

Abstract
Background
Cachexia is a formidable clinical challenge in pancreatic cancer. We assessed LY2495655 (antimyostatin antibody) plus standard‐of‐care chemotherapy in pancreatic cancer using cachexia status as a stratifier.
Methods
In this randomized, phase 2 trial, patients with stage II–IV pancreatic cancer were randomized to 300 mg LY2495655, 100 mg LY2495655, or placebo, plus physician‐choice chemotherapy from a prespecified list of standard‐of‐care regimens for first and later lines of care. Investigational treatment was continued during second‐line treatment. The primary endpoint was overall survival.
Results
Overall, 125 patients were randomized. In August 2014, 300 mg LY2495655 was terminated due to imbalance in death rates between the treatment arms; in January 2015, 100 mg LY2495655 treatment was terminated due to futility. LY2495655 did not improve overall survival: the hazard ratio was 1.70 (90% confidence interval, 1.1–2.7) for 300 mg vs. placebo and 1.3 (0.82–2.1) for 100 mg vs. placebo (recommended doses). Progression‐free survival results were consistent with the overall survival results. A numerically higher hazard ratio was observed in patients with weight loss (WL) of ≥5% (cachexia) than with <5% WL within 6 months before randomization. Subgroup analyses for patients stratified by WL in the 6 months preceding enrollment suggested that functional responses to LY2495655 (either dose) may have been superior in patients with <5% WL vs. patients with ≥5% WL. Among possibly drug‐related adverse events, fatigue, diarrhoea, and anorexia were more common in LY2495655‐treated than in placebo‐treated patients.
Conclusions
In the intention‐to‐treat analysis, LY2495655 did not confer clinical benefit in pancreatic cancer. Our data highlight the importance of assessing survival when investigating therapeutic management of cachexia and support the use of WL as a stratifier (independent of performance status).
Keywords: Myostatin, Cachexia, Pancreatic cancer, Muscle mass
Introduction
The overall prognosis for pancreatic cancer remains poor with a 5‐year survival rate of 7% (for all stages combined).1 Cachexia, a complex, multifactorial syndrome characterized by loss of skeletal muscle that is not reversible with nutritional support,2, 3 is prominent in many malignancies including pancreatic cancer4 and leads to decreased physical function, reduced tolerance to anticancer therapy, and decreased overall survival (OS).
Cachexia research has identified several promising drug targets, including myostatin, a member of the transforming growth factor‐β superfamily.5 Animal studies have demonstrated that myostatin is a highly conserved negative regulator of skeletal muscle mass.6, 7, 8, 9, 10, 11, 12, 13 In mouse cancer cachexia models, inhibition of the myostatin signalling pathway is associated with improvements in body weight, muscle volume, and physical performance.6, 14
The myostatin‐neutralizing mouse IgG1monoclonal antibody (mAb), LSN2478185, and its humanized derivative, LY2495655, significantly attenuated the loss of muscle mass and improved strength in mouse tumour models without affecting tumour growth.7 Importantly, co‐administration of these antibodies with gemcitabine did not reduce their strength preservation effects. In a multicentre phase 1 study, the safety and recommended doses of LY2495655 were assessed in 29 patients with advanced cancer; all doses of LY2495655 in the dose‐escalation phase (2, 7, 21, 70, 210, and 700 mg once every 2 weeks [Q2W]) and dose‐expansion phase (100 and 300 mg Q2W) appeared to be well‐tolerated with no significant safety concerns.15 The recommended doses of LY2495655 were 100 and 300 mg Q2W.
The goal of this clinical trial was to investigate the effects of LY2495655 plus standard‐of‐care chemotherapy regimen on OS, changes in muscle mass, and physical performance in locally advanced, inoperable, or metastatic pancreatic cancer. This was the first large, randomized trial with an emphasis on treating the cachexia syndrome in patients with pancreatic cancer using an especially comprehensive methodology to measure muscle mass and physical activity with functional assessments.
Patients and methods
Design and treatment
This multicentre, randomized, double‐blind, placebo‐controlled, phase 2 trial enrolled patients with locally advanced, inoperable, or metastatic pancreatic cancer (NCT01505530). Patients were randomized into three treatment arms: chemotherapy plus 300 mg LY2495655 (hereafter 300 mg), chemotherapy plus 100 mg LY2495655 (hereafter 100 mg), or chemotherapy plus placebo (hereafter placebo) (Supporting Information Appendix S1). Investigators chose either a single‐agent gemcitabine, gemcitabine plus erlotinib, or FOLFIRINOX as a first‐line treatment; therefore, chemotherapy was not assigned in the study but rather selected by the treating physicians based on patient age, performance, and co‐morbidities. Randomization was stratified by treatment regimen (gemcitabine vs. gemcitabine plus erlotinib vs. FOLFIRINOX), disease stage (II/III vs. IV), Eastern Cooperative Oncology Group (ECOG) performance status (PS) (0/1 vs. 2), and percentage of weight loss (WL) occurring within 6 months of enrollment (<5%vs. ≥5%). An assessment committee reviewed unblinded adverse event (AE) and serious AE data during an initial safety lead‐in and every 6 months thereafter. Protocols were approved by Ethical Review Boards, and signed informed consent was obtained prior to study procedures. Trials were compliant with the Declaration of Helsinki and other applicable guidelines.
