

Abstract
Background
A pharmacologic strategy for age‐related muscle weakness is desired to improve mortality and disability in the elderly. Angiotensin‐converting enzyme 2 (ACE2) cleaves angiotensin II into angiotensin 1‐7, a peptide known to protect against acute and chronic skeletal muscle injury in rodents. Since physiological aging induces muscle weakness via mechanisms distinct from other muscle disorders, the role of ACE2‐angiotensin 1‐7 in age‐related muscle weakness remains undetermined. Here, we investigated whether deletion of ACE2 alters the development of muscle weakness by aging and whether angiotensin 1‐7 reverses muscle weakness in older mice.
Methods
After periodic measurement of grip strength and running distance in male ACE2KO and wild‐type mice until 24 months of age, we infused angiotensin 1‐7 or vehicle for 4 weeks, and measured grip strength, and excised tissues. Tissues were also excised from younger (3‐month‐old) and middle‐aged (15‐month‐old) mice. Microarray analysis of RNA was performed using tibialis anterior (TA) muscles from middle‐aged mice, and some genes were further tested using RT‐PCR.
Results
Grip strength of ACE2KO mice was reduced at 6 months and was persistently lower than that of wild‐type mice (p < 0.01 at 6, 12, 18, and 24‐month‐old). Running distance of ACE2KO mice was shorter than that of wild‐type mice only at 24 months of age [371 ± 26 vs. 479 ± 24 (m), p < 0.01]. Angiotensin 1‐7 improved grip strength in both types of older mice, with larger effects observed in ACE2KO mice (% increase, 3.8 ± 1.5 and 13.3 ± 3.1 in wild type and ACE2KO mice, respectively). Older, but not middle‐aged ACE2KO mice had higher oxygen consumption assessed by a metabolic cage than age‐matched wild‐type mice. Angiotensin 1‐7 infusion modestly increased oxygen consumption in older mice. There was no difference in a wheel‐running activity or glucose tolerance between ACE2KO and wild‐type mice and between mice with vehicle and angiotensin 1‐7 infusion. Analysis of TA muscles revealed that p16INK4a, a senescence‐associated gene, and central nuclei of myofibers increased in middle‐aged, but not younger ACE2KO mice. p16INK4a and central nuclei increased in TA muscles of older wild‐type mice, but the differences between ACE2KO and wild‐type mice remained significant (p < 0.01). Angiotensin 1‐7 did not alter the expression of p16INK4a or central nuclei in TA muscles of both types of mice. Muscle ACE2 expression of wild‐type mice was the lowest at middle age (2.6 times lower than younger age, p < 0.05).
Conclusions
Deletion of ACE2 induced the early manifestation of muscle weakness with signatures of muscle senescence. Angiotensin 1‐7 improved muscle function in older mice, supporting future application of the peptide or its analogues in the treatment of muscle weakness in the elderly population.
Keywords: Muscle weakness, ACE2, Angiotensin 1‐7, p16INK4a
Introduction
Over the last few decades, skeletal muscle function has been recognized as a determinant of health condition and lifespan in the elderly population. Muscle weakness is a key component of two important age‐related pathological conditions, sarcopenia and frailty, both of which are closely associated with mortality and disability in the elderly, causing social and economic burden worldwide.1 Research efforts have been devoted to developing a therapeutic strategy for improving muscle weakness in the elderly. While physical training with nutritional support is the practical method for improving muscle weakness, its effect is limited because of impaired physical performance and fatigability of the fragile elderly population.2, 3 Thus, pharmacotherapy for age‐related muscle weakness is a suitable alternative option. However, to date, no drugs have been found to improve muscle weakness in medical practice.
The mechanisms underlying muscle weakness are multifactorial and may involve multiple molecular pathways. Accumulating evidence suggests that the renin‐angiotensin (RA) system, a main regulator of blood pressure, is involved in maintaining skeletal muscle homeostasis, making it a potential therapeutic target for muscle disorders.4 Burks et al. reported that angiotensin II type 1 receptor blockade with the drug losartan improved muscle regeneration after muscle injury in mouse models of Marfan syndrome and Duchenne muscular dystrophy (DMD).5 They also reported the preventive role of losartan against muscle regeneration after injury and immobilization‐induced disuse atrophy in ageing mice.6 Furthermore, another group reported that losartan treatment improved muscle strength and ameliorated fibrosis in a mouse model of congenital muscular dystrophy.7
The activation of the endogenous protective axis in the RA system is also a potential therapeutic target for muscle disorders. Angiotensin‐converting enzyme 2 (ACE2) cleaves angiotensin II to angiotensin 1‐7 (A1‐7), a heptapeptide shown to have a protective role in multiple pathologies by blocking over‐activation of the RA system. Recently, several studies have revealed that A1‐7 attenuates muscle dysfunction in animal model.8, 9, 10, 11, 12, 13 For example, https://www.ncbi.nlm.nih.gov/pubmed/?term=Acu%C3%B1a%20MJ%5BAuthor%5D&cauthor=true&cauthor_uid=24163134 et al. reported that A1‐7 decreases fibrosis and improves muscle function in DMD mice.10 In addition, the genetic deletion or pharmacological inhibition of the A1‐7 receptor Mas resulted in deteriorated muscular architecture and increased fibrosis with diminished muscle strength in DMD mice.10 Therefore, it is currently recognized that pharmacological inhibition of the RA system and activation of the A1‐7‐Mas pathway contribute similarly to the improvement of muscle disorders in rodents. Nevertheless, caution should be taken when applying the current concept of the RA system to the treatment of ageing‐associated muscle weakness, as the development of ageing‐associated muscle weakness is distinct from that of other muscle disorders. For example, repair from acute or chronic injury in most muscle disorders largely depends on the proliferative capacity of skeletal muscle stem cells or satellite cells. However, the role of satellite cells in the development of ageing‐associated muscle weakness remains controversial.14, 15, 16 In addition, the possible improvement of ageing‐associated muscle weakness by A1‐7, believed to protect against over‐activation of the RA system, remains unknown. To address this, we used ACE2 knockout (ACE2KO) and wild‐type mice to investigate (i) whether ACE2 plays a pivotal role in ageing‐associated changes in muscle function and (ii) whether A1‐7 improves muscle weakness in ageing mice.
Materials and methods
Experimental design
Experiments were performed using male ACE2KO mice (C57bl/6J background) and their wild‐type littermates. The mice were fed standard chow and water ad libitum. The study consists of two lines of assays: longitudinal assay for observing muscle function in terms of the temporal change over the lifetime and the effect of A1‐7 in older mice and cross‐sectional assay for single time‐point analysis using younger (3‐month‐old) and middle‐aged (15‐month‐old) mice (Figure 1). For the longitudinal assay, grip strength, running endurance, and body weight were measured at 3, 6, 12, 18, and 24 months of age. Thereafter, A1‐7 (4332‐v; PEPTIDE INSTITUTE, Osaka, Japan) in 0.9% saline or vehicle (0.9% saline) was infused for 4 weeks at a dose of 0.1 μg/kg body weight/min via a subcutaneously implanted osmotic mini‐pump (Alzet model 1004; Durect, Birmingham, AL, USA) according to the previous study.17 Three weeks after infusion, mice were subjected to an intraperitoneal glucose tolerance test (ipGTT), assessment of respiratory quotient using a metabolic cage, and assessment of physical activity using a running wheel. Finally, grip strength was measured, and tissues were extracted for further analysis. For the cross‐sectional assay, the ipGTT and assessment of respiratory quotient and grip strength were performed for experiments using middle‐aged (15‐month‐old) mice. Thereafter, in situ analysis of tibialis anterior (TA) muscles was performed, and tissues were extracted for further analysis. For experiments using young (3‐month‐old) mice, tissues were extracted for further analysis. We measured food intake in 8‐month‐old mice for 7 days consecutively as previously described.18, 19 Briefly, we separated the mice one per cage and gave them pre‐weighted amount of food. Seven days after, we weighed the remaining food and calculated their food intake. All experiments were approved by the Animal Care and Use Committee of Osaka University and were conducted in strict accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Figure 1.

The schematic overview of the experimental protocol. ACE2KO, angiotensin‐converting enzyme 2 knockout; A1‐7, angiotensin 1‐7; ipGTT, intraperitoneal glucose tolerance test; RT‐PCR, real‐time PCR.
Physical activity tests
Physical activity was assessed by measurement of forelimb grip strength and running endurance. Forelimb grip strength was measured using a recently modified method in order to improve validity and reliability.20 In brief, mice grasping the bar were pulled vertically by an inspector, and peak pull force was recorded on a digital force transducer (GPM‐100; Melquest, Toyama, Japan). The gauge recorded tension at the time the mouse released its forepaws from the bar. We performed six consecutive measurements for 2 days, and the average of 12 measurements was used to represent grip strength for each animal. All test sessions were performed during the afternoon hours of the light cycle (11 a.m. to 5 p.m.) in the vivarium where the animals were housed. To assess running endurance, we used a treadmill system (Exer‐3R; Columbus Instruments, Columbus, OH, USA). The procedure was performed as previously described.21 In brief, after acclimation to moderate treadmill running for 5 days, we measured the running time until mice became exhausted and stopped running on the sixth day. The total running distance was calculated by multiplying running time by the speed of the treadmill system.
RNA extraction, gene expression microarrays, and quantitative real‐time PCR‐based gene expression analysis
Total RNA was isolated from 0.1 g of mouse TA muscles with an RNeasy tissue fibrosis mini kit (74704; QIAGEN, Germany). Whole genome transcriptome profiling was assessed using a microarray system (SurePrint G3 Mouse 8 × 60K v2; Agilent, Santa Clara, CA, USA). For real‐time PCR (RT‐PCR), cDNA synthesis was performed with a ReverTra Ace qPCR RT kit (FSQ‐101; TOYOBO, Osaka, Japan) according to the manufacturer's instructions. All genes were evaluated by the SYBR green qPCR system (Applied Biosystems 7900HT Fast Real‐Time PCR System; Thermo Fisher Science, Waltham, MA, USA). Relative expression was calculated using the ΔΔCt method with normalization to constitutive genes, as indicated in each figure.
Evaluation of contractile properties of the tibialis anterior muscle in situ
Tibialis anterior muscle contractile properties were measured as previously described.20 Briefly, each mouse was anaesthetized and placed prone on a heated platform to maintain body core temperature at 37 °C. The sciatic nerve was carefully dissected, tied to a suture, and cut at the proximal end. The distal tendon of the TA muscle was tied at the muscle tendon junction using a 2–0 suture line, and the other end of the suture was tied to the load cell (UTA‐100GR; MinebeaMitsumi, Nagoya, Japan). The contractile properties of the TA muscle were measured using the load cell connected to a direct‐coupled amplifier (SC20AZ‐2; Labo support, Osaka, Japan), and the output was recorded using computer hardware (Power Lab; AD Instruments, Bella Vista, Australia). Optimal muscle length (Lo) and absolute twitch force (Pt) were determined with a resting tension between 10 and 20 g, when twitch force in response to 1 Hz supramaximal stimulation to the sciatic nerve was maximal. Maximal isometric tetanic force (Po) was determined in response to supramaximal stimulation between 50 and 200 Hz at Lo. Muscle fatigue was induced by repeated supramaximal stimulation once per second with 100 ms trains at a frequency of 100 Hz for 5 min. The data were recorded and analysed using Chart software version 5 (AD Instrument). The muscle cross‐sectional area was determined on the basis of a muscle density of 1.06 g/cm3 and a fibre length of the Lo ratio of 0.61.
