

Abstract
Background
Glucocorticoids (GC) play a major role in muscle atrophy. As skeletal muscle is a secretory organ, characterization of the muscle secretome elicited by muscle atrophy should allow to better understand the cellular mechanisms and to identify circulating biomarkers of this condition. Our project aimed to identify the changes in the muscle secretome associated with GC‐induced muscle atrophy and susceptible to translate into circulation.
Methods
We have identified the GC‐induced changes in the secretome of C2C12 muscle cells by proteomic analysis, and then, we have determined how these changes translate into the circulation of mice or human subjects exposed to high concentrations of GC.
Results
This approach led us to identify Serpina3n as one of the most markedly secreted protein in response to GC. Our original in vitro results were confirmed in vivo by an increased expression of Serpina3n in skeletal muscle (3.9‐fold; P < 0.01) and in the serum (two‐fold; P < 0.01) of mice treated with GC. We also observed increased levels of the human orthologue Serpina3 in the serum of Cushing's syndrome patients compared with healthy controls matched for age and sex (n = 9/group, 2.5‐fold; P < 0.01). An increase of Serpina3n was also demonstrated in muscle atrophy models mediated by GC such as cancer cachexia (four‐fold; P < 0.01), sepsis (12.5‐fold; P < 0.001), or diabetes (two‐fold; P < 0.01). In contrast, levels of Serpina3n both in skeletal muscle and in the circulation were reduced in several models of muscle hypertrophy induced by myostatin inhibition (P < 0.01). Furthermore, a cluster of data suggests that the regulation of muscle Serpina3n involves mTOR, an essential determinant of the muscle cell size.
Conclusions
Taken together, these data suggest that Serpina3n may represent a circulating biomarker of muscle atrophy associated to GC and, broadly, a reflection of dynamic changes in muscle mass.
Keywords: Serpina3n, Glucocorticoids, Muscle atrophy, Biomarker
Introduction
Lean body mass, in particular muscle mass, is an excellent predictive survival factor in many diseases. Indeed, low muscle mass is often associated with poor survival.1 Although the consequences of the muscle mass decline are clearly recognized, the diagnosis and the treatment of this condition remain challenging. Therefore, the identification of an early biomarker of the muscle atrophy process and a better knowledge of the mechanisms involved are deeply needed for a timely diagnosis and the development of new anti‐atrophic therapies. Ideally, this biomarker should be predictive and easily accessible in body fluids.
Emerging evidence has revealed that skeletal muscle has a secretory function. Therefore, analysis of its secretome provides a basis for understanding how this tissue communicates with other organs. One prominent example is interleukin (IL)‐6, which is released into circulation by contracting skeletal muscle and can regulate metabolic and inflammatory processes.2, 3 Besides IL‐6, several other myokines have been identified including IL‐8, IL‐15, insulin‐growth factor I (IGF‐I), follistatin‐like 1 (FSTL1), or fibroblast‐growth factor (FGF)‐21.3, 4, 5, 6 Moreover, secreted proteins may also reflect metabolic changes, which take place inside muscle cells. Indeed, myoblast differentiation is accompanied by dramatic changes in the secreted proteins profile such as increased expression of semaphorins, IGF‐I, matrix metalloproteinase (MMP)‐2, or several collagen types.7 However, changes of skeletal muscle secretome associated with muscle atrophy have not yet been investigated.
Characterization of the muscle secretome elicited by muscle atrophy should allow us to better understand the cellular mechanisms involved in this process and might lead to identify circulating biomarkers of this condition, as telopeptides are for bone loss.8 Glucocorticoids (GC) are recognized to play a major role in skeletal muscle atrophy,9, 10 either as drugs used to treat several medical conditions or as endocrine hormones released in response to many stress situations (e.g. sepsis, cancer, and insulinopenia). Indeed, the inhibition of GC action by a specific receptor antagonist (RU‐486) or by muscle‐specific invalidation of the GC receptor (GR) inhibits the muscle atrophy in these catabolic situations.11, 12, 13 Therefore, we hypothesized that skeletal muscle atrophy induced by GC is associated with specific alterations of the muscle secretome. The aim of our project was to identify the GC‐induced changes in the secretome of C2C12 muscle cells in culture and then to determine how these changes translate into the circulation of mice or human subjects exposed to high concentrations of GC. Having identified Serpina3n as a myokine released in parallel to muscle atrophy, we assessed its release in models of muscle hypertrophy to investigate whether its secretion reflects dynamic changes in muscle mass. Finally, we aimed to provide some clues regarding its mechanism of regulation parallel to changes in muscle mass.
Materials and methods
C2C12 cell culture
Mouse myoblasts from the muscle‐derived C2C12 cell line (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM) with GLUTAMAX ITM supplemented with 10% fetal bovine serum (FBS), 0.1% gentamycin, and 1% nonessential amino acids (all from Life Technologies, Inc., Carlsbad, California, USA) at 37°C in 5% CO2 incubator. When cells reached 70–80% confluence, the 10% FBS was replaced by 2% horse serum (HS) to induce myogenic differentiation. After 4 days of differentiation, cells were washed three times with phosphate buffered saline to reduce contaminating serum proteins, and cell atrophy was then induced by dexamethasone (DEX, Sigma‐Aldrich, MO, USA) at 10−6 M for 24 h in serum‐free conditions without phenol red and antibiotics. To assess the effects of IGF‐I treatment on cell atrophy‐induced by GC, differentiated cells were treated with LONG® R3 IGF‐I human recombinant (50 ng/mL, Sigma‐Aldrich) with or without DEX at 10−6 M for 48 h in 2% HS differentiation medium. Myotubes were also treated with rapamycin (Calbiochem, Merk Millipore, Germany) at 50 nM alone or in presence of DEX at 10−6 M for 24 h in 2% HS.
Adult human skeletal muscle cells (SkMDC) culture
Adult human skeletal muscle cells (from a 41‐year‐old donor, Cook Myosite, Pittsburgh, PA, USA) were cultured in growth medium consisting of DMEM with GLUTAMAX™, 20% FBS, 0.5% Ultroser G (Pall, Cergy, France), 0.1% gentamycin, and 1% nonessential amino acids at 37°C in a 5% CO2 incubator. SkMDC mean population doubling was determined at each passage. After 4 days of differentiation in DMEM supplemented with 1% FBS/2% HS, cells were treated with DEX at 10−6 M for 8 h.
Cell viability
Cell viability was determined using the CellTiter‐Glo® luminescent cell viability kit from Promega Corporation (Madison, WI, USA) according to the manufacturer's instructions. This method is based on the measurement of ATP production by the cells, proportional to the number of viable cells, detected by luciferin–luciferase reaction.
C2C12 and SkMDC myotube morphological analysis
C2C12 myotubes were photographed directly in culture plate without fixation using an AxioCam ERc5s digital camera coupled to an AxioVert.A1 microscope with the use of ZEN 2.3 software (Zeiss, Germany). Myotube diameter was measured on 75 myotubes in each condition from three independent experiments. For each myotube, three random measurements were performed along the length of the myotube (n = 3 measurements/myotube) using the ZEN 2.3 software, and the average of these three measurements was considered as one single value.
SkMDC myotubes were labelled with mouse monoclonal anti‐myosin heavy chain antibody (MF20, 1:50 dilution) and resolved with secondary antibodies conjugated to Alexa‐Fluor 488 (1:400 dilution, Invitrogen, Cergy‐Pontoise, France). Images were captured with a high‐resolution cooled digital XC30 camera coupled to a BX‐50 microscope (Olympus, Rungis, France) at a resolution of 0.64 μm/pixel. Myotube diameter was measured on 100 myotubes in each condition (from three independent experiments). For each myotube, five random measurements were performed along the length of the myotube using the image processing software ImageJ 1.47v, and the average of these five measurements was considered as one single value.
Collection of conditioned media
Conditioned media (CM) of C2C12 cells were collected after 24 h incubation in serum‐free medium. The CM was cleared by centrifugation (10 min at 300 g followed by 20 min at 2000 g) to discard dead cells, for example, apoptotic cell bodies and cell debris. Cleared CM were subsequently concentrated using an Amicon Ultra‐4 3 kDa cut‐off spin Column (Millipore, Watford, UK).
Nano‐LC‐MS/MS analysis
Proteins (80 μg) from CM of each condition (CTR vs. DEX, n = 3 independent experiments) were concentrated in the stacking gel of 1D electrophoresis gels (12% acrylamide) as described.14 Briefly, gels were stained in Coomassie Brilliant Blue G‐250 solution and excised lanes were reduced (DTT) and alkylated (iodoacetamide). Proteins were then digested by trypsin, and peptide mixtures were analysed in duplicate by online nanoflow liquid chromatography using the Ultimate 3000 RSLC (Dionex, Voisins le Bretonneux, France) and eluates being electrosprayed into a LTQVelos mass spectrometer for nano‐LC‐MS/MS analysis. Thermo Proteome Discoverer v1.3 was used for raw data file processing and MASCOT for database search (http://www.matrixscience.com). For protein identification, the Uniprot Mus musculus (07/2016, 51 518 seq) protein database was used; protein identification was considered valid if at least one peptide with a statistically significant Mascot score assigned it (with Mascot score ≥ 36 for P value < 0.05 with a false discovery rate at 1%). The acquired spectra were loaded into the Progenesis LC‐MS software (version 4.1, Nonlinear Dynamics, Newcastle, UK), and label‐free quantification was performed. Briefly, for each migration lane from the SDS‐PAGE, the profile data of the MS scans as well as MS/MS spectra were transformed to peak lists with Progenesis LC‐MS using a proprietary algorithm and then stored in peak lists comprising m/z and abundance. One sample was set as the reference, and the retention times of all other samples within the experiment were automatically aligned to create maximal overlay of the two‐dimensional feature maps. At this point, features with only one charge, with retention time windows lower than 6 s or with retention time lower than 20 min and higher than 80 min, were masked and excluded from further analyses. All remaining features were used to calculate a normalization factor for each sample that corrects for experimental variation. Samples were then allocated to their experimental group (CTR vs. DEX). For quantification, all unique validated peptides (with Mascot score ≥ 36 for P value < 0.05) of an identified protein were included, and the total cumulative abundance was calculated by summing the abundances of all peptides allocated to the respective protein.