LY2495655 was administered intravenously (IV) over 30 (±5) min every 14 days. Cycle 1 was 8 weeks and included four planned doses of study therapy (described in Supporting Information Appendix S2). Subsequent cycles were 4 weeks with two planned doses of treatment. FOLFIRINOX was dosed as described in Supporting Information Appendix S2. Patients continued treatment until disease progression, intolerable AEs, or patient/investigator decision. If the patient experienced disease progression on first‐line therapy or did not tolerate first‐line therapy, the patient could receive post‐first‐line chemotherapy of mFOLFOX6, 5‐fluorocuracil, capecitabine, gemcitabine‐based regimen, or best supportive care while continuing on study therapy or placebo.
Palpable tumour assessments were performed at the end of every treatment cycle; radiologic assessments were performed every 8 weeks. Muscle volume of the thigh and L4‐L5 region was measured as described.16 Functional assessments were performed every 8 weeks (6 min walk, hand grip strength, and timed up and go test) and at 4 weeks and every 8 weeks thereafter (stair climb) (Supporting Information Appendix S2).
Eligibility criteria
Males and females aged ≥18 years (ECOG PS ≤2) with histologically or cytologically diagnosed locally advanced (stage II/III) or metastatic (stage IV) pancreatic adenocarcinoma whose cancer was either not amenable to resection with curative intent or who had progression after prior surgery were eligible. Other inclusion criteria included measurable/nonmeasurable disease (RECIST version 1.117), an estimated life expectancy of ≥12 weeks, a requirement to perform all performance measures at baseline, and having received radiotherapy (where applicable) ≥4 weeks before study enrollment. Key exclusion criteria included completion of a prior study investigating a drug specifically targeting myostatin, central nervous system malignancies, orthopaedic or neurologic injury <6 months before randomization, prior systemic chemotherapy for unresectable/metastatic disease, endocrine pancreatic tumours or ampullary cancer, underlying muscle disease, and use of muscle‐building or performance‐enhancing medications. All patients provided written informed consent before study procedures.
Outcome measures
The primary endpoint was OS. Secondary outcomes included progression‐free survival (PFS), tumour response rate, duration of response, body composition, thigh muscle volume and recto‐femoral area, and physical performance outcomes (Supporting Information Appendix S2).
Statistical analysis
Enrollment of 120 patients provided 74% power with a type I error rate of 0.20 to detect 20% OS improvement (hazard ratio [HR] = 0.83) and 40% OS improvement (HR = 0.71) in low‐dose and high‐dose arms, respectively, assuming a median OS time of 6.5 months for the gemcitabine‐based stratum and 11 months for the FOLFIRINOX stratum.18, 19 This calculation was performed using a Bayesian Augmented Control model.
Efficacy analysis included the intent‐to‐treat population. Statistical analyses were conducted using Statistical Analysis Software® (SAS Institute Inc, Cary NC, USA). Time‐to‐event outcomes were estimated using the Kaplan–Meier method,20 and 90% confidence intervals (CIs) were estimated using the Greenwood method.21 A Cox proportional hazard model22 was used to estimate HR values and two‐sided 90% CI after adjusting for the aforementioned stratification factors as well as age, gender, and investigational site. Tumour response rates were compared between the treatment arms using a continuity‐corrected χ2 test, and the 90% CIs were constructed using the normal approximation to the binomial distribution.
Subgroup analyses of OS, PFS, and select functional and muscle outcomes were performed by baseline WL (<5% and ≥5%), ECOG PS (0–2), age, gender, and choice of first‐line chemotherapy (gemcitabine alone vs. gemcitabine plus erlotinib vs. FOLFIRINOX).
Treatment‐emergent adverse events (TEAEs) were defined as AEs that occurred or whose severity worsened after the first administration of any study drug regardless of study drug causality. The Medical Dictionary for Regulatory Activities version 13.0 (or higher) Lower Level Term was used to identify treatment emergence. TEAEs were graded using Common Terminology Criteria for Adverse Events version 4.02. Planned interim analyses were conducted to assess safety.
Results
Study population
Of 167 enrolled patients, 125 were randomized (41, 300 mg; 42, 100 mg; 41, placebo) to first‐line therapy (Figure 1). One patient assigned to 100 mg was not treated. Thirty‐six patients (28.8%) received second‐line therapy including two patients who did not complete one cycle of first‐line therapy (1 300 mg‐treated patient; 1 placebo‐treated patient). Five patients received third‐line therapy, and one received fourth‐line therapy.
Figure 1.