Intraperitoneal glucose tolerance test
Mice were fasted for 16 h and were anaesthetized with medetomidine (0.3 mg/kg), midazolam (4 mg/kg), and butorphanol (5 mg/kg). Blood was collected from the tip of the tail vein, and glucose levels were monitored with a glucometer at the indicated time points after intraperitoneal injection of glucose (2 g/kg body weight) for ipGTT. ACE2KO and wild‐type mice that received the same treatment sequentially underwent measurements on the same day.
Histochemistry
After excision, TA muscles of mice were immediately frozen in liquid nitrogen‐cooled isopentane, covered in embedding agent (Tissue‐Tek O.C.T. Compound; Sakura Finetek, Torrance, CA, USA), and stored at −80°C until cryosectioning (10 μm). Basic muscle morphology was assessed with haematoxylin and eosin staining, according to standard protocol. All stained, sectioned images were captured under a microscope (Keyence BZ‐X700; Keyence, Osaka, Japan) with a minimum of four fields of view per muscle section at ×20 magnification. All histological images were analysed using ImageJ software (ImageJ version 1.51; NIH, Bethesda, MD, USA) as previously described.22 We counted muscle fibre cross sections with central nuclei and normalized to the number of muscle fibres, as previously reported.23
Assessment of oxygen consumption, carbon dioxide elimination, and physical activity
Oxygen consumption and carbon dioxide elimination were measured using an O2/CO2 metabolism measuring system for small animals (MK‐5000RQ; Muromachi Kikai, Tokyo, Japan). Mice were placed in the calorimeter chambers and stabilized overnight, and we used data from the next light/dark cycle. In these chambers, mice were able to eat and drink ad libitum. The amount of exercise was measured using a wireless running wheel system (ENV‐044&SOF‐860; Med Associates Inc., St Albans City, VT, USA). We placed the running wheel in the ordinary mouse cage and counted the number of rotations of the running wheel.
Statistical analysis
All data are presented as the means ± SEM. Temporal changes of the physical tests in individual mice were analysed with a paired t‐test. Differences between two independent groups were analysed by Student's t‐tests. Differences among wild‐type mice with vehicle and A1‐7 and ACE2KO mice with vehicle and A1‐7 were analysed with two‐way ANOVA and Bonferroni testing to determine the difference between the genotypes and between the treatments. Difference in ACE2 expression among wild‐type mice with different ages was analysed with one‐way ANOVA and Bonferroni testing (Figure 6).
Results
Angiotensin‐converting enzyme 2 knockout mice exhibited more prominent age‐dependent declines in motor function than wild‐type mice
For the longitudinal assay, mice were fed a normal diet with periodic tests of physical strength until 24 months of age (Figure 1). There was no difference in grip strength between wild‐type and ACE2KO mice at 3 months of age (Figure 2A). ACE2KO mice had significantly reduced grip strength at 6 and 12 months of age compared with the previous time point, while wild‐type mice had reduced grip strength at 24 months of age, suggesting that age‐associated declines in muscle function progressed earlier in ACE2KO mice. The grip strength of 6‐month‐old ACE2KO mice was lower than that of wild‐type mice of the same age, and the difference remained significant until 24 months of age. Running distance in the treadmill test gradually decreased with age in both types of mice, and a difference between the groups was observed only at 24 months of age (Figure 2B). Body mass increased less during ageing in ACE2KO mice than in wild‐type mice. ACE2KO mice had lower body mass than wild‐type mice at 12 and 18 months of age, while the difference disappeared at 24 months of age because of reduction in body weight in the wild‐type mice (Figure 2C). During the whole experiments, 2 of the 33 ACE2KO mice and 5 of the 32 wild‐type mice died naturally before 24 months of age.
Figure 2.

Serial changes in (A,B) motor function and (C) body mass during the ageing process in angiotensin‐converting enzyme 2 knockout (ACE2KO) and wild‐type mice. (A) Forearm grip strength was measured six times per day for 2 days, and the data were averaged as individual values. (B) Running distance was measured with the treadmill test. The number of animals was 25 and 24 in wild‐type mice and ACE2KO mice, respectively. Difference from the previous test was analysed with a paired t‐test, and difference between the genotypes was analysed with a Student's t‐test. *p<0.01 vs. age‐matched ACE2KO mice, †p<0.05 vs. the previous test ‡p<0.01 vs. the previous test.
Angiotensin 1‐7 restored age‐associated muscle weakness in mice
As shown in Figure 3A, 4 weeks infusion of A1‐7 but not vehicle increased the grip strength of ACE2KO mice at 24 months of age. A1‐7 tended to increase the grip strength of wild‐type mice, but the difference did not reach statistical significance (P = 0.06). The percentage change of grip strength after A1‐7 infusion was higher than that after vehicle infusion in both ACE2KO and wild‐type mice (Figure 3B). Grip strengths of ACE2KO and wild‐type mice were similar after A1‐7 infusion and were equivalent to that in 6‐month‐old ACE2KO mice and 12‐month‐old wild‐type mice.
Figure 3.

Effect of A1‐7 infusion on grip strength of aged mice. Forearm grip strength was measured before and after infusion of A1‐7 (100 ng/kg/min) and vehicle for 4 weeks in 24‐month‐old mice. (A) Individual value before (pre) and after (post) infusion. Differences were analysed with a paired t‐test. (B) Percentage change after infusion. Differences were analysed with a Student's t‐test. ACE2KO, angiotensin‐converting enzyme 2 knockout; A1‐7, angiotensin 1‐7.