Animals
Six to 8‐week‐old male mice and rats were housed for 1 or 2 week(s) under standardized conditions of light (12:12 h light–dark cycle) and temperature (22 ± 2°C) before starting the experiments. They had access to standard chow pellets and water ad libitum. The Committee for Ethical Practices in Animal Experiments of the Université Catholique de Louvain approved the procedures.
Glucocorticoid treatment
FVB mice (n = 12, Janvier Breeding, Le‐Genest‐Saint‐Isle, France) were randomly allocated to one treatment group: DEX (n = 6) and CTR (n = 6). The GC‐treated group (DEX) received a daily subcutaneous injection of DEX (Aacidexam®; Organon, Oss, the Netherlands) at 5 mg/kg/day for 4 days, whereas the control (CTR) group received a daily subcutaneous injection of saline solution. Mice were sacrificed by decapitation, and blood was collected. For biochemical analysis, muscles [gastrocnemius (GA), tibialis anterior (TA), extensor digitorum longus (EDL) and soleus], liver, heart, spleen, testis, and adipose tissue were removed, weighted, deep‐frozen in liquid nitrogen, and stored at −80°C until further analysis. For histological analysis, one of the two GA muscles was mounted with tissue freezing medium (OCT), frozen in isopentane cooled on liquid nitrogen, and stored at −80°C. Serial cross‐sections (10 μm thick) were performed using a cryostat at −20°C.
Cancer cachexia
CD2F1 male mice (n = 16, Charles River Laboratories, Chatillon‐sur‐Chalaronne, France) were injected subcutaneously in the upper flank with C26 cells (1 × 106 cells in 0.1 mL saline, Cachexia group; n = 8) or a saline solution (CTR group; n = 8), as previously described.15 Eleven days after the injection of tumour cells, intracardiac blood and tissue samples were harvested following anaesthesia with isoflurane gas (Abbot, Ottignies, Belgium). GA muscles were weighted and frozen in liquid nitrogen. All of the samples were stored at −80°C.
Lipopolysaccharide‐induced sepsis
FVB mice (n = 12, Janvier Breeding) were randomly divided into two different groups: lipopolysaccharide (LPS) (n = 6) and CTR (n = 6). The LPS group was treated by a single intraperitoneal (ip) injection of LPS (750 μg/100 g of body weight; serotype 0127:B8, Sigma‐Aldrich), whereas the CTR group was injected with an equivalent volume of saline buffer. Twenty‐four hours after LPS injection, animals were sacrificed; blood was collected, and serum was stored at −80°C until analysis. GA and TA muscles were dissected, snap‐frozen in liquid nitrogen and stored at −80°C until processing. To assess the role of GC in the stimulation of Serpina3n, mice were injected with RU‐486, an antagonist of GC receptor. C57B1/6 wild‐type (n = 20, Janvier Breeding) mice were randomly divided into four different groups: CTR (n = 4), LPS (n = 6), RU‐486 (n = 4), and LPS + RU‐486 (n = 6). The LPS and LPS + RU‐486 groups were injected ip with LPS as described, whereas CTR and RU‐486 groups were ip injected with an equivalent volume of saline buffer. Fifteen minutes before LPS or saline injections, animals from RU‐486 and LPS + RU‐486 groups received a single ip injection of RU‐486 (5 mg/mice; Sigma‐Aldrich), whereas the two other groups received an equal amount of DMSO solution. Twelve hours after LPS injection, animals were sacrificed, and TA and EDL muscles were dissected, snap‐frozen in liquid nitrogen, and stored at −80°C until processing.
Streptozotocin‐induced diabetes
Wistar rats (n = 24, Janvier Breeding) were anaesthetised with a mixture of 100 mg/kg ketamine (Ketalar, Pfizer, New York, NY, USA) and 10 mg/kg xylazine hydrochloride (Rompun, Bayer, Leverkussen, Germany) administered by an ip injection, and diabetes was induced by injection of 60 mg streptozotocin (STZ) (freshly prepared in 0.01 M citrate buffer, pH 4.5)/kg body weight into the tail vein as previously described.16 The CTR group (n = 8) was injected with an equivalent volume of citrate buffer. Three days after STZ injection, diabetic animals were randomized in two groups: one was treated with insulin (STZ + INS group; n = 8), and the second group was not treated (STZ group; n = 8). To limit the risk of infection that might result from the large number of injections and manipulations, insulin was administrated by two implants (Linplant®, Linshin, Canada) placed in the interscapular region. Each implant delivered an average daily dose of 2 U of insulin per day. The untreated diabetic (STZ) and control animals (CTR) were sham‐operated. At the end of the 9 days infusion period, animals were sacrificed; blood and GA muscles were collected for analysis and stored at −80°C.
Muscle specific inactivation of FoxO
FoxO1/3/4‐floxed mice (CTR, n = 3) and muscle‐specific FoxO1/3/4 knockout mice (FoxO KO, n = 3) were generated as described17 and kindly provided by M. Sandri (VIMM, Padova, Italie). Blood, TA, and GA muscles were collected for analysis as described earlier.
Muscle specific inactivation of mTOR
mTOR‐floxed mice (CTR, n = 3) and muscle‐specific mTOR knockout mice (mTOR KO, n = 5) were generated as described.18, 19 Blood, TA, and GA muscles were collected for analysis as described earlier.
Myostatin inhibition‐induced muscle hypertrophy
FVB myostatin knockout (Mstn KO, n = 6) mice harbouring a constitutive deletion of the third Mstn exon20 and their control wild‐type littermates (n = 6); C57Bl/6 transgenic mice (mTr‐FS, n = 4) overexpressing the human Follistatin 288 (hFS288) short form specifically in skeletal muscle21 and their control wild‐type littermates (n = 4); and FVB mice treated by ip injection with sActRIIB‐Fc (n = 6) at a dose of 10 mg/kg twice a week for 14 days or with phosphate buffered saline (CTR, n = 6)22 were used. The sActRIIB‐Fc is a soluble ligand binding domain of Activin receptor type IIB fused to Fc domain IgG that induces Mstn inhibition. It was prepared as described23 and kindly provided by O. Ritvos (University of Helsinky, Finland). Blood, TA, and GA muscles were collected for analysis as described earlier.
Fibre cross‐sectional area
Cross‐sections of GA muscle were labelled with anti‐laminin‐α1 (L9393, Sigma‐Aldrich) to outline the fibres and resolved with corresponding secondary antibodies conjugated to Alexa‐Fluor 488 (Invitrogen, Cergy‐Pontoise, France). Images were captured with a high‐resolution cooled digital XC30 camera coupled to a BX‐50 microscope (Olympus, Rungis, France) at a resolution of 0.64 μm/pixel. Five fields, each containing 100 fibres, were analysed per muscle. Cross‐sectional area was determined for each fibre, using the image processing software ImageJ 1.47v.
mRNA analysis by real‐time quantitative PCR
Total RNA was isolated from the TA muscle using Tri Reagent (Sigma‐Aldrich) as described by the manufacturer. Recovery was 1 μg/mg of TA muscle. Reverse transcription and real‐time quantitative PCR were performed as previously described.24 Primers (Table 1) were tested to avoid primer dimers, self‐priming formation, or unspecific amplification, and GAPDH was used as the reporter gene. The primers were designed to have standardized optimal PCR conditions.
Table 1.
Primer sequences
| Gene | Primers 5′‐3′ | Accession no. | |
|---|---|---|---|
| Forward | Reverse | ||
| serpina3n | GACCTGTCTGCAATCACAGGA | TTTGGGGTTGGCTATCTTGGC | NM_009252.2 |
| serpina3 | GGCCTTTGCCACTGACTTTC | TCATGGGCACCATTACCCAC | NM_001085.4 |
| periostin | TTCATTGAAGGTGGCGATGGT | ACGGCCTTCTCTTGATCGTC | NM_001198765.1 |
| murf1 | TGTCTGGAGGTCGTTTCCG | ATGCCGGTCCATGATCACTT | AY059627 |
| atrogin‐1 | CCATCAGGAGAAGTGGATCTATGTT | GCTTCCCCCAAAGTGCAGTA | AF441120 |
| foxO3a | TCGTCTCTGAACTCCTTGCGT | TGGAGTGTCTGGTTGCCGT | NM_019740 |
| gapdh | TGCACCACCAACTGCTTA | GGATGCAGGGATGATGTTC | NM_001289726.1 |
Human subjects
Patients with proven Cushing's syndrome and healthy controls matched for age and sex were recruited from the Endocrine Clinic of the St‐Luc Academic Hospital (Brussels, Belgium). Exclusion criteria included pseudo‐Cushing, paraneoplastic or cyclic Cushing's syndrome, adrenocortical carcinoma, or pituitary irradiation during the last 6 months. All subjects underwent standard medical examination, and blood samples were collected from patients at the time of recruitment, in standardized conditions. This study (ClinicalTrials.gov identifier: NCT03229395) was approved by the local Ethical Committee of the Université Catholique de Louvain (Belgium) and was performed in accordance with the principles of the revised Declaration of Helsinki. All subjects were fully informed regarding study participation, and they provided written informed consent prior to inclusion in the study.