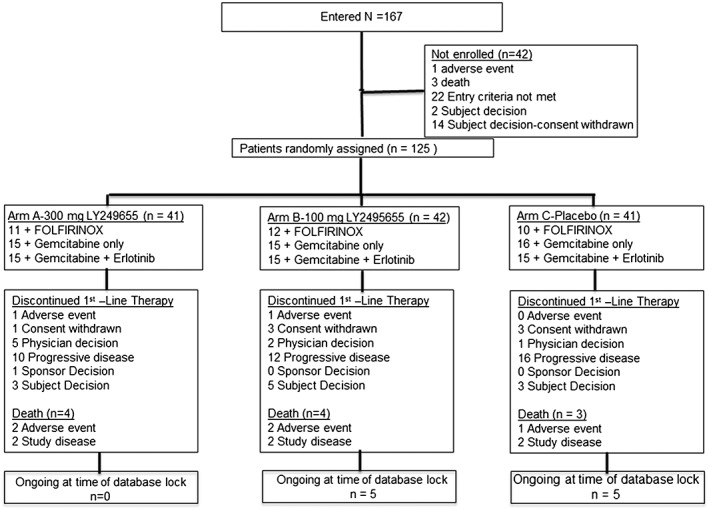
Patient flow diagram.
Of patients completing ≥1 cycles of first‐line therapy, 36.6%, 23.3%, and 22.0% received second‐line therapy in the 300 mg, 100 mg, and placebo arms, respectively (Table 1). No significant differences in baseline characteristics between treatment arms were observed, except for number of patients with WL ≥5% within the 6 months preceding enrollment, which was numerically higher in the 100 mg arm (Table 1). Baseline myostatin levels were similar across treatment arms and in men vs. women (Supporting Information Appendix S3).
Table 1.
Patient baseline characteristics
| Characteristic | 300 mg (n = 41) | 100 mg (n = 43) | Placebo (n = 41) |
|---|---|---|---|
| Gender, n (%) | |||
| Women | 16 (39) | 13 (30) | 15 (37) |
| Age, years | |||
| Mean (SD) | 65.0 (1.3) | 67.4 (10.7) | 68.4 (9.1) |
| Median (range) | 66.7 (44.8–82.0) | 66.7 (47.3–85.6) | 70.2 (46.6–84.5) |
| Race, n (%) | |||
| Asian | 0 (0) | 0 (0) | 1 (2.4) |
| Black or African American | 3 (7.3) | 1 (2.3) | 5 (12.2) |
| White | 38 (92.7) | 42 (97.7) | 35 (85.4) |
| Weight, kg | |||
| Mean (SD) | 72.6 (16.7) | 71.5 (19.5) | 71.2 (14.1) |
| Median (range) | 70.8 (41.6–112.7) | 66.1 (35.3–155.2) | 72.3 (43.9–115.0) |
| Lean mass, kg | |||
| Mean | 43.7 | 44.3 | 42.9 |
| Median (range) | 41.6 (21.7–72.1) | 45.8 (20.3–60.8) | 40.7 (24.7–68.9) |
| Fat mass, kg | |||
| Mean | 22.1 | 18.2 | 21.2 |
| Median (range) | 20.0 (1.6–44.7) | 16.9 (3.0–37.5) | 19.7 (6.7–43.0) |
| First‐line therapy, n (%) | |||
| Gemcitabine | 15 (36.6) | 15 (34.9) | 16 (39.0) |
| Gemcitabine + erlotinib | 15 (36.6) | 15 (34.9) | 15 (36.6) |
| FOLFIRINOX | 11 (26.8) | 12 (27.9) | 10 (24.4) |
| Second‐line therapy, n (%) | 15 (36.6) | 10 (23.3) | 9 (22.0) |
| Best supportive care | 2 (4.9) | 2 (4.7) | 4 (9.8) |
| Capecitabine | 6 (14.6) | 1 (2.3) | 2 (4.9) |
| Gemcitabine | 0 | 1 (2.3) | 0 |
| Gemcitasbine + erlotinib | 2 (4.9) | 3 (7.0) | 0 |
| mFOLFOX6 | 6 (14.6) | 3 (7.0) | 3 (7.3) |
| 5‐fluorouracil | 0 | 0 | 1 (2.4) |
| Disease stage at diagnosis, n (%) | |||
| II | 6 (14.6) | 1 (2.3) | 5 (12.2) |
| III | 7 (17.1) | 8 (18.6) | 7 (17.1) |
| IV | 28 (68.3) | 34 (79.1) | 29 (80.7) |
| ECOG PS, n (%) | |||
| 0 | 17 (41.5) | 16 (37.2) | 15 (36.6) |
| 1 | 23 (56.1) | 26 (60.5) | 25 (61.0) |
| 2 | 1 (2.4) | 1 (2.3) | 1 (2.4) |
| Weight loss within 6 months before enrollment, n (%) | |||
| <5% decrease | 19 (46.3) | 13 (30.2) | 18 (43.9) |
| ≥5% decrease | 22 (53.7) | 30 (69.8) | 23 (56.1) |
| Patients completing ≥1 cycles of first‐line therapy and receiving second‐line therapy, n (%) | 15 (36.6) | 10 (23.2) | 9 (22.0) |
ECOG, Eastern Cooperative Oncology Group; mFOLFOX6, folinic acid (leucovorin)‐fluorouracil‐oxaliplatin; PS, performance status; SD, standard deviation.
