

The symptoms of Clostridium difficile infection (CDI) are attributed largely to two C. difficile toxins, TcdA and TcdB. Significant efforts have been devoted to developing vaccines targeting both toxins through parenteral immunization routes.
KEYWORDS: Clostridium difficile infection, oral immunization, vaccine, nontoxigenic
ABSTRACT
The symptoms of Clostridium difficile infection (CDI) are attributed largely to two C. difficile toxins, TcdA and TcdB. Significant efforts have been devoted to developing vaccines targeting both toxins through parenteral immunization routes. However, C. difficile is an enteric pathogen, and mucosal/oral immunization would be particularly useful to protect the host against CDI, considering that the gut is the main site of disease onset and progression. Moreover, vaccines directed only against toxins do not target the cells and spores that transmit the disease. Previously, we constructed a chimeric vaccine candidate, mTcd138, comprised of the glucosyltransferase and cysteine proteinase domains of TcdB and the receptor binding domain of TcdA. In this study, to develop an oral vaccine that can target both C. difficile toxins and colonization/adhesion factors, we expressed mTcd138 in a nontoxigenic C. difficile (NTCD) strain, resulting in strain NTCD_mTcd138. Oral immunization with spores of NTCD_mTcd138 provided mice full protection against infection with a hypervirulent C. difficile strain, UK6 (ribotype 027). The protective strength and efficacy of NTCD_mTcd138 were further evaluated in the acute CDI hamster model. Oral immunization with spores of NTCD_mTcd138 also provided hamsters significant protection against infection with 2 × 104 UK6 spores, a dose 200-fold higher than the lethal dose of UK6 in hamsters. These results imply that the genetically modified, nontoxigenic C. difficile strain expressing mTcd138 may represent a novel mucosal vaccine candidate against CDI.
INTRODUCTION
Clostridium difficile is a spore-forming, anaerobic, and toxin-producing bacillus. It is the most common cause of nosocomial antibiotic-associated diarrhea and the etiologic agent of pseudomembranous colitis, with about 453,000 cases and 29,000 deaths yearly in the United States as reported by CDC in 2015 (1). Furthermore, a continual rise in severe C. difficile infections (CDI) has been observed worldwide (2, 3). CDI is transmitted through spores. C. difficile toxins (TcdA and TcdB) are the major virulent factors. The two toxins share similar domain structures, including the N-terminal glucosyltransferase domain (GT), the autocatalytic cysteine proteinase domain (CPD), the central translocation domain (TMD), and the C-terminal receptor binding domain (RBD) (4, 5). Standard therapy depends on treatment with vancomycin, metronidazole, or fidaxomicin. None of these is fully effective (6, 7). Moreover, an estimated 15 to 35% of those infected with C. difficile relapse following treatment (8, 9). Treatment of recurrent CDI is one of the major challenges in the field (10–12). Active vaccination is generally accepted as a logical and cost-effective approach to prevent CDI, but no vaccine effective at preventing primary and recurrent CDI is licensed (13, 14).
There are three C. difficile vaccines in different stages of clinical trials, including toxoids A and B from Sanofi (15), fusion protein (IC84) from Valneva (16), and genetically modified TcdA and TcdB from Pfizer (17). All three vaccine candidates target TcdA and TcdB or their RBDs and use parenteral routes for immunization. However, our published data show that anti-TcdA IgG, but not IgA, dramatically enhances TcdA-mediated cytotoxicity in vitro (18) and disease in vivo (19), raising safety concerns with parenteral immunization. In addition, C. difficile is an enteric pathogen, and mucosal/oral immunization would be particularly useful to protect the host against CDI, considering that the gut is the main site of disease onset and progression. Moreover, vaccines directed only against toxins do not target the cells and spores that transmit the disease and contribute to high-rate recurrent CDI.
Vaccination through the oral route has the advantage of inducing mucosal immunity (20, 21) and other multifarious advantages over traditional parenteral vaccines, including ease of administration, better patient compliance, needle-free painless delivery, and lower cost (22, 23). However, since the harsh acidic and proteolytic environment in the stomach can cause the vaccine subunit proteins to degrade, subunit-based oral vaccination is difficult to implement (24).
Previous studies have shown that asymptomatic colonization by nontoxigenic C. difficile strains tends to decrease the risk of CDI in humans (25). Nontoxigenic C. difficile strains have been shown to prevent fatal CDI in mice, hamsters, and piglets (26–28). Recently, we reported a novel C. difficile vaccine candidate (mTcd138) that targets both C. difficile toxins (29). To develop mucosal vaccines that can induce immune responses against toxins and C. difficile colonization, we engineered a nontoxigenic C. difficile strain to express mTcd138, i.e., strain NTCD_Tcd138, and our data indicate that NTCD_Tcd138 is a promising oral vaccine candidate against CDI. This is the first report on vaccines against CDI based on nontoxigenic C. difficile strains.
RESULTS
Expression of mTcd138 in NTCD.
Previously, we generated a fusion protein (mTcd138) that is comprised of the glucosyltransferase and cysteine domains of TcdB and the receptor domain of TcdA. To ensure that mTcd138 is atoxic, two point mutations were introduced into the glucosyltransferase domain of TcdB. Nontoxigenic C. difficile strain CCUG37785 (here referred to as NTCD) is a nontoxigenic strain (data not shown). To express mTcd138 in NTCD, the gene encoding mTcd138 was cloned in the Escherichia coli-C. difficile shuttle vector pRPF144, generating the plasmid pBL139. Plasmid pBL139 was further introduced into NTCD by conjugative transfer through an intermediate host, E. coli HB101 pRK24, resulting in strain NTCD_mTcd138. Western blot analysis showed that mTcd138 was expressed in NTCD_mTcd138 and was detected intracellularly, as well as in the supernatant of the bacterial culture (Fig. 1A).
FIG 1.