Western‐blots
Muscle proteins of GA muscle were homogenized in ice‐cold pH 7.0 buffer containing 20 mM Tris, 270 mM sucrose, 5 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM β‐glycerophosphate, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM DTT, 1% (vol/vol) Triton X‐100, and 10% protease inhibitor cocktail (Roche Applied Science, Belgium). Homogenates were centrifuged at 10 000 g for 10 min at 4°C, and supernatants were immediately stored at −80°C. Twenty micrograms of muscle proteins, secreted proteins of CM, or serum proteins were resolved by SDS‐polyacrylamide gel 12% electrophoresis and transferred to PVDF membranes. Membranes were probed with the following primary antibodies: anti‐Serpina3n (AF4709, R&D Systems, UK), anti‐Serpina3 (AF1295, R&D Systems, UK), anti‐Periostin (GTX100602, Genetex, Irvine, USA), and anti‐GAPDH (2118, Cell Signaling Technology, Leiden, the Netherlands). Membranes were then incubated with a horseradish peroxidase‐coupled secondary antibody (Cell Signaling Technology) and developed using Enhanced Chemiluminescence® Western Blotting Detection System Plus (GE Healthcare, Belgium). Developed films were scanned and analysed as previously described.25 For skeletal muscle extracts, signal intensity was normalized to GAPDH protein for skeletal muscle extracts and to total protein loading assessed by staining membranes using Coomassie blue for serum and MC extracts.
Statistics
Results are presented as means ± SE. Statistical analyses were performed using a one‐way or two‐way ANOVA followed by a Tukey test (multiple experimental groups) or unpaired t‐test (two experimental groups) to compare different conditions in in vitro and in vivo experiments. Statistical analyses were performed using GraphPad Prism (version 7), and significance was set at P < 0.05.
Results
Dexamethasone causes myotube atrophy in serum‐free conditions without affecting cell viability
In order to characterize our model of GC‐induced atrophy, we exposed C2C12 cells to DEX for 24 h in serum‐free conditions. In agreement with previous observations,26 DEX caused myotube atrophy illustrated by decreased myotube diameter (−28%; P < 0.05 with serum and −36%; P < 0.05 without serum) and increased expression of atrogenes such as MurF1, Atrogin‐1, and FoxO3a (Figure 1A, B, D). Despite the absence of serum, the myotube atrophy caused by DEX occurred without any major impact on cell viability (Figure 1C).
Figure 1.
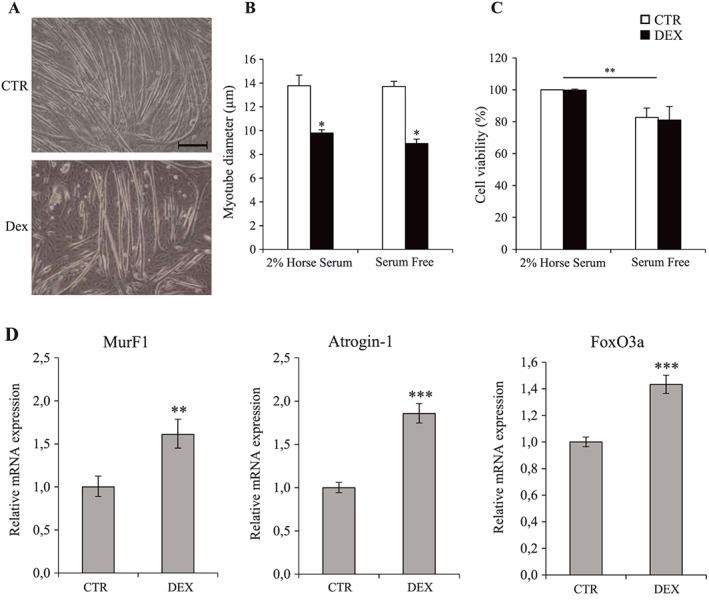
Dexamethasone induces myotube atrophy in serum‐free conditions without any major impact on cell viability. (A) Phase‐contrast representative images of CTR and DEX‐treated C2C12 cells. Myotube diameter (B), cell viability (C), and atrogenes mRNA expression (D) of C2C12 cells after 24 h of 10−6 M dexamethasone treatment in 2% horse serum or in serum‐free conditions. Scale bar corresponds to 100 μm. Results are means ± SE (n = 3). Statistical analysis was performed using two‐way ANOVA and Tukey post‐test (* P < 0.05 vs. CTR and ** P < 0.01 vs. 2% horse serum). DEX, dexamethasone.
Dexamethasone‐induced myotube atrophy is associated with increased Serpina3n release
To characterize the changes in C2C12 cells secretome induced by GC, proteins present in CM were identified and quantified by mass spectrometry. For each experiment (n = 3) and each condition (n = 2), we performed duplicates analysis of CM by LC‐MS/MS. A total number of 739 proteins were identified. Among these proteins, 26 were found to be differentially secreted between CTR and DEX conditions, with 15 proteins being more abundant in the presence of DEX (Table 2). Interestingly, Serpina3n was one of the most markedly secreted protein (fold change = 13.5) in response to DEX. RT‐qPCR performed on C2C12 extracts confirmed the stimulation of the expression of Serpina3n and periostin in the presence of DEX (Figure 2A, C). Western‐blots (WB) experiments on CM also showed an increased secretion of these two proteins (eight‐fold for Serpina3n and six‐fold for Periostin) (Figure 2B, D), confirming the results of LC‐MS/MS analysis.
Table 2.
Differentially secreted proteins between CTR and DEX‐treated C2C12 cells identified by nano‐LC‐MS/MS analysis
| Accession | Peptides | Score | ANOVA | Fold | Change | Protein | Gene GN |
|---|---|---|---|---|---|---|---|
| Q60590 | 2 (2) | 50.85 | 0.0025 | 16.9 | I | Alpha‐1‐acid glycoprotein 1 | Orm1 |
| G3X8T9 | 6 (6) | 295.96 | 0.0000 | 13.5 | I | Serine (Or cysteine) peptidase inhibitor, clade A, member 3N | Serpina3n |
| Q545R0 | 1 (1) | 23.1 | 0.0077 | 2.9 | I | Catenin (Cadherin associated protein), alpha 1 | Ctnna1 |
| Q61805 | 4 (4) | 209.28 | 0.0293 | 2.4 | I | Lipopolysaccharide‐binding protein | Lbp |
| P98064 | 2 (2) | 97.84 | 0.0006 | 2.1 | I | Mannan‐binding lectin serine protease 1 | Masp1 |
| D6RGQ0 | 7 (7) | 202.68 | 0.0018 | 1.8 | I | Complement factor H | Cfh |
| Q62009 | 28 (28) | 1369.95 | 0.0007 | 1.8 | I | Periostin | Postn |
| P47931 | 2 (2) | 107.81 | 0.0428 | 1.4 | I | Follistatin | Fst |
| Q9CR35 | 5 (5) | 152.27 | 0.0118 | 1.4 | I | Chymotrypsinogen B | Ctrb1 |
| F6SLR4 | 1 (1) | 20.1 | 0.0264 | 1.3 | I | Testican‐2 | Spock2 |
| B2RQQ8 | 6 (6) | 194.08 | 0.0244 | 1.3 | I | Collagen alpha‐2(IV) chain | Col4a2 |
| P62869 | 1 (1) | 45.96 | 0.0303 | 1.3 | I | Transcription elongation factor B polypeptide 2 | Tceb2 |
| P28301 | 6 (6) | 213.05 | 0.0257 | 1.3 | I | Lysyl oxidase | Lox |
| Q9JII6 | 1 (1) | 18.97 | 0.0302 | 1.2 | I | Alcohol dehydrogenase [NADP(+)] | Akr1a1 |
| P27773 | 11 (11) | 571.29 | 0.0176 | 1.1 | I | Protein disulfide‐isomerase A3 | Pdia3 |
| Q07797 | 1 (1) | 23.94 | 0.0354 | 5.2 | D | Galectin‐3‐binding protein | Lgals3bp |
| Q61554 | 4 (4) | 183.93 | 0.0481 | 1.9 | D | Fibrillin‐1 | Fbn1 |
| G5E8G2 | 1 (1) | 19.81 | 0.0290 | 1.8 | D | MCG124600 | Usp17ld |
| P07141 | 2 (2) | 140.75 | 0.0393 | 1.6 | D | Macrophage colony‐stimulating factor 1 | Csf1 |
| Q9WV91 | 1 (1) | 44.24 | 0.0408 | 1.5 | D | Prostaglandin F2 receptor negative regulator | Ptgfrn |
| Q3TNY9 | 11 (11) | 741.35 | 0.0088 | 1.5 | D | Biglycan | Bgn |
| P12032 | 3 (3) | 189.92 | 0.0489 | 1.5 | D | Metalloproteinase inhibitor 1 | Timp1 |
| B1B0C7 | 9 (9) | 349.69 | 0.0047 | 1.4 | D | Basement membrane‐specific heparan sulfate proteoglycan core protein | Hspg2 |
| P10518 | 3 (3) | 122.88 | 0.0341 | 1.4 | D | Delta‐aminolevulinic acid dehydratase | Alad |
| B1AYJ9 | 1 (1) | 70.83 | 0.0064 | 1.3 | D | Obg‐like ATPase 1 | Ola1 |
| Q3UQ28 | 15 (15) | 699.77 | 0.0147 | 1.3 | D | Peroxidasin homolog | Pxdn |
I or D = increase (I) or decrease (D) of protein secretion in CM of DEX‐treated C2C12 compared with CTR. DEX, dexamethasone.
Figure 2.
