

Introduction
Idiopathic pulmonary fibrosis (IPF) is characterized by excessive deposition of collagen in the lung, leading to chronically impaired gas exchange and death1-3. Oxidative stress is believed to be critical in this disease pathogenesis4-6, although exact mechanisms remain enigmatic. Protein S-glutathionylation (PSSG) is a posttranslational modification of proteins that can be reversed by glutaredoxin (GLRX)7. It remains unknown whether GLRX and PSSG play a role in lung fibrosis. Here, we explored the impact of GLRX and PSSG status on the pathogenesis of pulmonary fibrosis, using lung tissues from subjects with IPF, transgenic mouse models and direct administration of recombinant Glrx to airways of mice with existing fibrosis. We demonstrate that GLRX enzymatic activity was strongly decreased in fibrotic lungs, in accordance with increases in PSSG. Mice lacking Glrx were far more susceptible to bleomycin- or adenovirus encoding active transforming growth factor beta-1 (AdTGFB1)-induced pulmonary fibrosis, while conversely, transgenic overexpression of Glrx in the lung epithelium attenuated fibrosis. We furthermore showed that endogenous GLRX was inactivated through an oxidative mechanism and that direct administration of Glrx protein into airways augmented Glrx activity and reversed increases in collagen in mice with TGFB1- or bleomycin-induced fibrosis, even when administered to fibrotic, aged animals. Collectively, these findings suggest the therapeutic potential of exogenous GLRX in treating lung fibrosis.
Text
IPF is associated with excess oxidative stress and depletion of glutathione (GSH) in the lung lining fluid, sputum and serum has been observed5,6,8-10. Depletion of glutathione (GSH) in the lung lining fluid of subjects with IPF was first reported almost 30 years ago8, along with decreases in sputum and plasma GSH9,10. The observations of low GSH levels in individuals with IPF prompted studies to administer the GSH precursor, N-acetyl-L-cysteine, (NAC) in order to augment GSH in lung tissue11. Despite findings that NAC attenuated bleomycin-induced fibrosis in mice12,13, and promising results with NAC in an initial clinical trial14, a subsequent clinical trial failed to demonstrate a therapeutic effect of NAC in subjects with IPF15. One facet of GSH biochemistry that to date has not been considered in the pathogenesis of IPF is protein S-glutathionylation (PSSG), the covalent attachment of glutathione to reactive protein cysteine residues. Given the impact of GSH adduction on the size and charge of target proteins, PSSG is emerging as a regulatory mechanism whereby an oxidant signal is transduced in a (patho-) biological response5,16. Under physiological conditions, glutaredoxin (GLRX) deglutathionylates proteins, re-establishing the thiol group7.
The extent to which GLRX and PSSG are affected in the setting of fibrosis remains unknown. We therefore determined whether GLRX and PSSG are altered in lung tissues from subjects with IPF. GLRX mRNA and protein were slightly but statistically significantly lower in lung tissues from subjects with IPF compared to non-IPF subjects (Fig. 1a,b and Supplementary Tables 1,2). However, immunoprecipitation of GLRX from lung tissues revealed strongly lowered GLRX enzymatic activity in IPF lung tissues, as compared to non-IPF controls, despite an equivalent input of GLRX protein. (Fig. 1c, Supplementary Fig. 1a). Consistent with the lower GLRX activity, PSSG was notably higher in individuals with IPF (Fig. 1d). We also visualized patterns of PSSG in situ17, which also revealed strikingly higher reactivity in lung tissues from subjects with IPF, compared to non-IPF controls, and notably in cells resembling reactive type II epithelial cells (Fig. 1e and Supplementary Fig. 1b). Consistent with its role in de-glutathionylation, an inverse correlation between GLRX activity and PSSG was apparent across all subjects (Fig. 1f). Loss of GLRX and elevations in PSSG highly correlated with a lower diffusing capacity of carbon monoxide (DLCO) and forced vital capacity (FVC) across all subjects (Fig. 1g,h and Supplementary Fig. 1c,d).
Figure 1.
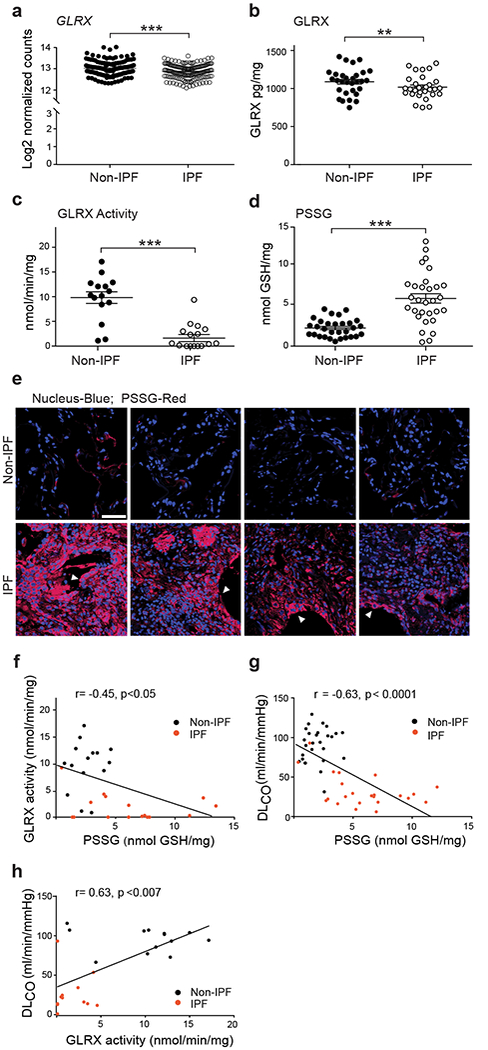
Lower glutaredoxin-1 (GLRX) enzymatic activity and higher protein S-Glutathionylation (PSSG) occur in lung tissues from subjects with IPF and correlate with disease severity. (a) mRNA expression of GLRX in lung tissue from subjects with IPF (n = 160) and without IPF (non-IPF; n = 132). *P < 0.05, Kruskal-Wallis test. Supplementary Table 1 shows subject demographics. (b) GLRX protein in lung tissues from individuals without IPF (Non-IPF; n = 30) and with IPF (n = 29). (c) GLRX enzyme activity following its immunoprecipitation from lung tissues from subjects with IPF (n = 16) and without IPF (non-IPF; n = 15). Results are expressed as nmol/min/mg of input protein. (d) PSSG in lung tissue from cases of IPF (n = 30) and non-IPF (n = 30). (e) In situ PSSG (red) in healthy lungs (n = 4) or lung tissue from subjects with IPF (n = 4). Scale bar, 50μm. White arrowheads, reactive type II pneumocytes. (f-h)Correlations between PSSG and GLRX (f), PSSG and DLCO (g), and GLRX activity and DLCO (h), in lungs of subjects with IPF and non-IPF controls. Supplementary Table 2 shows demographics of subject samples used in panels b-h. *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed Student’s t-test.
One target for PSSG important in pulmonary fibrosis is the death receptor, FAS18-20 through its role in augmenting epithelial apoptosis19. Epithelial cell apoptosis and impaired epithelial cell progenitor function are believed to be critical in fibrogenesis21-23. It is unknown whether FAS-SSG occurs in lungs of individuals with IPF and whether this correlates with disease parameters. Remarkably, in 17 out of 18 subjects evaluated, we detected robust elevations in FAS-SSG, while in healthy lungs we observed little to no detectable FAS-SSG (Supplementary Fig. 1e,f). Elevations in caspase-8 (CASP8) and caspase-3 (CASP3) activities were also apparent in lung tissues from subjects with IPF (Supplementary Fig. 1g,h) consistent with previous findings23 and correlated with FAS-SSG (Supplementary Fig. 1i). FAS-SSG and CASP3 showed a negative correlation with baseline FVC and DLCO in subjects with IPF as compared to controls (Supplementary Fig. 1j-m). Altogether, the lower GLRX activity and accompanying higher levels in PSSG, including FAS-SSG in lung tissues from individuals with IPF, and the observed correlations between these changes with reduced lung function, suggest a role of GLRX and S-glutathionylation in the pathogenesis of lung fibrosis.
We next assessed the impact of global ablation of Glrx on the development of pulmonary fibrosis and showed that bleomycin- or AdTGFB1-induced collagen deposition was greater in mice lacking Glrx, compared to wild-type (WT) littermates, (Fig. 2a,b and Supplementary Fig. 2a,b). As expected, elevations in overall PSSG and Fas-SSG induced by bleomycin or AdTGFB1 were more pronounced in mice lacking Glrx (Fig. 2c,d and Supplementary Fig. 2c), in association with greater activities of Casp3 (Fig. 2e and Supplementary Fig. 2d). The enhanced susceptibility to AdTGFB1-induced Casp3 activation and fibrosis observed in Glrx−/− mice was markedly lowered or absent in mice that also were Fas deficient (Lpr genotype, Supplementary Fig. 2a-d), demonstrating that the augmented susceptibility to lung fibrosis in Glrx−/− mice is due to Fas and is closely linked to Fas-SSG.
Figure 2.
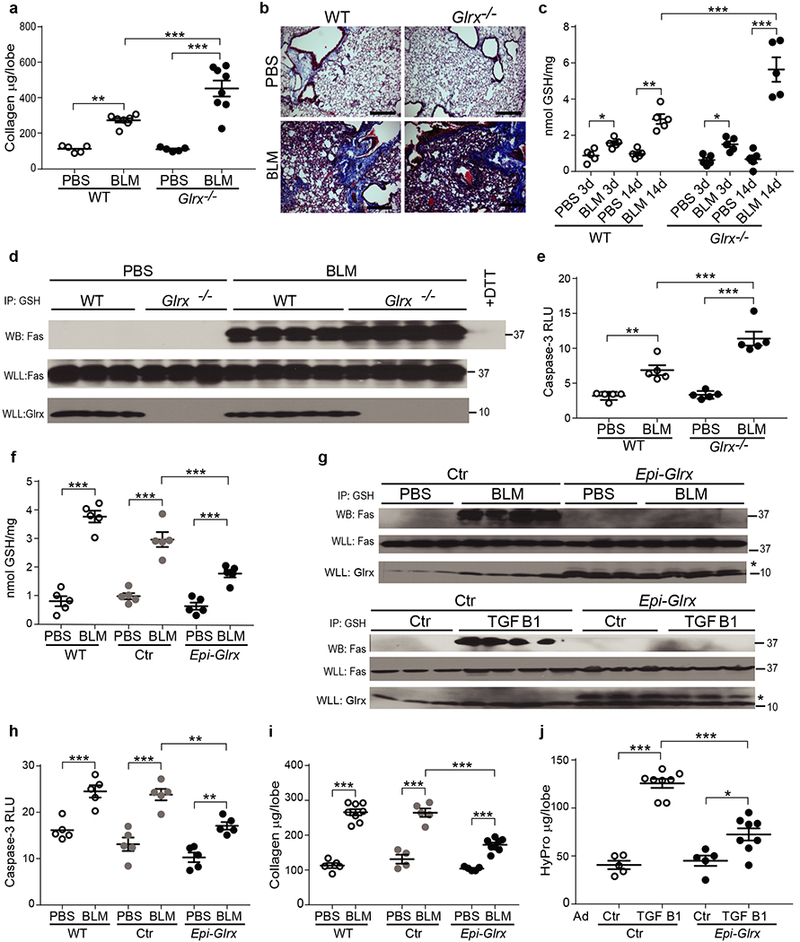
Bleomycin-induced lung fibrosis and protein S-glutathionylation are elevated in Glrx−/− mice, and lowered following transgenic overexpression of Glrx in lung epithelial cells. (a) Collagen in the right superior lobe, 21 days post administration of bleomycin (BLM) WT PBS n = 5, BLM n = 7, Glrx−/− PBS n = 5 & BLM n = 8, pooled from 2 independent experiments. (b) Masson trichrome staining in WT or Glrx−/− mice described in a. Shown are representative images. Scale bar, 50μm. (c) Overall PSSG in lungs from WT or Glrx−/− mice exposed to PBS or bleomycin (n = 5 mice/group). (d) Fas-SSG in lung tissues (WLL) from WT or Glrx−/− mice 21 days post bleomycin. + DTT: negative control, (e) Caspase 3 (Casp3) activity in lungs from WT or Glrx−/− mice 21 days post-bleomycin (n = 5 mice/group) pooled from 2 independent experiments, (f-j) Attenuation of bleomycin- or AdTGFB1-induced fibrosis, PSSG and Casp3 activation in mice overexpressing Glrx in airway epithelia (Epi-Glrx) compared to WT or Ctr groups. Total PSSG (f; n = 5 mice/group), Fas-SSG (g; Top panels), Casp3 activity (h; n = 5/group) or collagen content (I; Sircol, WT: PBS n = 5, BLM n = 8, Ctr PBS n = 4, BLM n = 5; Epi-Glrx n = 5, BLM n = 7 mice/group) in lung tissues. (g) Bottom panels; Assessment of Fas-SSG in Ctr or Epi-Glrx transgenic mice that received AdTGFB1 or AdCtr. Flag-GLRX confirms transgene expression. Representative blots reflect n = 3-4 mice/group. Ctr: CCSP-rtTA single transgenic mice fed dox. * denotes the Flag-Glrx transgene. j: Hydroxyproline in lungs from CCSP-rtTA/TetOP-Glrx bi-transgenic mice following delayed administration of dox. (ctr; n = 5, TGFB1 n = 8 mice/group). Shown are cropped Western blots. *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA, using Tukey’s post-hoc test.
Casp8 and Casp3 mediate degradation of Glrx and promote Fas-SSG and epithelial apoptosis19. Ablation of Casp8 in lung epithelial cells (Supplementary Fig. 3a) led to a robust attenuation of AdTGFB1-induced Casp8 and Casp3 activities in lung tissue (Supplementary Fig. 3b) and strongly attenuated AdTGFB1-induced Fas-SSG, fibrosis and mesenchymal gene expression (Supplementary Fig. 3c-e). Administration of adenovirus encoding Cre recombinase to Casp8loxP/loxP mice prior to administration of bleomycin similarly resulted in lower Casp8 and Casp3 activities, Fas-SSG and fibrosis (Supplementary Fig. 3f-h), in association with elevated mRNA expression of Cdh1 and lower expression of Vim and Acta2 mRNA (encoding E-cadherin, vimentin and alpha-smooth muscle actin, respectively, Supplementary Fig. 3i). Collectively these findings support the contribution of epithelial Casp8 in elevating Fas-SSG, impairing epithelial restitution, and enhancing fibrogenesis. These findings are in line with previous studies documenting that epithelial apoptosis20,21,24 and loss of epithelial progenitor function22 are critical drivers in the pathogenesis of pulmonary fibrosis, and strongly suggest that PSSG amplifies apoptosis.
Enhanced expression of GLRX leads to protein de-glutathionylation, and protects epithelial cells from apoptosis19. To address the impact of elevated expression of Glrx for the development of fibrosis, we created mice expressing TetOP-Glrx, which were crossed with CCSP-rtTA mice, to express Glrx in lung epithelial cells25. Mice expressing Glrx in the lung epithelium exposed to bleomycin showed diminished PSSG (Fig. 2f) and Fas-SSG (Fig. 2g, top panels) in lungs compared to WT littermates or mice expressing only CCSP-rtTA (Ctr). Furthermore, the bleomycin-mediated elevations in Casp3 activity (Fig. 2h) and fibrosis (Fig. 2i) were also lower in Glrx transgenic mice, as compared to the Ctr groups. We next delayed administration of doxycycline-containing food to CCSP-rtTA/TetOP-Glrx transgenic mice until 14 days after administration of AdTGFB1, a time point when elevations in collagen have been reported26. Delayed expression of epithelial Glrx still led to lower Fas-SSG (Fig. 2g, bottom panels) and fibrosis compared to the Ctr group, when evaluated 10 days thereafter (Fig. 2j).
We next determined whether direct administration of recombinant WT Glrx (Fig. 3a and Supplementary Fig. 4a-c) into airways of mice with existing fibrosis affected disease progression. Direct administration of WT Glrx into lungs of control mice resulted in elevations of Glrx in alveolar regions, with higher fluorescence being also observed within bronchial epithelia and cells within airspaces (Fig. 3b and Supplementary Fig.5). Administration of Glrx (6 μg every 3rd day) into airways of mice with existing bleomycin-induced fibrosis restored the attenuated Glrx activity (Fig. 3c) and reversed elevations in PSSG (Fig. 3d). Administration of Glrx reversed the bleomycin-mediated elevations in hydroxyproline over the next 13 days (Fig. 3e) and elevated pyridinoline crosslinks (Fig. 3f), indicative of enhanced degradation of collagen fibers. Glrx led to a reversal of the bleomycin-mediated elevations of hydroxyproline in a dose-dependent manner (Fig. 3g). Histopathological assessment of collagen fibers in parenchymal regions showed that Glrx attenuated the elevations in collagen induced by bleomycin at day 28, although some elevations in collagen and distortions in the tissue were still apparent within this timeframe (Fig. 3h, i). We next administered Glrx into lungs of mice 21 days post administration of AdTGFB1 (Fig. 3j, k), a time point when higher levels of hydroxyproline were detected (Fig. 3k). Administration of WT Glrx for the next 3 weeks reversed the elevated levels of hydroxyproline to control levels (Fig. 3k). In contrast, mutant Glrx lacking the N-terminal cysteine required for de-glutathionylating activity (Glrx Cys23Ser, Supplementary Fig. 4b) did not attenuate fibrosis (Fig. 3k).
Figure 3.
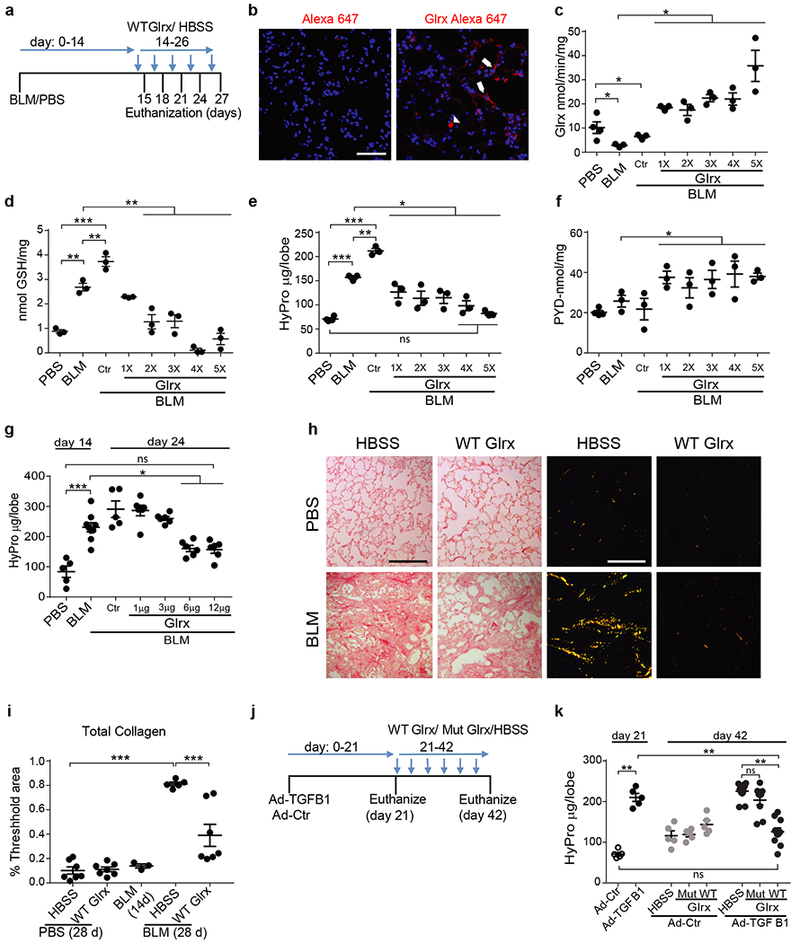
Direct administration of recombinant Glrx to the lung protects against the progression of, and reverses existing pulmonary fibrosis. (a) Schematic depicting experimental design. Glrx 1X-5X; number of administrations of Glrx (Ctr; HBSS vehicle). (b) Detection of Alexa 647 or Alexa labeled-Glrx (red) in lungs from control animals 18 hours post oropharyngeal administration. Optical slice = 0.1μm, Scale bar = 50μm. n = 3 mice/group. Arrows; labeled epithelial cells, Arrowheads; cells resembling macrophages. (c-f) Impact of sequential administrations of Glrx, or HBSS (ctr), on Glrx activity (c), PSSG (d), hydroxyproline (e) in mice exposed to bleomycin. (f) Pyridinoline (PYD) in lungs following administration of HBSS or Glrx. c-f n = 3/4 mice/group/time period. (g) Assessment of dose-dependence of Glrx administered starting on day 14 on bleomycin-induced elevations of hydroxyproline. Mice were euthanized on day 24. PBS n = 5, BLM n = 9, + Ctr n = 5, +1 / 3 / 6 and 12μg n = 6 mice/group. Shown are data pooled from two independent experiments. (h) Representative images showing total lung collagen via picrosirius red staining. Left: bright field images, Right: corresponding polarized light images. Scale bar =100 μm. (i) Quantification of total collagen in h. PBS/HBSS n = 7, PBS/Glrx n = 7, 14 day BLM n = 3, BLM/HBSS n = 5, BLM/Glrx n = 7. (j) Schematic depicting administration of Glrx into lungs of mice with existing AdTGFB1-induced fibrosis, (k) Hydroxyproline in the right superior lung lobe of mice of mice with AdTGFB1-induced fibrosis treated with WT or C23S Glrx. (21 days Ad-Ctr/TGFB1 n = 5, 42 days Ad-Ctr n = 5, 42 days AdTGFB1 HBSS n = 11, Mut Glrx n = 9 and WT Glrx n = 11 mice/group). Shown are data pooled from two independent experiments. * P < 0.05, ** p < 0.01, ***p < 0.001, ANOVA, using Tukey’s post-hoc test, ns: not significant.
Glrx has previously been shown to be inactivated via oxidation of structural cysteines outside of the active site27. In mice with bleomycin-induced fibrosis, or lungs from subjects with IPF, GLRX was S-glutathionylated (Supplementary Fig. 6a,b). S-glutathionylation of GLRX coincided with loss of activity and a higher PSSG content in lung tissue, suggesting that elevations in GLRX-SSG do not reflect catalytic activity, during which Cys23 of GLRX is transiently S-glutathionylated7, but instead point to oxidative inactivation. ELISA assays run under non-reducing conditions, showed slightly less immunoreactive GLRX in lungs from IPF subjects (Fig. 1b) or no change in lungs from mice with bleomycin-induced fibrosis (Supplementary Fig. 7a). However, in the presence of dithiotreitol (DTT), we detected slightly more immunoreactive GLRX in bleomycin-injured mice, or subjects with IPF compared to the respective control groups (Supplementary Fig. 6a,b), suggesting that in settings of fibrosis, GLRX was oxidatively modified. In support of this possibility, the loss of Glrx enzymatic activity in mice with bleomycin-induced fibrosis could be partially reversed by DTT (Supplementary Fig. 6c). Furthermore, direct administration of glutathione disulfide to recombinant Glrx resulted in S-glutathionylation of Glrx and a concomitant loss of enzymatic activity (Supplementary Fig. 6d), demonstrating that S-glutathionylation can result in inactivation of Glrx.
Pulmonary fibrosis manifests in individuals of ages 55 and older, suggesting that IPF is a disease associated with aging28,29. Redox mechanisms have been implicated to play a role in age-related pulmonary fibrosis6. We therefore addressed whether lower Glrx activity and higher PSSG levels were associated with the age-dependent enhanced susceptibility to pulmonary fibrosis. Compared to 3 month-old mice, Glrx activity was lower in lungs from 18 or 24 month-old mice (Fig. 4a), while conversely, PSSG was slightly but not significantly elevated with age in naïve animals (Fig. 4b). Bleomycin-induced fibrosis is enhanced and persistent in aged mice30. The lowered Glrx activity induced by bleomycin in 3 month-old mice was more pronounced in 18 month-old animals (Fig. 4c) and correlated with higher hydroxyproline content in lungs from 18 month-old compared to 3 month-old mice exposed to bleomycin (Fig. 4d). Overall Glrx levels only showed slight changes with age in naïve mice, or following administration of bleomycin (Supplementary Fig. 7a). Similarly, GLRX mRNA was marginally affected with age in healthy human lungs or in subjects with IPF, although a slight inverse correlation was detected between GLRX mRNA and increasing age in subjects with IPF (Supplementary Fig. 7b-c). Overall these findings suggest that the age-associated dysregulation of Glrx predominantly occurs post-translationally. Administration of recombinant Glrx into airways of 18 month-old animals starting 21 days post-administration of bleomycin led to higher Glrx activity in lung tissue, lowered PSSG, and led to lower collagen content evaluated 21 days later (Fig. 4e-g), along with lowered mRNA expression of Vim and Acta2, higher expression of Cdh1 (Fig. 4h) and lower Casp3 activity (Fig. 4i). Cys23Ser mutant Glrx did not affect Glrx activity, nor PSSG in lung tissue (Fig. 4e,f), marginally affected Cdh1, Acta2, and Vim (Fig. 4h), and did not affect Casp3 activity (Fig. 4i). Similar to findings in Fig. 3k, mutant Glrx did not affect hydroxyproline in aged mice with bleomycin-induced fibrosis (Fig. 4g), demonstrating that the catalytic activity of Glrx is required for its ability to attenuate pulmonary fibrosis. Glrx-mediated attenuation of Vim and Acta2 mRNAs along with elevations in Cdh1, suggest restoration of epithelial-mesenchymal balance but does not address how Glrx reverses existing fibrosis. We therefore addressed the collagen degrading activity of lung tissues treated with Glrx. WT Glrx (but not mutant Glrx) led to activation of collagenase activity in aged mice with bleomycin-induced fibrosis (Fig. 4j and Supplementary Fig. 8 a,b). These findings, along with the Glrx-mediated elevation in pyridinoline crosslinks (Fig. 3f) suggest a potential impact of Glrx on promoting degradation of extra cellular matrix (ECM).
Figure 4.
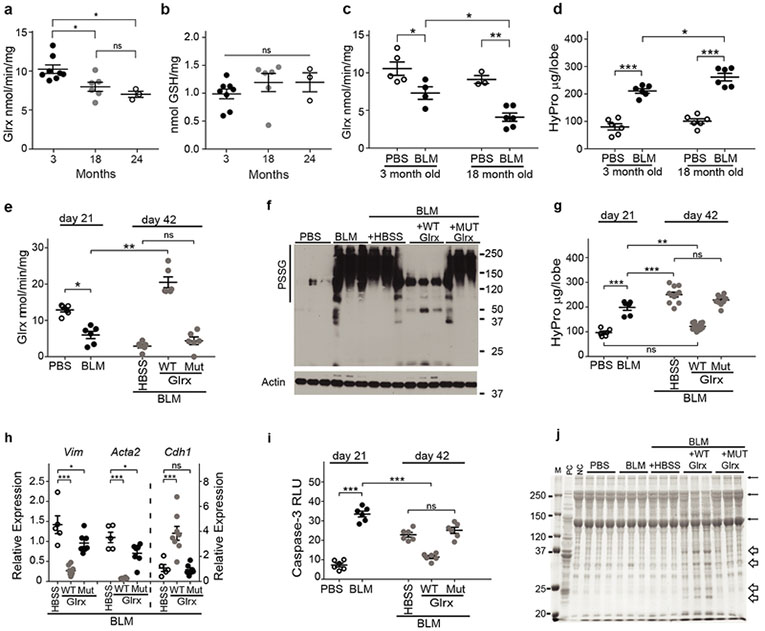
The aging-associated enhanced susceptibility to pulmonary fibrosis induced by bleomycin is associated with low Glrx activity and can be rescued following oropharyngeal administration of Glrx. Glrx activity (a) and PSSG (b) in lung tissues of naïve mice of different ages.(3 month n = 8, 18 month n = 6, 24 month n = 3). Glrx activity (c; 3 month PBS, n = 5, BLM n = 4, 18 month PBS n = 3, BLM n = 6) and hydroxyproline (d; n = 6) in 3 month or 18 month old mice exposed to PBS or bleomycin, (e-j) Impact of Glrx administration into airways of 18 month old mice with existing bleomycin-induced fibrosis. 21 days post-bleomycin, mice received HBSS (Ctr), WT Glrx (12 μg) or C23S Glrx (Mut, 12 μg) oropharyngeally, every 3rd day. Mice were euthanized on day 42.(e) Glrx activity n = 6 mice/group, (f) PSSG, via non-reducing Western blot analysis using an anti-GSH antibody, β-actin: loading control n = 3 mice/group, (g) Hydroxyproline (21 days PBS/BLM n = 6, 42 days HBSS n= 11, WT Glrx n = 14, and Mut Glrx n = 10). Shown is pooled data from 2 independent experiments, (h) Gene expression of Vim, Acta2 and Cdh1 lung tissue (HBSS n = 5, WT n = 8, Mut n = 7 mice/group). Data are expressed relative to mice exposed to PBS at 21 days. (i) Casp3 activity (n = 6 mice/group). RLU: Relative luminescence units. (j) Collagen degrading activity in lung lysates (n = 3 mice/group form a single analysis). Additional information is found in Supplementary Fig. 8a-b. M: marker, PC: positive control using collagen plus recombinant collagenase, and NC: negative control (rat tail collagen). α, β, and γ: the subunits of collagen. Bulky arrows indicate degradation products. *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA, using Tukey’s post-hoc test, ns: not significant.
Pulmonary fibrosis is an unrelenting progressive disease, associated with enhanced epithelial cell death, impaired epithelial progenitor function and consequently failure to restore normal epithelial barriers22,29,31. These processes are believed to stimulate an overpopulation of activated myofibroblasts and excessive deposition of ECM32. Current therapies have limited effectiveness to halt disease progression, which typically results in the death of subjects with IPF within 3-5 years from the time of diagnosis3,33,34. As was mentioned above, changes in glutathione redox homeostasis were first observed in subjects with IPF over 20 years ago8,10, yet a recent clinical trial with NAC showed no clear therapeutic benefit in subjects with IPF15. The present findings differ substantially from the prior observations. Unlike NAC, Glrx cannot be considered a non-specific antioxidant but rather is a member of a larger family of thiol disulfide oxidoreductases that regulates the oxidation state of protein thiols7,35. Alterations in protein thiol redox homeostasis occur in response to specific oxidative events, in a compartmentalized fashion, and affect selective amino acid residues16. Recent studies demonstrated that pharmacologic inhibition of non-phagocytic oxidase-4 (NOX4) reversed pulmonary fibrosis in aged mice30. NOX4 has been shown to produce H2O2 in the endoplasmic reticulum (ER)36 and to promote epithelial cell death during the development of bleomycin-induced fibrosis37. Our previous work showing that Fas-SSG occurred in association with ER-redox perturbations38 point to a potential role of ER-linked oxidative processes in pulmonary fibrosis31. Additionally, dysregulation of mitochondrial homeostasis, and associated increases in ROS, have been implicated in pulmonary fibrosis39,40. PSSG observed herein could be a potential consequence of NOX4- or mitochondrially-derived oxidants, although additional studies will be required to address these possibilities. PSSG has emerged as an oxidant-dependent post-translational modification of reactive protein cysteines that changes structure, function, and serves as a platform for GLRX-dependent restoration5,7. Herein, we demonstrate that the PSSG-GLRX redox axis is markedly altered in subjects with IPF, and in multiple murine models of fibrosis, based upon the observed elevations in PSSG and inactivation of GLRX.
The exact mechanistic details of inactivation of GLRX in settings of fibrosis are unclear. The present study shows that Glrx is oxidatively inactivated in association with S-glutathionylation, based upon the demonstration that the reducing reagent, DTT, partially restores the catalytic activity of Glrx immune-purified from fibrotic tissue, and that direct S-glutathionylation of recombinant Glrx inhibits its enzymatic activity (Supplementary Fig. 6c,d). The tissue-protective effect of exogenously administered Glrx may therefore be linked to the rescue of endogenous Glrx, suggested by rapid enhancement of Glrx activity in lung tissues even after a single administration of Glrx (Fig. 3c). However, additional studies will be required to formally test that scenario. Additional studies will also be required to elucidate the nature of inactivation of GLRX in lungs from subjects with IPF. Similarly the S-glutathionylated proteins that are targets for GLRX-mediated reduction during resolution of fibrosis and the cell types and locations in which they occur, will also require further investigation. The impact of Glrx on attenuating S-glutathionylation of Fas, and associated decreases in epithelial cell death19, along with the Glrx-mediated stimulation of collagen degrading activity suggest that Glrx exerts anti-fibrotic effects through multiple independent mechanisms. The present findings demonstrating that administration of recombinant Glrx into airways reverses existing fibrosis in aged mice, along with the observations that GLRX is inactivated in lungs from subjects with IPF suggest a potential novel treatment modality to halt progression of this devastating disease.
Online Methods:
Animals:
For all experiments, age matched male and female mice (C57BL6/NJ) were used. Bitransgenic mice carrying the rat Club cell secretory protein (CCSP) promoter 5’ to the open reading frame for the reverse tetracycline transactivator (CCSP-rtTA (Line 1, which in adult lung is expressed in bronchiolar and type II epithelial cells)41, plus seven tetracycline operon 5’ to the open reading frame for Cre recombinase (TetOP-Cre) mice were provided by Dr. Whitsett (University of Cincinnati)42. CCSP-rtTA+, TetOP-Cre+ mice were bred with mice carrying the Caspase8loxp allele43. Mice expressing CCSP-rtTA/TetOP-Cre/Casp8loxp/loxp were used to ablate Casp8 from lung epithelial cells, (denoted as ΔEpi-Casp8), by feeding mice doxycycline (Dox) containing chow (6 g/kg, Purina Diet Tech, St. Louis, MO) 3 days before the treatment of pro-fibrotic agents. Mice were maintained on Dox food until the completion of the experiment. Double transgenic littermates containing either containing CCSP-rtTA/TetOP-Cre or CCSP-rtTA/Casp8loxp/loxp transgenes (labeled as Ctr) fed Dox food were used as controls in the experiments. In selected experiments, caspase-8 was deleted from airway epithelial cells following oropharyngeal administration of Adenovirus expressing Cre recombinase (1 × 108 PFU, Vector Core, University of Iowa). Epithelial-specific transgenic mice overexpressing Glrx in lung epithelial cells (denoted as Epi-Glrx) were obtained by breeding CCSP-rtTA expressing mice with mice expressing the TetOP-FLAG Glrx construct25. The single transgenic littermates either containing CCSP-rtTA or TetOP-FLAG Glrx alleles (labeled as Ctr) fed Dox, were used as controls in the experiments. To overexpress Glrx, all mice were maintained on Dox-containing chow starting from 7 days before the instillation of bleomycin. Mice were euthanized 21 days post-administration of bleomycin. In selected experiments involving AdTGFbl, Dox food was withheld until 14 days after administration of AdTGFB1 and thereafter, mice were maintained on Dox food for an additional 10 days until euthanasia. Fas deficient (Lpr) mice44, and Glrx-deficient (Glrx−/−) mice were described previously45 and used to generate Lpr/Glrx−/− double knockouts. Un-manipulated three, eighteen and twenty four month old mice were euthanized, and lungs removed for the assessment of Glrx activity protein and PSSG content. Eighteen month old C57BL/6 mice were obtained from National Institute of Aging (NIA) and subjected to bleomycin-induced fibrosis. All animal use and treatment protocols were reviewed and approved by the Institutional Animal Care Committee (IACUC) of UVM.
Mouse models of fibrosis and administration of Glrx:
Bleomycin (5U/kg body weight, APP Pharmaceuticals, Schaumburg IL) or 5×108 PFU AdTGFB1223/225 (provided by Dr. Jack Gauldie, McMaster University) were administered oropharyngeally, as described previously17,38. PBS or Ad5 adenovirus (AdCtr, Vector Biolabs, Philadelphia, PA), were administered as respective controls. In selected experiments, recombinant Glrx proteins were administered oropharyngeally to mice various times post-administration of AdTGFB1 or bleomycin, as follows: 14 days post administration of bleomycin to 2-4 month old mice, some animals were euthanized to confirm fibrosis. Remaining animals received 6 μg Glrx oropharyngeally up to five times (i.e. Glrx 1X-5X), administered 3 days apart. 3 mice/group were euthanized 16 hours post-administration of Glrx on days 15, 18, 21, 24 or 27. As a control, mice received 5 administrations of HBSS and were euthanized on day 27 (+HBSS 5X). A separate dose-response study was done in which the Glrx administration dose was varied ranging from 1-12 μg of protein administered once every 3rd day starting on day 14 post-administration of bleomycin. Mice were euthanized on day 24.