Angiotensin‐converting enzyme 2 deletion and angiotensin 1‐7 did not affect tissue mass, glucose tolerance, and wheel‐running activities
We found that ACE2 deficiency did not alter the mass of the fast‐twitch TA muscle or slow and fast‐twitch gastrocnemius muscle at the age of 3, 15, and 25 months (Figure 4A). We previously reported that ACE2 deficiency exacerbated and A1‐7 infusion improved muscle insulin resistance induced by a high‐fat diet.17 However, ACE2 deficiency did not alter glucose tolerance in middle‐aged and older mice fed a normal diet (Figure 4B). ACE2 deficiency did not alter voluntary wheel‐running activity in older mice (Figure 4C). A1‐7 infusion also did not alter muscle mass, glucose tolerance, and the wheel‐running activity in older mice.
Figure 4.

Tissue weight, metabolic function, and physical activity in aged mice. (A) Wet weight of average tibialis anterior (TA) muscle and average gastrocnemius medialis (GM) muscle adjusted by body weight at the age of 3 months (n = 3–4), 15 months (n = 5), and 25 months (n = 8–9). (B) Intraperitoneal glucose tolerance test (ipGTT) was performed by intraperitoneal injection of glucose (2 g/kg body weight) under anaesthesia after 16 h of fasting at the age of 15 months (n = 6–7) and 25 months (n = 4–7). Left panel indicates serial change in blood glucose level, and right panel represents area under the curve (AUC). (C) Physical activity was assessed with a running wheel system at the age of 25 months. n = 3–6 in each group. (D) Oxygen consumption (left panel) and carbon dioxide elimination (right panel) were assessed with a metabolic cage at the age of 15 months (n = 6–7) and 25 months (n = 7–9). (E) Food consumption during 7 days in 15‐month‐old angiotensin‐converting enzyme 2 knockout (ACE2KO) and wild‐type (WT) mice. Differences between genotypes at the age of 3 and 15 months were analysed with Student's t‐test. Differences among groups at the age of 25 months were analysed with two‐way ANOVA to detect the difference between the genotypes and the difference between the treatments.
Angiotensin‐converting enzyme 2 deletion and angiotensin 1‐7 increased oxygen consumption in older mice
There was no difference in oxygen consumption and carbon dioxide elimination assessed by metabolic cages in middle‐aged ACE2KO and wild‐type mice (Figure 4D). In contrast, older ACE2KO mice had higher oxygen consumption and carbon dioxide elimination in both light and dark cycle than older wild‐type mice (Figure 4D). A1‐7 infusion to older mice increased oxygen consumption significantly in light cycle and non‐significantly (P = 0.07) in dark cycle. A1‐7 infusion did not increase carbon dioxide elimination in both light and dark cycle (Figure 4D). Food consumption measured in 8‐month‐old mice did not differ between the genotypes (Figure 4E).
A senescence‐associated gene, p16INK4a, increased in skeletal muscle of angiotensin‐converting enzyme 2 knockout mice
We performed RNA microarray analysis using TA muscles from middle‐aged mice to clarify alteration of the gene expression profile induced by ACE2 deletion. While middle‐aged ACE2KO mice had lower forelimb grip strength than age‐matched wild‐type mice, the contractile force of the TA muscle did not differ between ACE2 and wild‐type mice (Figure 5A and 5B). Fatigue of the TA muscle during repetitive contractions was more prominent in ACE2KO than wild‐type mice (Figure 5C). RNA microarray analysis revealed that, among the total 42 405 coding and non‐coding RNA analysed, only 14 genes in TA muscles of ACE2KO mice increased or decreased more than two‐fold compared with those of wild‐type mice (P < 0.01; Table 1). Representative genes including those with high false discovery rate (q‐value) were validated by RT‐PCR using different samples from 15‐month‐old mice (Npnt, Pdgfd, MYBPH, p16INK4a, and Ppp1r1a). Consequently, the results confirmed the difference between the genotypes, with the exception of MYBPH (Figure 6). Among the genes, the largest difference was observed in p16INK4a, known as an inducer of cellular senescence by promoting cell cycle arrest. We found that p53 and p21, which are involved in other cellular senescence pathways, were unaltered and decreased in ACE2KO mice compared with wild‐type mice, respectively. We also confirmed that p19, encoded by the same genetic locus as p16INK4a, was not altered by ACE2 deficiency. We did not find differences in expression of the tested genes in TA muscles of young mice at 3 months of age (Figure 6). In the older mice after vehicle infusion, the difference in expression between ACE2KO and wild‐type mice remained significant only for p16INK4a, which was significantly increased compared with middle‐aged mice. A1‐7 infusion did not alter expression of any of the tested genes in the older ACE2KO and wild‐type mice (Figure 6). Finally, ACE2 reduced significantly in 15‐month‐old wild‐type mice compared with 3‐month‐old mice, while the expression non‐significantly reincreased at 25 month. A1‐7 did not alter ACE2 expression in 25‐month‐old wild‐type mice.
Figure 5.
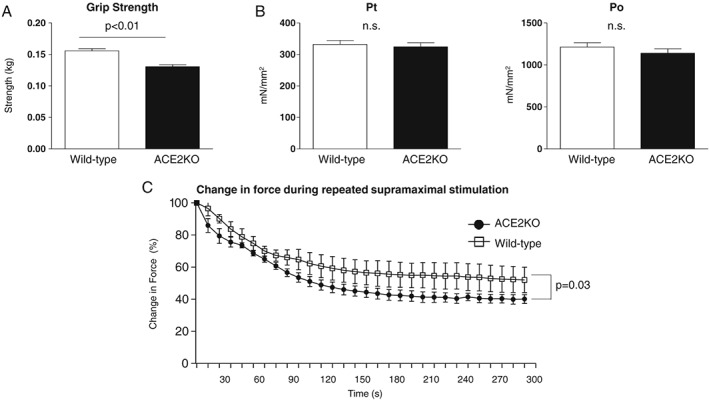
Grip strength and contractile force of tibialis anterior (TA) muscle in middle‐aged mice. (A) Forearm grip strength of 15‐month‐old mice. (B) Pt (absolute twitch force) and Po (maximal isometric tetanic force) in the TA muscles of 15‐month‐old mice. (C) Change in force during fatigue‐inducing conditions in the TA muscles. Values were compared by using Student's t‐test and are expressed as the mean ± SEM. n = 6 in each group.
Table 1.
List of genes with log2 fold change > |1| in expression and P < 0.01 between TA muscles of 15‐month‐old ACE2KO and wild‐type mice in microarray analysis
| Genbank accession | Gene symbol | Wild type | ACE2KO | Fold change (log2) | P‐value | q‐value |
|---|---|---|---|---|---|---|
| NM_033525 | Npnt | 38.3 ± 2.5 | 102.7 ± 3.4 | 1.42 | 0.0001 | 0.043 |
| XM_006509885 | Pdgfd | 152.8 ± 6.6 | 41.8 ± 3.4 | −1.87 | 0.0001 | 0.043 |
| NM_016749 | Mybph | 1305.1 ± 71.7 | 278.4 ± 100 | −2.23 | 0.0011 | 0.206 |
| NM_009877 | Cdkn2a(p16INK4a) | 27.5 ± 5.7 | 369.9 ± 47.1 | 3.75 | 0.0020 | 0.264 |
| NR_028261 | Rian | 425.0 ± 32.6 | 198.1 ± 5.4 | −1.1 | 0.0024 | 0.264 |
| NR_040707 | 1110046J04Rik | 650.9 ± 36.1 | 1349.9 ± 117.2 | 1.05 | 0.0047 | 0.429 |
| NM_021391 | Ppp1r1a | 634.4 ± 62.7 | 272.5 ± 2.52 | −1.22 | 0.0059 | 0.434 |
| NR_045633 | A230056J06Rik | 15.9 ± 3.6 | 41.4 ± 3.5 | 1.38 | 0.0069 | 0.434 |
| NM_001291046 | Nudt3 | 21.7 ± 1.8 | 44.6 ± 4.4 | 1.04 | 0.0085 | 0.434 |
| NM_007492 | Arx | 61.6 ± 10.3 | 127.5 ± 9.0 | 1.05 | 0.0086 | 0.434 |
| NM_173396 | Tgif2 | 148.0 ± 7.8 | 68.1 ± 15.2 | −1.12 | 0.0097 | 0.434 |
| XM_011247076 | Cd226 | 42.5 ± 3.3 | 102.3 ± 12.4 | 1.27 | 0.0098 | 0.434 |
| NM_009014 | Rad51b | 46.5 ± 5.9 | 17.6 ± 1.9 | −1.4 | 0.0098 | 0.434 |
| XR_406760 | Mus musculus predicted gene 5590 (Gm5590) | 44.9 ± 6.7 | 93.1 ± 8.1 | 1.05 | 0.0100 | 0.434 |
ACE2KO, angiotensin‐converting enzyme 2 knockout; TA, tibialis anterior.
n = 3, each group. P‐value and q‐value were calculated using a Student's t‐test and a Benjamini–Hochberg procedure, respectively.
Genes filled with grey were tested by quantitative real‐time PCR analysis.
Figure 6.

Gene expression of tibialis anterior muscle. Gene expression relative to GAPDH was analysed by quantitative real‐time PCR analysis of tibialis anterior muscles in mice at the age of 3 months (n = 5–8) and 15 months (n = 5–6) and at the age of 25 months (n = 6–9). Expression level was relative to that of 15‐month‐old wild‐type mice. MYBPH, p53, and p19 in which no differences were observed between 15‐month‐old ACE2KO and wild‐type mice were not tested in 25‐month‐old mice. For the genes except ACE2, differences between genotypes at the age of 3 and 15 months were analysed with Student's t‐test. Differences among groups at the age of 25 months were analysed with two‐way ANOVA to detect the difference between the genotypes and the difference between the treatments. For ACE2, the difference among wild‐type mice with different ages was analysed with one‐way ANOVA, and the difference between the treatments in 25‐month‐old mice was analysed with Student's t‐test. ACE2KO, angiotensin‐converting enzyme 2 knockout; A1‐7, angiotensin 1‐7; WT, wild‐type.
Central nuclei in tibialis anterior muscle fibres were frequently observed in angiotensin‐converting enzyme 2 knockout mice
Histological analysis of TA muscles revealed no significant differences in the cross‐sectional area of myofibres between middle‐aged ACE2KO and wild‐type mice (Figure 7A). There was also no increase in fibrosis and immune cell infiltration in TA muscles from ACE2KO mice. The only difference found in the histological analysis was an increase in central nuclei in muscle fibres from middle‐aged ACE2KO mice, which were rarely observed in wild‐type mice of the same age (Figure 7B). Central nuclei were not increased in the TA muscle from younger ACE2KO mice (Figure 7B). Central nuclei of myofibres increased in the older wild‐type mice compared with the middle‐aged mice (P < 0.01). The difference between the genotypes remained significant in the older mice (Figure 7B). A1‐7 infusion did not alter the cross‐sectional area or central nuclei in the older ACE2KO and wild‐type mice (Figure 7B).
Figure 7.

