

Abstract
Specific skeletal myopathy constitutes a common feature of heart failure, chronic obstructive pulmonary disease, and type 2 diabetes mellitus, where it can be characterized by the loss of skeletal muscle oxidative capacity. There is evidence from in vitro and animal studies that iron deficiency affects skeletal muscle functioning mainly in the context of its energetics by limiting oxidative metabolism in favour of glycolysis and by alterations in both carbohydrate and fat catabolic processing. In this review, we depict the possible molecular pathomechanisms of skeletal muscle energetic impairment and postulate iron deficiency as an important factor causally linked to loss of muscle oxidative capacity that contributes to skeletal myopathy seen in patients with heart failure, chronic obstructive pulmonary disease, and type 2 diabetes mellitus.
Keywords: Iron deficiency, Skeletal muscle, Oxidative capacity, Heart failure, Chronic obstructive pulmonary disease, Type 2 diabetes mellitus
Introduction
Specific skeletal myopathy represents an important pathophysiological feature of many chronic diseases and contributes to debilitating symptomatology. Indeed, derangements within skeletal muscle occur in such illnesses as rheumatoid arthritis, chronic kidney disease (CKD), chronic liver disease, heart failure (HF), chronic obstructive pulmonary disease (COPD), and type 2 diabetes mellitus (T2DM).1, 2, 3, 4, 5, 6 Pathophysiology of skeletal myopathy secondary to systemic disorders is multifactorial, being particularly complex in rheumatoid arthritis, CKD, and chronic liver disease where muscle structure and functioning may be influenced among others by a variety of circulating toxic metabolites, immune complexes, or enhanced muscle inflammatory signalling.7, 8, 9, 10 Instead, functional impairments of skeletal muscle seen in HF, COPD, and T2DM, such as decreased performance and decreased exercise capacity, seem to co‐exist with similar histological abnormalities and to result from comparable molecular pathomechanisms, most of which concerns energetics and yields in loss of muscle oxidative capacity.11
There are premises that iron plays a crucial role in skeletal muscle functioning, especially in the context of energy metabolism. Cellular oxidative metabolism strongly relies on iron availability, which is indispensable for both sufficient oxygen supply and effective substrate catabolism.12 It is worth noting that both iron overload and iron deficiency (ID) were proven to be detrimental for mitochondria that constitute cellular energy centres.13 Iron overload leads to the excessive formation and accumulation of reactive oxygen species whose harmful effects have already been described.14, 15 On the other hand, although numerous deleterious effects of ID on skeletal muscle have been summarized (for a detailed review, see Stugiewicz et al.16), they have not been discussed in the particular context of fuel selection and efficiency of specific catabolic routes for the energy production.
In this review, we aim to describe the possible molecular pathomechanisms of skeletal muscle energetic impairment, gathering evidence from in vitro and animal studies. Further, we postulate ID as an important causative factor of loss of muscle oxidative capacity that significantly contributes to skeletal myopathy seen in patients with HF, COPD, and T2DM and therefore may be considered as a co‐target in a therapeutic process.
Paragraph 1. Skeletal muscle energy metabolism
Energy‐consuming and multitasking skeletal muscle tissue possesses complex machinery, which integrates various pathways for the efficient adenosine triphosphate (ATP) production.17, 18, 19 The flexibility of usage of different energy sources that can be metabolized along distinct metabolic routes is inevitable for skeletal muscle to adapt to dynamic changes in their energy demand. The total amount of ATP stored in human skeletal muscles accounts for approximately 80 g and needs to be continuously re‐synthesized at the rate of its consumption.20 Muscles can take advantage of three main groups of energy substrates such as high‐energy phosphates (creatine phosphates), carbohydrates (glycogen and glucose), and lipids (triacylglycerol and free fatty acids).21, 22, 23, 24 Notably, endurance power of muscles is strongly related to their capacities to oxidize energy fuels in the process of mitochondrial oxidative phosphorylation (OXPHOS), which yields in ATP synthesis. Once oxygen flux to a muscle cell is not disturbed, OXPHOS can result in most efficient energy production, being fuelled by carbohydrate or fat as substrates.20 Both carbohydrate and lipid pathways branch to form four parallel pathways that converge on mitochondria.23 Thus, mitochondria integrate several metabolic routes, which result in the energy supply being essential for skeletal muscle functioning and capacity (Figure 1A).
Figure 1.
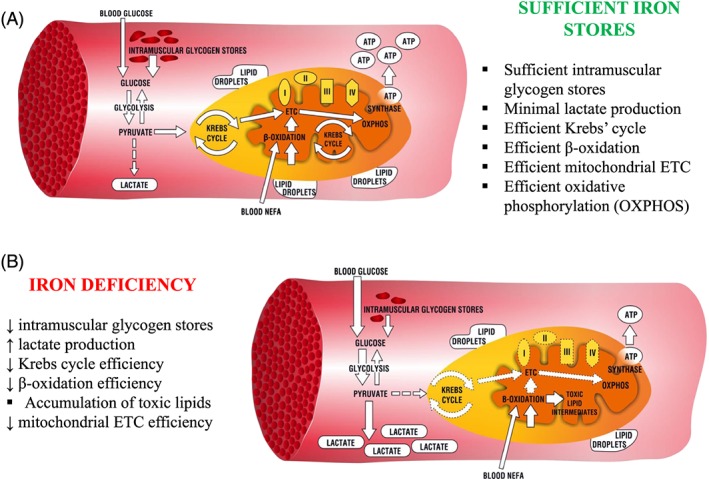
Energetic pathways in human skeletal myocytes (A) when iron stores are sufficient and (B) when changed upon iron deficiency. ETC, electron transport chain; ATP, adenosine triphosphate; I–IV, mitochondrial enzymatic complexes; NEFA, non‐esterified fatty acids.
Anaerobic vs. aerobic energy metabolism
Under limited oxygen availability, energy from carbohydrates in skeletal muscle is produced via anaerobic glycolysis, which results mostly in lactate production.25 The process has a net yield of 2 molecules of ATP per molecule of glucose. Therefore, it is not sufficiently powered to provide muscle with energy for a prolonged submaximal exercise. As soon as myocytes receive sufficient oxygen, they switch towards a more advanced form of energy acquisition, or OXPHOS, which is programmed as a natural continuation of glycolysis and is preceded by Krebs cycle.20 OXPHOS is conditioned by enzymatic complexes (complexes I–IV) of mitochondrial electron transport chain (ETC) that enables the generation of electrochemical gradient across the mitochondrial membrane essential for the synthesis of about 30–36 molecules of ATP. On the other hand, the efficiency of oxidative consumption of fats is estimated at 14 molecules of ATP.25
Carbohydrate energy metabolism in skeletal muscle
Catabolic processing of carbohydrates in skeletal muscle relies on two substrate sources, namely, intramuscular glycogen and blood glucose,22 which, depending on the oxygen availability, can be metabolized via anaerobic glycolysis in the cytoplasm or aerobic OXPHOS within mitochondrial enzymatic machinery. Notably, the substrates for mitochondrial oxidation at work intensities of around 80% of VO2max are initially supplied from glycogen droplets inside the muscle cells with no more than 20–30% of the fuel acquired from the capillaries.23, 26 During prolonged exercise, as soon as glycogen stores are depleted, the contribution of blood glucose becomes more appreciable, reaching close to 100% of muscle carbohydrate metabolism.22, 27 There are three points at which muscle glucose acquisition can be regulated: glucose delivery to the muscle cells, transmembrane transportation, and flux through the intracellular metabolism.22 The amount of glucose delivered to the muscular capillaries is usually referred as a resultant of blood flow and blood glucose concentration from which only the latter component has been proven to constitute a considerable limitation for glucose uptake during prolonged exercise.22, 28 Second rate‐limiting step of glucose acquisition is the permeability of the muscle cell membrane, which can be influenced by either extracellular stimuli, like contraction or insulin, or internal cellular factors, including metabolic status and Ca2+ signalling. All of the factors mentioned earlier have been proposed to affect either abundance or activity of the muscle‐specific glucose transporter (GLUT‐4).29, 30, 31 Finally, the final site of regulation of muscle glucose delivery is the flux of this monosaccharide through the metabolic routes, being mainly dependent on the activity of enzymes involved in glucose catabolism.