Interim analyses
Safety interim analyses were planned after completion of cycle 1 by 18 patients and every 6 months thereafter. Four interim analyses were performed by an internal assessment committee: December 2012 (safety), July 2013 (futility/safety, approximately 3 months after enrollment of the 45th patient), February 2014 (futility/safety, approximately 3 months after enrollment of the 80th patient), and August 2014 (safety). The assessment committee recommended continuation of the trial without modification after each of the first three interim analyses. At the August 2014 interim analysis (data lock: 31 July 2014), the committee noted an imbalance in deaths among patients with long‐term follow‐up who received 300 mg (31 deaths), 100 mg (19 deaths), and placebo (17 deaths). All deaths in this category were attributable to study disease and were not explained by any observed difference in AEs or other factors that were examined. Deaths on study drug and within 30 days of discontinuation were balanced across treatment arms. Consequently, LY2495655 300 mg treatment was discontinued. After the data cut‐off date (January 2015), LY2495655 was discontinued in all those receiving it during the extension phase because the primary objective was not met, and no improvement in assessed physical performance measures was observed.
Efficacy
There were 32 (78.0%), 30 (69.8%), and 25 (61.0%) deaths with 300 mg, 100 mg, and placebo, respectively (Table 2). Most deaths were disease‐related, and only a few were related to AEs. No significant OS benefit was observed with LY2495655 (HR = 1.7 [90% CI, 1.1–2.7] for 300 mg vs. placebo; HR = 1.3 [90% CI, 0.82–2.1] for 100 mg vs. placebo) (Figure 2A). Median OS was 8.0 months (90% CI, 6.0–10.0) for 300 mg, 9.8 months (90% CI, 5.9–13.5) for 100 mg, and 10.5 months (90% CI, 8.4–14.5) for placebo. Similarly, no significant PFS benefit was observed with LY2495655 (HR = 2.5 [90% CI, 1.6–4.0] for 300 mg vs. placebo; HR = 1.5 [90% CI, 1.0–2.3] for 100 mg vs. placebo) (Figure 2B). Median PFS was 4.9 months (90% CI, 3.3–6.0) for 300 mg, 6.9 months (90% CI, 5.4–7.9) for 100 mg, and 8.2 months (90% CI, 5.0–9.4) for placebo. Results of the preplanned Bayesian analysis are not presented due to the apparent lack of efficacy.
Table 2.
Patient death related to study disease and adverse event
| 300 mg (n = 41) | 100 mg (n = 43) | Placebo (n = 41) | |
|---|---|---|---|
| Death, n (%) | 32 (78) | 30 (70) | 25 (61) |
| Due to study disease | 29 (71) | 25 (58) | 23 (56) |
| Due to adverse event | 3 (7) | 5 (12) | 2 (5) |
Figure 2.
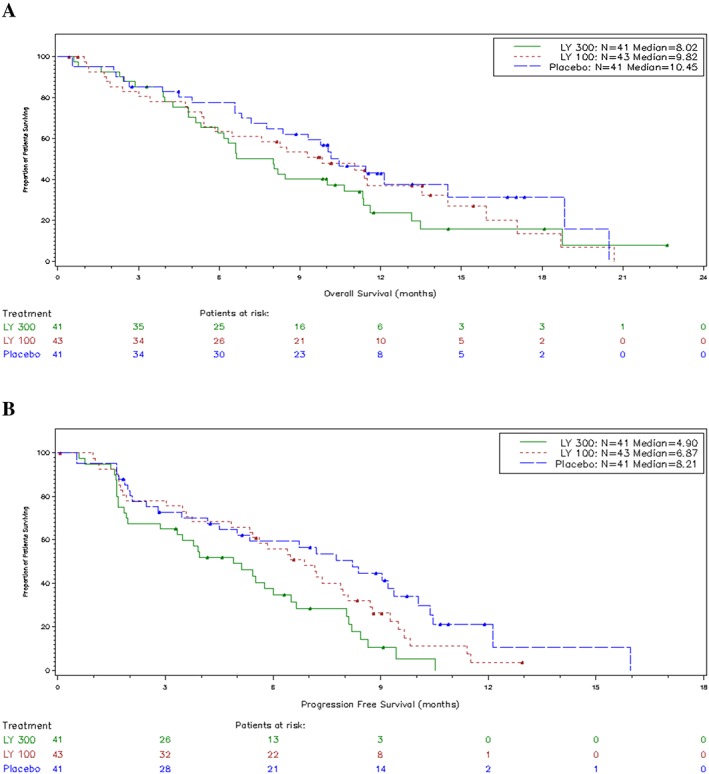
Overall survival (A) and progression‐free survival (B) by study arm.
Preplanned subgroup analyses demonstrated that the effect of LY495655 on OS may differ based on WL history. A numerically worse HR was observed for patients with ≥5% WL within 6 months before randomization (HR = 2.4 [90% CI, 0.49–2.0] for 300 mg vs. placebo; HR = 1.4 [90% CI, 0.81–2.5] for 100 mg vs. placebo) than for those with <5% WL within 6 months before randomization (HR = 0.99 [90% CI, 0.49–2.0] for 300 mg vs. placebo; HR = 0.87 [90% CI, 0.40–1.9] for 100 mg vs. placebo) (Table 3). No other interactions between prognostic factors and OS were observed (Table 3). Baseline characteristics were similar between groups of patients with <5% or ≥5% WL in the 6 months before enrollment (Supporting Information Appendix S4). In patients with ≥5% previous WL, baseline characteristics were similar across treatment groups (Supporting Information Appendix S5).