mTcd138 was expressed in nontoxigenic C. difficile CCUG37785 strain (NTCD), and oral immunization (IM) of mice with NTCD_mTcd138 spores induced protective responses. (A) Western blot analysis of supernatants and pellets of strains NTCD_mTcd138 and NTCD (mTcd138 as positive control) using anti-TcdA or anti-TcdB antibodies. (B) Oral immunization of mice with NTCD_mTcd138 spores induced mucosal and systemic toxin-specific antibody responses. Groups of C57 BL/6 mice (n = 10) were orally immunized with NTCD_mTcd138 (2 × 106 spores/immunization, 3 times at 14-day intervals). Sera and feces were collected after each immunization. Before use, feces were dissolved (0.1g/ml) in PBS containing proteinase inhibitors. Anti-TcdA/anti-TcdB IgG titers in sera and anti-TcdA/anti-TcdB IgA titers in sera or in feces were determined by ELISA (*, P < 0.05 versus 1st IM). (C) Oral immunization of mice with NTCD_mTcd138 or NTCD spores induced mucosal and systemic antibody responses against FliCD. Sera and feces were collected after each immunization. Anti-FliCD IgG or IgA titers in sera or feces were determined by ELISA (*, P < 0.05 versus 1st IM). (D) Antitoxin neutralizing titers of sera or feces from mice orally immunized with NTCD_mTcd138 spores. Vero cells were used to determine in vitro neutralizing activities of sera or feces. Bars show means ± standard deviations (SD). (*, P < 0.05 versus anti-TcdA; ns, no significance versus anti-TcdA). Student's unpaired t test was used for statistical significance.
Oral immunization of mice with NTCD_mTcd138 spores induced mucosal and systemic toxin-specific antibody responses.
Oral immunization of mice with NTCD_mTcd138 (2 × 106 spores per immunization, 3 times at 14-day intervals) induced both IgG and IgA antibody responses specific for both toxins in serum, as well as IgA antibodies specific for both toxins in feces (Fig. 1B). Most importantly, NTCD_mTcd138 immunization also induced neutralizing antibodies against both toxins (Fig. 1D). To determine whether NTCD or NTCD_mTcd138 immunization can induce anti-C. difficile responses, we generated a fusion protein containing the full lengths of C. difficile flagellin proteins FliC and FliD (designated FliCD; data not shown), and measured anti-FliCD antibody levels in sera and feces from NTCD- or NTCD_mTcd138-immunized mice. Interestingly, we found that in comparison with NTCD, NTCD_mTcd138 immunization could induce higher levels of anti-FliCD IgG/IgA responses in both serum and feces (Fig. 1C).
Oral immunization with NTCD_mTcd138 spores provided full protection to mice against infection with hypervirulent C. difficile strain UK6.
Protection efficacy of NTCD_mTcd138 was evaluated in a mouse model of CDI. After three oral immunizations (2 × 106 spores per mouse per immunization, 3 times at 14-day intervals), mice were challenged with 106 spores of C. difficile UK6, a hypervirulent strain of ribotype 027. In vehicle (phosphate-buffered saline [PBS])-immunized mice, significant disease symptoms, including weight loss (Fig. 2B) and severe diarrhea (Fig. 2C), were evident in all mice; 80% of mice succumbed by day 4 (Fig. 2A). In contrast, NTCD_mTcd138-immunized mice were fully protected and showed no signs of disease at any stage (Fig. 2). Interestingly, immunization with NTCD showed only slight, but not significant protection (Fig. 2A and B) against C. difficile UK6 challenge. NTCD_mTcd138-immunized mice secreted significantly smaller amounts of toxins compared to NTCD-only or PBS immunization groups (Fig. 3A and B). Fecal samples of NTCD_mTcd138-immunized mice contained significantly fewer UK6 spores than did PBS immunization groups (Fig. 3C). While spores of UK6 in feces of NTCD-immunized mice were fewer than those in PBS immunization control mice, there was a significant difference between the two groups (P = 0.0687) (Fig. 3C).
FIG 2.

Oral immunization with NTCD_mTcd138 spores provided full protection to mice against infection with hypervirulent C. difficile strain UK6. Groups of mice (n = 10) were orally immunized with spores of NTCD or NTCD_mTcd138 (2 × 106 spores in 200 μl PBS) or with PBS (200 μl) 3 times at 14-day intervals. Seven days after the third immunization, mice were given an antibiotic mixture in drinking water for 4 days, switched to regular water for 2 days, and given one dose of clindamycin (10 mg/kg) before infection with 106 C. difficile UK6 spores by gavage. Mice were monitored, and mouse survival (P = 0.495 between groups PBS and NTCD; P = 0.0002 between groups PBS and NTCD_mTcd138; P = 0.002 between groups NTCD and NTCD_mTcd138) (A), mean relative weight changes (P = 0.0004 between groups NTCD and NTCD_mTcd138; P = 0.2366 between groups PBS and NTCD) (B), and frequency of diarrhea (C) of different groups were recorded. Animal survivals were analyzed by Kaplan-Meier survival analysis with a log rank test of significance. Bars show means ± SD. (*, P < 0.05; ns, no significance, P > 0.05).
FIG 3.