Histological analysis of tibialis anterior muscle. (A) Representative pictures and (B) central nuclei per muscle fibre (%) and cross‐sectional area of single muscle fibres (μm2) of haematoxylin–eosin‐stained cross sections of tibialis anterior muscles in mice at the age of 3 and 15 months and at the age of 25 months. Circles indicate the centrally located nuclei. n = 4–6 in each group. Differences between genotypes at the age of 3 and 15 months were analysed with Student's t‐test. Differences among groups at the age of 25 months were analysed with two‐way ANOVA to detect the difference between the genotypes and the difference between the treatments. * P < 0.01 vs. 15‐month‐old wild‐type mice by a Student's t‐test. ACE2KO, angiotensin‐converting enzyme 2 knockout; A1‐7, angiotensin 1‐7; WT, wild‐type.
Discussion
In this study, we found that ACE2 deficiency resulted in the early manifestation of muscle weakness in mice. Comprehensive analysis of the TA muscle in middle‐aged (15‐month‐old) mice revealed that the senescence‐associated gene p16INK4a and central nuclei of muscle fibres were increased in ACE2KO mice. We found that a continuous A1‐7 infusion improved muscle strength in older ACE2KO and wild‐type mice without any alternation in the senescent phenotypes of skeletal muscle. To the best of our knowledge, this is the first study to show that ACE2 may influence the physiological process of skeletal muscle ageing and that A1‐7 may restore ageing‐associated muscle weakness.
Glucose intolerance and insulin resistance are closely related to ageing‐associated muscle weakness.24 We previously found that ACE2 deficiency exaggerated glucose tolerance and insulin resistance in mice fed a high‐calorie diet.17 However, in the present study, we did not find any differences in glucose tolerance between ACE2KO and wild‐type mice fed a normal diet until old age. A1‐7 infusion in older mice also did not alter glucose tolerance, suggesting that insulin sensitivity is not associated with the influence of ACE2 deficiency and A1‐7 infusion on muscle function.
We found that ACE2KO mice exhibited smaller body weights than wild‐type mice without affecting food consumption and muscle mass in lower limb at middle age. This implies the possibility that ACE2 deficiency alters metabolic function, leading to change in body composition that we did not measure in this study. However, the change in oxygen consumption between the genotypes in the older mice but not in the middle‐aged mice implicates that long‐term ACE2 deficiency somehow alters metabolic function, but the effect is not associated with the early manifestation of muscle senescence in ACE2KO mice. We found that A1‐7 infusion modestly increased oxygen consumption in older mice without affecting wheel‐running activity. Further investigation will be required to elucidate the precise mechanisms, but our findings suggest that A1‐7 increases systemic metabolism together with the improvement of muscle function in aged mice.
The only specific morphological finding observed was a prominent increase in central nuclei of muscle fibres from ACE2KO mice. It is well recognized that central nuclei appear during the regeneration process of injured muscle in several pathologies, including trauma and myopathies. Central nuclei in skeletal muscle also increase to a small extent during physiological ageing in mice, consistent with our finding that ~3% of muscle fibres had central nuclei in 25‐month‐old wild‐type mice.23 While muscle stem cells (i.e. satellite cells) are considered the origin of central nuclei in the regeneration process, the pathophysiological basis of central nuclei in aged muscle remains largely undetermined. It has been known that muscle denervation is associated with increased central nuclei.25 However, it is unlikely that decreased innervation, if existed in ACE2KO mice, solely caused the increased central nuclei in TA muscle, because decreased innervation should alter muscle size as well.25 Because of technical reasons, we used TA muscles as a representative fast‐twitch muscle for morphological and gene analysis instead of muscles that generate forearm grip strength. The discrepancy between the reduced forearm grip strength and the unaltered contractile force and muscle size of TA muscles in 15‐month‐old ACE2KO mice suggests that the magnitude of impact of ageing on muscle performance and morphology differs among muscles. Therefore, it is conceivable that the reduced grip strength in ACE2KO mice is not caused by the increased central nuclei in myofibres but by other deteriorations in muscle morphology of muscles promoting forearm grip strength.
While there were few group differences in gene clusters of TA muscles between middle‐aged ACE2KO and wild‐type mice in the RNA array analysis, the expression of a tumour suppressor protein, p16INK4a, also known as cyclin‐dependent kinase inhibitor 2A, was dramatically increased by ACE2 deficiency. The difference between the genotypes remained significant until old age. p16INK4a prevents mammalian cells from oncogenic stimuli by promoting retinoblastoma‐dependent cell cycle arrest, which also contributes to systemic ageing in mammals.26 In ageing skeletal muscle, p16INK4a is mainly expressed in satellite cells, or muscle stem cells, which surround the plasma membrane of muscle fibres. Senescent satellite cells expressing p16INK4a lose their ability to proliferate and differentiate upon muscle injury, yet it remains controversial if and how this dysfunction of satellite cells causes the development of sarcopenia.14, 15, 16 Recent findings in mice support the contribution of satellite cells to ageing‐associated changes in muscle fibrosis and myofibre maintenance14, 27 but not to the ageing‐associated loss of skeletal muscle size and strength, that is, sarcopenia.16 In contrast, Baker et al. reported that clearance of p16INK4a‐positive cells in skeletal muscle increased exercise ability in mice.15 It was also recently reported that p16INK4a is not a marker of senescence but has a causal role in the dysfunction of aged satellite cells.28 In this study, we found that ACE2 deficiency was associated with early induction of p16INK4a in skeletal muscle without alteration of other senescence genes, including p53 and p21. Further investigation is required to determine whether the increased p16INK4a seen in ACE2KO mice is a marker or inducer of organ ageing.
While continuous A1‐7 infusion improved grip strength in older ACE2KO and wild‐type mice, A1‐7 did not appear to reverse skeletal muscle ageing, as shown by the lack of alternation in p16INK4a expression and central nuclei with A1‐7 infusion. One explanation of this finding is that decreased A1‐7 caused by ACE2 deficiency induced skeletal muscle ageing, and A1‐7 improved the contractile function of muscle fibres but not the irreversible senescence of satellite cells and muscle fibres. Further investigation will be required to elucidate whether A1‐7 infusion to younger ACE2KO mice could restore the muscle senescence. Another possibility is that the accelerated skeletal muscle ageing seen in ACE2KO mice was induced via an A1‐7‐independent pathway. In addition to its function in the RA system, ACE2 interacts with the apelin system29 and functions as a collectrin homologue in intestinal tissue.30, 31 In this context, while we found that ACE2 expression reduced in TA muscles of middle‐aged wild‐type mice, it remains unclear whether the alternation in muscle ACE2 is responsible for age‐related muscle weakness. Further studies, including those that utilize mice lacking Mas, a receptor of A1‐7 or mice with tissue‐specific deletion of ACE2, will be required to further elucidate these possible mechanisms. The protective role of A1‐7 in skeletal muscle function has been shown with the use of several mouse models, including the chronic infusion of angiotensin II,11, 13 endotoxin exposure,12 disuse atrophy,8 and genetic induction of DMD.10 Underlying mechanisms proposed in these studies regarding the effects of A1‐7 include phosphorylation of AKT, prevention of the increase in atrogin‐1 and MuRF‐1,9, 11 reduction in phosphorylation of p38 MAPK, and inhibition of transforming growth factor‐β signalling.10, 12, 13 We did not find any influence of A1‐7 on these signalling pathways in TA muscles of aged mice (data not shown). We also found no change in mass and myofibril size of TA muscle by A1‐7 infusion. The improved grip strength and the lack of alteration in TA muscle might imply that the impact of A1‐7 is different among skeletal muscles. Further investigation will be required to elucidate the underlying mechanisms involved in the improvement of muscle weakness by A1‐7 in aged mice.
Despite the early manifestation of senescent phenotypes, ACE2KO mice exhibited a normal lifespan. This is in contrast to conventional mouse models of senescence, including klotho mice and a series of senescence‐accelerated mice, which die earlier than normal mice.32, 33 Whole‐body analysis of ageing phenotypes in ACE2KO mice will be required to clarify the mechanisms of early organ senescence with the normal lifespan. Finally, it is clinically relevant that A1‐7 improved ageing‐induced skeletal muscle weakness not only in ACE2KO mice but also in wild‐type mice. Currently, there is no established pharmacological therapy for the treatment of ageing‐associated muscle weakness. A1‐7 has been shown to play a protective role in multiple organs without any adverse effects in rodents.34, 35 Further investigation will be required to investigate a fascinating question whether a longer administration of A1‐7 would extend lifespan in older mice. Finally, our results provide an important basis for the future clinical application of A1‐7 and its analogues as safe drugs for the improvement of muscle weakness in the elderly population.
Conflict of Interest
All authors declare that they have no conflict of interest regarding the publication of this article.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.36
We are most grateful to Hikari Kitamura, Yuka Nakao, and Tomoko Sato for their excellent technical assistance. We thank Shuichi Araki (LMS Co., Ltd) and Mitsuhiro Abe (Unique Medical Co., Ltd) for their technical guidance. We also greatly appreciate the enthusiastic support from Kengo Nakagawa, because his special technique and knowledge were indispensable for our research.
Takeshita, H. , Yamamoto, K. , Nozato, S. , Takeda, M. , Fukada, S. , Inagaki, T. , Tsuchimochi, H. , Shirai, M. , Nozato, Y. , Fujimoto, T. , Imaizumi, Y. , Yokoyama, S. , Nagasawa, M. , Hamano, G. , Hongyo, K. , Kawai, T. , Hanasaki‐Yamamoto, H. , Takeda, S. , Takahashi, T. , Akasaka, H. , Itoh, N. , Takami, Y. , Takeya, Y. , Sugimoto, K. , Nakagami, H. , and Rakugi, H. (2018) Angiotensin‐converting enzyme 2 deficiency accelerates and angiotensin 1‐7 restores age‐related muscle weakness in mice. Journal of Cachexia, Sarcopenia and Muscle, 9: 975–986. 10.1002/jcsm.12334.
References
- 1. Cesari M, Landi F, Vellas B, Bernabei R, Marzetti E. Sarcopenia and physical frailty: two sides of the same coin. Front Aging Neurosci 2014;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beaudart C, Dawson A, Shaw SC, Harvey NC, Kanis JA, Binkley N, et al. Nutrition and physical activity in the prevention and treatment of sarcopenia: systematic review. Osteoporos Int 2017;28:1817–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoshimura Y, Wakabayashi H, Yamada M, Kim H, Harada A, Arai H. Interventions for treating sarcopenia: a systematic review and meta‐analysis of randomized controlled studies. J Am Med Dir Assoc 2017;18:553.e1–553.e16. [DOI] [PubMed] [Google Scholar]
- 4. Cabello‐Verrugio C, Morales MG, Rivera JC, Cabrera D, Simon F. Renin‐angiotensin system: an old player with novel functions in skeletal muscle. Med Res Rev 2015;35:437–463. [DOI] [PubMed] [Google Scholar]
- 5. Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF‐beta‐induced failure of muscle regeneration in multiple myopathic states. Nat Med 2007;13(2):204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burks TN, Andres‐Mateos E, Marx R, Mejias R, Van Erp C, Simmers JL, et al. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 2011;3(82):82ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elbaz M, Yanay N, Aga‐Mizrachi S, Brunschwig Z, Kassis I, Ettinger K, et al. Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy2J/dy2J mouse. Ann Neurol 2012;71:699–708. [DOI] [PubMed] [Google Scholar]
- 8. Morales MG, Abrigo J, Acuna MJ, Santos RA, Bader M, Brandan E, et al. Angiotensin‐(1‐7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis Model Mech 2016;9(4):441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cisternas F, Morales MG, Meneses C, Simon F, Brandan E, Abrigo J, et al. Angiotensin‐(1‐7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor‐dependent mechanism. Clin Sci 2015;128(5):307–319. [DOI] [PubMed] [Google Scholar]
- 10. Jose Acuna M, Pessina P, Olguin H, Cabrera D, Vio CP, Bader M, et al. Restoration of muscle strength in dystrophic muscle by angiotensin‐1‐7 through inhibition of TGF‐beta signalling. Hum Mol Genet 2014;23(5):1237–1249. [DOI] [PubMed] [Google Scholar]
- 11. Meneses C, Morales M, Abrigo J, Simon F, Brandan E, Cabello‐Verrugio C. The angiotensin‐(1‐7)/Mas axis reduces myonuclear apoptosis during recovery from angiotensin II‐induced skeletal muscle atrophy in mice. Pflugers Arch ‐ Eur J Physiol 2015;467(9):1975–1984. [DOI] [PubMed] [Google Scholar]
- 12. Morales MG, Olguin H, Di Capua G, Brandan E, Simon F, Cabello‐Verrugio C. Endotoxin‐induced skeletal muscle wasting is prevented by angiotensin‐(1‐7) through a p38 MAPK‐dependent mechanism. Clin Sci 2015;129(6):461–476. [DOI] [PubMed] [Google Scholar]
- 13. Morales MG, Abrigo J, Meneses C, Simon F, Cisternas F, Rivera JC, et al. The Ang‐(1‐7)/Mas‐1 axis attenuates the expression and signalling of TGF‐beta 1 induced by AngII in mouse skeletal muscle. Clin Sci 2014;127(3‐4):251–264. [DOI] [PubMed] [Google Scholar]
- 14. Liu W, Wei‐LaPierre L, Klose A, Dirksen RT, Chakkalakal JV. Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 2011;479(7372):232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu FJ, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med 2015;21(1):76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeda M, Yamamoto K, Takemura Y, Takeshita H, Hongyo K, Kawai T, et al. Loss of ACE 2 Exaggerates High‐Calorie Diet‐Induced Insulin Resistance by Reduction of GLUT4 in Mice. Diabetes 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ellacott KLJ, Morton GJ, Woods SC, Tso P, Schwartz MW. Assessment of Feeding Behavior in Laboratory Mice. Cell Metab 2010;12(1):10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uno K, Katagiri H, Yamada T, Ishigaki Y, Ogihara T, Imai J, et al. Neuronal pathway from the liver modulates energy expenditure and systemic insulin sensitivity. Science 2006;312(5780):1656–1659. [DOI] [PubMed] [Google Scholar]
- 20. Takeshita H, Yamamoto K, Nozato S, Inagaki T, Tsuchimochi H, Shirai M, et al. Modified forelimb grip strength test detects aging‐associated physiological decline in skeletal muscle function in male mice. Sci Rep 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Narkar VA, Downes M, Yu RT, Embler E, Wang Y‐X, Banayo E, et al. AMPK and PPAR delta Agonists are exercise mimetics. Cell 2008;134(3):405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rinnankoski‐Tuikka R, Silvennoinen M, Torvinen S, Hulmi JJ, Lehti M, Kivela R, et al. Effects of high‐fat diet and physical activity on pyruvate dehydrogenase kinase‐4 in mouse skeletal muscle. Nutr Metab 2012;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valdez G, Tapia JC, Kang H, Clemenson GD, Gage FH, Lichtman JW, et al. Attenuation of age‐related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci U S A 2010;107(33):14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Umegaki H. Sarcopenia and diabetes: Hyperglycemia is a risk factor for age‐associated muscle mass and functional reduction. J Diabetes Investig 2015;6(6):623–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu DX, Huang SK, Carlson BM. Electron microscopic study of long‐term denervated rat skeletal muscle. Anat Rec 1997;248(3):355–365. [DOI] [PubMed] [Google Scholar]
- 26. Munoz‐Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014;15(7):482–496. [DOI] [PubMed] [Google Scholar]
- 27. Keefe AC, Lawson JA, Flygare SD, Fox ZD, Colasanto MP, Mathew SJ, et al. Muscle stem cells contribute to myofibres in sedentary adult mice. Nat Commun 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sousa‐Victor P, Gutarra S, Garcia‐Prat L, Rodriguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014;506(7488):316–321. [DOI] [PubMed] [Google Scholar]
- 29. Sato T, Suzuki T, Watanabe H, Kadowaki A, Fukamizu A, Liu PP, et al. Apelin is a positive regulator of ACE2 in failing hearts. J Clin Invest 2013;123(12):5203–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camargo SM, Singer D, Makrides V, Huggel K, Pos KM, Wagner CA, et al. Tissue‐specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009;136(3):872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012;487(7408):477–U89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuroo M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45–51. [DOI] [PubMed] [Google Scholar]
- 33. Takeda T. Senescence‐accelerated mouse (SAM): a biogerontological resource in aging research. Neurobiol Aging 1999;20:105–110. [DOI] [PubMed] [Google Scholar]
- 34. Jiang F, Yang JM, Zhang YT, Dong M, Wang SX, Zhang Q, et al. Angiotensin‐converting enzyme 2 and angiotensin 1‐7: novel therapeutic targets. Nat Rev Cardiol 2014;11:413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Passos‐Silva DG, Verano‐Braga T, Santos RAS. Angiotensin‐(1‐7): beyond the cardio‐renal actions. Clin Sci 2013;124:443–456. [DOI] [PubMed] [Google Scholar]
- 36. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]