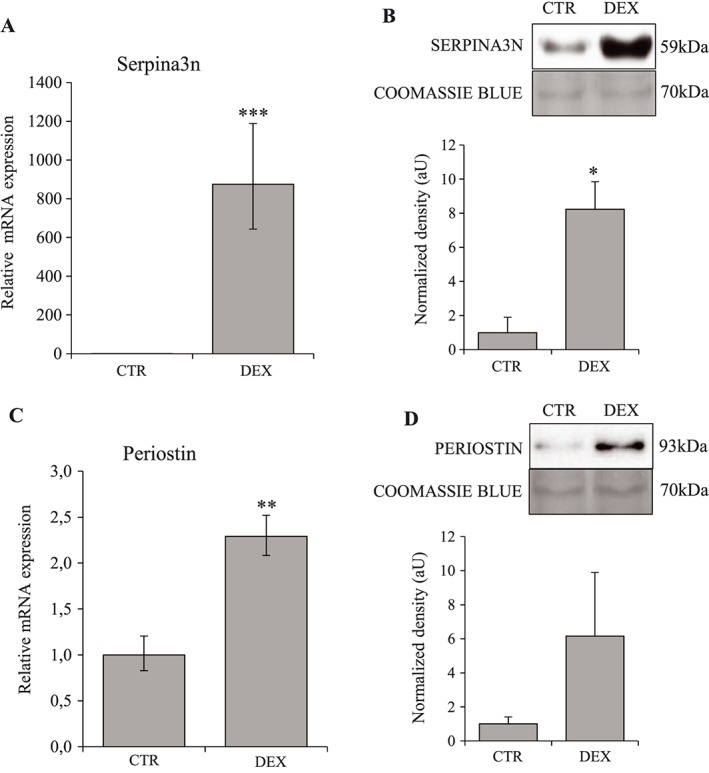
Dexamethasone stimulates Serpina3n and periostin expression and secretion by myotubes. C2C12 cells were treated 24 h with dexamethasone at 10−6 M in serum‐free conditions. Serpina3n (A) and periostin (C) mRNA levels in CTR and DEX‐treated cells. Serpina3n (B) and periostin (D) protein levels in conditioned media of CTR and DEX‐treated cells. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE (n = 3). Statistical analysis was performed using unpaired t‐test (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. CTR). DEX, dexamethasone.
Dexamethasone induces muscle atrophy together with increased muscle Serpina3n expression and release into circulation
As Serpina3n expression and secretion were greatly increased by GC in vitro, we investigated its expression in mice treated with DEX to demonstrate the in vivo relevance of our first observations. Muscle atrophy induced by GC was demonstrated by a decrease in muscle weight (−9% for GA, −6% for TA, −8% for EDL; P < 0.05) and in fibre cross‐sectional area (−27%; P = 0.001) (Figure 3). Moreover, as observed in vitro, GC‐induced muscle atrophy in vivo was associated with increased Serpina3n mRNA (1.5‐fold; P < 0.01) and protein abundance in skeletal muscle (3.9‐fold; P < 0.01) together with increased serum levels (two‐fold; P < 0.01) (Figure 4A–C). As skeletal muscle is not the only organ that expresses Serpina3n, we quantified Serpina3n protein abundance by WB also in liver, spleen, heart, testis, and adipose tissue. As illustrated in Figure 4D, whereas the spleen and testis Serpina3n expression increased in response to DEX (2.1‐ and 2.5‐fold, respectively; P < 0.05), the Serpina3n induction was the highest in the skeletal muscle (3.9‐fold; P < 0.01). Moreover, our WB experiments revealed that Serpina3n protein level was not changed in liver, the principal secretory organ of circulating proteins. These observations suggest therefore a crucial contribution of skeletal muscle to the increased Serpina3n serum levels in response to GC.
Figure 3.
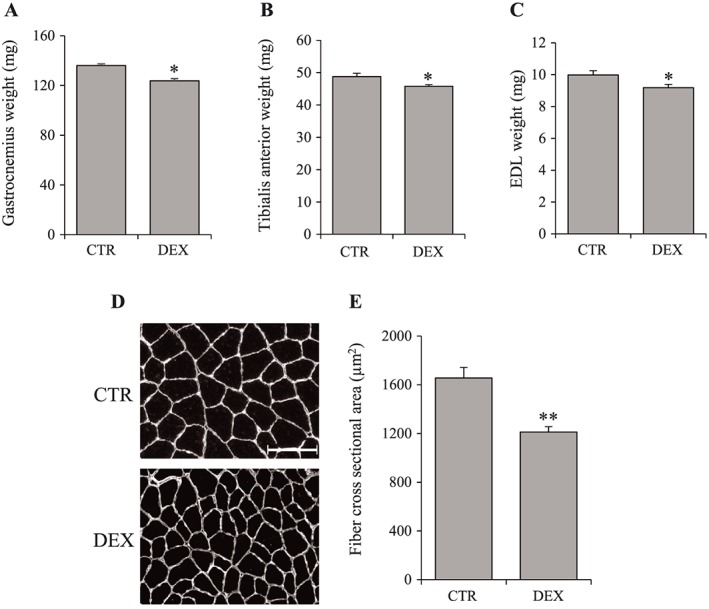
Dexamethasone induces muscle atrophy in vivo. Mice were treated with dexamethasone injection at 5 mg/kg/day for 4 days. Gastrocnemius (A), tibialis anterior (B), and EDL (C) weight of CTR and DEX‐treated mice. (D) Representative images of gastrocnemius muscle cross‐sections labelled for laminin‐1α to outline the fibres of CTR and DEX‐treated mice. (E) Measurements of fibre cross‐sectional area (average of 500 fibres per mice). Scale bar correspond to 60 μm. Results are means ± SE (n = 6/group). Statistical analysis was performed using unpaired t‐test (* P < 0.05, ** P < 0.01 vs. CTR). DEX, dexamethasone; EDL, extensor digitorum longus.
Figure 4.
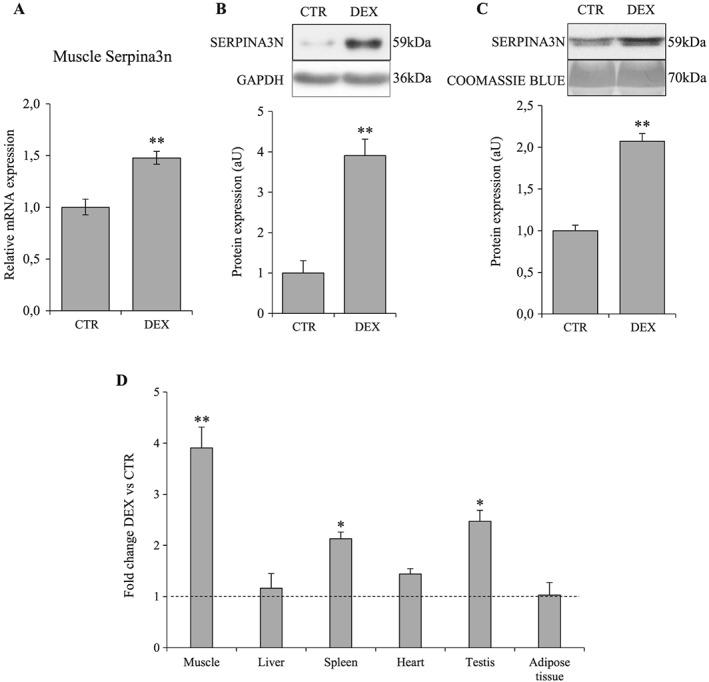
Dexamethasone increases Serpina3n expression in skeletal muscle and its secretion into circulation. Mice were treated with dexamethasone injection at 5 mg/kg/day for 4 days. Serpina3n mRNA levels in TA (A) and protein levels in gastrocnemius (B) muscles of CTR and DEX‐treated mice. (C) Serpina3n protein levels in serum of CTR and DEX‐treated mice. (D) Fold change of Serpina3n protein abundance in different tissues in response to dexamethasone treatment; dotted line corresponds to the CTR relative value. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE (n = 6/group). Statistical analysis was performed using unpaired t‐test (* P < 0.05, ** P < 0.01 vs. CTR). DEX, dexamethasone.
Muscle atrophy caused by cancer cachexia, sepsis, and diabetes is associated with increased muscle Serpina3n expression and release into circulation
To investigate the regulation of Serpina3n in other models of muscle atrophy requiring the action of GC, Serpina3n was quantified in skeletal muscle and in serum of animals with cancer cachexia, LPS‐induced sepsis, and STZ‐induced diabetes. We observed an increased expression of Serpina3n (mRNA and protein) in skeletal muscle in these three different models. Furthermore, increased circulating levels of Serpina3n protein were also observed in cachectic (four‐fold, P < 0.001), septic (12.5‐fold; P < 0.001), and diabetic mice (two‐fold; P < 0.01) (Figures 5, 6, and 7, respectively). Insulin treatment of diabetic mice restored Serpina3n to normal levels both in skeletal muscle and in the circulation, together with the restoration of muscle mass (Figure 7 and Supporting Information, Figure S1).
Figure 5.
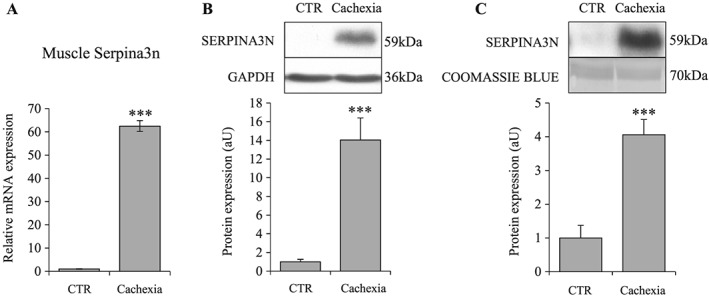
Cancer cachexia is associated with increased Serpina3n in skeletal muscle and in circulation. Serpina3n mRNA levels (A) and protein abundance (B) in gastrocnemius muscles of cachectic and CTR mice. (C) Serpina3n protein levels in circulation of cachectic and CTR mice. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE (n = 8/group). Statistical analysis was performed using unpaired t‐test (*** P < 0.001 vs. CTR).
Figure 6.