Table 3.
Overall survival analysis by subgroup (compared with placebo)
| Subgroup | N | 300 mg OS HR (90% CI) | 100 mg OS HR (90% CI) |
|---|---|---|---|
| Chemotherapy | |||
| Gemcitabine | 46 | 1.6 (0.80–3.3) | 0.93 (0.45–1.9) |
| Gemcitabine + erlotinib | 45 | 1.2 (0.59–2.3) | 1.4 (0.70–2.6) |
| FOLFIRINOX | 33 | 3.2 (1.0–9.9) | 1.7 (0.52–5.8) |
| ECOG PS | |||
| 0 | 43 | 1.3 (0.63–2.9) | 0.95 (0.41–2.2) |
| 1 or 2 | 82 | 1.6 (0.91–2.7) | 1.3 (0.78–2.3) |
| Disease stage | |||
| II/III | 34 | 1.5 (0.57–3.7) | 1.1 (0.41–3.1) |
| IV | 91 | 1.7 (1.0–2.8) | 1.4 (0.80–2.3) |
| Weight loss in last 6 months | |||
| <5% decrease | 50 | 0.99 (0.49–2.0) | 0.87 (0.40–1.9) |
| ≥5% decrease | 75 | 2.4 (1.3–4.2) | 1.43 (0.81–2.5) |
CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; HR, hazard ratio; OS, overall survival; PS, performance status.
Investigator‐reported objective tumour response rates were 22.0%, 25.6%, and 26.8% for 300 mg, 100 mg, and placebo, respectively. The median response duration was 5.9 (90% CI, 3.9–8.6) months for 300 mg, 8.0 (90% CI, 3.1–9.6) for 100 mg, and 9.2 (90% CI, 9.0–10.5) for placebo.
Changes in mass and functional evaluations from baseline to approximately 8 weeks are shown in Table 4. There were no significant improvements in muscle volume (as measured by both dual‐energy X‐ray absorptiometry and computed tomography scans) or functional tests between LY249655‐treated (100 or 300 mg) and placebo‐treated patients. Analysis of later visits was limited because of sample size but appeared to be consistent with the findings at the 8‐week assessment. The only significant difference observed between LY2495655 and placebo was with respect to improvement in 6 min walk from baseline to approximately 8 weeks. This difference was without adjusting for multiplicity and was only for the 300 mg dose (least squares means difference = 52.5 m; 90% CI, 3.9–101.1) but not for the 100 mg dose (least squares means difference = −32.9 m; 90% CI, −82.3–16.5).
Table 4.
Change from baseline to 8 weeks in mass and functional evaluation
| Evaluation |
300 mg – Placebo Difference (90% CI) |
100 mg – Placebo Difference (90% CI) |
|---|---|---|
| Thigh muscle, cm3 | 25.8 (−4.7–156.3) | 91.5 (−40.3–223.2) |
| Muscle area (CT), mm2 | 72.0 (−44.4–488.4) | 183.5 (−42.5–609.5) |
| Fat area (CT), cm2 | 9.9 (−9.9–29.6) | 10.7 (−9.5–30.9) |
| Lean mass (DXA), g | −718.5 (−36.6–699.7) | 546.7 (−0.90–94.2) |
| Fat mass (DXA), g | −119.7 (−75.6–242.0) | −868.4 (−12.8–49.6) |
| Hand grip, kg | 2.1 (−0.85–5.1) | 1.20 (−1.8–4.2) |
| Stair climbing power, Joule/sec | 7.2 (−22.4–36.7) | 7.5 (−22.9–37.9) |
| 6 min walk distance, m | 52.5 (3.9–101.1) | −32.9 (−82.3–16.5) |
| Timed up and go, sec | 1.7 (−3.8–0.47) | −0.20 (−2.0–2.4) |
| Pancreatic ADL, points | −2.6 (−5.2–0.11) | −3.0 (−5.7 to −0.26) |
ADL, activities of daily living; CI, confidence interval; CT, computed tomography; DXA, dual‐energy X‐ray absorptiometry.
Mass and functional evaluations by WL subgroups demonstrated that response to LY2495655 (vs. placebo) was better in patients with <5% WL (precachexia) than in those with ≥5% WL (cachexia). The evaluations in which these differences (which were not necessarily statistically significant) were observed included body mass measures (thigh muscle volume, lean mass, fat mass) and functional measurements (hand grip, 6 min walk distance) (Table 5).
Table 5.
Change from baseline to 8 weeks in mass and functional evaluation by previous weight loss
| Evaluation | 300 mg – Placebo | 100 mg – Placebo | ||
|---|---|---|---|---|
| <5% decrease | ≥5% decrease | <5% decrease | ≥5% decrease | |
| Thigh muscle, mL | 172 | −100 | 114 | 27 |
| Muscle area (CT), mm2 | 395 | −233 | 35 | 189.5 |
| Fat area (CT), mm2 | 433 | 1414 | 553 | 1410 |
| Lean mass (DXA), g | 88 | −1288 | 501 | 547 |
| Fat mass (DXA), g | 1202 | −502 | 1357 | 342 |
| Hand grip, kg | 3.7 | 1.0 | 4.2 | −1.5 |
| Stair climbing power,a mL Joule/sec | 23.4 | −14.8 | 5.1 | 7 |
| 6 min walk distance, m | 82 | 24 | 16 | −57 |
| Timed up and go, sec | 0.2 | −3.3 | 0.1 | −0.25 |
| Pancreatic ADL, points | −2.2 | −3.2 | −4.4 | 0.34 |
Bolded numbers indicate a better score in the <5% decrease group compared with ≥5% decrease group. ADL, activities of daily living; CT, computed tomography; DXA, dual‐energy X‐ray absorptiometry.
Evaluated at 12 weeks.
The pharmacokinetics of LY2495655 demonstrated a pattern of drug concentration–dependent nonlinearity. Population pharmacokinetics analysis revealed a half life for LY2495655 of approximately 12 to 23 days. Gemcitabine clearance was not affected by LY2495655 doses or by erlotinib doses. Erlotinib drug exposure appeared not to be affected by LY2495655 treatment. Based on the results from the target‐engagement model, almost complete target engagement (>95%) was achieved for both the 100 and 300 mg LY2495655 IV arms.
Safety
No dose‐exposure imbalances were observed between study arms (Supporting Information Appendix S6). Modest differences were observed between the study arms with respect to TEAEs and serious adverse events (Supporting Information Appendix S7); however, these differences did not demonstrate a clear pattern of toxicity relationship between treatment arms. Overall, 18 deaths (14.5%) were reported for patients on therapy, 27 deaths (21.8%) were reported for patients within 30 days of discontinuation, and 42 deaths (33.9%) were reported beyond 30 days of discontinuation. The majority of deaths were due to the underlying study disease. AE‐related deaths occurred in 7 (2 300 mg; 3 100 mg; 2 placebo), 3 (1 300 mg; 2 100 mg), and 0 patients on therapy, within 30 days of discontinuation, and beyond 30 days of discontinuation, respectively, but none were study drug‐related as determined by the investigator.
Two patients discontinued LY2495655 treatment due to AEs: 1 patient (300 mg arm) discontinued due to Grade 3 blood bilirubin increase during cycle 1 (unrelated to treatment), and a second patient (100 mg arm) discontinued due to Grade 2 hypersensitivity during cycle 1 (possibly treatment‐related).
The most common TEAEs regardless of causality were fatigue, diarrhoea, anorexia, nausea, decreased platelet count, anaemia, fever, vomiting, edema of the limbs, decreased neutrophil count, abdominal pain, constipation, dyspnea, and oral mucositis. Notable differences between the treatment arms in TEAE incidence were in the rates of decreased neutrophil count (39.0%, 23.8%, and 43.9% in the 300 mg, 100 mg, and placebo arms, respectively), oral mucositis (36.6%, 21.4%, and 22.0%, respectively), and other infections/infestations (2.4%, 7.1%, and 24.4%, respectively).
Among possibly study drug–related TEAEs, fatigue was more common in placebo‐treated than LY2495655‐treated patients (26.8%, 38.1%, and 51.2% in the 300 mg, 100 mg, and placebo arms, respectively), as were diarrhoea (26.8%, 23.8%, and 39.0%, respectively) and anorexia (17.1%, 16.7%, and 31.7%, respectively).
Discussion
In patients with stage II–IV unresectable pancreatic cancer receiving chemotherapy, LY2495655 administration did not improve OS compared with placebo. Indeed, compared with placebo, a trend toward poorer OS was observed with LY2495655. The median OS reported here (8.0, 9.8, and 10.5 months for the 300 mg, 100 mg, and placebo arms, respectively) seems similar to modern chemotherapy treatments for the placebo arm23, 24, 25, 26, 27; Chauffert et al. have reported OS of up to 13 months with the induction gemcitabine group (GEM: 1000 mg/m2 weekly for 7 weeks) vs. 8.6 months in the CHRT group (60 Gy, 2 Gy/fraction; concomitant 5‐fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) in patients with advanced pancreatic cancer.28 Although sample size may have limited the ability to demonstrate an interaction, with the exception of WL history, no other interactions between prognostic factors and OS were apparent.
The study's OS findings are consistent with results for the secondary endpoints, including PFS, tumour response rate, and duration of response. The median PFS observed here (4.9, 6.9, and 8.2 months for the 300 mg, 100 mg, and placebo arms, respectively) are also somewhat longer than historical values reported in gemcitabine‐treated patients.18, 19 The possibly longer OS and PFS observed here may be related to the use of FOLFIRINOX as an alternative regimen delivering a median OS of 11 months.26 It is also possible that this is related to between‐arm imbalances in the enrollment of early‐stage patients. Notably, we stratified patients by WL, which is unique to this trial. The observed association of WL category with outcome was independent of ECOG status and could potentially serve as an important variable in future pancreatic cancer research. Consistent with the observed lack of improvements in survival (OS and PFS) to study drug, we did not find clinically and statistically significant improvements with LY2495655 in the assessed physical performance measures (except for the 6 min walk with the 300 mg dose). However, it is still possible that subgroups of patients benefited from LY2495655. When AEs were considered, there were no obvious factors that could have influenced the findings. In the 300 mg, 100 mg, and placebo arms, respectively, rates of infections/infestations were 2.4%, 7.1%, and 21.4%, suggesting the possibility that LY2495655 may help prevent infection in these patients.