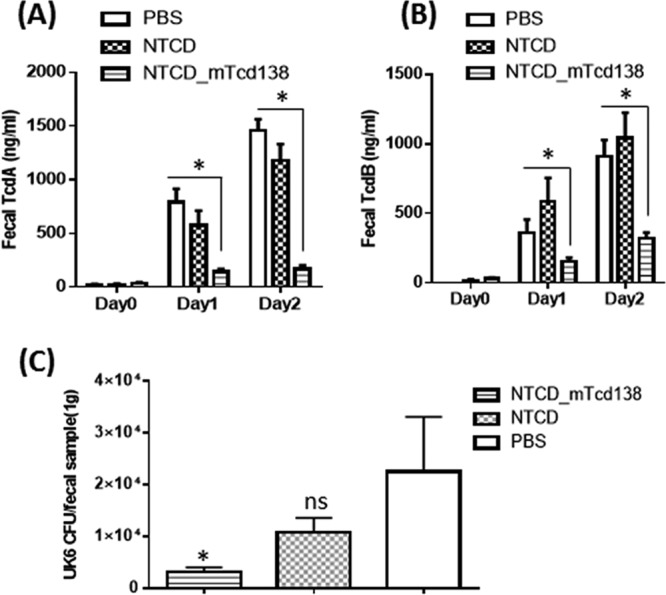
Fecal toxin levels and amounts of C. difficile UK6 spores in mice orally immunized with NTCD, NTCD_mTcd138 spores, or PBS followed by infection with C. difficile UK6 spores. Feces were collected on postinfection days 0, 1, and 2 and then dissolved (0.1 g/ml) in PBS with proteinase inhibitors. TcdA (A) or TcdB (B) levels in feces were determined by ELISA (*, P < 0.05 versus PBS or NTCD). (C) Fecal samples were collected at day 4 postchallenge for bacterial enumeration. An equal amount of 95% ethanol was mixed with each fecal sample for 30 min. Ethanol was removed by centrifugation, and pellets were diluted and spread on BHI-supplemented (BHIS) agar plates containing 10% taurocholic acid in an anaerobic chamber. After incubation for 48 h, the colonies on each plate were counted. The tcdB gene was amplified to distinguish toxigenic C. difficile (UK6) and nontoxigenic C. difficile strains. Bars represent means ± SD. (*, P < 0.05 versus PBS; ns, P > 0.05, i.e., no significance versus PBS; P = 0.0687 between groups NTCD and NTCD_mTcd138). Student's unpaired t test was used for statistical significance.
Oral immunization of hamsters with NTCD_Tcd138 spores induced protective responses against both toxins and infection with hypervirulent C. difficile strain UK6.
We further evaluated the immunogenicity and protection efficacy of NTCD_mTcd138 in hamsters. Oral immunization of hamsters with NTCD_mTcd138 (2 × 106 spores per hamster per immunization, 3 times at 14-day intervals) induced similar levels of anti-TcdA and anti-TcdB IgG antibodies in sera and feces (Fig. 4A and B). In addition, NTCD_mTcd138 immunization also induced significant anti-FliCD IgG antibodies in sera and feces. Similar to the results in mice, in comparison with NTCD, NTCD_mTcd138 immunization induced higher levels of anti-FliCD IgG responses in both sera and feces (Fig. 4C and D). We could not measure anti-TcdA/TcdB/FliCD IgA antibodies due to the lack of hamster-raised anti-IgA antibodies. But, importantly, neutralizing antibodies against both toxins were detected in both sera and feces (Fig. 4E and F). Hamsters are extremely sensitive to C. difficile infection and usually die within 2 to 3 days of infection at a dose of 100 spores. Therefore, the hamster is an ideal animal to test the strength of vaccine candidates against CDI. To evaluate the protection strength of NTCD_mTcd138, we challenged the immunized hamsters (2 × 106 spores of NTCD_mTcd138 or NTCD per hamster per immunization, 3 times at 14-day intervals) with C. difficile strain UK6 at 2 × 104 spores/hamster, which is 200-fold higher than the lethal C. difficile infection dose (100 spores). Oral immunization with NTCD_mTcd138 spores provided significant protection to hamsters in survival against such a high challenge dose in comparison with the PBS control group (P = 0.0458 between groups PBS and NTCD_mTcd138) (Fig. 5). Interestingly, in agreement with the results in mice (Fig. 2A and B), immunization of hamsters with NTCD-only spores also provided protection in survival, though not significant compared with the PBS control group (P = 0.0754 between groups PBS and NTCD) (Fig. 5A). Although the survival rate of the NTCD_mTcd138-immunized group was higher than that of the NTCD-immunized group, there was no significant difference in survival between the two groups (P = 0.089) (Fig. 5A). Both NTCD_mTcd138- and NTCD-immunized groups showed lower diarrhea rates than those in the PBS control group (Fig. 5B).
FIG 4.

Oral immunization of hamsters with NTCD_mTcd138 spores induced protective responses. Groups of golden Syrian hamsters (n = 10) were orally immunized with NTCD_mTcd138 at 2 × 106 spores 3 times at 14-day intervals. Sera and feces were collected after each immunization. Anti-TcdA/anti-TcdB IgG titers in sera and feces were determined by ELISA. (A, B) Immunization with NTCD_mTcd138 induced systemic toxin-specific antibody responses. (C, D) Immunization with NTCD_mTcd138 induced mucosal and systemic antibody responses against FliCD. (E, F) Vero cells were used to determine in vitro neutralizing activities of sera (E) or feces (F). The neutralizing titer is expressed as the maximum dilution of the sera that inhibits cell rounding caused by toxin at a given concentration. This given concentration is the minimum toxin dose causing cell rounding after a 16-hour of toxin exposure, i.e., 2.5 and 0.1 ng/ml for TcdA and TcdB, respectively. Bars stand for means ± SD. *, P < 0.05 versus 1st IM in panels A, B, C, and D; ns, no significance versus anti-TcdA, P > 0.05 in panels E and F. Student's unpaired t test was used for statistical significance.
FIG 5.