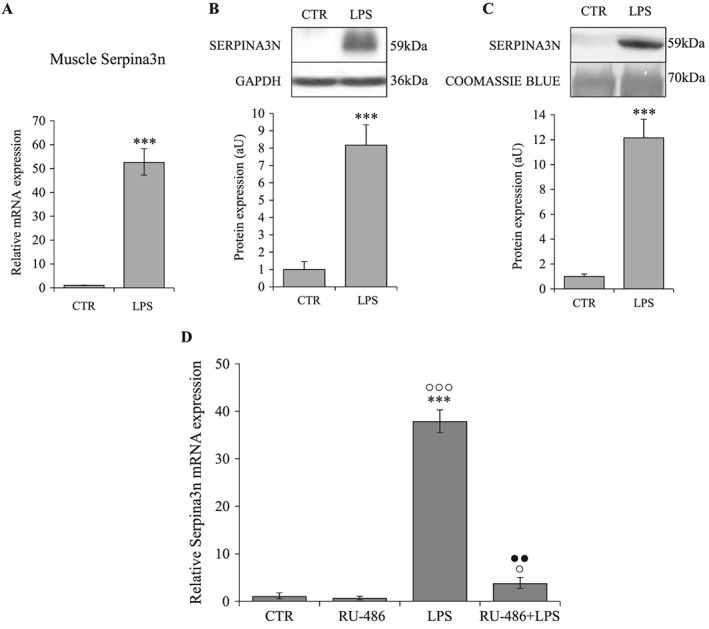
Sepsis is associated with increased Serpina3n in skeletal muscle and in circulation, and RU‐486 inhibits Serpina3n expression induced by LPS. Serpina3n mRNA levels in tibialis anterior (A) and protein abundance in gastrocnemius (B) muscles of LPS‐treated and CTR mice (n = 6/group). (C) Serpina3n protein levels in circulation of LPS‐treated and CTR mice. (D) Serpina3n mRNA levels in tibialis anterior muscles of CTR (n = 4), RU‐486 (n = 6), LPS (n = 4), and RU‐486 + LPS (n = 6) treated mice. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE. Statistical analysis was performed using unpaired t‐test or one‐way ANOVA and Tukey post‐test (*** P < 0.001 vs. CTR, ° P < 0.05, °°° P < 0.001 vs. RU‐486, ●● P < 0.01 vs. LPS). LPS, lipopolysaccharide.
Figure 7.

STZ‐induced diabetes is associated with increased Serpina3n in skeletal muscle and in circulation. Serpina3n mRNA levels (A) and protein abundance (B) in gastrocnemius muscles of CTR, STZ, and STZ + INS mice. (C) Serpina3n protein levels in serum of CTR, STZ, and STZ + INS mice. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE (n = 8/group). Statistical analysis was performed using one‐way ANOVA and Tukey post‐test (* P < 0.05, ** P < 0.01 vs. CTR; ° P < 0.05 vs. STZ + INS). STZ, streptozotocin.
Because the induction of Serpina3n is greater in cachexia and sepsis compared with GC treatment, we wondered whether proinflammatory cytokines might contribute to the increased Serpina3n expression in these conditions. Our in vitro experiments showed that TNF‐α did not induce neither significantly potentiate the GC‐induced expression of Serpina3n (Supporting Information, Figure S2). Furthermore, inhibition of GR activity by RU‐486 almost completely blunted the Serpina3n induction by LPS (Figure 6D). Taken together, these results demonstrate the predominant role of GC vs. proinflammatory cytokines in the regulation of Serpina3n expression even under inflammatory catabolic conditions.
Serpina3 expression increases in primary human muscle cells exposed to DEX and in serum of Cushing's syndrome patients
To extend to humans our results observed in mice, we measured the expression of human Serpina3, the orthologue of mice Serpina3n, in primary human muscle cells (SkMDC) exposed to DEX and in patients with overt Cushing's syndrome exposed to high concentrations of endogenous GC. As observed in murine C2C12 cells, DEX induced atrophy of human SkMDC cells after 8 h of treatment together with increased expression of the atrogenes MurF1 and Atrogin‐1 (Figure 8A–C). Moreover, mRNA expression of Serpina3 was also induced in SkMDC cells treated with DEX (4.4‐fold; P < 0.001) (Figure 8D). Quantification by WB of Serpina3 protein in serum of Cushing's patients demonstrated a clear increase of circulating Serpina3 levels by comparison with control subjects (2.5‐fold; P < 0.01) (Figure 8E).
Figure 8.
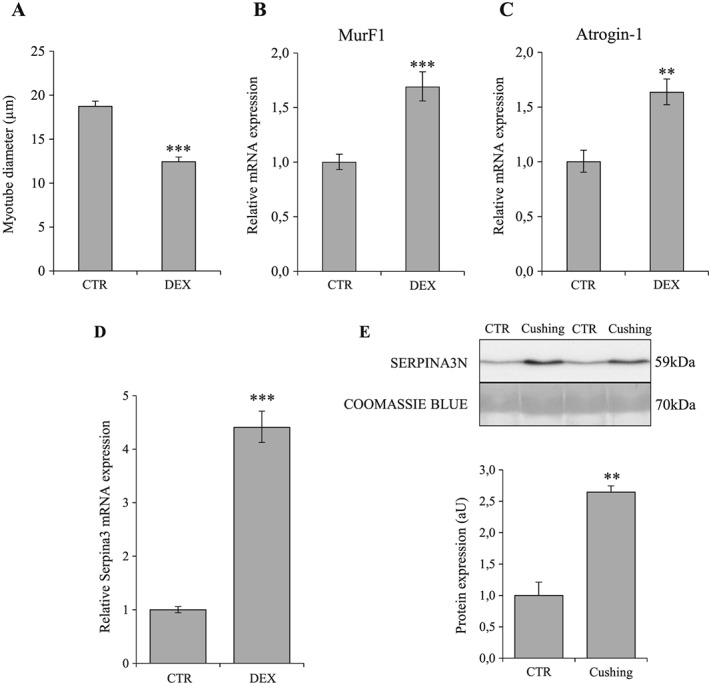
Exposure to glucocorticoids increases human Serpina3 expression in vitro and in vivo. Myotube diameter (A), Murf‐1 (B), Atrogin‐1 (C), and Serpina3n (D) mRNA levels in DEX‐treated and CTR human primary skeletal muscle cells SkMDC (n = 3 independent experiments). (B) Serpina3 protein levels in circulation of Cushing's and CTR patients (n = 9/group). For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE. Statistical analysis was performed using unpaired t‐test (** P < 0.01, *** P < 0.001 vs. CTR). DEX, dexamethasone.
Insulin‐growth factor‐I prevents glucocorticoids‐induced myotube atrophy and Serpina3n expression
To investigate whether reversing GC‐atrophy is associated with normalization of Serpina3n expression, C2C12 cells were treated for 48 h with IGF‐I, an anabolic hormone known to antagonize the atrophic action of GC. As expected, IGF‐I caused myotube hypertrophy (+11%; P < 0.05) and prevented muscle atrophy induced by GC (Figure 9A, B). More interestingly, IGF‐I blunted the increase of Serpina3n expression induced by DEX (Figure 9C). These observations support the hypothesis that Serpina3n expression reflects the dynamic changes of muscle mass.
Figure 9.
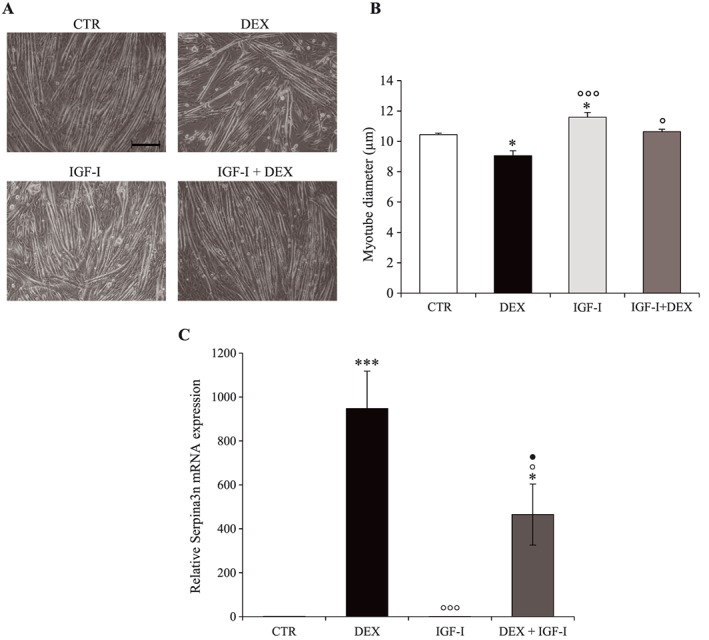
IGF‐I inhibits glucocorticoids‐induced myotube atrophy and Serpina3n mRNA expression. (A) Phase‐contrast representative images of CTR, DEX, IGF‐I, and IGF‐I + DEX treated C2C12 cells. Myotube diameter (B) and Serpina3n mRNA levels (C) of C2C12 cells after 48 h of dexamethasone (10−6 M) and/or IGF‐I (50 ng/mL). Scale bar corresponds to 100 μM. Results are means ± SE (n = 3). Statistical analysis was performed using one‐way ANOVA and Tukey post‐test (* P < 0.05, *** P < 0.001 vs. CTR, ° P < 0.05, °°° P < 0.001 vs. DEX, ● P < 0.05 vs. IGF‐I). DEX, dexamethasone; IGF‐I, insulin‐growth factor I.
mTOR inhibition increases Serpina3n expression in vitro and in vivo
Our next goal was to decipher the mechanisms responsible for the induction of Serpina3n in parallel to decrease in muscle mass. It is well established that the GC‐mediated muscle atrophy involves stimulation of FoxO and inhibition of mTOR,10 two major factors in the control of muscle mass. Although FoxO induction is responsible for the GC‐induced proteolysis through activation of autophagy and the ubiquitin‐proteasome pathway, muscle‐specific deletion of the main FoxO members (1, 3, 4) did not affect neither the Serpina3n mRNA expression (Figure 10A) nor the skeletal muscle mass (Supporting Information, Figure S3A). In contrast, muscle‐specific inactivation of mTOR caused an increased expression of Serpina3n in skeletal muscle (Figure 10B) together with marked muscle atrophy (Supporting Information, Figure S3B). The role of mTOR in the regulation of Serpina3n was also observed in vitro where rapamycin, a specific inhibitor of mTOR, potentiated the up‐regulation of Serpina3n expression by GC in C2C12 cells (Figure 10C). Our results suggest therefore that mTOR, but not FoxO, is a negative regulator of Serpina3n during muscle atrophy. Given the role of mTOR as a major regulator of muscle cell size, the regulation of Serpina3n by mTOR may explain why changes in Serpina3n evolves in opposite to changes in muscle mass.