These findings are in contrast to those from a randomized trial involving elderly patients who had a fracture from a fall before enrollment, wherein LY2495655 treatment increased lean body mass and possibly improved functional measures of muscle power.29 Another trial demonstrated that inhibition of myostatin with AMG 745 increased lean body mass and decreased fat mass after 29 days in men with prostate cancer who were undergoing antiandrogen therapy.30 In our study, we observed superior responses to LY2495655 in patients with <5% previous WL compared with patients with ≥5% previous WL. This finding was independent of ECOG PS. These data suggest that patients who are precachexic (defined by WL) at baseline fare better in terms of performance measures than cachexic patients. It is possible that those with more profound WL were generally too sick to benefit from LY2495655; additionally, LY2495655 may be more effective at preventing muscle wasting than reversing cachexia.
Myostatin can have conflicting effects on cachexia. In preclinical models, myostatin‐null mice that were injected with cancer cells were more sensitive to tumour‐induced cachexia than wild‐type mice.12 Although myostatin overexpression can cause cachexia in mice,31 it is not the sole mediator.12 Signalling via the activin type II receptor appears to play a key role in development and progression of cachexia,32 but myostatin is only one of multiple ligands for this receptor.33 The abnormal metabolism that is associated with cachexia is caused by a complex interplay of host‐derived and tumour‐derived factors.5, 34, 35, 36 Therefore, it is possible that inhibition of myostatin with LY2495655 caused a compensatory change in other factor(s) that negatively impacted this study population. For example, tumour biology may have forced a shift in search of amino acids that were no longer accessible from the muscle breakdown pathway. This study's observations suggest that multi‐modal interventions are likely needed to maximize care, and those interventions may be different or of different intensity depending on the existing level of WL. In this context, development of other cachexia‐modulating targets and combinatorial strategies are important areas for future research.
Additional analyses, including that of subgroups and exploratory biomarkers, are ongoing. These analyses may help determine optimal myostatin‐based treatment regimens according to clinical and molecular characteristics of patients. This is the first large, randomized trial to address the cachexia syndrome. Even though our results were negative, extensive effort from the patients and investigators in the secondary functionality endpoints was performed, thus helping to further develop functional readouts in pancreatic cancer.
In conclusion, although the study failed to meet its primary objectives, the results highlight the importance of including survival endpoints in studies addressing therapeutic management of cachexia. The data also support the use of WL in pancreatic cancer trials as a clinically important stratification factor that is independent of PS.
Conflict of interest
T.G., R.G., D.R., S.M., and S.S. have no conflicts of interest. B.K.L. and R.A.W. are employees of Eli Lilly and Company. H.T.W. was employed at Eli Lilly and Company at the time of this writing; she is currently employed at Ipsen, Cambridge, Massachusetts and has no conflict of interest.
Supporting information
Data S1. Supporting Information
Acknowledgements
This work was sponsored by Eli Lilly and Company. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.37
The authors thank Robert Panek and Nancy Sheridan of INC Research/InVentiv Health for their writing assistance.
Golan, T. , Geva, R. , Richards, D. , Madhusudan, S. , Lin, B. K. , Wang, H. T. , Walgren, R. A. , and Stemmer, S. M. (2018) LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. Journal of Cachexia, Sarcopenia and Muscle, 9: 871–879. 10.1002/jcsm.12331.