Lipid energy metabolism in skeletal muscle
Another two pathways for muscle energy metabolism rely on the lipid catabolism, which can be fuelled by intramuscular triglycerides or blood lipids. In general, in the main form of non‐esterified fatty acids (NEFA), lipid substrates are released from their stores in skeletal muscle or adipose tissue and transport to muscle mitochondria to be metabolized in the β‐oxidation process, which in turn yields in substrates for OXPHOS. The contribution of fatty acids to oxidative metabolism is essentially maximal at exercise intensities of 60% of VO2max, while at higher intensities being decreased.24, 32 Similarly to the order of carbohydrate substrate utilization, muscles primarily take an advantage from the intracellular lipid droplets, which remain in direct contact with the outer mitochondrial membrane. It allows muscle cell to circumvent the transport problems because of the low solubility of NEFA in the cytosol. Notably, lipid droplets comprise mainly intramuscular triglyceride whose concentration can adaptively increase in response to endurance training,23, 33 but also, according to the experimental evidence from human studies, it can accumulate in pathological conditions, contributing to the development of skeletal muscle insulin resistance.34, 35, 36, 37, 38
Interactions between carbohydrate and lipid metabolism in skeletal muscle
Crosstalk between carbohydrate and lipid metabolic routes in skeletal muscle has been proposed decades ago by Randle et al. and has been referred as ‘glucose‐fatty acid cycle’ or ‘Randle cycle’.39, 40 The original hypothesis has evolved over the years and in its current form postulates that the products of NEFA catabolism inhibit rate‐limiting enzymes of glycolysis, thus limiting glucose uptake and catabolism.41, 42 In general, Randle cycle should be considered as the biochemical mechanism that controls fuel selection, adjusting substrate supply and demand within skeletal muscle tissue, thus fine‐tuning hormonal regulation of substrate concentrations in the blood.42, 43 However, under conditions of disturbed energy status, an activation of the major sensor of cellular energy demand in skeletal muscle, AMP‐activated protein kinase, leads to abrogation of mechanisms of the glucose‐fatty acid cycle. Then, NEFA oxidation no longer inhibits the glucose uptake itself, while it may limit carbohydrate oxidative metabolism.44, 45
Paragraph 2. The importance of iron in the context of energy metabolism in skeletal muscle
Over recent years, there has been increasing interest in a role of iron metabolism in skeletal muscle functioning. Containing 10–15% of iron in the body, skeletal muscle mainly utilizes this micronutrient to build enzymes indispensable for oxidative metabolism, including myoglobin, which secures oxygen for a muscle cell as well as enzymes involved in substrate catabolism for OXPHOS.46, 47 Taking into consideration different extent of reliance on OXPHOS in a distinct type of muscle fibres, it should be noted that major amount of iron is present in slow ‘red’ fibres in which the oxidative energy production prevails. Thus, iron is of particular importance for muscles rich in red fibres, such as dorsal muscles, lower extremity extensors, the diaphragm, and intercostal muscles.48
Involvement of iron in Krebs cycle in skeletal muscle
Skeletal myocytes, like other mammalian cells, are attributed with two central regulators of cellular adaptive response to ID: IRP1 and IRP2 (for a detailed review, see Anderson et al.49 and Guo et al.50). Noteworthy, however, is that in a state of optimal or elevated intracellular iron, IRP1 switches from its transcriptional function towards enzymatic activity, being an important catalyst of Krebs cycle51 or series of reactions that intermediate between glycolysis and OXPHOS in the oxidative metabolism. Therefore, in non‐ID, environment IRP1 appears as a cytosolic isoform of aconitase that transforms citrate to isocitrate in Krebs cycle, which, in turn, constitutes a metabolic link between initial catabolism of either carbohydrate or fat and mitochondrial ETC, which leads to OXPHOS. Importantly, the second isoform of the aforementioned enzyme, which is referred as mitochondrial aconitase, is involved in Krebs cycle held in mitochondria. Because both cytosolic and mitochondrial aconitases contain iron–sulfur clusters (ISC), their activities decrease in a low iron state, because of either conversion to IRP1 in the case of cytosolic isoform51 or inactivation in the case of the mitochondrial enzyme.52
Involvement of iron in oxidative phosphorylation in skeletal muscle
As mentioned before, the efficiency of OXPHOS is directly related to the activities of four mitochondrial enzymatic complexes. Being embedded in the mitochondrial inner membrane, together they form the mitochondrial ETC that enables generation of electrochemical gradient needed for the final ATP synthesis.13, 25 It is worth mentioning that each of these enzymatic complexes contains iron in its structure in the form of either haem, which builds haem proteins (cytochromes) present in complexes III and IV, or ISC, which are the parts of ISC proteins in the complexes I, II, and III.13, 25 Because of its ability to exist in two interchangeable oxidative states [the reduced ferrous (Fe 2+) and the oxidized ferric (Fe 3+) forms], iron plays a central role in oxidation–reduction reactions (redox) carried out within mitochondrial ETC.13, 25 Because none of those aforementioned enzymes can efficiently function without iron atoms, this micronutrient is indispensable for the effective oxidative catabolism of both carbohydrates and fats.
Involvement of iron in fat catabolism
Iron‐containing prosthetic groups are not only present in the aforementioned enzymatic complexes but also account for the essential components of molecules that link initial catabolism of fatty acids to OXPHOS to enable the efficient energy acquisition in skeletal muscle. Indeed, an iron–sulfur enzyme, electron‐transferring‐flavoprotein dehydrogenase, was identified to be responsible for the transfer of products of fatty acids oxidation to the mitochondrial ETC.53, 54 Therefore, iron appears to be a microelement unique in its ubiquity in molecular systems of myocytes' energetics.
Possible importance of muscle‐specific regulation of iron metabolism
Taking into consideration the apparent role of iron availability in the efficient energetics of skeletal muscle, one should consider more in‐depth studies on the mechanisms orchestrating iron metabolism within myocytes. Although systemic iron homoeostasis has been extensively studied and can be reviewed elsewhere,55, 56, 57 local iron metabolism in skeletal muscle and its crosstalk with fuel selection and metabolism are still poorly understood. Because the expression of two main regulatory peptides, namely, hepcidin and hemojuvelin, has been confirmed in skeletal muscle,58, 59 therefore, the possibility of involvement of muscle‐specific regulation of iron metabolism in energy metabolism may worth to be discussed and further investigated.
Paragraph 3. Iron deficiency and metabolic alterations in skeletal muscle
Manifold deleterious effects of ID on skeletal muscle include a deranged selection of energy substrates and altered catabolic pathways. Experimental data from in vitro and animal studies indicate that skeletal muscle network of pathways for energy production is hampered by ID at different points of the distinct metabolic routes (Figure 1B).
Alterations in oxidative metabolism: oxidative‐to‐glycolytic shift
Affecting oxidative metabolism on several sites, ID rearranges skeletal muscle energy metabolism, limiting the contribution of the oxidative pathway in favour of glycolysis (Figure 2). Firstly, ID affects the morphology of mitochondria as the density of cristae of the mitochondrial inner membrane is decreased.60 Because these structures are responsible for binding of enzymes of mitochondrial ETC, such an alteration contributes to the mitochondrial oxidative inefficiency. Further, ID dramatically impairs OXPHOS, affecting both oxygen delivery and the final step of substrate catabolism within mitochondrial ETC. Notably, the concentration of myoglobin was decreased in predominantly slow‐ and mixed‐fibre skeletal muscle from iron‐deficient rats as compared with iron‐replete controls.61 ID also caused multifocal decoupling of mitochondrial ETC as the activities of I, II, and IV enzymatic complexes were decreased along with an inhibition of ISC protein maturation and decreased the concentration of cytochromes.62, 63, 64, 65, 66, 67, 68 Furthermore, Graber et al. suggested that reduced amount of intracellular haem could affect myoglobin content, as a haem synthesis inhibitor was able to reduce also a myoglobin level by 40% in rat skeletal muscle cells.69 Apart from derangements in OXPHOS, ID was reported to cause a decrease in level and activity of the key enzyme of Krebs cycle or mitochondrial aconitase, probably via post‐translational regulation, thus limiting the conversion of acetyl coenzyme A for OXPHOS. Finally, Finch et al. described in rat skeletal muscle links between ID and excessive lactate production that could result from impaired OXPHOS and consequent accumulation of product of enhanced glycolysis.70 All of these effects of ID may add to the general decrease in oxidative metabolism efficiency.
Figure 2.
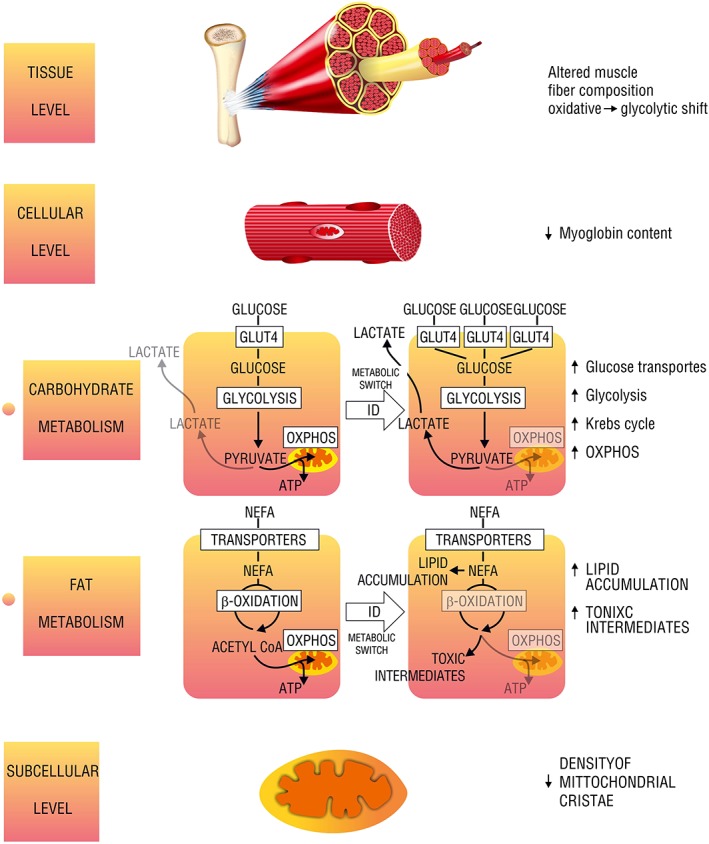
Different levels of muscle energetic alterations caused by iron deficiency. GLUT4, glucose transporter; NEFA, non‐esterified fatty acids; OXPHOS, oxidative phosphorylation; ID, iron deficiency; Acetyl‐CoA, acetyl coenzyme A; ATP, adenosine triphosphate.
Alterations in carbohydrate metabolism
The influence of ID on muscle carbohydrate uptake and utilization is multifaceted. Most of all, experimental data indicate an increased reliance on carbohydrate metabolism as reported for both mildly and severely iron‐deficient rats.71, 72, 73, 74 Several studies investigated the effects of ID on the glucose transporters of skeletal muscle and yielded in partially inconsistent results. In general, ID was proven to increase the expression of muscle glucose transporters. However, skeletal muscle of iron‐deficient rats demonstrated an increased expression of the muscle‐specific GLUT‐4,75 while experimental data from in vitro studies on the myocytes suggested an increase in expression of ubiquitously distributed transporter GLUT‐1. Concerning the intensity of carbohydrate metabolism, Barrientos et al. demonstrated that mice with skeletal muscle‐specific transferrin receptor knockout, which was interpreted as muscle‐specific ID, presented with an upregulation of genes involved in glycolysis along with an enhancement of gluconeogenesis in liver possibly due to increased muscle glucose demand.76
Alterations in fat metabolism
In general, ID is presumed to shift the skeletal muscle reliance from fat to glucose as the preferred metabolic substrate. Davis et al. have reported a significant decrease in the expression of several enzymes involved in the central pathway of muscle fat catabolism, β‐oxidation. Notably, the same study demonstrated an increase in expression of lipogenic genes, which leads to lipid accumulation in skeletal muscle.74 Indeed, an increased abundance of lipid droplets was reported in the skeletal muscles of ID rats.51, 77 Further, in transgenic mice lacking transferrin receptor, the β‐oxidation of fatty acids was impaired, with an accumulation of potentially toxic intermediates.76 It is worth noting that ID was also reported to increase lipid peroxidation in rats,78 leading to the severe cell damage. Therefore, the accumulation mentioned earlier due to ID may yield in the intensified detrimental effects.
The possible mechanism of impaired fuel metabolism in iron‐deficient skeletal muscle
Considering metabolic changes induced by ID in skeletal muscle, it can be concluded that low iron state increases non‐anaerobic glucose metabolism, thus improving glucose uptake and insulin sensitivity in this tissue. Indeed, ID is reported to improve insulin sensitivity in peripheral tissues of iron‐deficient animals71, 79, 80, 81 possibly through an enhanced expression of the glucose transporters,75, 82 but it also triggers less desirable metabolic adaptations, such as hyperglycaemia, hyperinsulinaemia, and hypertriglyceridaemia.73, 74, 77, 80, 83, 84 This phenomenon may result from the ID‐induced deficiencies of mitochondrial ETC, which in turn lead to ineffective carbohydrate and fat oxidative metabolism combined with compensatory increased glucose demand.
Paragraph 4. Links between iron deficiency, loss of oxidative capacity, and functional capacity in patients with chronic diseases accompanied by skeletal myopathy
The decline in muscle strength and quality has emerged as a common pathophysiological feature of HF, COPD, and T2DM, significantly aggravating symptoms and outcomes.1, 3, 85, 86, 87, 88, 89, 90 Skeletal myopathy that accompanies these diseases has been linked to the loss of skeletal muscle oxidative capacity, defined by the ability to oxidize nutrients to obtain energy.11 Macroscopically, structural changes that correlate with the decreased functional muscle capacity can be observed as a reduced muscle mass and volume measured in different body regions.3, 91, 92, 93 Among the skeletal muscle derangements occurred at various levels, from the macroscopic to subcellular, and reported in patients with HF, COPD, and T2DM, many may result from the loss of oxidative capacity.
Histological and ultrastructural alterations in skeletal muscle in chronic diseases
Histological examination of skeletal muscle in patients with HF reveals changes in fibre composition with an increased contribution of fast glycolytic fibres.85, 94 Because the fast muscle fibres rely mainly on anaerobic metabolism, this observation accounts for microscopic evidence on the decreased extent of OXPHOS within skeletal muscle, thus a shift in energy metabolism towards anaerobic route. Further, alterations in skeletal muscle seen at the cellular level involve mostly myocyte energetic centres, namely, mitochondria, and comprise not only a decreased total number and volume of these organelles but also diminished both mitochondrial volume density and surface density of mitochondrial cristae.85, 95 These structural modifications of cellular organelles may lead to severe limitation of oxidative ATP synthesis, which normally takes place exactly on mitochondria cristae. Indeed, the changes as mentioned earlier in mitochondria ultrastructure significantly correlate with decreased oxidative capacity of skeletal muscle,85, 95 suggesting a significant contribution of compromised muscle oxidative metabolism to exercise intolerance seen in patients with HF.
Analogous oxidative‐to‐glycolytic shifts within muscle fibres are observed in both patients with COPD96 and patients with T2DM.97 Skeletal muscle in COPD is characterized by a decreased mitochondrial content and decreased the fractional area of these organelles,98 whereas in T2DM, because muscle mitochondria present with smaller mean size and narrowed cristae, they most likely contribute to skeletal muscle dysfunction seen in diabetic patients.99, 100, 101 Therefore, both in COPD and in T2DM, the molecular centres of oxidative energy production exhibit structural alterations that diminish their ability to bind the oxidative enzymes.
Alterations of oxidative metabolism in skeletal muscle in chronic diseases
In general, oxidative metabolism in skeletal muscle in HF is limited, and the anaerobic glycolysis is enhanced. Concerning oxidative energetics, the efficiency of Krebs cycle in skeletal muscle of patients with HF is decreased because the activities of rate‐limiting enzymes, namely, citrate synthase (CS) and succinate dehydrogenase, are diminished.102, 103 These results suggest a limited substrate flux to OXPHOS and resultant inefficient ATP synthesis. Abnormal oxidative metabolism is coupled with a shift towards rapid energy sources such as high‐energy phosphates or glycolysis, which leads to intracellular acidification.85, 104
Abnormalities in oxidative metabolism of skeletal muscle have also been demonstrated in patients with COPD. The changes include a decrease in activity of CS of Krebs cycle105 and a diminished activity of enzymatic complex IV of mitochondrial ETC.106 Similarly, skeletal muscles in T2DM have decreased the activity of both CS and enzymatic complexes I and IV of mitochondrial ETC.99, 107 Importantly, in both COPD and T2DM, deranged oxidative metabolism coexists with increased activities of enzymes of anaerobic glycolysis.107, 108
Alterations of carbohydrate and fat metabolism in skeletal muscle in chronic diseases
Apart from the general decline in oxidative metabolism, skeletal muscle in HF demonstrates significant changes in fuel selection and their catabolism. For example, the glycogen content is decreased in skeletal muscle of patients with HF.2, 94 This alteration may contribute to the limitation of muscle functional capacity taking into consideration that mitochondrial oxidation is predominantly supplied from intramuscular glycogen droplets.23 On the other hand, fat catabolism is also compromised because skeletal myocytes in HF present with a diminished concentration of an essential enzyme mediating initial catabolism of fatty acids, 3‐hydroxyacyl‐coenzyme A‐dehydrogenase.94 Therefore, oxidative metabolism in skeletal muscle of patients with HF is disturbed not only because of abnormalities within mitochondria but also because of regarding inefficient initial catabolism of two main fuels, carbohydrates and fat. Also, Keith et al. reported that in patients with HF, plasma concentrations of lipid peroxidation products are increased,109 which possibly may result from abnormal mitochondrial functioning, namely, inefficient oxygen reduction coupled with the large accumulation of lipid intermediates being not effectively catabolized in the oxidative pathway. Skeletal muscle in patients with COPD demonstrates similar alterations as the amount of intramuscular glycogen is decreased.110 Furthermore, the accumulation of fat within skeletal muscle in COPD and the enhanced lipid peroxidation may suggest a deranged lipid oxidative metabolism.111, 112 Similarly, in T2DM, lipid accumulation occurs within skeletal muscle as a supposed consequence of impaired mitochondrial oxidative capacity and might contribute to the development of insulin resistance.113, 114, 115
The prevalence of iron deficiency in patients with chronic diseases
Iron deficiency has been recognized as a frequent comorbid condition in patients with HF with the prevalence estimated at 30–60% of those patients.116, 117, 118 In an international pooled cohort of >1500 European patients with HF, ID affected 50% of subjects.117, 119 Previously, we have gathered available evidence on the correlation between ID and impaired functional capacity in patients with HF, as well as on beneficial effects of iron supplementation on physical performance in patients with HF and ID, regardless of the presence of anaemia.16 Moreover, there is evidence on the improvement of myocardial function in patients with HF and ID after iron replacement therapy.120, 121, 122 Indeed, Gaber et al. reported that intravenous (i.v.) iron administration improved diastolic and systolic function as assessed using echocardiographic parameters, such as S′‐wave, E/E′ ratio, and peak systolic strain rate.121 In another small trial, iron repletion caused a correction of left ventricular end‐systolic dimension, left ventricular end‐diastolic dimension, left ventricular end diastolic posterior wall dimension, interventricular septal end diastolic dimension thickness, left ventricular mass index and left ventricular end systolic volume.120 Regarding COPD, several studies refer to the prevalence of ID. For example, among 113 patients with stable, moderately severe COPD, 18 were found to be iron deficient.123 Data from the study mentioned earlier indicate that iron‐deficient patients had more self‐reported exacerbations as well as a trend towards worse exercise tolerance.123 Notably, the higher severity of the disease has been correlated with the greater decline in lung function as assessed using FEV1%FVC.124 In other study, Horadagoda et al.125 detected ID in 38% of 94 consecutive patients with acute exacerbations of COPD. Further, ID has been linked to increased pulmonary artery pressure in a group of 75 non‐anaemic outpatients with COPD (subjects with COPD—41%).126 There are also data from the study of a prospective sample of 70 non‐anaemic patients with COPD where iron‐deficient subjects (48%) demonstrated lower pre‐training aerobic capacity and reduced training‐induced response in comparison with those with normal iron status.127 Concerning T2DM, there is a scarcity of data on the prevalence of ID. In general, a deranged iron metabolism seems to play an important role in the pathophysiology of diabetes.128, 129 Many studies have shown that iron overload contributes to diabetes mellitus,130, 131, 132 but there are also data from a large cohort study indicating that ID is not protective against T2DM.133 Furthermore, ID is highly prevalent (about 39%) in obese and overweight children and adolescents134 as well as in adult men and women.135, 136, 137 Indeed, ID is associated with the major risk factor for diabetes, obesity, which is causally related to the decrease in the ability of effective fat catabolism.136, 138, 139 Further, ID has been proposed to participate in obesity‐related inflammation.129 Importantly, there is an ongoing randomized placebo‐controlled clinical trial investigating the hypothesis that i.v. substitution with ferric carboxymaltose reduces HbA1c levels in patients with type 2 diabetes and ID, thereby improving metabolic status and quality of life.140
The mechanism of development of iron deficiency in patients with chronic diseases
The mechanism in which patients with chronic diseases develop ID is not fully understood. Regarding chronic illnesses assisted by inflammation, such as CKD, infections, cancer, and autoimmune diseases, the prevalent literature emphasizes the role of inflammatory‐driven up‐regulation of hepcidin, which presumably leads to functional ID or anaemia.141, 142, 143, 144, 145, 146 However, in case of HF, the mechanism seems to be different, because the level of hepcidin in patients with systolic HF is reported to be associated with low circulating pro‐inflammatory markers, being related neither to the presence of anaemia nor to haemoglobin level.147 Therefore, it has been proposed that in the initial phase of HF, hepcidin is high, which is probably related to its protective role against iron excess manifested by elevated serum ferritin.147 However, the prolonged up‐regulation of hepcidin inhibits iron absorption and release, thus leads to the development of ID with manifestation of its deleterious clinical consequences. In the end, ID represses hepcidin production to the bloodstream.147 It is worth noting that although the aforementioned mechanism has been postulated, it has been tested neither in the context of other factors influencing hepcidin expression nor in terms of time course or risk factors of ID development. In case of COPD and T2DM, there is lack of data proposing mechanism and indicating the time course of ID development.