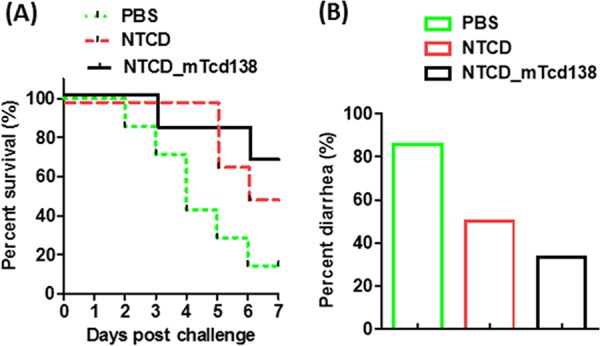
Oral immunization with NTCD_mTcd138 spores provided significant protection to hamsters against infection with virulent C. difficile strain UK6 at a dose 200-fold greater than the lethal infection dose. Groups of hamsters (n = 10) were orally immunized with NTCD or NTCD_mTcd138 (2 × 106 spores in 200 μl PBS) or PBS (200 μl) 3 times at 14-day intervals. Seven days after the third immunization, hamsters were given clindamycin (i.p., 40 mg/kg/day for 2 days), followed by infection with 2 × 104 C. difficile UK6 spores by gavage. Hamsters were monitored, and survivals (P = 0.0754 between groups PBS and NTCD; P = 0.0458 between groups PBS and NTCD_mTcd138; P = 0.0899 between groups NTCD and NTCD_mTcd138) (A) and diarrhea (B) were recorded. Animal survivals were analyzed by Kaplan-Meier survival analysis with a log rank test of significance.
DISCUSSION
CDI is transmitted through bacterial spores. Prevention is by limiting antibiotic use, hand washing, and terminal room cleaning in hospitals (30). Vaccination is a promising intervention approach that can provide long-term protection against CDI (31). No vaccine against CDI is currently licensed. Tremendous efforts have been devoted to developing vaccines targeting both toxins (32–34). However, vaccines directed only against toxins do not target the cells and spores that transmit the disease.
We recently reported a novel C. difficile vaccine candidate (mTcd138), which targets both C. difficile toxins (29) and was effective in prevention of CDI in both mice and hamsters through parenteral immunization routes. However, C. difficile is an enteric pathogen, and CDI starts and progresses in the intestine. Therefore, mucosal and especially oral immunization would be ideal. In addition, injectable vaccines induce good systemic immunity, but mucosal responses are often unsatisfactory, whereas mucosal vaccines provide both systemic and mucosal immunity (35). Oral vaccination is considered the easiest way to deliver immunogens; also, it is more acceptable to patients and reduces the need for highly trained personnel during mass immunization.
A major barrier for oral immunization is the harsh stomach and intestinal environment, which can degrade protein-based vaccines (24). C. difficile exists environmentally as spore forms. The unique multilayered structure of spores can allow them to survive under extreme temperatures, pHs, and humidity levels; in that regard, spores can serve as an effective oral delivery vehicle (36–38). By engineering nontoxigenic C. difficile strains to express mTcd138, i.e., NTCD_mTcd138, we combined two independent methods of CDI prevention in one treatment. Our data showed that oral immunization of mice/hamsters with NTCD_mTcd138 spores not only induced protective antibody responses to both toxins (Fig. 1 and 4) but also induced antibody responses to C. difficile flagellin proteins (e.g., FliC and FliD) (Fig. 1C and 4C and D), suggesting that NTCD_mTcd138 immunization could induce a protective antibody response to colonization factors of C. difficile. In support of this, fecal samples from NTCD/NTCD_mTcd138-immunized mice contain fewer spores of the infecting pathogenic strain than do a control (Fig. 3C).
Previous studies have shown that asymptomatic colonization by nontoxigenic C. difficile strains tends to decrease the risk of CDI in humans (25). Nontoxigenic C. difficile strains have been shown to prevent fatal CDI in mice, hamsters, and piglets (26–28), presumably by competing with toxigenic strains for colonization in intestine. However, it is not clear whether oral immunizations with NTCD/NTCD_mTcd138 spores provided colonization resistance against infection with C. difficile UK6 spores. In order to establish a mouse/hamster model of CDI, mice were pretreated with a mixture of six antibiotics (kanamycin, gentamicin, colistin, metronidazole, vancomycin, and clindamycin), and hamsters were pretreated with clindamycin, as described in Materials and Methods. We determined susceptibility of NTCD/NTCD_mTcd138 vegetative cells to these antibiotics as follows: the MICs of kanamycin, gentamicin, colistin, metronidazole, vancomycin, and clindamycin are 64, 16, 32, 0.5, 1, and 1 μg/ml, respectively. In further studies, we will evaluate whether NTCD/NTCD_mTcd138 survived in animals after antibiotic treatment.
In addition, in the hamster model of CDI, while immunizations with NTCD_mTcd138 provided significant protection against C. difficile UK6 challenge compared with the PBS control group (Fig. 5), there was no significant difference between NTCD_mTcd138 and NTCD groups (Fig. 5), which might be due to the high sensitivity of hamsters to CDI and the high challenge dose used (2 × 104 spores/hamster, which is 200-fold higher than the lethal C. difficile infection dose of 100 spores), which could hinder the effects of NTCD-mTcd138 immunization in hamsters. In future studies, we will further optimize the immunization dosages and examine immunization protection against other ribotypes of epidemic C. difficile strains, as well as whether the oral immunization alters the intestinal microbiome.
In summary, NTCD_mTcd138 is a promising oral vaccine candidate against CDI. In addition, the spore form of the immunogen is thermostable and does not need cold-chain production, storage, and transportation.
MATERIALS AND METHODS
Animals.
All studies conformed to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutes' Animal Care and Use Committees (IACUC) at the University of South Florida and Tufts University. Wild-type C57BL/6 mice and Golden Syrian hamsters were purchased from Charles River Laboratories.
Expression and purification of recombinant fusion protein mTcd138.
Bacterial strains and plasmids used in this study are detailed in Table 1. To generate mTcd138, the DNA sequences from C. difficile VPI 10463 (39) encoding the glucosyltransferase (GTD, amino acids [aa] 1 to 543) with 2 amino acid mutations (W102A and D288N) and cysteine proteinase (CPD, aa 543 to 767) domains of TcdB and the receptor binding domain (RBD) of TcdA were bridged with a linker (GGT GGC TCT GGT) sequence, synthesized by Geneart (Germany) and cloned between the BsrGI and EagI sites of the vector pHis1525 (Boca Scientific). mTcd138 was expressed in Bacillus megaterium and purified as described previously (29).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant details | Reference or source |
|---|---|---|
| C. difficile VPI 10463 | A known very-high-level toxin producer | 39 |
| C. difficile R20291 | Wild type; PCR ribotype 027 | 40 |
| C. difficile UK6 | An epidemic strain (provided by Dale Gerding and Abraham L. Sonenshein) isolated in the United Kingdom | 44 |
| CD2001 | Nontoxigenic C. difficile | Michel R. Popoff, Institut Pasteur |
| CCUG37780 | Michel Delmée (UCL, Belgium) to the Culture Collection of the University of Gothenburg (CCUG), Sweden | |
| CCUG37785 | ||
| CCUG37787 | ||
| Vector pHis1525 | Cloning vectors; the xylose operon is used as regulatory element | 41 |
| pRPF144 | Based on the pMTL960 backbone, subcloned into pBL139 | 42 |
| pBL139 | E. coli-C. difficile shuttle vector | 43 |
Expression and purification of recombinant fusion protein FliCD.
Gene sequences encoding FliC and FliD from C. difficile R20291 (40) were bridged with a linker (GGT GGC TCT GGT) sequence, synthesized by Geneart (Germany) and cloned between the BsrGI and EagI sites of the vector pHis1525 (41). FliCD was expressed in B. megaterium and purified as described previously (29).
Expression of mTcd138 in nontoxigenic strains.
The mTcd138 gene was amplified using pMA-mTcd138 as the template (synthesized by genscript), primers oND54 (CCCGAGCTCCTGCAGTAAAGGAGGTTTTTATGAGTTTAGTTAATAAGAAAACAG) and oND55 (CGCGGATCCTTACCCATATATCCCAGGGCTTTTTG), and Phusion Hi-Fidelity DNA polymerase (New England BioLabs). The fragment was cloned into pRPF144 (42) (kindly provided by Robert Fagan from University of Sheffield) using SacI and BamHI sites resulting in pBL139. pBL139 was introduced into the C. difficile nontoxigenic strains CD2001, CCUG37780, CCUG37785, and CCUG37787 by conjugation thorough an intermediate host, E. coli HB101 pRK24, as previously described (43).
Western blotting.
Nontoxigenic C. difficile strains were grown in brain heart infusion (BHI) medium in an anaerobic chamber at 37°C for 24 to 48 h, after which supernatant fluids were collected. C. difficile vegetative cell pellets were lysed in protein lysis buffer (distilled water [dH2O], 0.05 M Tris, 0.3 M NaCl, 0.5% Triton X-100, 2 mM EDTA, 0.4 mM Na3VO4, 2.5 mM leupeptin, 2.5 mM aprotinin, 2.5 mM 4-nitrophenyl 4-guanidinobenzoate hydrochloride [NPGB]). Protein concentration was measured using a bicinchoninic acid (BCA) protein assay (Thermo Scientific, Suwanee, GA). Protein extracts were subjected to 12% SDS-PAGE separation. Then, proteins were transferred onto the Nylon membrane. After blocking for 1 h at room temperature (RT) with 5% skim milk, the membrane was incubated overnight at 4°C with anti-toxin A and anti-toxin B antibodies (1:1,000). After washing with PBST (PBS with 0.05% Tween), the membrane was incubated with horseradish peroxidase-conjugated secondary goat anti-mouse antibody (catalog number ab97023, IgG, 1:3,000; Abcam, Cambridge, MA), the antibody-reactive bands were revealed by enhanced chemiluminescence detection on Hyperfilm (Thermo Fisher Scientific, Waltham, MA).
Preparation of C. difficile spores.
Sporulation of the C. difficile UK6 (BI/NAP1/027) (44) and NTCD_Tcd138 strains was induced in Clospore medium as described previously (45). Briefly, 20 ml of Columbia broth culture medium was inoculated into 500 ml of Clospore medium overnight, and the mixture was incubated without any agitation for 7 to 14 days at 37°C in an anaerobic incubator. The spore suspension was centrifuged at 7,000 rpm for 30 min, and the pellet was washed 5 times with sterile water and suspended in 10 ml of double-distilled water (ddH2O). The spore suspension was heated at 60°C for 20 min to kill vegetative cells and stored at 4°C. The spore concentration was determined by serial dilution on taurocholate cycloserine cefoxitin fructose agar (TCCFA) or BHI plates (46).
Mouse immunization and mouse model of C. difficile infection.
Western blot analysis showed that mTcd138 was expressed in nontoxigenic C. difficile strains CD2001, CCUG37780, CCUG37785, and CCUG37787 (data not shown). Strains CCUG37785, designated NTCD, and CCUG37785 expressing mTcd138, designated NTCD_mTcd138, were used in immunization experiments in animals. Female C57/BL6 mice were housed under the same conditions at a seminatural light cycle of 14 h/10 h (light/dark) in a specific pathogen-free (SPF) environment. Mice received water and food ad libitum. During immunizations and infection with C. difficile, mice were housed in infection rooms. Mice (n = 10) were immunized 3 times at 14-day intervals via oral administration with 2 × 106/100 μl spores of NTCD or NTCD_mTcd138. Control mice received the same amount of PBS. Sera were collected, and anti-TcdA/TcdB IgG titers were determined by enzyme-linked immunosorbent assay (ELISA). Fourteen days after the third immunization, immunized or control mice were given drinking water containing a mixture of six antibiotics including kanamycin (40 mg/kg of body weight), gentamicin (3.5 mg/kg), colistin (4.2 mg/kg), metronidazole (21.5 mg/kg), and vancomycin (4.5 mg/kg) for 5 days and then received autoclaved water for 2 days, followed by a single dose of clindamycin (10 mg/kg) intraperitoneal injection before challenge with 106 C. difficile UK6 spores/mouse via oral gavage as described previously (47). After infection, mice were monitored daily for a week for survival, weight changes, diarrhea, and other symptoms of the disease. Diarrhea was defined as wet tails and loose or watery feces. The death data included the numbers of mice who died after infection and those euthanized if the weight loss was greater than 20%.
Hamster immunization and hamster model of C. difficile infection.
Golden Syrian female hamsters were housed individually in cages under the same conditions at a light cycle of 14 h/10 h (light/dark) in an SPF environment. Hamsters received water and food ad libitum. During immunizations and infection with C. difficile, hamsters were housed in infection rooms. Hamsters were orally immunized with 2 × 106/100 μl spores of NTCD or NTCD_mTcd138 3 times at 14-days intervals. Control hamsters were immunized with the same volume of PBS. Sera were collected from the jugular vein, and anti-TcdA/TcdB IgA or IgG titers were determined by ELISA. Two weeks after the third immunization, hamsters were intraperitoneally (i.p.) administered one dose of clindamycin at 30 mg/kg followed by oral challenge with 2 × 104 C. difficile UK6 spores 5 days later. The hamsters were monitored for 7 days for diarrhea and other disease symptoms.
ELISA for determination of antitoxin and anti-FliCD IgA and IgG.
ELISAs were performed as previously described (29). Briefly, Costar 96-well ELISA plates were coated with 100 μl/well of TcdA (0.5 μg/ml), TcdB (0.5 μg/ml), or FliCD (0.5 μg/ml) at 4°C overnight. Following washing of the unbound material, plates were blocked with 300 μl of blocking buffer (PBS + 5% dry milk) at room temperature for 2 h. After washing, 100 μl of 10-fold diluted sera or fecal samples was added into each well of the plates and incubated for 1.5 h at room temperature. Following washing with PBS, 100 μl of mouse IgG-horseradish peroxidase (HRP) (1:3,000) or mouse IgA-HRP (1:3,000) was added to each well and incubated for 30 min to 1 h. Subsequent to a washing step with PBS, substrate tetramethylbenzidine (TMB) was added to allow color development at room temperature for 5 to 30 min. The reaction was stopped by addition of H2SO4 to each well, and the optical density (OD) values at 450 nm were recorded by a spectrophotometer. Antitoxin and anti-FliCD IgG or IgA titers of a given sample (the titer of serum or fecal sample from immunized mice/hamsters was defined as the dilution factor at which the OD450 nm is greater than or equal to 2-fold that of serum or fecal sample from nonimmunized mice/hamsters).
Quantification of C. difficile spores from mouse feces.
Fecal samples were collected on day 4 postinfection. Fifty milligrams of feces was dissolved in 500 μl sterile MilliQ water for 16 h at 4°C and then treated with 500 μl of purified ethanol (Sigma-Aldrich) for 1 h at room temperature to kill vegetative cells. Samples were vortexed, serially diluted, and plated onto selective medium supplemented with taurocholate (0.1%, wt/vol), cefoxitin (16 μg/ml), and l-cycloserine (250 μg/ml). The plates were incubated anaerobically at 37°C for 48 h, colonies were counted, and results were expressed as the CFU/gram of feces on day 4. The tcdB gene was amplified to distinguish toxigenic C. difficile UK6 and nontoxigenic C. difficile strains.
Quantitation of C. difficile toxins in mouse feces.
After challenge with C. difficile spores, feces were collected and dissolved in PBS (0.1g/ml) containing a protease inhibitor cocktail, and the supernatants were collected after centrifugation and used for determination of TcdA/TcdB concentrations by ELISA. Briefly, 96-well Costar microplates were coated with 100 μl of anti-TcdA antibody (1 μg/ml) and anti-TcdB antibody (1 μg/ml) overnight in PBS at 4°C. On the next day, each well was blocked with 300 μl of blocking buffer (PBS + 5% dry milk) at RT for 2 h. Next, standards and samples were added to each well (100 μl) in duplicate and incubated for 90 min at 25°C. After another set of washings, HRP-chicken anti-C. difficile TcdA/TcdB (1:5,000 dilution in PBS; Gallus Immunotech, Shirley, MA) was added to wells for 30 min at RT. A final set of 3 washings preceded the addition of the TMB microwell peroxidase substrate for 20 min at RT in the dark. The reaction was stopped with 2 N H2SO4, and the absorbance was measured using a plate reader at 450 nm.
Neutralizing assays.
Vero cells were used to assess neutralizing activities of serum samples. The neutralizing titer is defined as the maximum dilution of the samples that blocks cell rounding caused by toxin at a given concentration. This given concentration is the minimum dose of the toxin that causes all cells to round after a 16-hour exposure to the toxin, i.e., 2.5 and 0.1 ng/ml for TcdA and TcdB, respectively.
Statistical analysis.
Animal survival was analyzed by Kaplan-Meier survival analysis with a log rank test of significance. When comparing results for two groups, Student's unpaired t test was used for statistical significance; when comparing the results of more than two groups, one-way analysis of variance (ANOVA) with post hoc analysis by Bonferroni tests was used. Results are expressed as means ± standard errors of means. Differences were considered statistically significant if P values were <0.05 (*). All statistical analyses were performed using GraphPad Prism software.
ACKNOWLEDGMENTS
We thank Robert Fagan from University of Sheffield, United Kingdom, for generously providing expression vectors. We also thank all Sun lab members for support and discussions.
This work was supported in part by National Institutes of Health grants to X.S. (K01-DK092352, R21-AI113470, R03-DK112004, R01-AI132711) and A.L.S. (R01-GM042219).
Author contributions: X.S. conceived and designed the project. A.L.S. participated in the design of the project. Y.W., S.W., L.B., C.L., K.Z., Z.D., and X.J. performed experiments and data analysis. S.W., Y.W., X.S., A.L.S., Z.D., and S.T. wrote and revised the manuscript. All authors read and approved the final manuscript.
