: pcDNA3.1-Flag-p53-S249 | This paper | N/A |
| Plasmids: pcDNA3.1-Flag-p53-K280 | (Girardini et al., 2011) | N/A |
| Plasmids: pcDNA3.1-p53-K280-4M (33A,46A,81A 315A) | (Girardini et al., 2011) | N/A |
| Plasmids: pcDNA3.1-p53-K280-4M-RS (33A,46A,81A 315A & S249) | This paper | N/A |
| N/A | ||
| Plasmids: pET-30a-His-p53-S249A250 | This paper | N/A |
| Plasmids: GST-Rb(379-928) | Jiandong Chen | Moffitt Cancer Center |
| Plasmids: pcDNA3.1-p53-4M (33A,46A,81A,315A) | (Girardini et al., 2011) | N/A |
| Plasmids: pcDNA3.1-p53-4M-RS (33A,46A,81A 315A & S249) | This paper | N/A |
| Plasmids: pcDNA3.1-p53-4M-D249 (33A,46A,81A 315A & D249) | This paper | N/A |
| Plasmids: pcDNA3.1-Flag-Pin1 | (Lee et al., 2009) | N/A |
| Plasmids: plenti6/V5-p53-WT | Addgene | Addgene Plasmid #22945 |
| Plasmids: plenti6/V5-p53-S249 | Addgene | Addgene Plasmid #22935 |
| Plasmids: plenti6/V5-p53-H273 | Addgene | Addgene Plasmid #22934 |
| Plasmids: pEGFP-N1-p53-RS(S249) | This paper | N/A |
| Plasmids:pcDNA-3.1-HA-c-Myc | (Wang et al., 2015) | N/A |
| Plasmids:pcDNA-3.1-4XFlag-FBW7a | (Liu et al., 2010) | N/A |
| Software and Algorithms | ||
| PrimerX | Carlo Lapid | http://www.bioinformatics.org/primerx/ |
| Bio-Rad Image Lab | Bio-Rad | N/A |
| Bio-Rad CFX Manager | Bio-Rad | N/A |
| ImageJ | NIH Image | N/A |
| Other | ||
Highlights.
p53-RS is phosphorylated by CDK4/Cyclin D1 in HCC cells and primary HCC tissues.
PIN1 binds to phosphorylated p53-RS and mediates its nuclear localization.
p53-RS binds to c-Myc in the nucleus of HCC cells and increases its activity.
p53-RS renders HCC cells more sensitive to CDK4 inhibitor.
ACKNOWLEDGMENTS
We thank Wei Gu, Ping Wang and Jiandong Chen for offering plasmids, Kunping Lu for the Pin1 plasmid and Pin1i ATRA, Tong Wu for HCC cells, Mary Price for flow cytometry, Luis Marrero for time-lapse microscopy, and Wenjuan Liao for artwork. Hua Lu and Shelya X Zeng were supported in part by NIH-NCI grants R01CA095441, R01CA172468, R01CA127724, R21CA190775, and R21 CA201889.
Footnotes
Competing Interests
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adhikary S, and Eilers M (2005). Transcriptional regulation and transformation by Myc proteins. Nature reviews. Molecular cell biology 6, 635–645. [DOI] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC and Knudsen ES (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews. Drug discovery 14, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayub A, Ashfaq UA, and Haque A (2013). HBV Induced HCC: Major Risk Factors from Genetic to Molecular Level. BioMed research international. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Staller P, and Eilers M (1998). Control of cell proliferation by Myc. Trends in cell biology 8, 202–206. [DOI] [PubMed] [Google Scholar]
- Bressac B, Kew M, Wands J, and Ozturk M (1991). Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350, 429–431. [DOI] [PubMed] [Google Scholar]
- Brosh R, and Rotter V (2009). When mutants gain new powers: news from the mutant p53 field. Nature reviews. Cancer 9, 701–713. [DOI] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, and Mendell JT (2008). Widespread microRNA repression by Myc contributes to tumorigenesis. Nature genetics 40, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, and Palmiter RD (1989). Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 59, 1145–1156. [DOI] [PubMed] [Google Scholar]
- Dai MS, Arnold H, Sun XX, Sears R, and Lu H (2007). Inhibition of c-Myc activity by ribosomal protein L11. The EMBO journal 26, 3332–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Sun XX, and Lu H (2010). Ribosomal protein L11 associates with c-Myc at 5 S rRNA and tRNA genes and regulates their expression. The Journal of biological chemistry 285, 12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Banerjee A, Chandra PK, and Chakravarty R (2007). Pin1-HBx interaction: a step toward understanding the significance of hepatitis B virus genotypes in hepatocarcinogenesis. Gastroenterology 133, 727–728; author reply 728–729. [DOI] [PubMed] [Google Scholar]
- Farrell AS, Pelz C, Wang X, Daniel CJ, Wang Z, Su Y, Janghorban M, Zhang X, Morgan C, Impey S., et al. (2013). Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Molecular and cellular biology 33, 2930–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart TL, and Bouchard MJ (2010). The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. Journal of virology 84, 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. (2011). A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer cell 20, 79–91. [DOI] [PubMed] [Google Scholar]
- Goh AM, Coffill CR, and Lane DP (2011). The role of mutant p53 in human cancer. The Journal of pathology 223, 116–126. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, and Simon MC (2007). HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer cell 12, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouas D, Shi H, and Hainaut P (2009). The aflatoxin-induced TP53 mutation at codon 249 (R249S): biomarker of exposure, early detection and target for therapy. Cancer letters 286, 29–37. [DOI] [PubMed] [Google Scholar]
- Gouas DA, Shi H, Hautefeuille AH, Ortiz-Cuaran SL, Legros PC, Szymanska KJ, Galy O, Egevad LA, Abedi-Ardekani B, Wiman KG, et al. (2010). Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: interaction with hepatitis B virus X protein. Carcinogenesis 31, 1475–1482. [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, and White RJ (2005). c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nature cell biology 7, 311–318. [DOI] [PubMed] [Google Scholar]
- Hansen LJ, Tennant BC, Seeger C, and Ganem D (1993). Differential activation of myc gene family members in hepatic carcinogenesis by closely related hepatitis B viruses. Molecular and cellular biology 13, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, and Harris CC (1991). Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350, 427–428. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Schwank J, Staib F, Wang XW, and Harris CC (2007). TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 26, 2166–2176. [DOI] [PubMed] [Google Scholar]
- Johnson DG, and Walker CL (1999). Cyclins and cell cycle checkpoints. Annual review of pharmacology and toxicology 39, 295–312. [DOI] [PubMed] [Google Scholar]
- Jung JK, Arora P, Pagano JS, and Jang KL (2007). Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer research 67, 5771–5778. [DOI] [PubMed] [Google Scholar]
- Junk DJ, Vrba L, Watts GS, Oshiro MM, Martinez JD, and Futscher BW (2008). Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia 10, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, and Lu H (2001). A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Molecular cell 7, 283–292. [DOI] [PubMed] [Google Scholar]
- Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, and Vogelstein B (1992). Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 256, 827–830. [DOI] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, and Orkin SH (2010). A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremsdorf D, Soussan P, Paterlini-Brechot P, and Brechot C (2006). Hepatitis B virus-related hepatocellular carcinoma: paradigms for viral-related human carcinogenesis. Oncogene 25, 3823–3833. [DOI] [PubMed] [Google Scholar]
- Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, and Sabapathy K (2012). Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer cell 22, 751–764. [DOI] [PubMed] [Google Scholar]
- Liao JM, and Lu H (2013). ChIP for identification of p53 responsive DNA promoters. Methods Mol Biol 962, 201–210. [DOI] [PubMed] [Google Scholar]
- Liao P, Wang W, Shen M, Pan W, Zhang K, Wang R, Chen T, Chen Y, Chen H, and Wang P (2014). A positive feedback loop between EBP2 and c-Myc regulates rDNA transcription, cell proliferation, and tumorigenesis. Cell death & disease 5, e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, and Kaldis P (2013). Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Y, Zhou Q, Gui SY, and Li X (2002). The point mutation of p53 gene exon7 in hepatocellular carcinoma from Anhui Province, a non HCC prevalent area in China. World journal of gastroenterology 8, 480–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Fisher RP, Bailey P, and Levine AJ (1997). The CDK7-cycH-p36 complex of transcription factor IIH phosphorylates p53, enhancing its sequence-specific DNA binding activity in vitro. Molecular and cellular biology 17, 5923–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP, and Zhou XZ (2007). The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nature reviews. Molecular cell biology 8, 904–916. [DOI] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nature reviews. Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Ozturk M (1991). p53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet 338, 1356–1359. [DOI] [PubMed] [Google Scholar]
- Pang R, Lee TK, Poon RT, Fan ST, Wong KB, Kwong YL, and Tse E (2007). Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 132, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Qi LN, Bai T, Chen ZS, Wu FX, Chen YY, De Xiang B, Peng T, Han ZG, and Li LQ (2015). The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China : role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver international : official journal of the International Association for the Study of the Liver 35, 999–1009. [DOI] [PubMed] [Google Scholar]
- Rivadeneira DB, Mayhew CN, Thangavel C, Sotillo E, Reed CA, Grana X, and Knudsen ES (2010). Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 138, 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Nakamura M, Wulf G, Liou YC, and Lu KP (2001). Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nature cell biology 3, 793–801. [DOI] [PubMed] [Google Scholar]
- Sherr CJ (1996). Cancer cell cycles. Science 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- Soderholm JF, Bird SL, Kalab P, Sampathkumar Y, Hasegawa K, Uehara-Bingen M, Weis K, and Heald R (2011). Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS chemical biology 6, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staib F, Hussain SP, Hofseth LJ, Wang XW, and Harris CC (2003). TP53 and liver carcinogenesis. Human mutation 21, 201–216. [DOI] [PubMed] [Google Scholar]
- Sun XX, Dai MS, and Lu H (2008). Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. The Journal of biological chemistry 283, 12387–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Terradillos O, Billet O, Renard CA, Levy R, Molina T, Briand P, and Buendia MA (1997). The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 14, 395–404. [DOI] [PubMed] [Google Scholar]
- Wang W, Liao P, Shen M, Chen T, Chen Y, Li Y, Lin X, Ge X, and Wang P (2015). SCP1 regulates c-Myc stability and functions through dephosphorylating c-Myc Ser62. Oncogene. [DOI] [PubMed] [Google Scholar]
- Wei S, Kozono S, Kats L, Nechama M, Li W, Guarnerio J, Luo M, You MH, Yao Y, Kondo A, et al. (2015). Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nature medicine 21, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. (2011). Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nature chemical biology 7, 285–295. [DOI] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, and Nakayama KI (2004). Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal 23, 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SM, Zhou H, Chen RC, Wang YF, Chen F, Zhang CG, Zhen Y, Yan JH, and Su JH (1998). Sequencing of p53 mutation in established human hepatocellular carcinoma cell line of HHC4 and HHC15 in nude mice. World journal of gastroenterology 4, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ES, and Means AR (2007). PIN1, the cell cycle and cancer. Nature reviews. Cancer 7, 381–388. [DOI] [PubMed] [Google Scholar]
- Yin L, Ghebranious N, Chakraborty S, Sheehan CE, Ilic Z, and Sell S (1998). Control of mouse hepatocyte proliferation and ploidy by p53 and p53ser246 mutation in vivo. Hepatology 27, 73–80. [DOI] [PubMed] [Google Scholar]
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, and Del Sal G (2002). The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419, 853–857. [DOI] [PubMed] [Google Scholar]
- Zarkowska T, and Mittnacht S (1997). Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. The Journal of biological chemistry 272, 12738–12746. [DOI] [PubMed] [Google Scholar]
- Zhou X, Hao Q, Liao P, Luo SW, Zhang MH, Hu GH, Liu HB, Zhang YW, Cao B, Baddoo M., et al. (2016). Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.