Iron deficiency as a potential cause of impaired skeletal muscle energetics in chronic diseases
According to the so‐called muscle hypothesis, abnormalities occurring in structure and functioning of skeletal muscles include significant metabolic derangements and are to be directly responsible for impaired exercise capacity in patients with HF.148 Similar intrinsic metabolic alterations within skeletal muscle with possible important impact on exercise capacity are observed in COPD and T2DM [see above]. Physiologically, metaboreceptors (the kind of muscle afferents) are stimulated by products of skeletal muscle work, which in turn inform the brain stem on the level of muscle activity. The response to such metaboreflex involves sympathetic activation and consequent ventilatory, haemodynamic, and physiological adaptations to meet the muscle nutritional requirements.149, 150, 151 Abnormally large metaboreceptor response, however, contributes to the experience of dyspnoea and leads to the disordered exercise physiology. Such phenomenon has already been demonstrated in HF,152, 153 and it has been postulated in COPD154 where it needs further in‐depth investigation. Although the excessive activation of ergoreceptors has not been evaluated in T2DM, its contribution to exercise intolerance may be hypothesized as skeletal muscle in diabetes has the oxidative capacity limited with the accumulation of anaerobic metabolites.
It is worth noting that ID has been postulated as an important causative factor that can significantly contribute to the loss of skeletal muscle oxidative capacity seen in patients with HF, COPD, or T2DM.11 Therefore, it is possible that ID exerts its detrimental effects by limiting the oxidative metabolism and consequent accumulation of anaerobic metabolites, which in turn contributes to the exaggerated ergoreflex response, thus to exercise limitation. Skeletal myopathy that occurs in response to ID and resultant energetic impasse may constitute a potential pathophysiological link between disturbed iron status and diminished exercise capacity in patients with aforementioned chronic diseases. It is worth noting that ID predominantly damages skeletal muscle energetics, which is reported to be disturbed at similar points in certain chronic syndromes (Figure 3). However, there is still a lack of direct experimental or clinical evidence to support this hypothesis.
Figure 3.
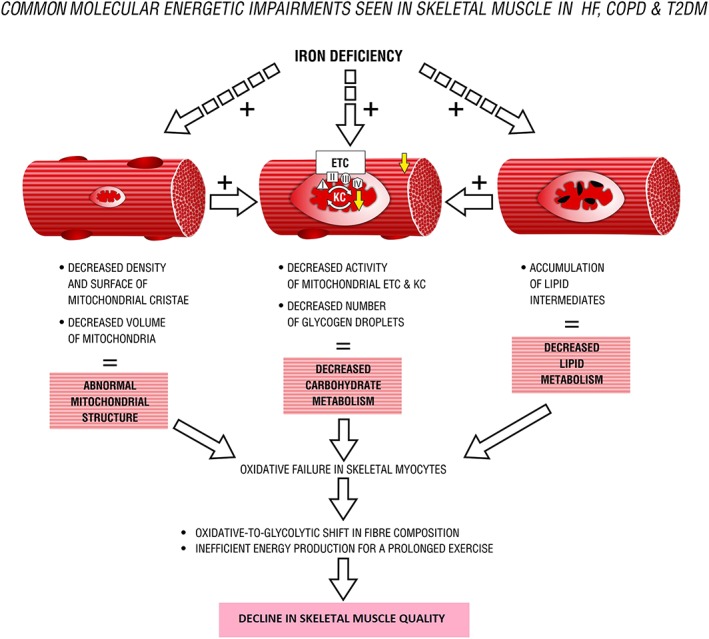
Common molecular energetic impairments seen in skeletal muscle in heart failure (HF), chronic obstructive pulmonary disease (COPD), and type 2 diabetes mellitus (T2DM). ETC, electron transport chain; KC, Krebs cycle; I–IV, mitochondrial enzymatic complexes.
To date, mechanistic studies on ID and resultant decreased exercise performance have been performed predominantly in animal models, while data regarding the relationship between ID and skeletal muscle dysfunction in iron‐deficient anaemic human subjects are limited and inconsistent (for a detailed review, see Stugiewicz et al.16). With regard to oxidative capacity, although there is lack of mechanistic studies investigating the effects of iron supplementation on skeletal muscle energetics, clinical findings show that iron administration to iron‐deficient both untrained subjects and athletes improves muscle energetic efficiency.155, 156 Therefore, further studies are needed to establish the pathophysiological links between ID and skeletal muscle dysfunction observed in HF, COPD, and T2DM, to step forward in co‐targeting of muscle abnormalities in a therapeutic process.
Iron therapy in chronic diseases
It is worth noting that to date iron therapy has already been tested in anaemic and/or iron‐deficient patients. Particularly, i.v. iron replacement, as compared with oral iron or inactive controls, is reported to be effective in improving both haemoglobin levels together with reduction in blood transfusion rates and quality of life in anaemic adults without CKD.157With regard to iron‐deficient patients with HF, irrespective of concomitant anaemia, i.v. iron is associated with an improvement in exercise capacity, clinical status, quality of life, and significant reduction in the risk of hospitalizations for worsening HF.158, 159 Based on two clinical trials (FAIR‐HF and CONFIRM‐HF), i.v. ferric carboxymaltose has been recommended for treatment of ID in symptomatic patients with HF and left ventricular ejection fraction <45%.159, 160, 161 Importantly, in acutely decompensated HF, ID has been also recognized as a highly prevalent comorbidity that should be monitored (especially regarding not stationary character of iron status in those patients) and managed.162 Considering other therapeutic approaches, because myocardium of HF patients has decreased iron content, which may significantly contribute to the existing mitochondrial dysfunction,163 novel myocardial‐targeted therapies should be designed and developed. In case of COPD, data on iron therapy are limited.164 In a small retrospective study, i.v. iron together with erythropoiesis‐stimulating agents has been reported to improve anaemia and ID and that has been associated with a significant improvement in self‐assessed shortness of breath.164 Regarding T2DM, in the ongoing clinical trial, i.v. iron is tested in the context of metabolic status and quality of life in iron‐deficient patients with T2DM.140 Apparently, iron therapy has been investigated more deeply in HF patients, and there is a need of more in‐depth studies on iron replacement in other diseases, such as COPD or T2DM.
Of note, several current therapeutic strategies aim to target muscle mitochondrial energetics in order to enhance metabolic efficiency, and an optimal iron supply may play an important role in an adequate response to such attempts. For example, the utilization of trimetazidine as a metabolic modulator that acts by re‐programming muscle metabolism towards the enhanced glucose oxidative catabolism has been tested in animal models.165, 166 Importantly, the aforementioned agent is reported to induce a fast‐to‐slow shift in fibre composition and to restore oxidative phenotype of muscle.165, 166 Similarly, another muscle‐targeted compound, namely, acylated‐ghrelin, is proposed to normalize mitochondrial oxidative capacity.167 Thus, both the two molecules mentioned earlier are meant to increase muscle oxidative capacity, which in turn is crucially dependent on optimal iron availability. In case of other experimental attempt, Inoue et al. has reported beneficial effects of exercise train on ageing mice skeletal muscle in the context of improvement metabolic and mitochondrial impairments.168 Taking into consideration the fundamental role of iron in both exercise capacity16 and mitochondrial functioning, it may be hypothesized that undisturbed iron metabolism may be of particular importance for the adequate response to the therapy.
Conclusions
Evidence gathered from animal and in vitro studies indicates that ID damages skeletal muscle energetics at different levels. Skeletal muscle dysfunction causally linked to the impaired cellular energy metabolism has been recognized as an important pathophysiological feature in chronic diseases, such as HF, COPD, and T2DM. Although the unfavourable influence of ID on the energy metabolism has been postulated in the syndromes mentioned earlier, the hypothesis of whether abnormal iron homoeostasis contributes to the skeletal muscle derangements still needs to be verified. Therefore, further studies are indispensable to investigate the clinical correlations between ID and skeletal muscle dysfunction in chronic diseases.
Conflict of interest
Wrocław Medical University received an unrestricted grant from Vifor Pharma outside the submitted work. M.K. reports financial support from Vifor Pharma for travel and accommodation for scientific meeting. W.B. reports personal fees from Vifor Pharma, outside the submitted work. P.P. reports personal fees from Vifor Pharma, personal fees from AMGEN, outside the submitted work. E.A.J. reports personal fees from Vifor Pharma and FRESENIUS, outside the submitted work. All the other authors report no conflict of interest related to the content of this manuscript.
Acknowledgements
This research was financially supported by the National Science Centre (Kraków, Poland) grant allocated on the basis of the decision number DEC‐2012/05/E/NZ5/00590. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.169
Dziegala, M. , Josiak, K. , Kasztura, M. , Kobak, K. , von Haehling, S. , Banasiak, W. , Anker, S. D. , Ponikowski, P. , and Jankowska, E. (2018) Iron deficiency as energetic insult to skeletal muscle in chronic diseases. Journal of Cachexia, Sarcopenia and Muscle, 9: 802–815. 10.1002/jcsm.12314.
References
- 1. Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole‐Wilson PA. Symptoms and quality of life in heart failure: the muscle hypothesis. Br Heart J 1994;72:S36–S399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gosker HR, Wouters EF, van der Vusse GJ, Schols AM. Skeletal muscle dysfunction in chronic obstructive pulmonary disease and chronic heart failure: underlying mechanisms and therapy perspectives. Am J Clin Nutr 2000;71:1033–104747. [DOI] [PubMed] [Google Scholar]
- 3. Volpato S, Bianchi L, Lauretani F, Lauretani F, Bandinelli S, Guralnik JM, et al. Role of muscle mass and muscle quality in the association between diabetes and gait speed. Diabetes Care 2012;35:1672–16799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers—update 2014. J Cachexia Sarcopenia Muscle 2014;5:261–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dasarathy S. Cause and management of muscle wasting in chronic liver disease. Curr Opin Gastroenterol 2016;32:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roshanravan B, Gamboa J, Wilund K. Exercise and CKD: skeletal muscle dysfunction and practical application of exercise to prevent and treat physical impairments in CKD. Am J Kidney Dis 2017;69:837–85252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, et al. Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 2017;8:735–74747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verzola D, Bonanni A, Sofia A, Montecucco F, D'’Amato E, Cademartori V, et al. Toll‐like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J Cachexia Sarcopenia Muscle 2017;8:131–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huffman KM, Jessee R, Andonian B, Davis BN, Narowski R, Huebner JL, et al. Molecular alterations in skeletal muscle in rheumatoid arthritis are related to disease activity, physical inactivity, and disability. Arthritis Res Ther 2017;19:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhanji RA, Narayanan P, Allen AM, Malhi H, Watt KD. Sarcopenia in hiding: the risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology 2017;66:2055–206565. [DOI] [PubMed] [Google Scholar]
- 11. Leermakers PA, Gosker HR. Skeletal muscle mitophagy in chronic disease. Curr Opin Clin Nutr Metab Care 2016;19:427–43333. [DOI] [PubMed] [Google Scholar]
- 12. Beard JL. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr 2001;131:568S–579S, discussion 580S. [DOI] [PubMed] [Google Scholar]
- 13. Galy B, Ferring‐Appel D, Sauer SW, Kaden S, Lyoumi S, Puy H, et al. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab 2010;12:194–201. [DOI] [PubMed] [Google Scholar]
- 14. Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 2013;61:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puntarulo S. Iron, oxidative stress and human health. Mol Asp Med 2005;26:299–312. [DOI] [PubMed] [Google Scholar]
- 16. Stugiewicz M, Tkaczyszyn M, Kasztura M, Banasiak W, Ponikowski P, Jankowska EA. The influence of iron deficiency on the functioning of skeletal muscles: experimental evidence and clinical implications. Eur J Heart Fail 2016;18:762–77373. [DOI] [PubMed] [Google Scholar]
- 17. Hänninen O, Atalay M. Oxidative metabolism in skeletal muscle In Oxidative Stress Skelet. Muscle. Basel: Birkhäuser Basel; 1998. p 29–42. [Google Scholar]
- 18. Westerblad H, Bruton JD, Katz A. Skeletal muscle: energy metabolism, fiber types, fatigue and adaptability. Exp Cell Res 2010;316:3093–30999. [DOI] [PubMed] [Google Scholar]
- 19. Bottinelli R, Reggiani C. Human skeletal muscle fibres: molecular and functional diversity. Prog Biophys Mol Biol 2000;73:195–262. [DOI] [PubMed] [Google Scholar]
- 20. Atalay M, Hänninen OOP. Muscle energy metabolism. Encycl Life Support Syst Physiol Maint 2010;4:26–47. [Google Scholar]
- 21. Spriet LL. Anaerobic metabolism in human skeletal muscle during short‐term, intense activity. Can J Physiol Pharmacol 1992;70:157–16565. [DOI] [PubMed] [Google Scholar]
- 22. Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Phys TherPhysiology 2005;20:260–27070. [DOI] [PubMed] [Google Scholar]
- 23. Hoppeler H. Skeletal muscle substrate metabolism. Int J Obes Relat Metab Disord 1999;23:S7–S1010. [DOI] [PubMed] [Google Scholar]
- 24. Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. Role of fatty acid uptake and fatty acid β‐oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta Mol Cell Biol Lipids 1801;2010:1–22. [DOI] [PubMed] [Google Scholar]
- 25. Murray R, Granner D, Rodwell V. Harper's Illustrated Biochemistry, 27th ed. New York: The McGraw‐Hill Companies; n.d. . [Google Scholar]
- 26. Katz A, Broberg S, Sahlin K, Wahren J. Leg glucose uptake during maximal dynamic exercise in humans. Am J Physiol Endocrinol Metab 1986;251: E65, E70. [DOI] [PubMed] [Google Scholar]
- 27. Williams BD, Plag I, Troup J, Wolfe RR. Isotopic determination of glycolytic flux during intense exercise in humans. J Appl Physiol 1995;78:483–490. [DOI] [PubMed] [Google Scholar]
- 28. Ahlborg G, Felig P, Hagenfeldt L, Hendler R, Wahren J. Substrate turnover during prolonged exercise in man. Splanchnic and leg metabolism of glucose, free fatty acids, and amino acids. J Clin Invest 1974;53:1080–109090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coderre L, Kandror KV, Vallega G, Pilch PF. Identification and characterization of an exercise‐sensitive pool of glucose transporters in skeletal muscle. J Biol Chem 1995;270:27584–275888. [DOI] [PubMed] [Google Scholar]
- 30. Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5' AMP‐activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998;47:1369–137373. [DOI] [PubMed] [Google Scholar]
- 31. Terada S, Muraoka I, Tabata I. Changes in [Ca2+]i induced by several glucose transport‐enhancing stimuli in rat epitrochlearis muscle. J Appl Physiol 2003;94:1813–1820. [DOI] [PubMed] [Google Scholar]
- 32. Sahlin K, Tonkonogi M, Soderlund K. Energy supply and muscle fatigue in humans. Acta Physiol Scand 1998;162:261–2666. [DOI] [PubMed] [Google Scholar]
- 33. Li Y, Xu S, Zhang X, Yi Z, Cichello S. Skeletal intramyocellular lipid metabolism and insulin resistance. Biophys Reports 2015;1:90–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 1999;48:1113–11199. [DOI] [PubMed] [Google Scholar]
- 35. Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, et al. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 2001;50:2579–258484. [DOI] [PubMed] [Google Scholar]
- 36. Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance‐trained athletes. J Clin Endocrinol Metab 2001;86:5755–576161. [DOI] [PubMed] [Google Scholar]
- 37. Jiménez‐Caballero PE, Mollejo‐Villanueva M, Alvarez‐Tejerina A. Mitochondrial encephalopathy due to complex I deficiency. Brain tissue biopsy findings and clinical course following pharmacological. Rev Neurol 2008;47:27–30. [PubMed] [Google Scholar]
- 38. Anastasiou CA, Kavouras SA, Lentzas Y, Gova A, Sidossis LS, Melidonis A. Diabetes mellitus is associated with increased intramyocellular triglyceride, but not diglyceride, content in obese humans. Metabolism 2009;58:1636–164242. [DOI] [PubMed] [Google Scholar]
- 39. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty‐acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (London, England) 1963;1:785–7899. [DOI] [PubMed] [Google Scholar]
- 40. Randle PJ. Fuel selection in animals. Biochem Soc Trans 1986;14:799–806. [DOI] [PubMed] [Google Scholar]
- 41. Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 2000;49:677–68383. [DOI] [PubMed] [Google Scholar]
- 42. Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 1998;14:263–28383. [DOI] [PubMed] [Google Scholar]
- 43. Frayn KN, Arner P, Yki‐Järvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem 2006;42:89–103. [DOI] [PubMed] [Google Scholar]
- 44. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. AJP Endocrinol Metab 2009;297:E578–E59191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol 2006;574:95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boulton FE. The myoglobin content of human skeletal muscle. Br J Haematol 1973;25:281. [PubMed] [Google Scholar]