We declare that we have no potential conflicts of interest.
REFERENCES
- 1.Lessa FC, Winston LG, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:2369–2370. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Donoghue C, Kyne L. 2011. Update on Clostridium difficile infection. Curr Opin Gastroenterol 27:38–47. doi: 10.1097/MOG.0b013e3283411634. [DOI] [PubMed] [Google Scholar]
- 3.Khan MA, Hays JP, Elabbasy MT, Al-Mogbel MS. 2017. Rise of Clostridium difficile infections: an overview. Rev Med Microbiol 28:152–157. doi: 10.1097/MRM.0000000000000111. [DOI] [Google Scholar]
- 4.Jank T, Aktories K. 2008. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16:222–229. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Pruitt RN, Lacy DB. 2016. Toward a structural understanding of Clostridium difficile toxins A and B. Front Cell Infect Microbiol 2:28. doi: 10.3389/fcimb.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. 2007. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 45:302–307. doi: 10.1086/519265. [DOI] [PubMed] [Google Scholar]
- 7.Koo HL, Garey KW, Dupont HL. 2010. Future novel therapeutic agents for Clostridium difficile infection. Expert Opin Investig Drugs 19:825–836. doi: 10.1517/13543784.2010.495386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbut F, Richard A, Hamadi K, Chomette V, Burghoffer B, Petit JC. 2000. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J Clin Microbiol 38:2386–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonna I, Welsby PD. 2005. Pathogenesis and treatment of Clostridium difficile infection. Postgrad Med J 81:367–369. doi: 10.1136/pgmj.2004.028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gough E, Shaikh H, Manges AR. 2011. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 53:994–1002. doi: 10.1093/cid/cir632. [DOI] [PubMed] [Google Scholar]
- 11.Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 44:354–360. doi: 10.1097/MCG.0b013e3181c87e02. [DOI] [PubMed] [Google Scholar]
- 12.Senior K. 2013. Faecal transplantation for recurrent C difficile diarrhoea. Lancet Infect Dis 13:200–201. doi: 10.1016/S1473-3099(13)70052-5. [DOI] [PubMed] [Google Scholar]
- 13.Rebeaud F, Bachmann MF. 2012. Immunization strategies for Clostridium difficile infections. Expert Rev Vaccines 11:469–479. doi: 10.1586/erv.12.18. [DOI] [PubMed] [Google Scholar]
- 14.Kelly CP, Kyne L. 2011. The host immune response to Clostridium difficile. J Med Microbiol 60:1070–1079. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 15.de Bruyn G, Saleh J, Workman D, Pollak R, Elinoff V, Fraser NJ, Lefebvre G, Martens M, Mills RE, Nathan R, Trevino M, van Cleeff M, Foglia G, Ozol-Godfrey A, Patel DM, Pietrobon PJ, Gesser R, H-030-012 Clinical Investigator Study Team. 2016. Defining the optimal formulation and schedule of a candidate toxoid vaccine against Clostridium difficile infection: a randomized phase 2 clinical trial. Vaccine 34:2170–2178. doi: 10.1016/j.vaccine.2016.03.028. [DOI] [PubMed] [Google Scholar]
- 16.Sheldon E, Kitchin N, Peng Y, Eiden J, Gruber W, Johnson E, Jansen KU, Pride MW, Pedneault L. 2016. A phase 1, placebo-controlled, randomized study of the safety, tolerability, and immunogenicity of a Clostridium difficile vaccine administered with or without aluminum hydroxide in healthy adults. Vaccine 34:2082–2091. doi: 10.1016/j.vaccine.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Bezay N, Ayad A, Dubischar K, Firbas C, Hochreiter R, Kiermayr S, Kiss I, Pinl F, Jilma B, Westritschnig K. 2016. Safety, immunogenicity and dose response of VLA84, a new vaccine candidate against Clostridium difficile, in healthy volunteers. Vaccine 34:2585–2592. doi: 10.1016/j.vaccine.2016.03.098. [DOI] [PubMed] [Google Scholar]
- 18.He X, Sun X, Wang J, Wang X, Zhang Q, Tzipori S, Feng H. 2009. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect Immun 77:2294–2303. doi: 10.1128/IAI.01577-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steele J, Mukherjee J, Parry N, Tzipori S. 2013. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 207:323–330. doi: 10.1093/infdis/jis669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alignani D, Maletto B, Liscovsky M, Ropolo A, Moron G, Pistoresi-Palencia MC. 2005. Orally administered OVA/CpG-ODN induces specific mucosal and systemic immune response in young and aged mice. J Leukoc Biol 77:898–905. doi: 10.1189/jlb.0604330. [DOI] [PubMed] [Google Scholar]
- 21.Sarti F, Perera G, Hintzen F, Kotti K, Karageorgiou V, Kammona O, Kiparissides C, Bernkop-Schnurch A. 2011. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials 32:4052–4057. doi: 10.1016/j.biomaterials.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK. 2013. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm 447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramirez JEV, Sharpe LA, Peppas NA. 2017. Current state and challenges in developing oral vaccines. Adv Drug Delivery Rev 114:116–131. doi: 10.1016/j.addr.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uddin MJ, Gill HS. 2018. From allergen to oral vaccine carrier: a new face of ragweed pollen. Int J Pharm doi: 10.1016/j.ijpharm.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding DN. 1998. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 351:633–636. doi: 10.1016/S0140-6736(97)08062-8. [DOI] [PubMed] [Google Scholar]
