

Platelets are increasingly recognized to play a role in the complications of Streptococcus pneumoniae infections. S. pneumoniae expresses neuraminidases, which may alter glycans on the platelet surface.
KEYWORDS: Streptococcus pneumoniae, NanA, platelets, sialic acid, desialylation
ABSTRACT
Platelets are increasingly recognized to play a role in the complications of Streptococcus pneumoniae infections. S. pneumoniae expresses neuraminidases, which may alter glycans on the platelet surface. In the present study, we investigated the capability of pneumococcal neuraminidase A (NanA) to remove sialic acid (desialylation) from the platelet surface, the consequences for the platelet activation status and reactivity, and the ability of neuraminidase inhibitors to prevent these effects. Our results show that soluble NanA induces platelet desialylation. Whereas desialylation itself did not induce platelet activation (P-selectin expression and platelet fibrinogen binding), platelets became hyperreactive to ex vivo stimulation by ADP and cross-linked collagen-related peptide (CRP-XL). Platelet aggregation with leukocytes also increased. These processes were dependent on the ADP pathway, as inhibitors of the pathway (apyrase and ticagrelor) abrogated platelet hyperreactivity. Inhibition of NanA-induced platelet desialylation by neuraminidase inhibitors (e.g., oseltamivir acid) also prevented the platelet effects of NanA. Collectively, our findings show that soluble NanA can desialylate platelets, leading to platelet hyperreactivity, which can be prevented by neuraminidase inhibitors.
INTRODUCTION
Streptococcus pneumoniae is a major cause of community-acquired pneumonia and invasive infections (1). S. pneumoniae is known to activate platelets via different mechanisms, including through engagement of the immune receptor FcγRIIA and the integrin αIIbβ3 and by activation of Src and Syk tyrosine kinases (2). A role for Toll-like receptor 2 (TLR-2) in pneumococcus-induced platelet activation has also been proposed (3), but with contradictory results (4). Thrombocytopenia, which may result from platelet activation, is an independent predictor of mortality in severe pneumonia (5). Whether platelet desialylation by pneumococcal neuraminidase A (NanA) is another mechanism of platelet activation by S. pneumoniae is still unknown.
The importance of platelets in processes beyond hemostasis and coagulation is increasingly appreciated (6). Activated platelets release different immune molecules, modulate the response of leukocytes, and contribute to the formation of neutrophil extracellular traps (NETs) that trap bacteria in the circulation, avoiding their further dissemination (7, 8). Furthermore, platelet activation may also increase the risk for cardiovascular complications, which are common during S. pneumoniae infections (9).
Three genes encode neuraminidase (NA) activity in S. pneumoniae—nanA, nanB, and nanC—among which only nanA is expressed by all pneumococci. The NanA protein is located at the cell surface of pneumococci, where it is involved in the facilitation of mucosal colonization (10, 11). Platelet surface glycoproteins (GPs) are decorated with the N-acetylneuraminic acid (Neu5Ac) type of sialic acid (12), about 60% of which is susceptible to cleavage by neuraminidase (13). Desialylation of platelets exposes galactose residues and leads to platelet apoptosis, phagocytosis, and clearance from the circulation by the Ashwell-Morel receptor (AMR) in the liver (14, 15). Grewal et al. showed in a mouse model of pneumococcal sepsis that removal of desialylated platelets by the AMR protected against disseminated intravascular coagulation (16).
Although NanA is well studied as a surface virulence factor (11, 17, 18), its effect on platelet activation and function remains unclear, as well as whether neuraminidase inhibitors, such as oseltamivir acid, are able to prevent these effects (19). