

Abstract
Optical assays of synaptic strength would greatly facilitate studies of neuronal transmission and its dysregulation in disease. Here we introduce a genetic toolbox for all-optical interrogation of synaptic electrophysiology (‘synOptopatch’) via mutually exclusive expression of a channelrhodopsin actuator and an archaerhodopsin-derived voltage indicator. Optically induced activity in the channelrhodopsin-expressing neurons generated excitatory and inhibitory post-synaptic potentials which were optically resolved in the reporter-expressing neurons. We further developed a yellow spine-targeted Ca2+ indicator to localize optogenetically triggered synaptic inputs. We demonstrated synOptopatch recordings in cultured rodent neurons and in acute rodent brain slice. In synOptopatch measurements of primary rodent cultures, acute ketamine administration suppressed disynaptic inhibitory feedbacks, mimicking the effect of this drug on network function in both rodents and humans. We localized this action of ketamine to excitatory synapses onto interneurons. These results establish an in vitro all-optical model of disynaptic disinhibition, a synaptic defect hypothesized in schizophrenia-associated psychosis.
Introduction
Changes in synaptic strength underpin learning and memory, are the mechanism by which most neuropeptides1 and neuromodulators2 act, and contribute to the pathogenesis of many disorders. To study synaptic transmission, synaptic plasticity and their impairment in disease, one would like a rapid, reliable and quantitative means to probe the strength of synapses in a neural circuit.
Such tools have remained elusive due to the difficulty of performing simultaneous electrophysiological measurements on defined pairs of neurons. Moreover, whole-cell patch clamp measurements can dialyze cellular components, possibly interfering with plasticity mechanisms.3 It is also technically challenging to maintain patch clamp connections for long enough to probe long term plasticity.
Pairing of optogenetic actuation with fluorescence imaging4 could offer a facile probe of synaptic function. The technical challenges are to ensure that optogenetic stimuli are delivered only to the presynaptic cells, that readouts come only from the postsynaptic cells, and that there is no optical crosstalk between stimulus and readout wavelengths.
In the Optopatch technique,5 cells co-expressed a blue light-activated channelrhodopsin, CheRiff, and a red light-excited voltage indicator, QuasAr. This technique enabled high-speed measurements of neuronal excitability in cultured neurons6, acute brain slice, and peripheral nerves in vivo7. However, due to co-expression of actuator and reporter it was often difficult to distinguish synaptically mediated potentials from spurious optical stimulation of postsynaptic neurites that crossed the stimulus zone8 (Supplementary Fig. 1a).
Here we describe an approach to achieve mutually exclusive expression of optogenetic actuators in presynaptic cells and voltage reporters in postsynaptic cells (synOptopatch). We further developed a novel spine-targeted yellow Ca2+ indicator, spine-jRGECO1a, which we combined with simultaneous synOptopatch measurements to probe the relation between synaptic Ca2+ and membrane voltage during sub- and supra-threshold activity. The synOptopatch technique functioned in primary cultured neurons and in acute brain slice. We developed pharmacological and genetic techniques to specify the sub-type of the presynaptic and postsynaptic cells. The complete set of synOptopatch constructs is described in Supplementary Table 1 and is available on Addgene.
We then used synOptopatch to explore in vitro the mechanism of action of ketamine, a non-specific NMDAR antagonist. In healthy subjects acute sub-anesthetic ketamine induced symptoms that mimicked both positive and negative symptoms of schizophrenia9. In rodents and in healthy humans ketamine induced elevated hippocampal glutamate and cortical hyperexcitability10,11,12,13. In acute brain slices, ketamine decreased excitatory postsynaptic potential (EPSP) amplitudes in pyramidal neurons14, as expected for an NMDAR blocker, but also decreased the amplitude of disynaptic inhibition.15 The mechanism by which a blocker of excitatory neurotransmission enhances network excitability has not been conclusively established.16
We found that ketamine had a dramatic dis-inhibitory effect in cultured neuronal networks. Combinations of Optopatch and synOptopatch measurements established that the dominant action of ketamine was to block excitatory-to-inhibitory synapses. These results demonstrate that synOptopatch can be used to dissect complex synaptically mediated phenomena.
Results
Cre-activated and Cre-inactivated lentiviral constructs for exclusive expression of QuasAr2 and CheRiff
To image membrane voltage we used QuasAr2-Citrine containing a triple repeat of the Kir2.1 membrane trafficking signal17,18. When co-expressed with the channelrhopsin, CheRiff, in cultured rat hippocampal neurons, this GEVI reported optically evoked action potentials with signal-to-noise ratio (SNR) of 39 ± 3 in a 500 Hz bandwidth (n = 12 neurons, all statistics are mean ± s.e.m. unless specified; Supplementary Fig. 1b).
We developed a Cre-recombinase19 based system for mutually exclusive expression of CheRiff-CFP and QuasAr2-Citrine (Fig. 1a). Cre-on CheRiff-CFP comprised a double-floxed inverse ORF (DIO) flanked by parallel double lox (loxP and lox2272) sites20. Cre-off QuasAr2-Citrine was flanked by a lox variant, FAS, which does not show cross-reactivity with loxP or lox2272 sites21 (Fig. 1a; Supplementary Fig. 2). Co-transduction of DIO Cre-on CheRiff-CFP, FAS Cre-off QuasAr2-Citrine and low-titer Cre virus led to mutually exclusive expression of the actuator and reporter (Fig. 1b).
Figure 1. All-optical assay of synaptic function.
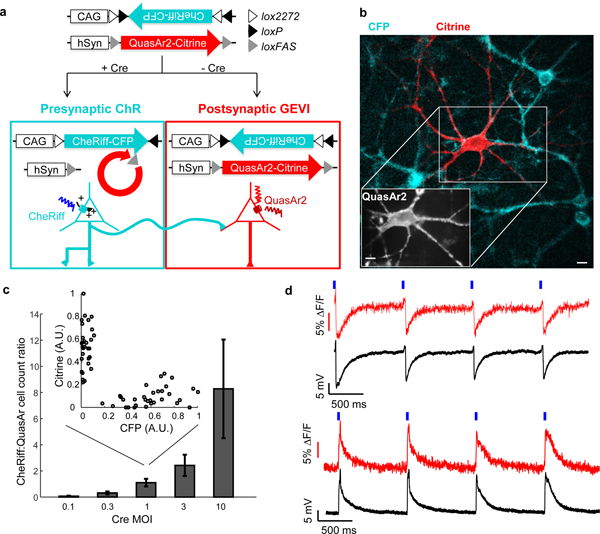
(a) Scheme for Cre recombinase-mediated exclusive expression of Cre-on CheRiff and Cre-off QuasAr2. (b) Green: presynaptic cell expressing CheRiff-CFP. Red: postsynaptic cell expressing QuasAr2-Citrine (Citrine fluorescence; Inset: QuasAr2 fluorescence). Scale bars: 10 μm. (c) Ratio of cells expressing CheRiff to QuasAr2 as a function of MOI of AAV virus encoding Cre. Error bars represent standard deviation calculated by bootstrap method. Inset: At MOI = 1, approximately equal numbers of neurons expressed CheRiff or QuasAr2, but no cells expressed both (n = 63 neurons). (d) Optical initiation and monitoring of primarily inhibitory (top) and primarily excitatory (bottom) postsynaptic potentials. These measurements were performed without synaptic blockers and may contain minor contributions from both excitatory and inhibitory inputs (e.g. slight uptick in top recording). Blue, 10 ms blue light stimulation; red, whole-cell single-trial unfiltered fluorescence; black, patch-clamp recording.
Titration of the Cre virus tuned the ratio of actuator- to reporter-expressing neurons, from 0.07 at a Cre titer of 0.1 MOI to 8.3 at a Cre titer of 10 MOI, allowing control of the relative sizes of the pre- and post-synaptic populations (Fig. 1c). We proceeded with a Cre titer of 1 MOI, corresponding to a 1.1:1 ratio of actuator- to reporter-expressing neurons. Mean expression levels per expressing neuron of the CheRiff-CFP and QuasAr2-Citrine depended on the titers of the corresponding lentiviruses, but did not depend on the titer of the Cre virus, suggesting that a single copy of the Cre virus was likely sufficient to activate all CheRiff and inactivate all QuasAr2 (Supplementary Fig. 2g). In analysis of 324 neurons across a range of titers of each of the three constructs, we did not observe any cells co-expressing CheRiff-CFP and QuasAr2-Citrine, confirming the orthogonality of the gene expression system (Supplementary Fig. 2h).
SynOptopatch enables all-optical measures of synaptic transmission
We delivered wide-field flashes of blue light (488 nm, 20 – 120 mW/cm2, 10 ms, repeated at 1 Hz) to evoke spikes in the CheRiff-expressing neurons. The blue illumination covered a circular area 280 μm in diameter, typically encompassing 2.6 ± 0.3 CheRiff-expressing neurons (n = 10 fields of view), though not every CheRiff-expressing neuron necessarily synapsed onto each postsynaptic cell. We recorded the postsynaptic responses in the QuasAr2-expressing neurons via red excitation (640 nm, 400 W/cm2, 500 Hz frame rate) and near-infrared fluorescence. Simultaneous manual patch clamp measurements provided ground-truth on the postsynaptic potentials in n = 10 neurons (Fig. 1d).
We observed a variety of responses, including purely excitatory, purely inhibitory, and mixed excitatory/inhibitory post-synaptic potentials (PSPs), with close correspondence of the optical and electrical traces (Supplementary Fig. 3a,b) reflecting the linearity and speed (1.2 ms response time) of the QuasAr2 GEVI5. The mean EPSP amplitude was (22 ± 10 mV, n = 6 neurons) and the mean IPSP amplitude was (−6 ± 2 mV, n = 4 neurons). The mean slope of the F vs. V relation was 45 ± 6% ΔF/F per 100 mV, where the error represents the standard error of a fit to n = 10 cells (Supplementary Fig. 3c). Shot noise and camera noise together contributed a mean noise of 0.8% ΔF/F (corresponding to 1.8 mV) in a 500 Hz bandwidth.
To test for optical crosstalk, we measured the effect of blue light on QuasAr2 in cultures not expressing a channelrhodopsin (Supplementary Fig. 3d-f). QuasAr2 did not generate detectable photocurrent under red light (400 W/cm2), blue light (120 mW/cm2), or both. The blue light led to fluorescence transients of ~1.5% ΔF/F, which were readily distinguished from true postsynaptic responses by their rapid rise and fall concurrent with the blue stimulus.
To test the long-term stability of the cultures, we optogenetically evoked and probed PSPs before and after a 2 hr. interval during which the sample sat on the microscope stage. We did not observe a systematic change in the PSP response (n = 14 neurons, Supplementary Fig. 3g). To test for photodamage, we illuminated a sample with continuous red light at 400 W/cm2. The signal photobleached to 50% of initial intensity in 29 min, but we did not observe a change in the PSP waveform during a 30 min exposure. (Supplementary Fig. 3h,i).
The synOptopatch technique enabled repeated measurements on the same cells over several days. Chronic incubation with gabazine (20 μM, 48 hrs) followed by wash-out led to a homeostatic increase in the ratio of inhibitory to excitatory PSPs (IPSP:EPSP amplitude, 0.080 ± 0.03 before gabazine vs 0.23 ± 0.05 after gabazine, n = 9 neurons, p = 0.04, two-sided paired-sample t-test), whereas control plates showed no significant change in PSP over 48 hrs (IPSP:EPSP amplitude, 0.091 ± 0.03 before vs 0.099 ± 0.01 after 48 hrs, n = 5 neurons, p = 0.76, two-sided paired-sample t-test Supplementary Fig. 4).
Resolving presynaptic cell types with pharmacology
Neural circuits involve many types of synapses, and thus we sought to isolate specifically excitatory vs. inhibitory signals. SynOptopatch measurements in n = 403 cultured rat hippocampal neurons revealed pure EPSPs in n = 301 neurons (75%), pure IPSPs in n = 17 neurons (4%), and a mixed excitatory and inhibitory response in n = 85 neurons (21%, Fig. 2a). We hypothesized that the mixed responses were due to co-activation of excitatory and inhibitory pre-synaptic neurons. Indeed we occasionally observed postsynaptic responses with intermittent inhibitory components, indicative of stochastic activation of an inhibitory presynaptic neuron (Fig. 2b). Targeted stimulation of individual presynaptic neurons using patterned blue light (Methods) resolved mixed postsynaptic responses into individual excitatory and inhibitory components, confirming the presence of multiple presynaptic neurons (Fig. 2c).
Figure 2. Pharmacological dissection of synaptic transmission and genetic tagging of inhibitory neurons.
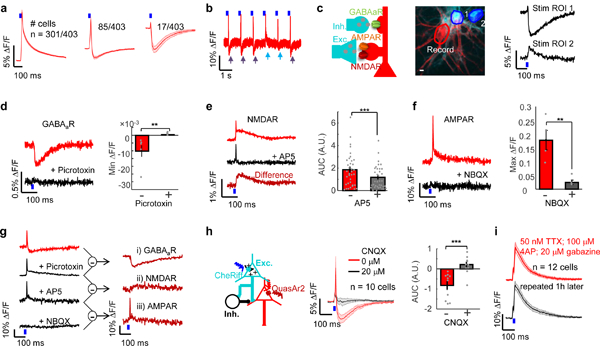
(a) In the absence of drugs, cells showed purely excitatory, mixed excitatory/inhibitory, and purely inhibitory PSPs. (b) Example single-cell single-trial trace showing intermittent optically evoked IPSPs (purple arrows) and IPSP failures (blue arrows). (c) Left: Schematic showing excitatory and inhibitory synaptic inputs into a single cell. Middle: green: CheRiff-CFP, red: QuasAr2-Citrine, blue: DMD masks for patterned blue light stimulation. Right: Stimulation of ROI 1 evoked a pure IPSP and of ROI 2 evoked a pure EPSP. Two-trial average. Scale bar 10 μm. (n = 3 times; representative data are shown) (d) Optogenetically triggered IPSP (red) was blocked by picrotoxin (50 μM; black; five-trial average). Bottom: quantification of minimum of ΔF/F before and after addition of picrotoxin. (−0.99% ± 0.37% vs −0.02% ± 0.02%, n = 6 neurons, **p = 0.04, two-sided paired-sample t-test) (e) NMDAR blocker AP5 (50 μM) blocked slow component of EPSP (red: before; black: after AP5 addition; dark red: difference; five-trial average). Bottom: quantification of maximum of ΔF/F before and after addition of NBQX and AP5. (1.86 ± 0.25 vs 1.19 ± 0.17, n = 36 neurons, ***p = 2×10−6, two-sided paired-sample t-test). (f) AMPAR blocker NBQX (20 μM) blocked EPSPs (red: before; black: after NBQX addition; five-trial average). Bottom: quantification of maximum of ΔF/F before and after addition of NBQX. (17.94% ± 3.58% vs 2.48% ± 0.08%, n = 4 neurons, **p = 0.035, two-sided paired-sample t-test) (g) Pharmacological dissection of a mixed EPSP/IPSP. The cell was recorded following sequential addition of picrotoxin (50 μM), AP5 (50 μM) and NBQX (20 μM). Five-trial average. (n = 3 times; representative data are shown) (h) Left: Schematic showing network inhibition. Middle: In cells with mixed excitatory/inhibitory PSPs, blockade of excitatory transmission with CNQX (20 μM; black) relieved inhibition (red). Right: quantification of AUC before and after addition of CNQX. (−0.80 ± 0.24 vs 0.19 ± 0.17, n = 10 neurons, ***p = 0.001, two-sided paired-sample t-test). (i) 50 nM TTX, 100 μM 4-AP allowed detection of monosynaptic transmission (red). Signals were stable after 1 h under repeated measurements (black; n = 12 neurons). All shaded error bars and error bars, s.e.m.
We sought a pharmacological means to isolate the contributions of distinct presynaptic cell types and postsynaptic receptors to the PSP. Picrotoxin (50 μM) eliminated the inhibitory components of the PSP (Fig 2d), AP5 (50 μM) eliminated the slow NMDAR-dependent part of the EPSP while preserving the fast AMPAR-dependent component (Fig. 2e), and NBQX (20 μM) eliminated both the fast and slow excitatory components (Fig. 2f). In neurons with mixed presynaptic inputs, sequential application of each of these three blockers isolated the respective contributions to the PSP (Fig. 2g).
We ascribed the suppression by NBQX—an AMPAR blocker—of the putative NMDAR-dependent slow component of the EPSP to the voltage-dependent Mg2+ block of NMDARs, i.e. without AMPAR-mediated depolarization, the NMDARs did not activate. Optogenetic induction of presynaptic spikes in a medium containing 0 mM Mg2+, NBQX (20 μM) and picrotoxin (50 μM) restored the slow NMDAR component of the PSP, but not the fast AMPAR component, confirming this hypothesis (Supplementary Fig. 5). While many other blockers could be used to achieve finer segmentation of postsynaptic responses by receptor sub-types, the above examples demonstrate the broad flexibility of synOptopatch for dissecting mechanisms of synaptic transmission.
We next probed whether IPSPs were primarily driven by direct optogenetic stimulation of inhibitory neurons vs. an E ➔ I disynaptic mechanism (Fig. 2h). In cells that showed mixed excitatory/inhibitory PSPs, addition of CNQX (20 μM), an AMPAR blocker, blocked both the excitatory and inhibitory components of the PSP in 8 of 10 neurons tested, establishing that the IPSP was predominantly driven through a disynaptic (or polysynaptic) mechanism (Fig. 2h).
To measure purely monosynaptic PSPs, we adapted a protocol previously developed for optogenetic mapping of synaptic connections in brain slice22. TTX (50 nM) blocked network activity, 4-AP (100 μM), a non-specific potassium channel blocker, enabled the membrane potential to float, and gabazine (20 μM) blocked inhibitory signals. Optogenetic stimulation then evoked large monosynaptic EPSPs which were stable for > 1 hr (n = 12 neurons, Fig. 2i).
Resolving active spines with spine-jRGECO1a
We next sought to use spine-localized Ca2+ imaging to probe the specific synapses activated during optogenetically induced synaptic transmission. The GCaMP6s genetically encoded Ca2+ indicator has previously been used to probe Ca2+ accumulation in individual synaptically activated dendritic spines under high magnification imaging23, but this indicator is not spectrally compatible with one-photon optogenetic stimulation. Red-shifted Ca2+ indicators are spectrally compatible with optogenetic stimulation24, but under wide-field imaging conditions it was difficult to separate spine-specific signals from the much brighter background from the parent dendrite.
To measure Ca2+ in dendritic spines, we developed spine-enriched versions of the jRGECO1a24 and GCaMP6s Ca2+ indicators. Dendritic spines contain high concentrations of filamentous actin, which has been widely used as a spine marker25. The calponin homology (CH) domain of rat Utrophin (Utr, amino acids 1–261), binds to actin filaments26 and at low concentration does not perturb their dynamics. jRGECO1a and GCaMP6s were fused to CH-Utr. Expression was driven by a transcriptional regulatory system that used negative feedback to minimize levels of untargeted probe (Fig. 3a). High-resolution images showed that neurons expressing the complete construct had lower levels of dendritic background than did neurons expressing a construct lacking the transcriptional regulatory system (Supplementary Fig. 6)27. Immunostaining for PSD-95 confirmed spine localization of the reporter (Supplementary Fig. 6). We compared the Ca2+ response of the jRGECO1a-Utr and cytosolic jRGECO1a in HEK cells. Ca2+ transients induced by addition of ionomycin (10 μM) were indistinguishable between the two constructs (ΔF/F 1.6 ± 0.1 vs. 1.5 ± 0.07, n = 26 cells for jRGECO1a-Utr, n = 29 for cytosolic jRGECO1a, p = 0.4, student’s t-test). Supplementary Fig. 6). For both the spine-jRGECO1a and spineGCaMP6s indicators, fluorescence movies of spontaneous activity showed striking flickering patterns throughout the dendritic arbor, indicative of asynchronous activation of individual spines by synaptic inputs, as well as near-synchronous activation of many spines by back-propagating action potentials (Supplementary Videos 1 and 2).
Figure 3. Simultaneous presynaptic optogenetic stimulation, spine Ca2+ imaging and somatic voltage imaging.
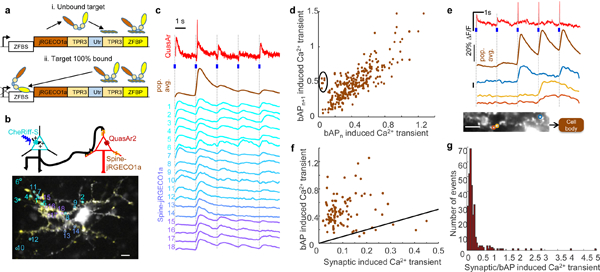
(a) Design of spine-targeted jRGECO1a. (i) TPR3 linkers linked jRGECO1a, Utrophin (Utr), and a zinc finger DNA binding protein (ZFBP). (ii) Upon saturation of actin binding of spine-jRGECO1a, unbound protein accumulated in the nucleus where it bound the zinc finger binding site (ZFBS) downstream of the transcriptional start site and thereby blocked transcription. (b) Top: Schematic showing three color imaging with blue light excitable soma-localized CheRiff in presynaptic cells, yellow light excitable spine-jRGECO1a and red light excitable QuasAr2 co-expressed in postsynaptic cells. Bottom: spine-jRECO1a channel overlaid with active spines colored in yellow. Active spines are circled and numbered with correspondingly colored traces in (c). (c) Blue: 10 ms blue light stimulation of soma-localized CheRiff. Red: QuasAr2 fluorescence. Orange: population average of spine-jRGECO1a fluorescence over all the spines. Cyan-purple: spine-jRGECO1a fluorescence in individual spines. Dashed lines mark stimulus times. (d) Scatter plot of spine Ca2+ transients (ΔF) in pairs of successive bAPs. Transient amplitudes were predominantly correlated between events (R2 = 0.66, n = 100 spines, 3 bAPs) but some spines showed failures on some events (circled). (e) bAP failure in spines. Blue: 10 ms blue light stimulation of soma-localized CheRiff. Red: QuasAr2 fluorescence; orange: population average of spine-jRGECO1a over all the spines. Bottom: three spines on the same dendritic branch showed occasional bAP failures. (f) Scatter plot of bAP induced Ca2+ transients vs. PSP induced Ca2+ transients (R2 = 0.005, n = 100 spines). Black line indicates slope 1, corresponding to equal magnitude. (g) Histogram of the ratio of Ca2+ transient amplitudes driven by bAPs vs. synaptic events. All scale bars 15 μm.
We then sought to combine optogenetic stimulation of presynaptic inputs with simultaneous imaging of dendritic spine Ca2+ and somatic voltage. This measurement required several modifications to the synOptopatch constructs and to the optical setup. We developed a bicistronic construct to drive co-expression of spine-jRGECO1a and QuasAr2-dark Citrine5 (a non-fluorescent Citrine variant, Supplementary Fig. 7a, Methods). To minimize spurious activation of CheRiff by the yellow light used for jRGECO1a imaging, we fused CheRiff with a trafficking motif derived from KV2.1, to localize expression to the soma28 and we used a digital micromirror device (DMD) to pattern the yellow illumination to span the dendritic arbor of the postsynaptic cell while avoiding presynaptic CheRiff-expressing somas (Fig. 3b, Methods). A dichroic beam splitter in the emission path separated the QuasAr2 from the spine-jRGECO1a emission, sending each to a separate camera (Supplementary Fig. 7b, Methods).
Wide-field blue light stimuli (488 nm, 200 mW/cm2, 10 ms duration, repeated at 0.5 Hz) triggered presynaptic spikes, which in turn evoked somatic PSPs (detected with QuasAr2 fluorescence) and synchronous Ca2+ transients in a subset of postsynaptic spines (Fig. 3b, c, Supplementary Fig. 8a,b). On average, we observed 8 ± 3 spines activated per stimulus (mean ± s.d, n = 5 neurons, Supplementary Fig. 8b, Methods). Occasionally, blue light stimulation triggered postsynaptic APs, which led to brief spikes in QuasAr2 fluorescence and concurrent whole-cell Ca2+ transients (Fig. 3c, d, Supplementary Fig. 8a,b).
Simultaneous recording of spine Ca2+ and membrane voltage enabled detailed explorations of single-spine dynamics under both sub- and supra-threshold conditions. As expected, there was a positive correlation between the total synaptic Ca2+ transient and somatic PSP amplitude (Supplementary Fig. 8c, n = 14 events, R2 = 0.42, p = 0.01). Due to the simultaneous activation of multiple spines it was not possible to apportion the PSP voltage to contributions from individual spines.
We next studied how the back-propagating APs (bAPs) modulated spine Ca2+ levels. We observed strong bAP-to-bAP correlation in the bAP-induced Ca2+ amplitude at the level of single spines (Fig. 3d, R2 = 0.66, n = 100 spines). Remarkably, a small portion of spines (4 of 100) showed striking absences of Ca2+ transients for some, bAPs, while responding to others (Fig. 3e). While the mechanism underlying these failures is not known, we can rule out bAP failure in the parent dendritic branches because we observed bAP activation of spines distal to spines with bAP failures (Fig. 3e, Supplementary Fig. 8d).
Finally, we studied the correlation between synaptically induced spine Ca2+ transients and bAP-induced transients. The correlation was much weaker than the bAP-to-bAP correlation, (R2 = 0.005, n = 100 spines) presumably reflecting the different mechanisms driving synaptic vs. bAP spine Ca2+ transients (Fig. 3f). For a small fraction of spines (6 of 100) the synaptic Ca2+ transient exceeded the magnitude of the bAP transient, again possibly reflecting bAP failure in a subset of spines (Fig. 3f, g). These intriguing observations illustrate how the combination of synOptopatch with spine-jRGECO1a provides a flexible platform for explorations of the interaction of voltage and calcium in single-spine dynamics.
SynOptopatch in acute brain slices
We next sought to measure somatic PSPs in acute brain slices. To minimize non-specific GEVI fluorescence from neuropil, we previously developed a somatically localized QuasAr variant with improved trafficking in vivo. This variant, QuasAr2s, reported action potentials in acute brain slices with high signal-to-noise ratio17.
We validated synOptopatch in visual cortex, using an Rbp4-Cre29 driver to target expression of virally delivered Cre-on QuasAr2s-Citrine to a subset of L5 neurons. Cre-off CheRiff-CFP was expressed broadly, but not in the QuasAr2s-expressing neurons (Fig. 4,Methods).
Figure 4. SynOptopatch detected EPSPs and IPSPs in acute mouse brain slice.
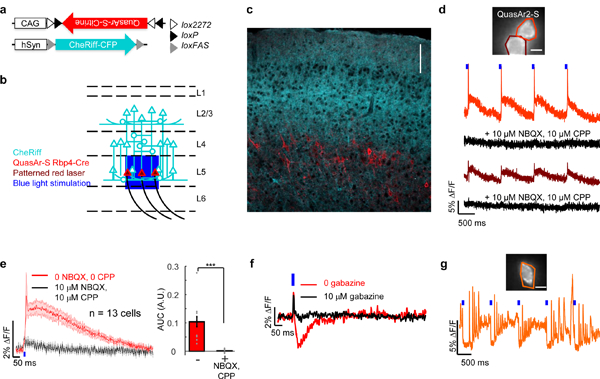
(a), (b) Schematic showing experimental design. Rbp4-Cre mice were injected with hSyn-Cre-off CheRiff-CFP and CAG-Cre-on soma-localized QuasAr (QuasAr-S) leading to QuasAr-S expression in Layer 5 pyramidal neurons and CheRiff expression throughout the cortex. (c) Confocal image of a fixed brain slice. Scale bar 100 μm. (n = 3 times; representative data are shown) (d) Top: Fluorescence of QuasAr-S. Bottom: fluorescence traces of the two circled cells before (red) and after (black) addition of NBQX (10 μM) and CPP (10 μM). (n = 13 cells; representative data are shown) (e) Average PSPs of 13 cells before (red) and after NBQX and CPP (black). Shaded error bars, s.e.m. Right: quantification of AUC before and after addition of NBQX and CPP (0.1 ± 0.017 A.U. vs 0.002 ± 0.002 A.U., n = 13 neurons, ***p = 1×10−4, two-sided paired-sample t-test). (f) IPSP before (red) and after 10 μM gabazine (black). Five-trial average. (n = 1 in 14 cells) (g) Single-trial IPSP detected under elevated (5 mM) extracellular K+ and 10 μM NBQX, 10 μM AP5. Scale bars in (d, g): 10 μm. (n = 8 cells; representative data are shown). All statistics are mean ± s.e.m.
To minimize the background fluorescence in the QuasAr channel, the red illumination was patterned with a DMD to illuminate expressing somas only (Methods)30. Wide-field blue light stimulation (50 mW/cm2, 5 ms, repeated at 1 Hz, 330 μm on a side) induced EPSPs, and sometimes spikes, that were readily detected on a single-trial basis (Fig. 4d). Addition of NBQX (10 μM) and CPP (10 μM), eliminated the fluorescence transients in the QuasAr2-expressing cells, confirming that the signals were due to synaptic transmission (Fig. 4d, e).
Only one in 14 cells showed an IPSP, which was eliminated by gabazine (Fig. 4f). We hypothesized that the rarity of inhibitory signals arose because the reversal potential for Cl-, the main ion transported by GABAa receptors, was close to the resting potential in most cells. In patch clamp measurements, the membrane potential and intracellular chloride concentration are typically set by the patch pipette to reveal inhibition31. Lacking control of these parameters, we instead increased the extracellular K+ concentration from 2.5 to 5 mM, raising the resting voltage by an estimated 14 mV32. Under high K+, NBQX (10 μM) and CPP (10 μM), blue light stimulation induced clear IPSPs in 8 of 10 cells (Fig. 4g, Supplementary Fig. 9). Of these responding cells, some (5 of 8) also showed spontaneous activity, which was transiently suppressed by blue light stimulation. These results demonstrate the feasibility of all-optical assays for inhibitory transmission in acute brain slice.
SynOptopatch dissection of ketamine-induced disinhibition
We used the robust disynaptic inhibition in cultured networks (Fig. 2h) to explore the mechanism of ketamine-induced disinhibition. In cultured rat hippocampal neurons, we measured PSPs before and after applying 50 μM ketamine. For cells with purely excitatory inputs, ketamine suppressed the slow NMDAR component (AUC decreased from 1.08 ± 0.08 to 0.73 ± 0.05 A.U., n = 143 neurons, p = 2×10−6; Fig. 5a, b), as expected for an NMDAR blocker. Strikingly, in cells with mixed excitatory and inhibitory inputs, ketamine largely abolished the IPSP (AUC increased from −6.1 ± 0.8 to 0.7 ± 1.2 A.U., n = 56 neurons, p = 3×10−6; Fig. 5c, d). Thus acute ketamine administration in vitro largely suppressed inhibitory feedbacks, consistent with data from acute brain slices15.
Figure 5. Mechanistic analysis of ketamine-induced disinhibition.
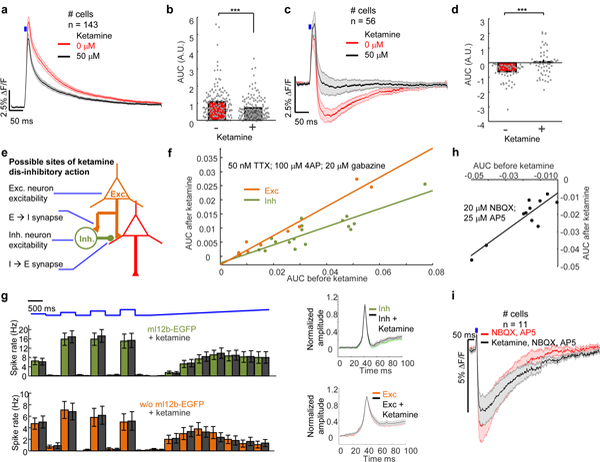
(a) For cells with pure EPSPs (red), ketamine (50 μM, black) decreased the slow component of the EPSP (n = 143 neurons). (b) Quantification of the AUC in (a) (***p = 2×10−6, two-tailed t-test). (c) For cells with mixed EPSP and IPSP, the mean IPSP decreased after ketamine addition (n = 56 neurons). (d) Quantification of AUC in (b). (***p = 3×10−6, two-tailed t-test). (e) Possible sites of ketamine disinhibitory action: glutamatergic synapses onto inhibitory neurons; intrinsic excitability of excitatory or inhibitory neurons; GABAergic synapses. (f) Under conditions of monosynaptic excitatory transmission (50 nM TTX, 100 μM 4-AP, 20 μM gabazine), ketamine (50 μM) induced a larger decrement in the EPSP in inhibitory neurons (green, n = 17 neurons) than in excitatory neurons (orange, n = 10 neurons). ANCOVA analysis comparing the slopes, p = 0.001. (g) Left: In inhibitory neurons (green) the mean firing rate during a series of optogenetic stimuli was 12.9 ± 2.2 Hz before ketamine and 13.4 ± 2.2 Hz after ketamine (n = 27 neurons, p = 0.86). In excitatory neurons (orange) the mean firing rate was 5.1 ± 1.1 Hz before ketamine and 5.1 ± 1.1 Hz after ketamine (n = 22 neurons, p = 0.98). Right: In inhibitory neurons (green) the mean AP width before ketamine was 6.9 ± 0.3 ms, and after ketamine it was 6.8 ± 0.3 ms (n = 27 neurons, p = 0.81, two-sided paired-sample t-test). In excitatory neurons (orange) the mean AP width before ketamine was 9.3 ± 0.5 ms, and after ketamine it was 8.2 ± 0.8 ms (n = 22 neurons, p = 0.30, two-sided paired-sample t-test). (h) Effect of ketamine on inhibitory transmission. IPSPs were probed in the presence of NBQX (20 μM) and AP5 (25 μM). Regression fit of AUC before and after ketamine of inhibitory synaptic transmission (slope = 0.72 ± 0.12, R2 =0.8, n = 11 neurons). (i) Red: IPSP before ketamine. Black: IPSP after ketamine. All shaded error bars and error bars, s.e.m. All statistics are mean ± s.e.m.
We reasoned that ketamine-induced disinhibition could come from: 1) blockade of E-to-I synapses; 2) decreased intrinsic excitability of inhibitory neurons; 3) increased excitability of excitatory neurons; or 4) blockade of I-to-E synapses (Fig. 5e). We conducted optogenetic experiments to test each hypothesis independently.
To determine the identity of the postsynaptic neurons we used an enhancer derived from the mouse Dlx1 and Dlx2 transcription factors, mI12b, to drive expression of eGFP in inhibitory neurons33,34,35. We confirmed via immunostaining, Optopatch, and synOptopatch measurements that the enhancer drove expression in an inhibitory sub-population (Supplementary Fig. 10, Methods).36,37
To study the effect of ketamine on monosynaptic excitatory transmission, we performed measurements in the presence of TTX and 4-AP (to block polysynaptic transmission) and gabazine (to block inhibitory transmission). We used mI12b-eGFP to distinguish postsynaptic responses in excitatory vs. inhibitory neurons and measured each postsynaptic cell before and after ketamine addition. Ketamine blocked E-to-I transmission significantly more strongly than E-to-E transmission, as measured by the slope of the plot of postsynaptic AUC after ketamine vs. before ketamine (inhibitory postsynaptic neurons, slope, 0.32 ± 0.05, n = 10 neurons; excitatory postsynaptic neurons, slope, 0.52 ± 0.04, n = 17 neurons; ANCOVA analysis of inhibitory vs. excitatory postsynaptic neurons, p = 0.001, Fig. 5f). These findings are consistent with the prevailing model that E-to-I transmission is a strong site of ketamine action.
To study the effect of ketamine on intrinsic excitability, we co-expressed CheRiff and QuasAr2-dark Citrine in the same cells (the ‘Optopatch’ configuration), and used mI12b-eGFP to identify neuron sub-types. Under a wide variety of optogenetic stimulus patterns, ketamine did not significantly affect spike rate or action potential waveform in either inhibitory or excitatory neurons (Fig. 5g). Thus ketamine is unlikely to exert its disinhibitory effect by modulating intrinsic excitability of either excitatory or inhibitory neurons.
To investigate the effects of ketamine on GABAergic synapses, we added NBQX and AP5 to block excitatory transmission. Regression fit showed ketamine slightly but significantly increased the AUC of GABAergic synaptic transmission (n = 11 neurons, R2 = 0.8, p = 0.0002) (Fig. 5h), by slowing the kinetics of GABAergic synaptic transmission (Fig. 5i). This effect is consistent with prior observations in acute slice38, but is of the wrong sign and too small to account for the overall dis-inhibitory effect of ketamine.
Together, these results show that the disinhibitory action of ketamine is primarily via blockade of E-to-I transmission. More importantly these results demonstrate a robust in vitro all-optical assay of disynaptic inhibition, a core circuit function that is hypothesized to be dysregulated in schizophrenia-associated psychosis16.
Discussion
The combined voltage and Ca2+ measurements revealed intriguing phenomena which merit further exploration. While the mechanism of bAP failure in dendritic spines is unknown, we speculate that this effect may arise in spines where the neck electrical resistance is large compared to the membrane resistance of the spine head, leading to significant attenuation of the bAP amplitude in the spine, and therefore failure to activate voltage-gated Ca2+ channels.
A specific action of ketamine on excitatory-to-inhibitory synapses has been hypothesized16, but to our knowledge, our experiments are the first to localize the disinhibitory effect to these synapses. Though PV-interneurons express more NR2A and NR2C than do pyramidal neurons39,40, heterologously expressed NMDAR subunits show little difference in ketamine sensitivity between subunits41. Furthermore, the concentration of ketamine used in this study, 50 μM, was far beyond the IC50 reported for heterologously expressed NMDARs (0.5 – 0.7 μM)41. Thus the selectivity for E-to-I synapses likely comes from the fact that NMDARs contribute more to the EPSP in fast-spiking interneurons than in excitatory neurons42. These findings highlight the importance of measuring the functional effects of channel block in the neuronal context and not just on heterologously expressed channels.
We anticipate that the synOptopatch toolkit will be useful across a range of neuroscience applications, though some applications remain challenging. The synOptopatch constructs reported EPSPs and spikes in human induced pluripotent stem cell-derived neurons (Supplementary Fig. 11), though the SNR was lower than in primary neurons due to low expression of the optogenetic constructs, and will likely require further improvements for use in disease modeling applications. Direct measurements of spine voltage in primary or stem cell-derived neurons will require improvements in sensitivity of the voltage indicator.
The acute slice measurements open the door to all-optical circuit mapping. In the present work, the GEVI expression was set by expression of the Cre recombinase, while the CheRiff expression was excluded from the Cre-expressing cells but not otherwise genetically targeted. A Cre-off-Flp-on GEVI combined with Cre-on-CheRiff would allow independent genetic specification of pre- and post-synaptic cell types. We expect that synOptopatch in vivo could open doors to explore synaptic plasticity in learning and memory.
Online Methods
Design of synOptopatch
We tried several strategies for mutually exclusive expression of CheRiff and QuasAr2. We found that electroporation of two populations of neurons with QuasAr2 or CheRiff and mixed co-culture led to insufficient survival of expressing neurons. We thus sought to take advantage of Cre recombinase-controlled gene expression.
Cre-on constructs.
Double lox sites loxP and lox2272 were adopted from the vector pCAG-FLEX-fwd[Chrimson-tdT] (Addgene #59137). The vector was linearized by double digestion with Nhel and Kpnl and purified by the GeneJET gel extraction kit (Fermentas). Inverted CheRiff-CFP cDNA was generated by PCR amplification and then combined with the pCAG-FLEX backbone by Gibson ligation. The FLEX-DIO-CheRiff-CFP construct was then amplified by PCR and cloned into a modified FCK lentivirus backbone (Addgene #51694), FCAG backbone, in which the original CaMKII promoter was replaced by a CAG promoter.
Cre-off constructs.
We tried several strategies to create a Cre-off construct. We first created a Cre-off QuasAr2-Citrine by flanking forward QuasAr2-Citrine sequence with parallel double lox sites loxP and lox2272. Upon lentiviral delivery to cultured rat hippocampal neurons, either with or without co-infection with a Cre-expressing virus, the constructs individually showed the anticipated Cre-dependent expression (Supplementary Fig. 2a-c). We then used lentiviral vectors to co-infect neurons with both constructs, and delivered Cre virus at low multiplicity of infection (MOI) to activate CheRiff and inactivate QuasAr2 in a subset of the neurons. However, we observed many neurons that co-expressed actuator and reporter (Supplementary Fig. 2d,e).
We hypothesized that the co-expression might be due to the presence of loxP and lox2272 sites in both Cre-on and Cre-off constructs, leading to Cre-mediated cross-reactions between the two constructs. We then turned to orthogonal FAS lox sites. FAS lox sites were adopted from the vector pAAV-Ef1a-FAS-ChETA-TdTomato-WPRE-pA (Addgene #37089). The vector was linearized by double digestion with Ascl and Nhel and purified by the GeneJET gel extraction kit (Fermentas). QuasAr2-Citrine cDNA was generated by PCR amplification and combined with the Ef1a-FAS backbone via Gibson ligation. The sequence of FAS lox sites and QuasAr2 and Citrine was then amplified by PCR and cloned into a modified FCK lentivirus backbone (Addgene #51694), FSYN backbone, in which the original CaMKII promoter was replaced by a hSyn promoter.
When QuasAr2-Citrine was co-expressed with either mI12b-EGFP or spine-jRGECO1a, the Citrine fluorescence had spectral overlap with the other fluorescent marker. We found that the presence of the citrine tag was beneficial to trafficking. To keep the structural elements of the Citrine tag while eliminating its fluorescence we mutated the Citrine chromophore from GYG to GGG using site-directed mutagenesis (Agilent) to create a dark Citrine construct.
Cre constructs.
Cre cDNA segments were generated from the template of pCAG-Cre (Addgene #13775) and cloned into FSYN backbone.
Virus production, primary neuronal culture and viral transduction
Experiments in cultured neurons were primarily performed with home-made lentiviral vectors to facilitate rapid testing of many constructs. Experiments in acute brain slices were primarily performed with commercially produced AAV2/9 vectors.
Low titer lentivirus production of synOptopatch.
Plasmids encoding Cre-off QuasAr2-Citrine, Cre-off QuasAr2-dark Citrine and Cre-on CheRiff-CFP were used to produce low titer lentivirus according to published methods43. Briefly, low passage number HEK293T cells (ATCC, #CRL-11268) were plated onto gelatin-coated (Stemcell technologies, #07903) 10 cm dishes. When HEK cells reached 80% confluence, the medium was exchanged to a serum-free DMEM medium. After 0.5–1 hours, cells were transfected using polyethylenimine (PEI; Sigma 408727). 6.22 μg of the vector plasmid, 4 μg of the 2nd generation packaging plasmid psPAX2 (Addgene #12260), and 1.78 μg of viral entry protein VSV-G plasmid pMD2.G (Addgene #12259) were mixed into 540 μL serum-free DMEM, and 16 μL of 1 mg/mL PEI were added in the end. The mixture was incubated at room temperature for 10 min, and added dropwise to the plate. After 4 hours, the medium was exchanged back to 10 mL DMEM10. The supernatant was harvested at 36 hours post transfection, and another 10 mL DMEM10 were added to the cells and incubated for another 24 hours. At 60 hours post transfection, the supernatant was harvested again and combined with the first batch of supernatant, centrifuged 5 min at 500 g, and filtered through a 0.45 μm filter (EMD Millipore, #SE1M003M00). The un-concentrated virus was tested with Lenti-X™ GoStix™ (Clontech, #631243), aliquoted and stored at −80 °C for neuronal transduction.
AAV Cre virus.
High titer AAV2/9 virus with hSyn Cre-GFP at a titer of 5.54×1013 GC/mL was obtained from UPenn Vector Core. High titer AAV2/9 virus with hSyn Cre at a titer of 2.30×1013 GC/mL was obtained from the Gene Transfer Vector Core at Massachusetts Eye and Ear Infirmary & Schepens Eye Research Institute (MEEI), Harvard Medical School.
Primary neuronal culture and viral transduction.
All procedures involving animals were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at Harvard University. Mouse experiments were performed on strain C57BL/6. Rat experiments were performed on strain sprague dawley.
Hippocampal neurons from P0 rat pups were dissected and cultured in NBActiv4 medium (Brainbits) at a density of 40,000 cm−2 on glass-bottom dishes (InVitro Scientific) or pre-treated dishes (Ibidi 81156) pre-coated with poly-d-lysine (Sigma P7205) and matrigel (BD biosciences 356234). At 2–4 hours post plating, AAV or lenti Cre virus at MOI = 1 and Cre-on CheRiff-CFP low-titer lentivirus at MOI = 5 (typically 200 μL) were added into the neurons. At 1 day in vitro (DIV), plating medium with virus was aspirated and glia cells were plated on top of the neurons at a density of 7000 cm-2. By DIV 5, glia grew into a monolayer and 2 μM AraC was added into the neuronal culture medium to inhibit glial growth. At DIV 5–7, 1 mL of the culture medium was removed and saved for later use. Cre-off QuasAr2-Citrine low-titer lentivirus at MOI = 5 (typically 200 μL) was added into the neurons. 24 hours after adding the virus, the medium was replaced with the 1 mL saved medium and 1 mL fresh medium.
The titer of AAV or lenti Cre virus was determined by titration in neurons as shown in Supplementary Figure 2c. The titer of low titer lentivirus was evaluated by Lenti-X GoStix (Clontech).
Imaging and electrophysiology in primary neurons
Imaging apparatus for primary neurons.
Experiments were conducted on a home-built inverted fluorescence microscope equipped with 405 nm, 488 nm, 532 nm, 561 nm, 594 nm, and 640 nm laser lines and a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0). Beams from lasers were combined using dichroic mirrors and sent through an acousto-optic tunable filter (AOTF; Gooch and Housego TF525–250-6–3-GH18A) for temporal modulation of intensity of each wavelength. The beams were then expanded and sent either to a DMD (Vialux, V-7000 UV, #9515) for spatial modulation or sent directly into the microscope (to avoid power losses associated with the DMD). The beams were focused onto the back-focal plane of a 60× water immersion objective, numerical aperture 1.2 (Olympus UIS2 UPlanSApo 60×/1.20 W, for primary neurons). For CFP, Citrine and QuasAr2, fluorescence emission was separated from laser excitation using a quad-band dichroic mirror (Semrock, Di03-R405/488/532/635-t1–25×36). Imaging of fluorescent proteins was performed at illumination intensities of 2–4 W/cm2. Imaging of QuasAr2 direct fluorescence was performed at an illumination intensity of 400 W/cm2. Stimulation of CheRiff was performed at an illumination intensity of 20–120 mW/cm2.
The optimal camera frame rate entails a balance of signal-to-noise ratio (favoring a slower rate when camera electronic readout noise is significant), field of view (favoring a slower rate) and temporal resolution (favoring a faster rate). We found that at room temperature, all action potentials had a full-width and half-maximum > 5 ms, leading to an optimal frame rate of 500 Hz. For measurements at elevated temperatures one may need to image faster.
Imaging of primary culture.
Measurements were performed on primary cultures at DIV 14–21. Experiments were conducted in XC solution containing 125 mM NaCl, 2.5 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 15 mM HEPES, 30 mM glucose (pH 7.3) and adjusted to 305–310 mOsm with sucrose. Experiments were performed at 23 °C under ambient atmosphere.
For resolving presynaptic cell types with pharmacology, synaptic blockers were added to the imaging medium. The blockers were NBQX (20 μM, Tocris, #1044), CNQX (20 μM, Tocris, #0190), D-AP5 (25 μM, Tocris, #0106), gabazine (SR-95531, 20 μM, Tocris), and picrotoxin (50 μM, Tocris, #1128). For probing monosynaptic transmission, TTX (50 nM, Tocris, #1078) and 4-AP (100 μM, Tocris, #0940) were added to the imaging medium.
For probing the effects of ketamine, 50 μM ketamine (Zoetis) was added to the imaging medium.
Simultaneous electrophysiology recording and fluorescence imaging.
Filamented glass micropipettes (WPI) were pulled to a tip resistance of 5–10 MΩ, and filled with internal solution containing 125 mM potassium gluconate, 8 mM NaCl, 0.6 mM MgCl2, 0.1 mM CaCl2, 1 mM EGTA, 10 mM HEPES, 4 mM Mg-ATP, 0.4 mM Na-GTP (pH 7.3); adjusted to 295 mOsm with sucrose. Pipettes were positioned with a Sutter MP285 manipulator. Whole-cell current clamp recordings were acquired using a patch clamp amplifier (A-M Systems, Model 2400), filtered at 5 kHz with the internal filter and digitized with a National Instruments PCIE-6323 acquisition board at 10 kHz.
Simultaneous whole-cell patch clamp and fluorescence recordings were acquired on a home-built, inverted epifluorescence microscope, described above.
Immunostaining of mI12b labeled neurons
For experiments on primary culture, primary cultures were fixed and stained using primary mouse anti Gad67 (Millipore MAB5406) and secondary goat anti mouse 594 (Abcam ab150116) antibodies. The immunostaining followed a protocol described previously7.
Optopatch measurement of mI12b labeled neurons
Neurons were transfected with lentivirus encoding Cre-off CheRiff-CFP, Cre-off QuasAr2-dark Citrine and mI12b-EGFP.
Synaptic blockers (NBQX, D-AP5, and gabazine) were added to block network activity. Cells were stimulated with 500 ms blue light (1 Hz) of increasing intensity (20 to 120 mW/cm2) for four seconds and ramp blue light of increasing intensity (0 to 120 mW/cm2) for another four seconds, while firing patterns were recording under continuous red illumination.
Simultaneous imaging of postsynaptic spine-jRGECO1a and QuasAr2 with optogenetic presynaptic stimulation
Construction of a bicistronic construct with spine-jRGECO1a and QuasAr2.
To avoid spectral overlap of spine-jRGECO1a and the Citrine tag of QuasAr2, we used QuasAr2-dark Citrine. We used the porcine teschovirus-1 (P2A) sequence to co-express spine-jRGECO1a and QuasAr2-dark Citrine.
Gene delivery.
Cre-on soma-localized CheRiff-CFP was lentivirally delivered to presynaptic cells and the sparseness was controlled by the MOI of Cre virus (MOI = 0.5). The bicistronic construct with spine-jRGECO1a and QuasAr2-dark Citrine was delivered to neurons via calcium phosphate, as previously described5. Neurons expressing the reporters and not the actuator were selected for measurement.
Imaging apparatus.
We modified the imaging apparatus for simultaneous spine-jRGECO1a and QuasAr2 imaging. We used a 40× silicone oil UPLSAPO objective, numerical aperture 1.25 (Olympus UIS2 UPLSAPO 40×/1.25). For CFP, spine-jRGECO1a and QuasAr2, fluorescence emission was separated from laser excitation using a quad-band dichroic mirror (Chroma, ZT405/488/561/640rpc). The 488 nm and the 561 nm light were sent through the DMD (Vialux, V-7000 UV, #9515). Stimulation of soma-localized CheRiff was performed at an illumination intensity of 200 mW/cm2. Imaging of spine-jRGECO1a was performed by using the DMD to pattern 561 nm light onto the postsynaptic cell while avoiding presynaptic CheRiff-expressing somata. The 561 nm light was at an intensity of 0.4 W/cm2. Imaging of QuasAr2 direct fluorescence was performed with 640 nm light at an intensity of 400 W/cm2.
A 640 nm dichroic beam splitter (Semrock, FF640-FDi01–25×36) in the emission path separated the QuasAr2 from the spine-jRGECO1a emission, sending each to a separate camera, a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0) for voltage imaging and an EMCCD camera (Andor iXonEM+ DU-897E) for Ca2+ imaging (20 Hz).
Virus production, hiPSC-derived neuron culture and viral transduction
Concentrating synOptopatch lentivirus.
Lenti-X concentrator (Clontech, 631231) was used to concentrate low titer synOptopatch lentivirus. Concentrated lentivirus was then used for hiPSC-derived neuron experiments.
High titer lentivirus encoding Cre.
For experiments in hiPSC-derived neurons, high titer lentivirus with Cre driven by a hSyn promoter were produced by Alstem LLC with a titer of 2.60×109 IFU/mL.
HiPSC-derived neuron culture and viral transduction.
To achieve orthogonal expression of actuator and reporter, we first tried lentiviral delivery of Cre-independent CheRiff and QuasAr2 to separate pools of hiPSCs in the stem cell state, followed by mixing and replating at the progenitor state. However, after 28 days of differentiation we only detected weak expression of both constructs, likely a consequence of gene silencing during differentiation. We then tried the Cre-dependent synOptopatch approach that had worked in primary neurons and achieved robust and non-overlapping expression of reporter and actuator (Supplementary Fig. 10g).
Neuronal differentiation of human stem cells was carried out as previously described44 with the following modification. Human iPSCs with TetO-NGN2-PURO were plated onto plastic dishes. Doxycline (2 μg/mL) was added at 1 day (DIV 1) after plating to induce NGN2 expression. Puro was added at DIV 2 to kill the cells which did not express NGN2. On DIV 4, differentiated neurons were replated at a density of 80,000 cm−2 on preestablished rat glial monolayers grown on 8-well ibidi dishes (ibidi, 80826) in neural basal medium with B27. On DIV 10, lentivirus of Cre at MOI = 1 was added into the medium and incubated for one day. On DIV 14, concentrated lentiviruses of Cre-on CheRiff and Cre-off QuasAr2 at MOI = 5 were added and incubated for one day. 50% medium exchanges were done every 3–4 days. Two days before imaging, 200 nM all trans-retinal was added into the medium.
Imaging in hiPSC-derived neurons
Imaging apparatus for hiPSC-derived neurons.
We used the same imaging apparatus as for primary neurons, but a 40× water immersion objective, numerical aperture 1.2 (Zeiss C-apochromat 40×/1.2 W). Stimulation of CheRiff was performed at a higher illumination intensity of 400 smW/cm2.
Imaging of hiPSC-derived neurons.
For hiPSC-derived neurons, measurements were performed at DIV30 – 42. Experiments were conducted in XC medium at 37 °C controlled by a Tokai Hit stage top incubator (Tokai Hit, WSKM).
For potentiating synaptic transmission of hiPSC-derived neurons, CTZ (50 μM, Tocris, # 0713) was added to the imaging medium.
Virus production, acute brain slices and viral injection
AAV virus preparation.
FAS Cre-off CheRiff-CFP construct was cloned into an AAV vector, AAV2/9-hSyn-WPRE-SVPA, for custom AAV production. Cre-on soma-localized QuasAr2 was cloned into an AAV vector, AAV2/9-CAG-WPRE-SVPA, for custom AAV production.
All custom AAV production was by the Gene Transfer Vector Core at Massachusetts Eye and Ear Infirmary & Schepens Eye Research Institute (MEEI), Harvard Medical School.
Virus injection for acute slices measurement.
AAV2/9 hSyn-Cre-off CheRiff-CFP (5.55×1013 GC/mL) and AAV2/9 CAG-FLEX QuasAr2S-Citrine (2.09×1012 GC/mL) were mixed in a 1:2 volume ratio for virus injection.
Rbp4-Cre+/− mice were crossed with wild-type C57BL/6 mice. Pups were cryo-anesthetized at P0-P2 and immobilized dorsal side up under a stereotaxic microscope. Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting Digital Mouse Stereotaxic Instrument). Pups were injected in the left hemisphere, 0.9 mm lateral and 0.7 mm anterior to lambda. Starting at a depth of 0.6 mm beneath the surface of the skull, virus injections (40 nL, 5 nL/s) were performed at 0.1 mm increments as the pipette was withdrawn. Pups were placed back in their home cage once they were awake.
Genotyping.
Genotyping for Rbp4 was performed with the PCR primer pairs: Cre 5’: 5’ TAT CTC ACG TAC TGA CGG TG 3’ and Cre 3’: 5’ AGA CTA ATC GCC ATC TTC CAG C 3’ to yield a 500 bp band from Cre.
Acute slice preparation.
Acute brain slices were prepared from P16–P28 Rbp4-Cre+/− mice. The mice were anesthetized by isoflurane and then perfused with carbogen (95% O2, 5% CO2)-saturated ice-cold slicing solution with the following composition (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.5 CaCl2, 7 MgCl2, 11.6 Na-ascorbate, and 3.1 Na-pyruvate. Mice were then decapitated and the brains were rapidly coronally sliced with 300 μm thickness on a vibratome (Leica VT 1200S).
Slices were incubated for 45 min at 34 °C in a carbogenated artificial CSF (ACSF) with the following composition (in mM): 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2. The osmolarity of all solutions was adjusted to 300–310 mOsm and the pH was maintained at 7.3 under constant bubbling with carbogen.
Imaging in acute brain slices
Imaging apparatus for acute slices.
Experiments were conducted on a home-built upright fluorescence microscope equipped with 488 nm, and 640 nm laser lines and a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0)30. Briefly, lasers beams were combined using dichroic mirrors, sent through an acousto-optic tunable filter (Gooch and Housego 48058–2.5-.55) for intensity modulation, and then expanded and focused onto the back-focal plane of a 20× water immersion objective, numerical aperture 1.0 (Olympus XLUMPLFLN 20×/1.0 W). Both 488 nm light and 640 nm light could go through an alternative optical path containing a digital micromirror device (DMD, Vialux, V-7000 UV, #9515) for patterned illumination. 640 nm light was patterned to only illuminate the somas of neurons, while 488 nm light was targeted to the whole field at intensity of 50–100 mW/cm2.
For fast data acquisition, a small field of view around the cell of interest was chosen at the center of the camera to achieve a frame rate of 500 frames per second.
Imaging acute slices.
For acute slices, measurements were conducted in ACSF at 23 °C under ambient atmosphere. The slice was immobilized in a Warner Instruments RC-27LD flow chamber using a slice anchor (Warner Instruments, SHD-27LH/2). ACSF, perfused with carbogen, was flowed through the chamber at a rate of 2 mL/minute and recycled through a flow pump (Fisher scientific, 13–876-2).
To confirm that fluorescence transients arose from synaptic transmission, synaptic blockers were added to the imaging medium. The blockers were NBQX (10 μM, Tocris, #1044), (R)-CPP (10 μM, Tocris, #0247), and gabazine (SR-95531, 10 μM, Tocris).
Confocal imaging
Acute slices were fixed and confocal fluorescence imaging was performed on an Olympus FV1000 confocal microscope at the Harvard Center for Brain Sciences microscope facility.
Data analysis
Data were analyzed with homemade code written in MATLAB.
Data analysis for primary culture.
Fluorescence intensities from raw movies were extracted using a maximum likelihood pixel weighting algorithm described in Ref45. Traces showing spontaneous spikes or PSPs were rejected (~5% of the data). The remaining traces were then averaged by aligning with each blue light stimulation.
We used several parameters to classify synaptic inputs. First, we calculated the area under the curve (AUC) of the fluorescence trace corresponding to the PSP. Next we calculated maximum height of the PSP (Amp). Cells that had AUC < 0 and Amp below the noise floor were classified as having purely inhibitory inputs; cells with AUC < 0 and Amp above the noise floor were classified as having mixed inputs; cells with AUC > 0 were classified as having purely excitatory inputs.
Data analysis for simultaneous spine Ca2+ and soma voltage recordings.
Spines were identified by the amplitude of the fluorescence fluctuations at each pixel. A threshold was selected to identify up to 100 spines per neuron. Synaptically activated spines were identified by the following criteria: Ca2+ transients among the top 15% of all spines; above the noise level; and aligned with presynaptic stimulation. Fluorescence traces were corrected for photobleaching.
Data analysis for voltage recordings in hiPSC-derived neurons and acute brain slices.
A region of interest comprising the cell body and adjacent neurites was manually defined, and fluorescence intensities were calculated from the unweighted mean of pixel values within the region of interest. Background fluorescence from a cell-free region was subtracted from the baseline fluorescence of the cell. Traces were then corrected for photobleaching, and averaged by aligning with each blue light stimulation.
Statistics
All error ranges represent standard error of the mean, unless otherwise specified. For the same neurons before and after drug manipulation, paired sample t-test was used. For two-sample comparisons of a single variable, student’s t-test was used. Comparisons of ketamine effects on excitatory monosynaptic transmission for inhibitory vs excitatory postsynaptic neurons were made using a one-way ANOVA analysis. Analysis of ketamine effect on GABAergic synaptic transmission was made by linear regression fit. Probabilities of the null hypothesis p < 0.05 were judged to be statistically significant.
Supplementary Material
Acknowledgements
We thank V. Joshi, K. Williams, M. Lee and S. Begum for technical assistance. We thank V. Parot for help with analysis of spine Ca2+ data, S. Turney and the Harvard Center Brain Science (CBS) for loan of optical equipment, B. Sabatini for Rbp4-Cre mice, and C. Werley and L. Williams for the mI12b plasmid. This work was supported by the Howard Hughes Medical Institute and a grant from the Gordon and Betty Moore Foundation. YA was supported by a long term fellowship from the Human Frontiers Science Program and by a postdoctoral fellowship from the Edmund and Lili Safra center for Brain Sciences. DBA and ESJ were supported by NIH grant NS-089491. KE and RN were supported by the Stanley Center and NIMH grants (5U01MH105669–04 and 1U01MH115727–01).
Competing financial interests
AEC and KE are founders of Q-State Biosciences. LZF and AEC have filed a patent related to synOptopatch.
Footnotes
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
The homemade MATLAB code are available from the corresponding author on reasonable request.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
References
- 1.van den Pol AN Neuropeptide Transmission in Brain Circuits. Neuron 76, 98–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tritsch NX & Sabatini BL Dopaminergic Modulation of Synaptic Transmission in Cortex and Striatum. Neuron 76, 33–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuner T & Augustine GJ A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Emiliani V, Cohen AE, Deisseroth K & Häusser M All-Optical Interrogation of Neural Circuits. J. Neurosci. 35, 13917–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 1–34 (2014). doi: 10.1038/nmeth.3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werley CA et al. All-Optical Electrophysiology for Disease Modeling and Pharmacological Characterization of Neurons. Curr. Protoc. Pharmacol. 11.20.1–11.20.24 (2017). doi: 10.1002/cpph.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou S et al. Genetically Targeted All-Optical Electrophysiology with a Transgenic Cre-Dependent Optopatch Mouse. J. Neurosci. 36, 11059–11073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shemesh OA et al. Temporally precise single-cell-resolution optogenetics. Nat. Neurosci. 20, 1796–1806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krystal Karper LP., Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, DS Charney., J. H. Subanesthetic effects of the NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine effects. Arch Gen Psychiatry 51, 199–214 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Kraguljac N et al. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol. Psychiatry 1–8 (2016). doi: 10.1038/mp.2016.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson ME, Homayoun H & Moghaddam B NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc. Natl. Acad. Sci. U. S. A. 101, 8467–72 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Lazzaro V et al. Ketamine increases human motor cortex excitability to transcranial magnetic stimulation. J. Physiol. 547, 485–96 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamm JP, Peterka DS, Gogos JA & Yuste R Altered Cortical Ensembles in Mouse Models of Schizophrenia. Neuron 94, 153–167.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izumi Y & Zorumski CF Metaplastic effects of subanesthetic ketamine on CA1 hippocampal function. Neuropharmacology 86, 273–281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grunze HCR et al. NMDA-Dependent Modulation of CA1 Local Circuit Inhibition. J. Neurosci. 76, 2034–2043 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisman JE et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 31, 234–242 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adam Y et al. All-optical electrophysiology reveals brain-state dependent changes in hippocampal subthreshold dynamics and excitability. bioRxiv 281618 (2018). doi: 10.1101/281618 [DOI] [Google Scholar]
- 18.Gradinaru V et al. Molecular and Cellular Approaches for Diversifying and Extending Optogenetics. Cell 141, 154–165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branda CS & Dymecki SM Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Developmental Cell 6, 7–28 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Atasoy D, Aponte Y, Su HH & Sternson SM A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 28, 7025–7030 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders A, Johnson CA & Sabatini BL Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Front. Neural Circuits 6, 47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petreanu L, Mao T, Sternson SM & Svoboda K The subcellular organization of neocortical excitatory connections. Nature 457, 1142–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen T-W et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dana H et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer M, Kaech S, Knutti D & Matus A Rapid actin-based plasticity in dendritic spines. Neuron 20, 847–854 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Burkel BM, Von Dassow G & Bement WM Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822–832 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gross G et al. Recombinant Probes for Visualizing Endogenous Synaptic Proteins in Living Neurons. Neuron 78, 971–985 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker CA, Elyada YM, Parra-Martin A & Bolton M Cellular resolution circuit mapping in mouse brain with temporal-focused excitation of soma-targeted channelrhodopsin. Elife 5, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerfen CR, Paletzki R & Heintz N GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adam, Y. et al. All-optical electrophysiology in awake behaving rodents. Submiss. (2017).
- 31.Edwards FA, Konnerth A & Sakmann B Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch‐clamp study. J. Physiol. 430, 213–249 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dingledine R Brain slices. (Springer Science & Business Media, 2013). [Google Scholar]
- 33.Potter GB et al. Generation of Cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol. Cell. Neurosci. 40, 167–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poitras L, Ghanem N, Hatch G & Ekker M The proneural determinant MASH1 regulates forebrain Dlx1/2 expression through the I12b intergenic enhancer. Development 134, 1755–1765 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Colasante G et al. Rapid Conversion of Fibroblasts into Functional Forebrain GABAergic Interneurons by Direct Genetic Reprogramming. Cell Stem Cell 17, 719–734 (2015). [DOI] [PubMed] [Google Scholar]
- 36.McCormick D.a, Connors BW, Lighthall JW & Prince D. a. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J. Neurophysiol. 54, 782–806 (1985). [DOI] [PubMed] [Google Scholar]
- 37.Kwan AC & Dan Y Dissection of cortical microcircuits by single-neuron stimulation in vivo. Curr. Biol. 22, 1459–1467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNally JM, McCarley RW, McKenna JT, Yanagawa Y & Brown RE Complex receptor mediation of acute ketamine application on in vitro gamma oscillations in mouse prefrontal cortex: Modeling gamma band oscillation abnormalities in schizophrenia. Neuroscience 199, 51–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinney JW A Specific Role for NR2A-Containing NMDA Receptors in the Maintenance of Parvalbumin and GAD67 Immunoreactivity in Cultured Interneurons. J. Neurosci. 26, 1604–1615 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xi D, Keeler B, Zhang W, Houle JD & Gao WJ NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: Application of laser microdissection technique. J. Neurosci. Methods 176, 172–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paoletti P & Neyton J NMDA receptor subunits: function and pharmacology. Current Opinion in Pharmacology 7, 39–47 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Jones RSG & Bühl EH Basket-like interneurones in layer II of the entorhinal cortex exhibit a powerful NMDA-mediated synaptic excitation. Neurosci. Lett. 149, 35–39 (1993). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 43.Geraerts M, Micheils M, Baekelandt V, Debyser Z & Gijsbers R Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med. 7, 1299–1310 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Nehme AR et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 23, 2509–2523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D & Cohen AE Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat. Methods 9, 90–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.