- 47. Robach P, Cairo G, Gelfi C, Bernuzzi F, Pilegaard H, Vigano A, et al. Strong iron demand during hypoxia‐induced erythropoiesis is associated with down‐regulation of iron‐related proteins and myoglobin in human skeletal muscle. Blood 2007;109:4724–473131. [DOI] [PubMed] [Google Scholar]
- 48. Ohira Y, Gill SL. Effects of dietary iron deficiency on muscle fiber characteristics and whole‐body distribution of hemoglobin in mice. J Nutr 1983;113:1811–18188. [DOI] [PubMed] [Google Scholar]
- 49. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta, Mol Cell Res 1823;2012:1468–148383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo B, Yu Y, Leibold EA. Iron regulates cytoplasmic levels of a novel iron‐responsive element‐binding protein without aconitase activity. J Biol Chem 1994;269:24252–2426060. [PubMed] [Google Scholar]
- 51. Ross KL, Eisenstein RS. Iron deficiency decreases mitochondrial aconitase abundance and citrate concentration without affecting tricarboxylic acid cycle capacity in rat liver. J Nutr 2002;132:643–65151. [DOI] [PubMed] [Google Scholar]
- 52. Liew Y‐F, Shaw N‐S. Mitochondrial cysteine desulfurase iron‐sulfur cluster S and aconitase are post‐transcriptionally regulated by dietary iron in skeletal muscle of rats. J Nutr 2005;135:2151–21588. [DOI] [PubMed] [Google Scholar]
- 53. Ruzicka FJ, Beinert H. A new iron‐sulfur flavoprotein of the respiratory chain. A component of the fatty acid beta oxidation pathway. J Biol Chem 1977;252:8440–84455. [PubMed] [Google Scholar]
- 54. Zhang J, Frerman FE, Kim J‐JP. Structure of electron transfer flavoprotein‐ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc Natl Acad Sci 2006;103:16212–162177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell 2010;142:24–38. [DOI] [PubMed] [Google Scholar]
- 56. Zhang A‐S. Control of systemic iron homeostasis by the hemojuvelin–hepcidin axis. Adv Nutr 2010;1:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finberg KE. Regulation of systemic iron homeostasis. Curr Opin Hematol 2013;20:208–21414. [DOI] [PubMed] [Google Scholar]
- 58. Polonifi A, Politou M, Kalotychou V, Xiromeritis K, Tsironi M, Berdoukas V, et al. Iron metabolism gene expression in human skeletal muscle. Blood Cells Mol DisBlood Cells, Mol Dis 2010;45:233–2377. [DOI] [PubMed] [Google Scholar]
- 59. Gkouvatsos K, Wagner J, Papanikolaou G, Sebastiani G, Pantopoulos K. Conditional disruption of mouse HFE2 gene: maintenance of systemic iron homeostasis requires hepatic but not skeletal muscle hemojuvelin. Hepatology 2011;54:1800–18077. [DOI] [PubMed] [Google Scholar]
- 60. Cartier LJ, Ohira Y, Chen M, Cuddihee RW, Holloszy JO. Perturbation of mitochondrial composition in muscle by iron deficiency. Implications regarding regulation of mitochondrial assembly. J Biol Chem 1986;261:13827–1383232. [PubMed] [Google Scholar]
- 61. Hagler L, Askew EW, Neville JR, Mellick PW, Coppes RI, Lowder JF. Influence of dietary iron deficiency on hemoglobin, myoglobin, their respective reductases, and skeletal muscle mitochondrial respiration. Am J Clin Nutr 1981;34:2169–217777. [DOI] [PubMed] [Google Scholar]
- 62. Finch CA, Miller LR, Inamdar AR, Person R, Seiler K, Mackler B. Iron deficiency in the rat. Physiological and biochemical studies of muscle dysfunction. J Clin Invest 1976;58:447–45353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maguire JJ, Davies KJA, Dallman PR, Packer L. Effects of dietary iron deficiency on iron–sulfur proteins and bioenergetic functions of skeletal muscle mitochondria. Biochim Biophys Acta Bioenerg 1982;679:210–22020. [DOI] [PubMed] [Google Scholar]
- 64. Mackiler B, Grace R, Finch CA. Iron deficiency in the rat: effects of oxidative metabolism in distinct types of skeletal muscle. Pediatr Res 1984;18:499–500. [DOI] [PubMed] [Google Scholar]
- 65. Ackrell BA, Maguire JJ, Dallman PR, Kearney EB. Effect of iron deficiency on succinate‐ and NADH‐ubiquinone oxidoreductases in skeletal muscle mitochondria. J Biol Chem 1984;259:10053–100599. [PubMed] [Google Scholar]
- 66. Puig S, Vergara SV, Thiele DJ. Cooperation of two mRNA‐binding proteins drives metabolic adaptation to iron deficiency. Cell Metab 2008;7:555–56464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zick M, Rabl R, Reichert AS. Cristae formation—linking ultrastructure and function of mitochondria. Biochim Biophys Acta, Mol Cell Res 1793;2009:5–19. [DOI] [PubMed] [Google Scholar]
- 68. Merrill JF, Thomson DM, Hardman SE, Hepworth SD, Willie S, Hancock CR. Iron deficiency causes a shift in AMP‐activated protein kinase (AMPK) subunit composition in rat skeletal muscle. Nutr Metab (Lond) 2012;9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Graber SG, Woodworth RC. Myoglobin expression in L6 muscle cells. Role of differentiation and heme. J Biol Chem 1986;261:9150–91544. [PubMed] [Google Scholar]
- 70. Finch CA, Gollnick PD, Hlastala MP, Miller LR, Dillmann E, Mackler B. Lactic acidosis as a result of iron deficiency. J Clin Invest 1979;64:129–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brooks GA, Henderson SA, Dallman PR. Increased glucose dependence in resting, iron‐deficient rats. Am J Phys 1987;253:E461–E4666. [DOI] [PubMed] [Google Scholar]
- 72. Linderman JK, Dallman PR, Rodriguez RE, Brooks GA. Lactate is essential for maintenance of euglycemia in iron‐deficient rats at rest and during exercise. Am J Phys 1993;264:E662–E6677. [DOI] [PubMed] [Google Scholar]
- 73. Linderman JK, Brooks GA, Rodriguez RE, Dallman PR. Maintenance of euglycemia is impaired in gluconeogenesis‐inhibited iron‐deficient rats at rest and during exercise. J Nutr 1994;124:2131–21388. [DOI] [PubMed] [Google Scholar]
- 74. Davis MR, Rendina E, Peterson SK, Lucas EA, Smith BJ, Clarke SL. Enhanced expression of lipogenic genes may contribute to hyperglycemia and alterations in plasma lipids in response to dietary iron deficiency. Genes Nutr 2012;7:415–42525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mehdad A, Campos NA, Arruda SF, Siqueira EM. Iron deprivation may enhance insulin receptor and Glut4 transcription in skeletal muscle of adult rats. J Nutr Health Aging 2015;19:846–85454. [DOI] [PubMed] [Google Scholar]
- 76. Barrientos T, Laothamatas I, Koves TR, Soderblom EJ, Bryan M, Moseley MA, et al. Metabolic catastrophe in mice lacking transferrin receptor in muscle. EBioMedicine 2015;2:1705–171717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sherman AR, Guthrie HA, Wolinsky I, Zulak IM. Iron deficiency hyperlipidemia in 18‐day‐old rat pups: effects of milk lipids, lipoprotein lipase, and triglyceride synthesis. J Nutr 1978;108:152–16262. [DOI] [PubMed] [Google Scholar]
- 78. Knutson MD, Walter PB, Ames BN, Viteri FE. Both iron deficiency and daily iron supplements increase lipid peroxidation in rats. J Nutr 2000;130:621–6288. [DOI] [PubMed] [Google Scholar]
- 79. Farrell PA, Beard JL, Druckenmiller M. Increased insulin sensitivity in iron‐deficient rats. J Nutr 1988;118:1104–1109. [DOI] [PubMed] [Google Scholar]
- 80. Borel MJ, Beard JL, Farrell PA. Hepatic glucose production and insulin sensitivity and responsiveness in iron‐deficient anemic rats. Am J Phys 1993;264:E380–E39090. [DOI] [PubMed] [Google Scholar]
- 81. Han D‐H, Hancock CR, Jung SR, Higashida K, Kim SH, Holloszy JO. Deficiency of the mitochondrial electron transport chain in muscle does not cause insulin resistance. PLoS One 2011;6:e19739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Potashnik R, Kozlovsky N, Ben‐Ezra S, Rudich A, Bashan N. Regulation of glucose transport and GLUT‐1 expression by iron chelators in muscle cells in culture. Am J Phys 1995;269:E1052–E10588. [DOI] [PubMed] [Google Scholar]
- 83. Amine E, Desilets E, Hegsted D. Effect of dietary fats on lipogenesis in iron deficiency anemic chicks and rats. J Nutr 1976;106:405–411. [Google Scholar]
- 84. Yamagishi H, Okazaki H, Shimizu M, Izawa T, Komabayashi T. Relationships among serum triacylglycerol, fat pad weight, and lipolysis in iron‐deficient rats. J Nutr Biochem 2000;11:455–46060. [DOI] [PubMed] [Google Scholar]
- 85. Zizola C, Schulze PC. Metabolic and structural impairment of skeletal muscle in heart failure. Heart Fail Rev 2013;18:623–63030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigaré R, et al. An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014;189:e15–e6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schols AMWJ, Broekhuizen R, Weling‐Scheepers CA, Wouters EF. Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr 2005;82:53–599. [DOI] [PubMed] [Google Scholar]