Figure 10.
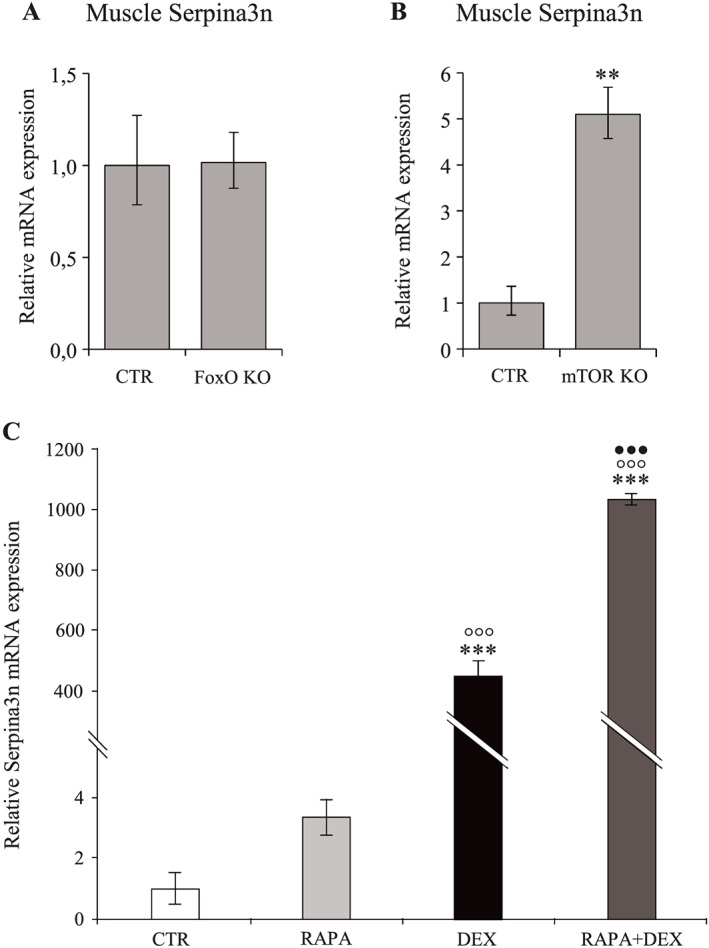
Muscle‐specific deletion of mTOR induces muscle Serpina3n expression while FoxO deletion does not affect it. Serpina3n mRNA levels in tibialis anterior muscle of FoxO KO and CTR mice (A, n = 3/group), and mTOR KO and CTR mice (B, n = 3 and 5/group, respectively). Serpina3n mRNA levels (C) of C2C12 cells after 24 h of dexamethasone (10−6 M) and/or RAPA (50 nM) treatment (n = 3 independent experiments). Results are means ± SE. Statistical analysis was performed using unpaired t‐test or one‐way ANOVA and Tukey post‐test (** P < 0.01, *** P < 0.001 vs. CTR, °°° P < 0.05 vs. RAPA, ••• P < 0.001 vs. DEX). DEX, dexamethasone; KO, knockout.
Muscle hypertrophy is associated with decreased muscle Serpina3n expression and release into circulation
Having demonstrated a strong link between the increase of circulating Serpina3n and muscle atrophy, we wondered whether muscle hypertrophy could be associated with decreased Serpina3n expression. Therefore, we quantified muscle Serpina3n expression in three models of muscle hypertrophy induced by Mstn inhibition (Supporting Information, Figure S4). As seen in Figure 11A, Serpina3n mRNA was decreased in Mstn KO (−41%, P < 0.01), mTr‐FS (−68%; P < 0.01), and sActRIIB‐Fc treated mice (−31%, P < 0.05), indicating that muscle hypertrophy is associated with decreased Serpina3n expression. Moreover, we observed also decreased protein levels of Serpina3n in skeletal muscle and in serum of Mstn KO mice (Figure 11B, C, respectively). Taken together, these observations suggest that muscle synthesis of Serpina3n evolves in parallel with changes in skeletal muscle mass.
Figure 11.
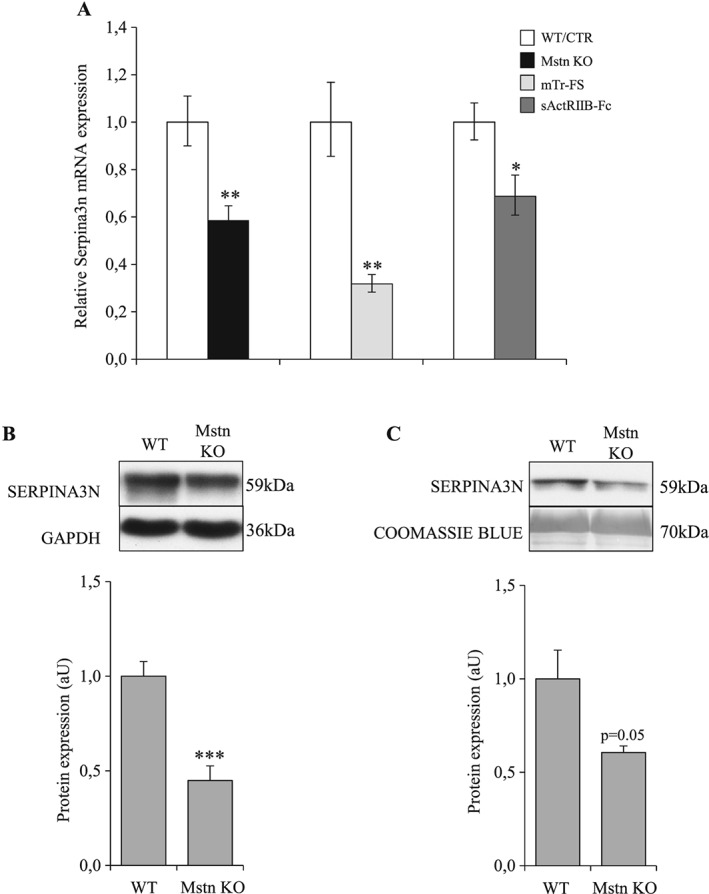
Muscle hypertrophy caused by myostatin inhibition is associated with decreased Serpina3n expression and secretion. (A) Serpina3n mRNA levels in tibialis anterior muscle of Mstn KO and WT mice (n = 6/group), mTr‐FS (Follistatin), and WT mice (n = 4/group) and sActRIIB‐treated and saline‐treated mice (n = 6/group). (B) Serpina3n protein abundance in gastrocnemius muscle of Mstn KO and WT mice. (C) Serpina3n protein abundance in circulation of Mstn KO and WT mice. For western‐blotting, 20 μg of proteins were loaded in each lane for each sample. Results are means ± SE. Statistical analysis was performed using unpaired t‐test (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. CTR or WT). Mstn KO, myostatin knockout; WT, wild‐type.
Discussion
Characterization of changes in muscle secretome associated with GC‐induced muscle atrophy led us to the identification of Serpina3n as one of the most markedly secreted protein in response to DEX. An increased expression of Serpina3n in skeletal muscle and in the serum of mice treated with DEX confirmed our in vitro results. Moreover, we also observed increased levels of Serpina3, the human orthologue of the mouse Serpina3n, in the serum of patients with overt Cushing's syndrome. The increase of Serpina3n in muscle and in serum was also demonstrated in various murine models of muscle atrophy mediated by GC such as cancer cachexia, sepsis, or diabetes. In contrast, levels of Serpina3n both in skeletal muscle and in the circulation were reduced in several models of muscle hypertrophy induced by Mstn inhibition. Taken together, these data suggest that Serpina3n may represent a circulating biomarker of muscle atrophy associated to GC and, broadly, a reflection of dynamic changes in muscle mass.
Murine Serpina3n belongs to the serine protease inhibitors family and represents the mouse orthologue of the human α1‐antichymotrypsin (ACT or Serpina3). Although human Serpina3 is coded by a single gene, repeated duplication has resulted in a cluster of 13 closely related genes at the same locus in mice. Of these, Serpina3n was identified to be an extracellular inhibitor of several proteases such as granzyme B, cathepsins G/B/L, and leucocyte elastase27, 28 in addition to trypsin and chymotrypsin. Serpina3n is highly expressed in brain, testis, lung, heart, spleen, liver, and skeletal muscle.29, 30 In the skeletal muscle, Serpina3n is mainly localized around the myofiber and co‐localized with laminin.31 Previous works have reported the induction of Serpina3n in C2C12 muscle cells in response to GC exposure,32 and in animal models of cancer cachexia33 and muscle dystrophy or injury.31 However, its secretion by muscle cells into the circulation had not yet been reported.
Serpina3n is ubiquitously expressed and not restricted to skeletal muscle.29, 30 However, our observations suggest that increase in circulating levels of Serpina3n associated with muscle atrophy results mainly from increased muscle secretion. Indeed, the increase of Serpina3n protein expression caused by GC is much higher in skeletal muscle than in other organs. Furthermore, the increase of Serpina3n gene expression, which we observed in response to GC in skeletal muscle, was not observed in the liver, the principal secretory organ of circulating proteins. These two observations suggest therefore the major contribution of skeletal muscle to the increase in circulating levels Serpina3n during muscle atrophy.
The consequences of the increased Serpina3n secretion in response to GC and, more generally, to muscle catabolic stimuli are not known. It has been demonstrated that Serpina3n reduces the rate of aortic rupture and death in a mouse model of abdominal aortic aneurysm. On the other hand, topical administration of Serpina3n accelerates tissue repair and wound healing in a mouse diabetic model.28, 34 Moreover, Serpina3n could attenuate neuropathic pain and prevent inflammatory‐mediated neurodegeneration.35, 36 The biological action of the secreted Serpina3n and its role in the skeletal muscle development and homeostasis are still poorly defined. Nevertheless, two studies support a protective role of Serpina3n towards skeletal muscle, in particular in the Duchenne muscular dystrophy (mdx) model and in acute muscle injury, two situations characterized by increased muscle Serpina3n expression, as we observed in our in vitro and in vivo models of atrophy. In absence of specific receptor on muscular membrane, the protective effect of Serpina3n seems related to its antiprotease activity towards extracellular proteases, such elastase. Indeed, in the mdx mouse model, increased levels of elastase protein and activity decreased myoblast proliferation, increased cell death, and decreased myoblast differentiation, fusion, and myotube growth, underlying the loss of regenerative capacity observed in this condition.37 Furthermore, in the same model, skeletal muscle‐specific overexpression of Serpina3n, which blocks the activity of skeletal muscle proteases, in particular elastase, improves sarcolemma integrity and stability, leading to less myofiber degeneration and fibrosis.31 This protective effect of Serpina3n in the mdx model is illustrated by a restoration of running capacity and reduction in creatine kinase serum levels. Therefore, taking into account these observations, we speculate that Serpina3n induction in GC‐induced muscle atrophy might be protective by reducing destructive protease activity and by maintaining the regenerative capacity of skeletal muscle.
The molecular mechanisms responsible for the induction of Serpina3n during GC‐induced atrophy are still unknown. The first hypothesis is a direct action of activated GR on the Serpina3n gene transcription. Indeed, as described by Lannan et al.,38 GR may directly stimulate Serpina3 transcription by binding to the glucocorticoid response element present in the Serpina3 promotor. A second hypothesis is an indirect mechanism involving an intermediary factor. Considering that the changes in skeletal muscle mass caused by GC and by Mstn inhibition involve the enzyme mTOR39, 40 and the transcription factor FoxO,41 these two molecules represent potential candidates for the regulation of Serpina3n. Although the role of FoxO is undisputed in the muscle atrophy caused by GC, our results obtained in mice harbouring a muscle‐specific deletion of FoxO‐1‐3‐4 exclude FoxO as a significant actor in the regulation of the Serpina3n. In contrast, several evidences lead us to consider the role of mTOR in the regulation of the Serpina3n. First, as we observed, IGF‐I, a major mTOR activator, inhibits the increase of Serpina3n expression together with the atrophy caused by DEX. Second, rapamycin, a specific inhibitor of mTOR, potentiates the up‐regulation of Serpina3n expression by GC. Third, as demonstrated in mice harbouring, a muscle‐specific deletion of mTOR, absence of mTOR is associated with increased Serpina3n mRNA together with skeletal muscle atrophy. Finally, the decrease of Serpina3n observed in our three models of Mstn inhibition might result from the increase of mTOR activity, previously reported to contribute to the muscle hypertrophy induced by Mstn inhibition.39 All of these results suggest therefore that mTOR blockade up‐regulates the expression of Serpina3n, whereas mTOR activation down‐regulates Serpina3n.
Many catabolic situations characterized by muscle atrophy are associated with the release of several proinflammatory cytokines (TNF‐α, IL1‐β, IL‐6 …), making these molecules potential players in the atrophic process. Because the induction of Serpina3n is greater in cachexia and sepsis compared with GC treatment, we wondered whether these proinflammatory cytokines might contribute to the increased Serpina3n expression in these conditions. While it has been described that TNF‐α could induce Serpina3 expression in astrocytes, hepatocytes, and lung cells,38, 42, 43 our in vitro results show that TNF‐α did not induce Serpina3n expression neither potentiate its induction by GC. Furthermore, inhibition of GR activity by RU‐486 almost completely blunted the Serpina3n induction by LPS. Taken together, these results demonstrate the predominant role of GC vs. proinflammatory cytokines in the regulation of Serpina3n expression, even in inflammatory catabolic conditions.
Although the identification of Serpina3n was achieved in serum‐free conditions, its release in conditioned medium in response to GC does not reflect a disruption of the sarcolemma. Indeed, cell viability was not impaired by GC. Furthermore, the presence of a signal peptide allows it secretion according to the classical pathway into conditioned medium as well as into circulation, as we observed. In this regard, Serpina3n should be distinguished from other markers of muscle injury, such as creatine kinase.44
The strength of our observations stems in part from the fact that we have been able to demonstrate the increase in Serpina3n not only in vitro but also in experimental animal models as well in human pathological conditions characterized by skeletal muscle atrophy.9 Furthermore, the demonstration of increased Serpina3n levels in the circulation makes this protein a potential candidate biomarker of muscle atrophy. In addition, the increase of Serpina3n muscle expression observed in mdx mice and after cardiotoxin injury indicates that it might be useful as a broad marker of myopathy.31 Finally, besides Serpina3n, other proteins have been identified by our original secretomic analysis and will deserve future attention.
One obvious limitation of this study is the small number of subjects. Before considering Serpina3n as a potential biomarker of muscle atrophy useful in clinical settings, additional studies are clearly mandatory not only in larger human populations but also in more diverse populations characterized by muscle atrophy of different origins. Therefore, its interest as a biomarker of the muscle atrophy process will have to be delineated in future prospective studies, in particular in muscle atrophy models mediated by GC such as cancer cachexia, sepsis, or diabetes.
In summary, our work demonstrates that muscle and circulating Serpina3n level are increased in several models of GC‐mediated muscle atrophy such as cancer cachexia, sepsis, or diabetes and decreased in models of muscle hypertrophy caused by Mstn inhibition. Moreover, we also observed an increase of human Serpina3 levels in the serum of patients exposed to high concentrations of endogenous GC. Furthermore, a cluster of data suggests that the regulation of muscle Serpina3n involves mTOR, an essential determinant of the muscle cell size. To our knowledge, our study is the first to show that GC increase muscle expression and circulating levels of Serpina3n in mouse and in humans in parallel to muscle atrophy. Therefore, Serpina3n could represent a new potential biomarker of GC‐mediated muscle atrophy.
Conflict of interest
The authors have declared that no conflict of interest exists.
Supporting information
Figure S1 Cachexia, LPS‐induced sepsis and STZ‐induced diabetes are associated with muscle atrophy. Gastrocnemius weight of cachectic vs CTR mice (A, n = 8/group), LPS vs CTR mice (B, n = 6/group), and STZ or STZ + INS vs CTR rats (C, n = 6/group). Results are means ± SE. Statistical analysis was performed using unpaired t‐test or 1‐way ANOVA and Tukey posttest (*** p < 0.001 vs CTR, ○○○ p < 0.001 vs STZ + INS).
Figure S2 TNF‐α does not induce neither significantly potentiate the GC‐induced expression of Serpina3n. Serpina3n mRNA levels of C2C12 cells after 48 h of dexamethasone (10‐6 M) and/or TNF‐α (10 ng/ml) treatment. Results are means ± SE. Statistical analysis was performed using 1‐ way ANOVA and Tukey posttest (*** p < 0.001 vs CTR, *** p < 0.05 vs TNF‐α).
Figure S3 mTOR inhibition, but not FoxO, are associated with muscle atrophy. Gastrocnemius weight of FoxO KO vs CTR mice (A, n = 3/group), and mTOR KO vs CTR mice (B, n = 3 and 5/group respectively). Results are means ± SE. Statistical analysis was performed using unpaired t‐test (** p < 0.01 vs CTR).
Figure S4 Muscle hypertrophy caused by Myostatin (Mstn) inhibition. Weight of GC muscle in Mstn KO and WT mice (n = 6/group), and of TA in mTr‐FS (Follistatin) and WT mice (n = 4/group) and in sActRIIB‐treated and saline‐treated mice (n = 6/group). Results are means ± SE. Statistical analysis was performed using unpaired t‐test (*** p < 0.001 vs CTR or WT).
Acknowledgements
We thank Pr M. Sandri (Venetian Institute of Molecular Medicine, Padova, Italy), Dr. L. Grobet (Department of Morphology and Pathology, University of Liege, Belgium), and Dr. S. J. Lee (Department of Molecular Biology and Genetics, The Johns Hopkins University School of Medicine, Baltimore, MD, USA) for the genetically modified mice. We thank Pr O. Ritvos for sActRIIB‐Fc soluble receptor. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.45
This work was funded by grants from Fonds de Recherche Clinique to MG and the Saint‐Luc Foundation to JPT (Cliniques Universitaires Saint‐Luc, Belgium) and a grant from the FRS‐FNRS/Télévie to LB, NMD and JPT (InterCachectomics Consortium).
Gueugneau, M. , d'Hose, D. , Barbé, C. , de Barsy, M. , Lause, P. , Maiter, D. , Bindels, L. B. , Delzenne, N. M. , Schaeffer, L. , Gangloff, Y.‐G. , Chambon, C. , Coudy‐Gandilhon, C. , Béchet, D. , and Thissen, J.‐P. (2018) Increased Serpina3n release into circulation during glucocorticoid‐mediated muscle atrophy. Journal of Cachexia, Sarcopenia and Muscle, 9: 929–946. 10.1002/jcsm.12315.