References
- 1. Society AC . Cancer Facts & Figures 2016. Atlanta: American Cancer Society; 2016. [Google Scholar]
- 2. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 3. Muscaritoli M, Anker SD, Argiles J, Aversa Z, Bauer JM, Biolo G, et al. Consensus definition of sarcopenia, cachexia and pre‐cachexia: joint document elaborated by Special Interest Groups (SIG) "cachexia‐anorexia in chronic wasting diseases" and "nutrition in geriatrics". Clin Nutr 2010;29:154–159. [DOI] [PubMed] [Google Scholar]
- 4. Mueller TC, Burmeister MA, Bachmann J, Martignoni ME. Cachexia and pancreatic cancer: are there treatment options? World J Gastroenterol 2014;20:9361–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013;10:90–99. [DOI] [PubMed] [Google Scholar]
- 6. Murphy KT, Chee A, Gleeson BG, Naim T, Swiderski K, Koopman R, et al. Antibody‐directed myostatin inhibition enhances muscle mass and function in tumor‐bearing mice. Am J Physiol Regul Integr Comp Physiol 2011;301:R716–R726. [DOI] [PubMed] [Google Scholar]
- 7. Smith RC, Cramer MS, Mitchell PJ, Capen A, Huber L, Wang R. Myostatin neutralization results in preservation of muscle mass and strength in preclinical models of tumor‐induced muscle wasting. Mol Cancer Ther 2015;14:1661–1670. [DOI] [PubMed] [Google Scholar]
- 8. Whittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun 2003;300:965–971. [DOI] [PubMed] [Google Scholar]
- 9. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF‐beta superfamily member. Nature 1997;387:83–90. [DOI] [PubMed] [Google Scholar]
- 10. Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 2007;3:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 2004;350:2682–2688. [DOI] [PubMed] [Google Scholar]
- 12. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 13. Reisz‐Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha‐Hikim I, Hogue A, et al. Lower skeletal muscle mass in male transgenic mice with muscle‐specific overexpression of myostatin. Am J Physiol Endocrinol Metab 2003;285:E876–E888. [DOI] [PubMed] [Google Scholar]
- 14. Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jameson GS, Von Hoff DD, Weiss GJ, Richards DA, Smith DA, Becerra C. Safety of the antimyostatin monoclonal antibody LY2495655 in healthy subjects and patients with advanced cancer. J Clin Oncol 2012;30(suppl): abstr 2516. [Google Scholar]
- 16. Shen W, Punyanitya M, Wang Z, Gallagher D, St‐Onge MP, Albu J, et al. Total body skeletal muscle and adipose tissue volumes: estimation from a single abdominal cross‐sectional image. J Appl Physiol 2004;97:2333–2338. [DOI] [PubMed] [Google Scholar]
- 17. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 18. Oettle H, Richards D, Ramanathan RK, van Laethem JL, Peeters M, Fuchs M, et al. A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann Oncol 2005;16:1639–1645. [DOI] [PubMed] [Google Scholar]
- 19. Saif MW, Oettle H, Vervenne WL, Thomas JP, Spitzer G, Visseren‐Grul C, et al. Randomized double‐blind phase II trial comparing gemcitabine plus LY293111 versus gemcitabine plus placebo in advanced adenocarcinoma of the pancreas. Cancer J 2009;15:339–343. [DOI] [PubMed] [Google Scholar]
- 20. Kaplan EL, Meier P. Nonparametric estimation of incomplete observations. JASA 1958;53:457–481. [Google Scholar]
- 21. Greenwood M. The natural duration of cancer. Rep Public Health Med Subj 1926;33:1–26. [Google Scholar]
- 22. Cox DR. Regression models and life‐tables. J Royal Stat Soc: Series B 1972;74:187–122. [Google Scholar]
- 23. Lorgis V, Chauffert B, Gentil J, Ghiringhelli F. Influence of localization of primary tumor on effectiveness of 5‐fluorouracil/leucovorin combined with irinotecan and oxaliplatin (FOLFIRINOX) in patients with metastatic pancreatic adenocarcinoma: a retrospective study. Anticancer Res 2012;32:4125–4130. [PubMed] [Google Scholar]
- 24. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cardenes HR, Moore AM, Johnson CS, Yu M, Helft P, Chiorean EG, et al. A phase II study of gemcitabine in combination with radiation therapy in patients with localized, unresectable, pancreatic cancer: a Hoosier Oncology Group study. Am J Clin Oncol 2011;34:460–465. [DOI] [PubMed] [Google Scholar]
- 26. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–1825. [DOI] [PubMed] [Google Scholar]
- 27. Herman JM, Wild AT, Wang H, Tran PT, Chang KJ, Taylor GE, et al. Randomized phase III multi‐institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: final results. J Clin Oncol 2013;31:886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chauffert B, Mornex F, Bonnetain F, Rougier P, Mariette C, Bouché O, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5‐FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000‐01 FFCD/SFRO study. Ann Oncol 2008;19:1592–1599. [DOI] [PubMed] [Google Scholar]
- 29. Becker C, Lord SR, Studenski SA, Warden SJ, Fielding RA, Recknor CP, et al. Myostatin antibody (LY2495655) in older weak fallers: a proof‐of‐concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol 2015;3:948–957. [DOI] [PubMed] [Google Scholar]
- 30. Padhi D, Higano CS, Shore ND, Sieber P, Rasmussen E, Smith MR, et al. Pharmacological inhibition of myostatin and changes in lean body mass and lower extremity muscle size in patients receiving androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 2014;99:E1967–E1975. [DOI] [PubMed] [Google Scholar]
- 31. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Sci 2002;296:1486–1488. [DOI] [PubMed] [Google Scholar]
- 32. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 33. de Caestecker M. The transforming growth factor‐beta superfamily of receptors. Cytokine Growth Factor Rev 2004;15:1–11. [DOI] [PubMed] [Google Scholar]
- 34. Bachmann J, Ketterer K, Marsch C, Fechtner K, Krakowski‐Roosen H, Büchler MW, et al. Pancreatic cancer related cachexia: influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 2009;9:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 2009;89:381–410. [DOI] [PubMed] [Google Scholar]
- 36. Mendes MC, Pimentel GD, Costa FO, Carvalheira JB. Molecular and neuroendocrine mechanisms of cancer cachexia. J Endocrinol 2015;226:R29–R43. [DOI] [PubMed] [Google Scholar]
- 37. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information