Alternatively, mice were administered AdTGFB1 or AdCtr, and 21 days thereafter received either HBSS (vehicle), WT Glrx (6 μg) or C23S Glrx (6 μg) once every 3rd day. Mice were euthanized at day 42 for assessment of hydroxyproline. In 18 month old mice that received bleomycin, Glrx or HBSS administration were delayed until 3 weeks post-bleomycin, in order to address the impact of Glrx on existing fibrosis, and mice were euthanized on day 42. A dose of 6 μg of Glrx (with the exception of the dose response study) was administered to 2-4 month old adult mice, while a 12 μg dosing regimen used in 18 month old mice, corresponding to approximate doses of 250 μg/kg body weight.
Human subjects:
De-identified formalin-fixed paraffin-embedded or frozen lung tissue from subjects diagnosed with idiopathic pulmonary fibrosis on the basis of appropriate clinic-radiographic features and usual interstitial pneumonia on histopathologic examination, or normal tissues were obtained from the National Institutes of Health Lung Tissue Research Consortium or from the Department of Pathology and Laboratory Medicine, University of Vermont. Demographics of the study population are provided in Supplementary Table 1. The Institutional Review Board at the University of Vermont certified that the study did not constitute human subjects research.
The Lung Genomics Research Consortium (LGRC) Cohort46 was used to determine expression of the GLRX gene. Expression of the GLRX gene was measured at the probe-level (probe ID: A_23_P69908), from the Agilent-014850 Whole Human Genome Microarray 4×44K G4112F (Agilent, Santa Clara, CA), used in the LGRC cohort as previously described46. Data from individuals with idiopathic pulmonary fibrosis (IPF) and controls were used for this analysis. The gene expression data is available on the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE47460. R software (R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria) was used for GLRX expression analysis. The R stats package version 3.0.1 was implemented. GLRX Gene expression in lung tissue was available for 160 individuals with IPF and 132 controls from the LGRC cohort; demographic and physiologic characteristics are presented in Supplementary Table 2. Differences in gender composition were seen between individuals with IPF and Control (69% vs. 50% males, respectively). Given this gender imbalance, a linear model was used to control for the effect of gender in GLRX gene expression between the two conditions. Gender did not have an effect on GLRX expression in this model. To address whether GLRX expression changes in normal human lung with age, RNAseq data from the Broad Institute’s Genotype-Tissue Expression (GTEx) Project was analyzed (GTEx Analysis V6p; URL: http://www.gtexportal.org/home/datasets; file: GTEx_Analysis_v6p_RNA-seq_RNA-SeQCv1.1.8_gene_rpkm.gct.gz). Applying custom perl scripts (perl v5.18.2), data from 320 normal human lung samples were parsed and assigned corresponding age bins and sex values based on separate sample annotation files (see URL above; file: GTEx_Data_V6_Annotations_SampleAttributesDS.txt; file: GTEx_Data_V6_Annotations_SubjectPhenotypesDS.txt). Gene expression data for GLRX (ENSG00000173221.9), reported as Reads Per Kilobase of transcript per Million mapped reads (RPKM), was extracted for each sample using a second custom perl script, followed by statistical analysis (linear regression analysis or the Kruskal-Wallis test) in Prism v7.0a.
Assessment of Fibrosis:
Lung sections were stained with Masson’s trichrome reagent to stain collagen. Lung sections were imaged using an Olympus BX50 Light Microscope with Qlmaging Retiga 2000R digital camera. Total lung collagen was measured in the right middle lobe of the lung after overnight digestion with 10 mg/ml pepsin in 0.5 M acetic acid using the Sircol Assay (Biocolor) as directed by the manufacturer. Hydroxyproline content was assessed in the right superior lung lobe47. The amount of collagen in tissue sections was quantified using picrosirius red and imaging under polarized light48. Paraffin-embedded whole lung tissues (5 μm sections) were processed and stained in 0.1% Sirius red F3BA (Pfaltz & Bauer, S03695) in saturated picric acid. Monochromatic images were taken using an Olympus SZX12 dissecting microscope, in order to reduce selection bias. Square grids were randomly generated to select 10 parenchymal regions. Grids containing edges, blank spaces, airways, or debris, were disregarded. Images of thus randomly selected regions were acquired using an Olympus BX50 light microscope at ×20 magnification with circularly polarized light (Olympus). Overall collagen content was assessed using MetaMorph software48. The data are expressed as a ratio of the colored pixels representing collagen fibers to the total area, and reflect the average of 10 images/animal.
Western Blot Analysis:
Lung tissues were lysed in buffer containing 20mM Tris∙HCl (pH 7.5), 130mM NaCl, and 1% NP-40 or 50mM Tris∙HCl (pH 7.5), 150mM NaCl, 0.2% SDS, 0.5% Triton ×100 and 0.5% NP-40 with 20mM N-Ethyl Maleimide (NEM). Samples were pelleted via centrifugation, and following protein quantitation of the supernatant, samples were re-suspended in loading buffer with dithiothreitol (DTT), and resolved by SDS-PAGE. Proteins were transferred to PVDF and membranes were probed using a standard immunoblotting protocol using the following primary antibodies: FAS (Millipore, BD biosciences), GLRX (Virogen), and β-Actin (Sigma). Uncropped Western blots are provided in Supplementary Figures 9-13.
Densitometric Analysis:
Densitometric analyses were performed utilizing BioRad® VersaDoc™ Imager Software. Western Blot x-ray films were imaged and rectangles of identical size were traced around each band and assessed for density (intensity/mm2). Values were normalized to corresponding β-Actin bands following background subtraction.
Quantitative RT-PCR for epithelial and mesenchymal markers:
RNA was extracted, DNAse treated using RNEasy columns (Qiagen) and reverse transcribed into cDNA using MMLV RT (Gibco-BRL) according to manufacturer’s instructions. Mesenchymal (Vim and Acta2) and epithelial (Cdh1) gene expression were determined by quantitative RT-PCR using an Applied Biosystem CFX1000 with 25ng of cDNA per reaction and SYBR green for individual target genes. The relative expression was normalized using the expression of Cyclophilin mRNA by delta-delta Ct method. Primer sequences and accession codes are as follows: Vimentin (Vim, gene ID: 12550) Fwd: TGAAGGAAGAGATGGCTCGT, Rev: TCCAGCAGCTTCCTGTAGGT, Alpha-SMA (Acta2, gene ID: 11475) Fwd: CGCTGTCAGGAACCCTGAGA, Rev: CGAAGCCGGCCTTACAGA and E-cadherin (Cdh1, gene ID: 12550) Fwd: ACTGTGAAGGGACGGTCAAC, Rev: GGAGCAGCAGGATCAGAATC.