- 88. Tkaczyszyn M, Drozd M, Węgrzynowska‐Teodorczyk K, Flinta I, Kobak K, Banasiak W, et al. Depleted iron stores are associated with inspiratory muscle weakness independently of skeletal muscle mass in men with systolic chronic heart failure. J Cachexia Sarcopenia Muscle 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle 2017;8:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Doehner W, Turhan G, Leyva F, Rauchhaus M, Sandek A, Jankowska EA, et al. Skeletal muscle weakness is related to insulin resistance in patients with chronic heart failure. ESC Hear Fail 2015;2:85–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Piepoli MF, Kaczmarek A, Francis DP, Davies LC, Rauchhaus M, Jankowska EA, et al. Reduced peripheral skeletal muscle mass and abnormal reflex physiology in chronic heart failure. Circulation 2006;114:126–13434. [DOI] [PubMed] [Google Scholar]
- 92. Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, et al. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation 1992;85:1364–137373. [DOI] [PubMed] [Google Scholar]
- 93. Bernard S, LeBlanc P, Whittom F, Carrier G, Jobin J, Belleau R, Maltais F. Peripheral muscle weakness in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;158:629–63434. [DOI] [PubMed] [Google Scholar]
- 94. Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long‐term heart failure. Circulation 1990;81:518–52727. [DOI] [PubMed] [Google Scholar]
- 95. Drexler H, Riede U, Münzel T, König H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation 1992;85:1751–17599. [DOI] [PubMed] [Google Scholar]
- 96. Gosker HR, Zeegers MP, Wouters EFM, Schols AMWJ. Muscle fibre type shifting in the vastus lateralis of patients with COPD is associated with disease severity: a systematic review and meta‐analysis. Thorax 2007;62:944–9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gaster M, Staehr P, Beck‐Nielsen H, Schrøder HD, Handberg A. GLUT4 is reduced in slow muscle fibers of type 2 diabetic patients: is insulin resistance in type 2 diabetes a slow, type 1 fiber disease? Diabetes 2001;50:1324–13299. [DOI] [PubMed] [Google Scholar]
- 98. Gosker HR, Hesselink MKC, Duimel H, Ward KA, Schols AMWJ. Reduced mitochondrial density in the vastus lateralis muscle of patients with COPD. Eur Respir J 2007;30:73–799. [DOI] [PubMed] [Google Scholar]
- 99. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–295050. [DOI] [PubMed] [Google Scholar]
- 100. Bianchi L, Volpato S. Muscle dysfunction in type 2 diabetes: a major threat to patient'’s mobility and independence. Acta Diabetol 2016;53:879–88989. [DOI] [PubMed] [Google Scholar]
- 101. Kaushik S, Singh R, Cuervo AM. Autophagic pathways and metabolic stress. Diabetes Obes MetabDiabetes, Obes Metab 2010;12:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mettauer B, Zoll J, Sanchez H, Lampert E, Ribera F, Veksler V, et al. Oxidative capacity of skeletal muscle in heart failure patients versus sedentary or active control subjects. J Am Coll Cardiol 2001;38:947–95454. [DOI] [PubMed] [Google Scholar]
- 103. Mancini DM, Coyle E, Coggan A, Beltz J, Ferraro N, Montain S, et al. Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 1989;80:1338–134646. [DOI] [PubMed] [Google Scholar]
- 104. Mangner N, Weikert B, Bowen TS, Sandri M, Höllriegel R, Erbs S, et al. Skeletal muscle alterations in chronic heart failure: differential effects on quadriceps and diaphragm. J Cachexia Sarcopenia Muscle 2015;6:381–39090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Picard M, Godin R, Sinnreich M, Baril J, Bourbeau J, Perrault H, et al. The mitochondrial phenotype of peripheral muscle in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008;178:1040–10477. [DOI] [PubMed] [Google Scholar]
- 106. Naimi AI, Bourbeau J, Perrault H, Baril J, Wright‐Paradis C, Rossi A, Taivassalo T, Sheel AW, Rabøl R, Dela F, Boushel R. Altered mitochondrial regulation in quadriceps muscles of patients with COPD. Clin Physiol Funct Imaging 2010;31:124–131. [DOI] [PubMed] [Google Scholar]
- 107. Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J 1999;13:2051–206060. [DOI] [PubMed] [Google Scholar]
- 108. Green HJ, Bombardier E, Burnett M, Iqbal S, D'’Arsigny CL, O'’Donnell DE, Ouyang J, Webb KA. Organization of metabolic pathways in vastus lateralis of patients with chronic obstructive pulmonary disease. AJP Regul Integr Comp Physiol 2008;295:R935–R94141. [DOI] [PubMed] [Google Scholar]
- 109. Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, et al. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol 1998;31:1352–13566. [DOI] [PubMed] [Google Scholar]
- 110. Jakobsson P, Jorfeldt L, von Schenck H. Insulin resistance is not exhibited by advanced chronic obstructive pulmonary disease patients. Clin Physiol 1995;15:547–55555. [DOI] [PubMed] [Google Scholar]
- 111. Maddocks M, Shrikrishna D, Vitoriano S, Natanek SA, Tanner RJ, Hart N, et al. Skeletal muscle adiposity is associated with physical activity, exercise capacity and fibre shift in COPD. Eur Respir J 2014;44:1188–119898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Boots AW, Haenen GRMM, Bast A. Oxidant metabolism in chronic obstructive pulmonary disease. Eur Respir J Suppl 2003;46:14s–27s. [DOI] [PubMed] [Google Scholar]
- 113. Schmitz‐Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin‐stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 1999;274:24202–2421010. [DOI] [PubMed] [Google Scholar]
- 114. He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 2001;50:817–82323. [DOI] [PubMed] [Google Scholar]
- 115. Nielsen J, Mogensen M, Vind BF, Sahlin K, Hojlund K, Schroder HD, Ortenblad N. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. AJP Endocrinol Metab 2010;298:E706–E71313. [DOI] [PubMed] [Google Scholar]
- 116. Cohen‐Solal A, Damy T, Terbah M, Kerebel S, Baguet J‐P, Hanon O, et al. High prevalence of iron deficiency in patients with acute decompensated heart failure. Eur J Heart Fail 2014;16:984–99191. [DOI] [PubMed] [Google Scholar]
- 117. Klip IT, Comin‐Colet J, Voors AA, Ponikowski P, Enjuanes C, Banasiak W, Lok DJ, Rosentryt P, Torrens A, Polonski L, van Veldhuisen DJ, van der Meer P, Jankowska EA. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013;165:575–582.e3. [DOI] [PubMed] [Google Scholar]
- 118. Yeo TJ, Yeo PSD, Ching‐Chiew Wong R, Ong HY, Leong KTG, Jaufeerally F, Sim D, Santhanakrishnan R, Lim SL, M Y Chan M, Chai P, Low AF, Ling LH, Ng TP, Richards AM, Lam CS. Iron deficiency in a multi‐ethnic Asian population with and without heart failure: prevalence, clinical correlates, functional significance and prognosis. Eur J Heart Fail 2014;16:1125–113232. [DOI] [PubMed] [Google Scholar]
- 119. Klip IT, Jankowska EA, Enjuanes C, Voors AA, Banasiak W, Bruguera J, Rozentryt P, Polonski L, van Veldhuisen DJ, Ponikowski P, Comin‐Colet J, van der Meer P. The additive burden of iron deficiency in the cardiorenal‐anaemia axis: scope of a problem and its consequences. Eur J Heart Fail 2014;16:655–66262. [DOI] [PubMed] [Google Scholar]
- 120. Usmanov RI, Zueva EB, Silverberg DS, Shaked M. Intravenous iron without erythropoietin for the treatment of iron deficiency anemia in patients with moderate to severe congestive heart failure and chronic kidney insufficiency. J Nephrol n.d.;21:236–24242. [PubMed] [Google Scholar]
- 121. Gaber R, Kotb NA, Ghazy M, Nagy HM, Salama M, Elhendy A. Tissue Doppler and strain rate imaging detect improvement of myocardial function in iron deficient patients with congestive heart failure after iron replacement therapy. Echocardiography 2012;29:13–188. [DOI] [PubMed] [Google Scholar]
- 122. Núñez J, Monmeneu JV, Mollar A, Núñez E, Bodí V, Miñana G, et al. Left ventricular ejection fraction recovery in patients with heart failure treated with intravenous iron: a pilot study. ESC Hear Fail 2016;3:293–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Nickol AH, Frise MC, Cheng H‐Y, McGahey A, McFadyen BM, Harris‐Wright T, et al. A cross‐sectional study of the prevalence and associations of iron deficiency in a cohort of patients with chronic obstructive pulmonary disease. BMJ Open 2015;5:e007911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Watson L, Vonk JM, Löfdahl CG, Pride NB, Pauwels RA, Laitinen LA, et al. Predictors of lung function and its decline in mild to moderate COPD in association with gender: results from the Euroscop study. Respir Med 2006;100:746–75353. [DOI] [PubMed] [Google Scholar]
- 125. Horadagoda C, Dinihan T, Roberts M, Kairaitis K. Body composition and micronutrient deficiencies in patients with an acute exacerbation of chronic obstructive pulmonary disease. Intern Med J 2017;47:1057–106363. [DOI] [PubMed] [Google Scholar]
- 126. Plesner LL, Schoos MM, Dalsgaard M, Goetze JP, Kjøller E, Vestbo J, et al. Iron deficiency in COPD associates with increased pulmonary artery pressure estimated by echocardiography. Heart Lung Circ 2017;26:101–1044.27372430 [Google Scholar]
- 127. Barberan‐Garcia A, Rodríguez DA, Blanco I, Gea J, Torralba Y, Arbillaga‐Etxarri A, et al. Non‐anaemic iron deficiency impairs response to pulmonary rehabilitation in COPD. Respirology 2015;20:1089–109595. [DOI] [PubMed] [Google Scholar]
- 128. Altamura S, Kopf S, Schmidt J, Müdder K, da Silva AR, Nawroth P, et al. Uncoupled iron homeostasis in type 2 diabetes mellitus. J Mol Med 2017;95:1387–139898. [DOI] [PubMed] [Google Scholar]
- 129. Wang X, Fang X, Wang F. Pleiotropic actions of iron balance in diabetes mellitus. Rev Endocr Metab Disord 2015;16:15–23. [DOI] [PubMed] [Google Scholar]