References
- 1. Prado CM, Cushen SJ, Orsso CE, Ryan AM. Sarcopenia and cachexia in the era of obesity: clinical and nutritional impact. Proc Nutr Soc 2016;75:188–198. [DOI] [PubMed] [Google Scholar]
- 2. Steensberg A, van Hall G, Osada T, Sacchetti M, Saltin B, Klarlund Pedersen B. Production of interleukin‐6 in contracting human skeletal muscles can account for the exercise‐induced increase in plasma interleukin‐6. J Physiol 2000;529:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pedersen BK. Muscle as a secretory organ. Compr Physiol 2013;3:1337–1362. [DOI] [PubMed] [Google Scholar]
- 4. Plomgaard P, Halban PA, Bouzakri K. Bimodal impact of skeletal muscle on pancreatic beta‐cell function in health and disease. Diabetes Obes Metab 2012;13:78–84. [DOI] [PubMed] [Google Scholar]
- 5. Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, et al. Follistatin‐like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric‐oxide synthase‐dependent mechanism. J Biol Chem 2008;283:32802–32811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hojman P, Pedersen M, Nielsen AR, Krogh‐Madsen R, Yfanti C, Akerstrom T, et al. Fibroblast growth factor‐21 is induced in human skeletal muscles by hyperinsulinemia. Diabetes 2009;58:2797–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henningsen J, Rigbolt KT, Blagoev B, Pedersen BK, Kratchmarova I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol cell proteomics: MCP 2010;9:2482–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stastna M, Van Eyk JE. Secreted proteins as a fundamental source for biomarker discovery. Proteomics 2012;12:722–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pirlich M, Biering H, Gerl H, Ventz M, Schmidt B, Ertl S, et al. Loss of body cell mass in Cushing's syndrome: effect of treatment. J Clin Endocrinol Metab 2002;87:1078–1084. [DOI] [PubMed] [Google Scholar]
- 10. Schakman O, Kalista S, Barbe C, Loumaye A, Thissen JP. Glucocorticoid‐induced skeletal muscle atrophy. Int J Biochem Cell Biol 2013;45:2163–2172. [DOI] [PubMed] [Google Scholar]
- 11. Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest 2009;119:3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schakman O, Dehoux M, Bouchuari S, Delaere S, Lause P, Decroly N, et al. Role of IGF‐I and the TNFalpha/NF‐kappaB pathway in the induction of muscle atrogenes by acute inflammation. Am J Physiol Endocrinol Metab 2012;303:E729–E739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB j: official publication of the Federation of American Societies for Experimental Biology 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Theron L, Gueugneau M, Coudy C, Viala D, Bijlsma A, Butler‐Browne G, et al. Label‐free quantitative protein profiling of vastus lateralis muscle during human aging. Mol cell proteomics: MCP 2014;13:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bindels LB, Neyrinck AM, Claus SP, Le Roy CI, Grangette C, Pot B, et al. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J 2016;10:1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dehoux M, Van Beneden R, Pasko N, Lause P, Verniers J, Underwood L, et al. Role of the insulin‐like growth factor I decline in the induction of atrogin‐1/MAFbx during fasting and diabetes. Endocrinology 2004;145:4806–4812. [DOI] [PubMed] [Google Scholar]
- 17. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 2004;24:9508–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 2009;187:859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grobet L, Pirottin D, Farnir F, Poncelet D, Royo LJ, Brouwers B, et al. Modulating skeletal muscle mass by postnatal, muscle‐specific inactivation of the myostatin gene. Genesis 2003;35:227–238. [DOI] [PubMed] [Google Scholar]
- 21. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 2001;98:9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hulmi JJ, Oliveira BM, Silvennoinen M, Hoogaars WM, Pasternack A, Kainulainen H, et al. Exercise restores decreased physical activity levels and increases markers of autophagy and oxidative capacity in myostatin/activin‐blocked mdx mice. Am J Physiol Endocrinol Metab 2013;305:E171–E182. [DOI] [PubMed] [Google Scholar]
- 23. Hoogaars WM, Mouisel E, Pasternack A, Hulmi JJ, Relizani K, Schuelke M, et al. Combined effect of AAV‐U7‐induced dystrophin exon skipping and soluble activin Type IIB receptor in mdx mice. Hum Gene Ther 2012;23:1269–1279. [DOI] [PubMed] [Google Scholar]
- 24. Dehoux MJ, van Beneden RP, Fernandez‐Celemin L, Lause PL, Thissen JP. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett 2003;544:214–217. [DOI] [PubMed] [Google Scholar]
- 25. Schakman O, Kalista S, Bertrand L, Lause P, Verniers J, Ketelslegers JM, et al. Role of Akt/GSK‐3beta/beta‐catenin transduction pathway in the muscle anti‐atrophy action of insulin‐like growth factor‐I in glucocorticoid‐treated rats. Endocrinology 2008;149:3900–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato AY, Richardson D, Cregor M, Davis HM, Au ED, McAndrews K, et al. Glucocorticoids induce bone and muscle atrophy by tissue‐specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology 2017;158:664–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horvath AJ, Irving JA, Rossjohn J, Law RH, Bottomley SP, Quinsey NS, et al. The murine orthologue of human antichymotrypsin: a structural paradigm for clade A3 serpins. J Biol Chem 2005;280:43168–43178. [DOI] [PubMed] [Google Scholar]
- 28. Ang LS, Boivin WA, Williams SJ, Zhao H, Abraham T, Carmine‐Simmen K, et al. Serpina3n attenuates granzyme B‐mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis 2011;2:e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horvath AJ, Forsyth SL, Coughlin PB. Expression patterns of murine antichymotrypsin‐like genes reflect evolutionary divergence at the Serpina3 locus. J Mol Evol 2004;59:488–497. [DOI] [PubMed] [Google Scholar]
- 30. Coronado MJ, Brandt JE, Kim E, Bucek A, Bedja D, Abston ED, et al. Testosterone and interleukin‐1beta increase cardiac remodeling during coxsackievirus B3 myocarditis via serpin A 3n. Am J Physiol Heart Circ Physiol 2012;302:H1726–H1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tjondrokoesoemo A, Schips T, Kanisicak O, Sargent MA, Molkentin JD. Genetic overexpression of Serpina3n attenuates muscular dystrophy in mice. Hum Mol Genet 2016;25:1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome‐wide analysis of glucocorticoid receptor‐binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A 2012;109:11160–11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shum AM, Fung DC, Corley SM, McGill MC, Bentley NL, Tan TC, et al. Cardiac and skeletal muscles show molecularly distinct responses to cancer cachexia. Physiol Genomics 2015;47:588–599. [DOI] [PubMed] [Google Scholar]
- 34. Hsu I, Parkinson LG, Shen Y, Toro A, Brown T, Zhao H, et al. Serpina3n accelerates tissue repair in a diabetic mouse model of delayed wound healing. Cell Death Dis 2014;5:e1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vicuna L, Strochlic DE, Latremoliere A, Bali KK, Simonetti M, Husainie D, et al. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell‐derived leukocyte elastase. Nat Med 2015;21:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haile Y, Carmine‐Simmen K, Olechowski C, Kerr B, Bleackley RC, Giuliani F. Granzyme B‐inhibitor serpina3n induces neuroprotection in vitro and in vivo. J Neuroinflammation 2015;12:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arecco N, Clarke CJ, Jones FK, Simpson DM, Mason D, Beynon RJ, et al. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci Rep 2016;6:24708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lannan EA, Galliher‐Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFalpha coregulate gene expression in vitro and in vivo. Endocrinology 2012;153:3701–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab 2009;296:E567–E572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodriguez J, Vernus B, Toubiana M, Jublanc E, Tintignac L, Leibovitch S, et al. Myostatin inactivation increases myotube size through regulation of translational initiation machinery. J Cell Biochem 2011;112:3531–3542. [DOI] [PubMed] [Google Scholar]
- 41. Kang SH, Lee HA, Kim M, Lee E, Sohn UD, Kim I. Forkhead box O3 plays a role in skeletal muscle atrophy through expression of E3 ubiquitin ligases MuRF‐1 and atrogin‐1 in Cushing's syndrome. Am J Physiol Endocrinol Metab 2017;312:E495–E507. [DOI] [PubMed] [Google Scholar]
- 42. Kasza A, Bugno M, Koj A. Long‐term culture of HepG2 hepatoma cells as a model for liver acute phase response during chronic inflammation. Effects of interleukin‐6, dexamethasone and retinoic acid. Biol Chem Hoppe Seyler 1994;375:779–783. [DOI] [PubMed] [Google Scholar]
- 43. Machein U, Lieb K, Hull M, Fiebich BL. IL‐1 beta and TNF alpha, but not IL‐6, induce alpha 1‐antichymotrypsin expression in the human astrocytoma cell line U373 MG. Neuroreport 1995;6:2283–2286. [DOI] [PubMed] [Google Scholar]
- 44. Ohlendieck K. Novel proteomic biomarkers for skeletal muscle diseases. Biomark Med 2017;11:409–412. [DOI] [PubMed] [Google Scholar]
- 45. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia, Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cachexia, LPS‐induced sepsis and STZ‐induced diabetes are associated with muscle atrophy. Gastrocnemius weight of cachectic vs CTR mice (A, n = 8/group), LPS vs CTR mice (B, n = 6/group), and STZ or STZ + INS vs CTR rats (C, n = 6/group). Results are means ± SE. Statistical analysis was performed using unpaired t‐test or 1‐way ANOVA and Tukey posttest (*** p < 0.001 vs CTR, ○○○ p < 0.001 vs STZ + INS).
Figure S2 TNF‐α does not induce neither significantly potentiate the GC‐induced expression of Serpina3n. Serpina3n mRNA levels of C2C12 cells after 48 h of dexamethasone (10‐6 M) and/or TNF‐α (10 ng/ml) treatment. Results are means ± SE. Statistical analysis was performed using 1‐ way ANOVA and Tukey posttest (*** p < 0.001 vs CTR, *** p < 0.05 vs TNF‐α).
Figure S3 mTOR inhibition, but not FoxO, are associated with muscle atrophy. Gastrocnemius weight of FoxO KO vs CTR mice (A, n = 3/group), and mTOR KO vs CTR mice (B, n = 3 and 5/group respectively). Results are means ± SE. Statistical analysis was performed using unpaired t‐test (** p < 0.01 vs CTR).
Figure S4 Muscle hypertrophy caused by Myostatin (Mstn) inhibition. Weight of GC muscle in Mstn KO and WT mice (n = 6/group), and of TA in mTr‐FS (Follistatin) and WT mice (n = 4/group) and in sActRIIB‐treated and saline‐treated mice (n = 6/group). Results are means ± SE. Statistical analysis was performed using unpaired t‐test (*** p < 0.001 vs CTR or WT).