- 26.Songer JG, Jones R, Anderson MA, Barbara AJ, Post KW, Trinh HT. 2007. Prevention of porcine Clostridium difficile-associated disease by competitive exclusion with nontoxigenic organisms. Vet Microbiol 124:358–361. doi: 10.1016/j.vetmic.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 27.Nagaro KJ, Phillips ST, Cheknis AK, Sambol SP, Zukowski WE, Johnson S, Gerding DN. 2013. Nontoxigenic Clostridium difficile protects hamsters against challenge with historic and epidemic strains of toxigenic BI/NAP1/027 C. difficile. Antimicrob Agents Chemother 57:5266–5270. doi: 10.1128/AAC.00580-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villano SA, Seiberling M, Tatarowicz W, Monnot-Chase E, Gerding DN. 2012. Evaluation of an oral suspension of VP20621, spores of nontoxigenic Clostridium difficile strain M3, in healthy subjects. Antimicrob Agents Chemother 56:5224–5229. doi: 10.1128/AAC.00913-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang YK, Yan YX, Kim HB, Ju X, Zhao S, Zhang K, Tzipori S, Sun X. 2015. A chimeric protein comprising the glucosyltransferase and cysteine proteinase domains of toxin B and the receptor binding domain of toxin A induces protective immunity against Clostridium difficile infection in mice and hamsters. Hum Vaccin Immunother 11:2215–2222. doi: 10.1080/21645515.2015.1052352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler M, Olson A, Drekonja D, Shaukat A, Schwehr N, Shippee N, Wilt TJ. 2016. Early diagnosis, prevention, and treatment of Clostridium difficile: update. Agency for Healthcare Research and Quality (US), Rockville, MD. [PubMed] [Google Scholar]
- 31.Kociolek LK, Gerding DN. 2016. Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat Rev Gastroenterol Hepatol 13:150–160. doi: 10.1038/nrgastro.2015.220. [DOI] [PubMed] [Google Scholar]
- 32.Tian JH, Fuhrmann SR, Kluepfel-Stahl S, Carman RJ, Ellingsworth L, Flyer DC. 2012. A novel fusion protein containing the receptor binding domains of C. difficile toxin A and toxin B elicits protective immunity against lethal toxin and spore challenge in preclinical efficacy models. Vaccine 30:4249–4258. doi: 10.1016/j.vaccine.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 33.Donald RG, Flint M, Kalyan N, Johnson E, Witko SE, Kotash C, Zhao P, Megati S, Yurgelonis I, Lee PK, Matsuka YV, Severina E, Deatly A, Sidhu M, Jansen KU, Minton NP, Anderson AS. 2013. A novel approach to generate a recombinant toxoid vaccine against Clostridium difficile. Microbiology 159:1254–1266. doi: 10.1099/mic.0.066712-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao S, Ghose-Paul C, Zhang K, Tzipori S, Sun X. 2014. Immune-based treatment and prevention of Clostridium difficile infection. Hum Vaccin Immunother 10:3522–3530. doi: 10.4161/21645515.2014.980193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Premanand B, Prabakaran M, Kiener TK, Kwang J. 2013. Recombinant baculovirus associated with bilosomes as an oral vaccine candidate against HEV71 infection in mice. PLoS One 8:e55536. doi: 10.1371/journal.pone.0055536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knecht LD, Pasini P, Daunert S. 2011. Bacterial spores as platforms for bioanalytical and biomedical applications. Anal Bioanal Chem 400:977–989. doi: 10.1007/s00216-011-4835-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhou ZW, Gong ST, Li XM, Yang YY, Guan RL, Zhou S, Yao SW, Xie YQ, Ou ZY, Zhao JH, Liu ZG. 2015. Expression of Helicobacter pylori urease B on the surface of Bacillus subtilis spores. J Med Microbiol 64:104–110. doi: 10.1099/jmm.0.076430-0. [DOI] [PubMed] [Google Scholar]
- 38.Tang ZL, Sun HC, Chen TJ, Lin ZP, Jiang HY, Zhou XY, Shi CB, Pan HJ, Chang OQ, Ren PL, Yu JY, Li XR, Xu J, Huang Y, Yu XB. 2017. Oral delivery of Bacillus subtilis spores expressing cysteine protease of Clonorchis sinensis to grass carp (Ctenopharyngodon idellus): induces immune responses and has no damage on liver and intestine function. Fish Shellfish Immunol 64:287–296. doi: 10.1016/j.fsi.2017.03.030. [DOI] [PubMed] [Google Scholar]
- 39.Akerlund T, Persson I, Unemo M, Noren T, Svenungsson B, Wullt M, Burman LG. 2008. Increased sporulation rate of epidemic clostridium difficile type 027/NAP1. J Clin Microbiol 46:1530–1533. doi: 10.1128/JCM.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malten M, Hollmann R, Deckwer WD, Jahn D. 2005. Production and secretion of recombinant Leuconostoc mesenteroides dextransucrase DsrS in Bacillus megaterium. Biotechnol Bioeng 89:206–218. doi: 10.1002/bit.20341. [DOI] [PubMed] [Google Scholar]
- 42.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493. doi: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouillaut L, McBride SM, Sorg JA. 2011. Genetic manipulation of Clostridium difficile. Curr Protoc Microbiol Chapter 9:Unit 9A.2. doi: 10.1002/9780471729259.mc09a02s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Killgore G, Thompson A, Johnson S, Brazier J, Kuijper E, Pepin J, Frost EH, Savelkoul P, Nicholson B, van den Berg RJ, Kato H, Sambol SP, Zukowski W, Woods C, Limbago B, Gerding DN, McDonald LC. 2008. Comparison of seven techniques for typing international epidemic strains of Clostridium difficile: restriction endonuclease analysis, pulsed-field gel electrophoresis, PCR-ribotyping, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, amplified fragment length polymorphism, and surface layer protein A gene sequence typing. J Clin Microbiol 46:431–437. doi: 10.1128/JCM.01484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez J, Springthorpe VS, Sattar SA. 2011. Clospore: a liquid medium for producing high titers of semi-purified spores of Clostridium difficile. J AOAC Int 94:618–626. [PubMed] [Google Scholar]
- 46.Burns DA, Heap JT, Minton NP. 2010. Clostridium difficile spore germination: an update. Res Microbiol 161:730–734. doi: 10.1016/j.resmic.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]