- 130. Wilson JG, Lindquist JH, Grambow SC, Crook ED, Maher JF. Potential role of increased iron stores in diabetes. Am J Med Sci 2003;325:332–3399. [DOI] [PubMed] [Google Scholar]
- 131. Jiang R, Manson JE, Meigs JB, Ma J, Rifai N, Hu FB. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA 2004;291:711–717. [DOI] [PubMed] [Google Scholar]
- 132. Tuomainen TP, Nyyssönen K, Salonen R, Tervahauta A, Korpela H, Lakka T, et al. Body iron stores are associated with serum insulin and blood glucose concentrations. Population study in 1,013 eastern Finnish men. Diabetes Care 1997;20:426–4288. [DOI] [PubMed] [Google Scholar]
- 133. Aregbesola A, Voutilainen S, Virtanen JK, Mursu J, Tuomainen T‐P. Body iron stores and the risk of type 2 diabetes in middle‐aged men. Eur J Endocrinol 2013;169:247–25353. [DOI] [PubMed] [Google Scholar]
- 134. Pinhas‐Hamiel O, Newfield RS, Koren I, Agmon A, Lilos P, Phillip M. Greater prevalence of iron deficiency in overweight and obese children and adolescents. Int J Obes 2003;27:416–4188. [DOI] [PubMed] [Google Scholar]
- 135. Chambers EC, Heshka S, Gallagher D, Wang J, Pi‐Sunyer FX, Pierson RN. Serum iron and body fat distribution in a multiethnic cohort of adults living in New York City. J Am Diet Assoc 2006;106:680–6844. [DOI] [PubMed] [Google Scholar]
- 136. Yanoff LB, Menzie CM, Denkinger B, Sebring NG, McHugh T, Remaley AT, et al. Inflammation and iron deficiency in the hypoferremia of obesity. Int J Obes 2007;31:1412–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ausk KJ, Ioannou GN. Is obesity associated with anemia of chronic disease? A population‐based study. Obesity 2008;16:2356–236161. [DOI] [PubMed] [Google Scholar]
- 138. Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab 2013;17:329–34141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Cepeda‐Lopez AC, Osendarp SJ, Melse‐Boonstra A, Aeberli I, Gonzalez‐Salazar F, Feskens E, et al. Sharply higher rates of iron deficiency in obese Mexican women and children are predicted by obesity‐related inflammation rather than by differences in dietary iron intake. Am J Clin Nutr 2011;93:975–98383. [DOI] [PubMed] [Google Scholar]
- 140. Schindler C, Birkenfeld AL, Hanefeld M, Schatz U, Köhler C, Grüneberg M, et al. Intravenous ferric carboxymaltose in patients with type 2 diabetes mellitus and iron deficiency: CLEVER trial study design and protocol. Diabetes Ther 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Biophys Acta, Gen Subj 1790;2009:682–69393. [DOI] [PubMed] [Google Scholar]
- 142. Xu Y, Ding X, Zou J, Liu Z, Jiang S, Chen Y. Serum hepcidin in haemodialysis patients: associations with iron status and microinflammation. J Int Med Res 2011;39:1961–19677. [DOI] [PubMed] [Google Scholar]
- 143. Jha V, Jairam A, Aggarwal P, Kohli H, Gupta K, Sakhuja V, et al. Iron status, inflammation and hepcidin in ESRD patients: the confounding role of intravenous iron therapy. Indian J Nephrol 2010;20:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Abdel‐Khalek MA, El‐Barbary AM, Essa SA‐M, Ghobashi AS. Serum hepcidin: a direct link between anemia of inflammation and coronary artery atherosclerosis in patients with rheumatoid arthritis. J Rheumatol 2011;38:2153–21599. [DOI] [PubMed] [Google Scholar]
- 145. van Eijk LT, Kroot JJC, Tromp M, van der Hoeven JG, Swinkels DW, Pickkers P. Inflammation‐induced hepcidin‐25 is associated with the development of anemia in septic patients: an observational study. Crit Care 2011;15:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Rumjon A, Sarafidis P, Brincat S, Musto R, Malyszko J, Bansal SS, et al. Serum hemojuvelin and hepcidin levels in chronic kidney disease. Am J Nephrol 2012;35:295–304. [DOI] [PubMed] [Google Scholar]
- 147. Jankowska EA, Malyszko J, Ardehali H, Koc‐Zorawska E, Banasiak W, von Haehling S, et al. Iron status in patients with chronic heart failure. Eur Heart J 2013;34:827–83434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Piepoli MF, Coats AJS. The “skeletal muscle hypothesis in heart failure” revised. Eur Heart J 2013;34:486–4888. [DOI] [PubMed] [Google Scholar]
- 149. Scott AC, Wensel R, Davos CH, Georgiadou P, Kemp M, Hooper J, et al. Skeletal muscle reflex in heart failure patients: role of hydrogen. Circulation 2003;107:300–3066. [DOI] [PubMed] [Google Scholar]
- 150. Scott A, Wensel R, Davos C, Georgiadou P, Ceridavies L, Coats A, et al. Putative contribution of prostaglandin and bradykinin to muscle reflex hyperactivity in patients on Ace‐inhibitor therapy for chronic heart failure. Eur Heart J 2004;25:1806–181313. [DOI] [PubMed] [Google Scholar]
- 151. Mark AL, Victor RG, Nerhed C, Wallin BG. Microneurographic studies of the mechanisms of sympathetic nerve responses to static exercise in humans. Circ Res 1985;57:461–4699. [DOI] [PubMed] [Google Scholar]
- 152. Piepoli M, Clark AL, Volterrani M, Adamopoulos S, Sleight P, Coats AJ. Contribution of muscle afferents to the hemodynamic, autonomic, and ventilatory responses to exercise in patients with chronic heart failure: effects of physical training. Circulation 1996;93:940–95252. [DOI] [PubMed] [Google Scholar]
- 153. Grieve DA, Clark AL, McCann GP, Hillis WS. The ergoreflex in patients with chronic stable heart failure. Int J Cardiol 1999;68:157–16464. [DOI] [PubMed] [Google Scholar]
- 154. Nakamoto FP, Neder JA, Maia J, Andrade MS, Silva AC. Skeletal muscle ergoreflex overactivity is not related to exercise ventilatory inefficiency in non‐hypoxaemic patients with COPD. Eur J Appl Physiol 2007;101:705–71212. [DOI] [PubMed] [Google Scholar]
- 155. Zhu YI, Haas JD. Altered metabolic response of iron‐depleted nonanemic women during a 15‐km time trial. J Appl Physiol 1998;84:1768–177575. [DOI] [PubMed] [Google Scholar]
- 156. Hinton PS, Sinclair LM. Iron supplementation maintains ventilatory threshold and improves energetic efficiency in iron‐deficient nonanemic athletes. Eur J Clin Nutr 2007;61:30–399. [DOI] [PubMed] [Google Scholar]
- 157. Clevenger B, Gurusamy K, Klein AA, Murphy GJ, Anker SD, Richards T. Systematic review and meta‐analysis of iron therapy in anaemic adults without chronic kidney disease: updated and abridged Cochrane review. Eur J Heart Fail 2016;18:774–78585. [DOI] [PubMed] [Google Scholar]
- 158. Jankowska EA, Tkaczyszyn M, Suchocki T, Drozd M, von Haehling S, Doehner W, et al. Effects of intravenous iron therapy in iron‐deficient patients with systolic heart failure: a meta‐analysis of randomized controlled trials. Eur J Heart Fail 2016;18:786–79595. [DOI] [PubMed] [Google Scholar]
- 159. von Haehling S, Jankowska E, Anker SD. The management of co‐morbidities in patients with heart failure–iron deficiency. Int Cardiovasc Forum J 2017;10. [Google Scholar]
- 160. Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009;361:2436–244848. [DOI] [PubMed] [Google Scholar]
- 161. Ponikowski P, van Veldhuisen DJ, Comin‐Colet J, Ertl G, Komajda M, Mareev V, et al. Beneficial effects of long‐term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J 2015;36:657–66868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Van Aelst LNL, Abraham M, Sadoune M, Lefebvre T, Manivet P, Logeart D, et al. Iron status and inflammatory biomarkers in patients with acutely decompensated heart failure: early in‐hospital phase and 30‐day follow‐up. Eur J Heart Fail 2017;19:1075–10766. [DOI] [PubMed] [Google Scholar]
- 163. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, et al. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail 2017;19:522–53030. [DOI] [PubMed] [Google Scholar]
- 164. Silverberg DS, Mor R, Weu MT, Schwartz D, Schwartz IF, Chernin G. Anemia and iron deficiency in COPD patients: prevalence and the effects of correction of the anemia with erythropoiesis stimulating agents and intravenous iron. BMC Pulm Med 2014;14:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Molinari F, Pin F, Gorini S, Chiandotto S, Pontecorvo L, Penna F, et al. The mitochondrial metabolic reprogramming agent trimetazidine as an “exercise mimetic” in cachectic C26‐bearing mice. J Cachexia Sarcopenia Muscle 2017;8:954–97373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Song M, Chen F, Li Y, Zhang L, Wang F, Qin R, et al. Trimetazidine restores the positive adaptation to exercise training by mitigating statin‐induced skeletal muscle injury. J Cachexia Sarcopenia Muscle 2018;9:106–11818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Barazzoni R, Gortan Cappellari G, Palus S, Vinci P, Ruozi G, Zanetti M, et al. Acylated ghrelin treatment normalizes skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rat chronic heart failure. J Cachexia Sarcopenia Muscle 2017;8:991–9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Inoue A, Cheng XW, Huang Z, Hu L, Kikuchi R, Jiang H, et al. Exercise restores muscle stem cell mobilization, regenerative capacity and muscle metabolic alterations via adiponectin/AdipoR1 activation in SAMP10 mice. J Cachexia Sarcopenia Muscle 2017;8:370–38585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]