Production of recombinant GLRX and assessment of GLRX activity:
Mouse Glrx was subcloned into PGEX-2T vector (GE Healthcare). N-terminal Cys was mutated to serine, using site directed mutagenesis (Agilent). Both constructs were verified by sequencing. Both constructs were grown in BL21 Gold competent E. Coli (GE Healthcare). Glrx-GST was isolated using GSH-sepharose affinity purification (Sigma). After removal of the GST tag with thrombin (Sigma), lipopolysaccharide was removed via endotoxin removal columns (Sigma). Purity of the final product was confirmed via Coomassie blue gels. Enzymatic activity of Glrx was determined in reaction mix containing 137 mM Tris-HCI (pH 8.0), 0.5 mM GSH, 1.2 units GSSG reductase (Roche), 0.35 mM NADPH, 1.5 mM EDTA (pH 8.0) and 2.5 mM Cys-SO3. The reaction was allowed to proceed at 30°C and NAPDH consumption was followed spectrophotometrically at 340 nm. Data are expressed as units, where 1 unit equals the oxidation of 1 μM NADPH/min/mg protein49. In experiments designed to address the direct impact of S-glutathionylation on enzymatic activity of Glrx, Glrx was incubated with 50 mM GSSG at 37 °C for 30 min, following removal of excess GSSG using Micro BioSpin 6 Columns (Biorad). The enzymatic activity of reduced or oxidized Glrx was then evaluated in reaction mix containing 137 mM Tris-HCI (pH 8.0 ), 0.5 mM GSH, 1.2 units GSSG reductase (Roche), 0.35 mM NADPH, 1.5 mM EDTA (pH 8.0)and 20 μM di-eosin glutathione disulfide50. The secondary structures of wild-type and C23S Glrx were evaluated via circular dichroism (CD) spectroscopy. Samples were prepared in Dulbecco’s phosphate buffered saline (2.7 mM KCI, 138 mM NaCI, 1.5 mM KH2PO4, 8.06 mM Na2HPO4, pH 7.4). CD spectral data were collected in the 250 to 195 nm range using a Jasco J-815 spectropolarimeter with a scan rate of 20 nm/min, a bandwidth of 1.0 nm, and an integration time of 8 s. The CD data acquired in the 250 to 200 nm range were analyzed by the program Beta Structure Selection (BeStSel) to investigate the secondary structure of each variant. The reported error estimates represent the root-mean-square deviation between secondary structure contributions measured using X-ray crystallography and those estimated from BeStSel analysis of the CD spectra for a reference dataset of 73 proteins51. CD analysis confirms that the secondary structure of Glrx in aqueous solution is essentially unchanged by the C23S substitution, and functional differences between the two variants must arise from a C23S induced change to a different fundamental property of the protein. Recombinant WT Glrx was labeled with AlexaFluor 647 (Invitrogen) and its distribution in lung tissue was determined 18 hours post-oropharyngeal administration. Fresh frozen lung sections were prepared and presence of labeled Glrx was detected via confocal laser scanning microscopy (Zeiss).
In select experiments, lung tissues were homogenized in lysis buffer containing 0.137M Tris-HCI, 0.13M NaCI, and 1% NP-40, and Glrx was immunoprecipitated, using anti-GLRX antibody (Virogen). The activity of immunoprecipitated Glrx was determined as described above. To verify that equal amounts of Glrx were utilized, Western blots were performed on immunoprecipitated samples. In order to address whether Glrx was inactivated via an oxidative mechanism, Glrx was immunoprecipitated from lung tissues, and samples were incubated with 500 μM DTT for 30 min at room temp. Samples were washed in buffer containing 0.137M Tris-HCI, 0.13M NaCI, and 1% NP-40, then washed in buffer containing 0.137 Tris-HCI, 0.5M NaCI, and 0.1875 EDTA, and GLRX activity assessed as described. Results are expressed as nmol/min/mg of input protein. As a control, pre-immune IgG was used instead of GLRX specific antibody, and background values obtained were subtracted.
Caspase assays:
Lung tissues were homogenized, and Caspase-3 and −8 activities were measured using Caspase-GloR assay kits (Promega). Data are expressed as arbitrary units after normalization to protein content.
In situ detection of protein S-glutathionylation:
PSSG was detected in situ in paraffin-embedded lung tissues, following deparaffinization, rehydration, and permeabilization of tissues in the presence of the thiol-blocking reagent NEM. Slides were next incubated with streptavidin to block endogenous biotinylated proteins. Specimens were thereafter subjected to Glrx-catalyzed cysteine derivatization, by incubating tissues in reaction mix containing Glrx, GSSG reductase, GSH, and NADPH for 30 minutes at room temperature17. Slides were washed and incubated with biotin-conjugated NEM followed by reaction with Alexa Fluor-conjugated streptavidin. As a control, replicate slides were incubated with reaction mix lacking Glrx. Any resultant background reactivity was subtracted from labeling observed in the presence of Glrx. Images were acquired using a Zeiss LSM 510 META Confocal Laser Scanning Imaging System. Images were captured at 40× magnification in oil immersion. The image files were converted to Tiff format using Adobe Photoshop CS5. Brightness and contrast were adjusted equally in all images. Fluorescent Images in Fig. 1 were quantified using Image J software. For assessment of PSSG (red) three regions were randomly selected per subject and intensity values were pooled from non-IPF and IPF samples (n=4). These values were normalized to the intensity of DAPI (blue-nuclei).
Detection of overall PSSG, FAS-SSG, and GLRX-SSG:
Overall protein S-glutathionylation in lung tissue was assessed quantitatively following tissue homogenization, precipitation of proteins with 0.6% sulfosalicylic acid, and release of GSH with 1% NaBH4. GSH was quantitated using the 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB)–GSSG reductase recycling method52. To detect S-glutathionylation of FAS or GLRX, lung lysates were prepared (50 mM Tris, pH 7.4, 150 mM NaCI, 0.25% SDS, 1% NP-40, 0.5% CHAPS, and 20 mM N-ethylmaleimide with protease inhibitor cocktail [Sigma-Aldrich]), and protein content was equalized. 2 μg/ml antiglutathione antibody (American Diagnostica) was added to immunoprecipitate S-glutathionylated proteins using protein G agarose beads. Samples were analyzed by immunoblotting using anti-FAS (Millipore) or anti-GLRX (Virogen) antibodies. As a control, a portion of the lysate was treated with 50 mM DTT to reduce glutathionylated proteins, and these samples were purified through columns (Bio-Rad) to remove DTT before subsequent IP.
Assessment of collagen-degrading activity
Lung lysates (2.5 μg) were individually incubated with 50 μg of collagen type I (354236, Corning Inc., NY) in a total reaction volume of 40 μl with Tris-HCl buffer (0.01 M; pH-7.4) at 37 °C for 24 h. The reaction was terminated by adding 20 μl of sample buffer containing 4% SDS, 0.25 M dithiothreitol and 1 M urea, and boiled for 7 min. Samples were loaded onto 7.5% SDS –polyacrylamide gels. Gel was stained with Coomassie Brilliant blue R-250 (0.25%) to visualize the bands. Collagen alone incubated without lung lysates served as negative control and collagen incubated with 2.5 μg of standard purified collagenase (C8176, Sigma, St. Louis, MO) served as positive control for the collagenolytic activity. The quantification of protein expression was performed by densitometry using ImageJ software (imagej.nih.gov/ij/). In addition, lung tissues were hydrolyzed with 6M HCL overnight at 110°C. Lysates were cooled, then neutralized utilizing NaOH. Samples were assayed for pyridinoline (PYD) content, according to manufacturer’s instructions (Quidel, San Diego CA). Results are expressed as nmoles PYD per μg protein.
Image Processing:
Digital images were acquired by scanning X-ray film on a photo scanner (perfection 5000; EPSON). Photoshop (CS5; Adobe) and Illustrator (CS5; Adobe) were used to assemble the figures. Samples are run on the same gel. In some cases, lanes were assembled for consistency as indicated by vertical black line. When required, brightness and contrast were adjusted equally in all lanes.
Statistics:
Data from mouse studies were analyzed by one-way analysis of variance (ANOVA) using Tukey’s test to adjust for multiple comparisons. The two-tailed Student’s t-test when comparisons were made between two experimental groups. Center values of all plots represent mean values and error bars represent standard error of the mean. With the exception of studies involving Caspase-8LoxP mice involving bleomycin and Adeno-Cre (Supplementary Fig. 3f-i), and collagenase activity (Fig. 4j), all experiments were repeated at least once in separate groups of mice. Data from multiple independent experiments were averaged. Total animal numbers in each group are denoted in each figure legend. Random samples from two experiments were selected for biochemical (PSSG, western blots, collagen degradation gels & caspase assays) and mRNA analyses. Human data are presented as mean ± standard deviation or median [inter-quartile range] and were analyzed with unpaired t-tests. GLRX expression was evaluated using the Kruskal-Wallis test. The P value for GLRX represents probe-level statistics unadjusted for multiple comparisons. Correlation plots for GLRX expression and age in IPF and control lungs were generated with the R ggplot2 package, and Spearman correlation coefficients determined. Correlations between lung function and Western Blot densitometry data were performed using Pearson correlation coefficients on combined IPF and non-IPF subject groups. Subject quality of life was assessed by the St. George’s Respiratory Questionnaire (SGRQ)53. The data were analyzed using JMP® Pro 10 (SAS Institute Inc., Cary, NC, USA) and Graphpad Prism (Version 7.04). P values < 0.05 were regarded as statistically significant. No data were excluded from the analyses.
Life Sciences Reporting Summary:
Detailed information on experimental design and reagents can be found in the Life Sciences Reporting Summary accompanying this manuscript.
Data availability:
The gene expression data is available on the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE47460.
Supplementary Material
Acknowledgements:
This work was funded by NIH R35HL135828, R01HL079331, R01 HL060014, and R03 HL095404 (YMWJ-H), an ATS unrestricted grant, and NIH R01HL122383 (VA), Parker B. Francis fellowships (VA and JvdV), NIH R01HL05646 (AvdV), T32 HL076122 (DHM, and JTJ), NCRR 1S10RR019246 (DJT), American Heart Association 16GRNT27660006, NIH R01DK103750, and 1UL1TR001430 (MMB), NIH R01HL133013 and R03 AG051857 (RM), P30GM103532 (CGI), FAMRI Young Clinical Scientist Award 113393 and 1K01HL125474-01 (JGV) and U01HL108642 and RC2 HL101715 (NK). Analysis of biological specimens and data provided by the Lung Tissue Research Consortium (LTRC) was supported by R03 HL095404 (YMWJ-H). We would like to thank J. Whitsett, University of Cincinnati for generously providing the CCSP-rtTA, TetOP-Cre mice, J. Gauldie for providing AdTGFB1, N. Bishop for help with imaging of GLRX and collagen, JM. Siddesha for technical support, and P. Vacek for statistical support.
Footnotes
Conflict of Financial Interest
YMWJ-H and VA hold patents entitled: “Treatments Involving Glutaredoxins and Similar Agents”, “Treatments of oxidative stress conditions” (United States Patent No. 8,679,811 and 9,907,828 YMW J-H and VA), and “Detection of Glutathionylated Proteins” (U.S. Patent No. 8,877,447; YMW J-H)
References:
- 1.du Bois RM Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 9, 129–140 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Barkauskas CE & Noble PW Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am J Physiol Cell Physiol 306, C987–996 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinnula VL & Myllarniemi M Oxidant-antioxidant imbalance as a potential contributor to the progression of human pulmonary fibrosis. Antioxid Redox Signal 10, 727–738 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Janssen-Heininger YM, et al. Regulation of apoptosis through cysteine oxidation: implications for fibrotic lung disease. Ann. N. Y. Acad. Sci. 1203, 23–28 (2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurundkar A & Thannickal VJ Redox mechanisms in age-related lung fibrosis. Redox Biol 9, 67–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA & Shelton MD Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal 10, 1941–1988 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantin AM, Hubbard RC & Crystal RG Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 139, 370–372 (1989). [DOI] [PubMed] [Google Scholar]
- 9.Beeh KM, et al. Glutathione deficiency of the lower respiratory tract in subjects with idiopathic pulmonary fibrosis. Eur. Respir. J. 19, 1119–1123 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Rahman I, et al. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 27, 60–68 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Meyer A, Buhl R & Magnussen H The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 7, 431–436 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Shahzeidi S, Sarnstrand B, Jeffery PK, McAnulty RJ & Laurent GJ Oral N-acetylcysteine reduces bleomycin-induced collagen deposition in the lungs of mice. Eur. Respir. J. 4, 845–852 (1991). [PubMed] [Google Scholar]
- 13.Hagiwara SI, Ishii Y & Kitamura S Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am. J. Respir. Crit. Care Med. 162, 225–231 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Demedts M, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 353, 2229–2242 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr. & Raghu G Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2093–2101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janssen-Heininger YM, et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aesif SW, et al. In situ analysis of protein S-glutathionylation in lung tissue using glutaredoxin-1-catalyzed cysteine derivatization. Am. J. Pathol. 175, 36–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwano K, et al. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Invest. 104, 13–19 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anathy V, et al. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Biol. 184, 241–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagimoto N, et al. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am. J. Respir. Cell Mol. Biol. 17, 272–278 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Sisson TH, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 181, 254–263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang J, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 22, 1285–1293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korfei M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 838–846 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degryse AL, et al. TGFbeta signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol 300, L887–897 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anathy V, et al. Glutaredoxin-1 attenuates S-glutathionylation of the death receptor fas and decreases resolution of Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 189, 463–474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sime PJ, Xing Z, Graham FL, Csaky KG & Gauldie J Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashemy SI, Johansson C, Berndt C, Lillig CH & Holmgren A Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J. Biol. Chem. 282, 14428–14436 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Selman M & Pardo A Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 189, 1161–1172 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Mora AL, Rojas M, Pardo A & Selman M Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov 16, 810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hecker L, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 6, 231ra247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kropski JA & Blackwell TS Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Invest. 128, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thannickal VJ & Horowitz JC Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 3, 350–356 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King TE Jr., et al. A phase 3 trial of pirfenidone in subjects with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Richeldi L, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2071–2082 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Lillig CH, Berndt C & Holmgren A Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Wu RF, Ma Z, Liu Z & Terada LS Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 30, 3553–3568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carnesecchi S, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15, 607–619 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anathy V, et al. Oxidative processing of latent Fas in the endoplasmic reticulum controls the strength of apoptosis. Mol. Cell. Biol. 32, 3464–3478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bueno M, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest. 125, 521–538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mora AL, Bueno M & Rojas M Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Invest. 127, 405–414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Online Methods References:
- 41.Perl AK, Zhang L & Whitsett JA Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am. J. Respir. Cell Mol. Biol. 40, 1–3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perl AK, et al. Conditional recombination reveals distinct subsets of epithelial cells in trachea, bronchi, and alveoli. Am. J. Respir. Cell Mol. Biol. 33, 455–462 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salmena L, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17, 883–895 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA & Nagata S Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356, 314–317 (1992). [DOI] [PubMed] [Google Scholar]
- 45.Ho YS, et al. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 43, 1299–1312 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauer Y, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 52, 217–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woessner JF Jr. The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch. Biochem. Biophys. 93, 440–447 (1961). [DOI] [PubMed] [Google Scholar]
- 48.Fonseca C, Taatjes DJ, Callas P, Ittleman F & Bovill EG The effects of aging on the intimal region of the human saphenous vein: insights from multimodal microscopy and quantitative image analysis. Histochem. Cell Biol. 138, 435–445 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Reynaert NL, Wouters EF & Janssen-Heininger YM Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am. J. Respir. Cell Mol. Biol. 36, 147–151 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coppo L, Montano SJ, Padilla AC & Holmgren A Determination of glutaredoxin enzyme activity and protein S-glutathionylation using fluorescent eosin-glutathione. Anal. Biochem. 499, 24–33 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Micsonai A, et al. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 112, E3095–3103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahman I, Kode A & Biswas SK Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1, 3159–3165 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Jones PW, Quirk FH, Baveystock CM & Littlejohns P A self-complete measure of health status for chronic airflow limitation. The St. George’s Respiratory Questionnaire. Am. Rev. Respir. Dis. 145, 1321–1327 (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression data is available on the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE47460.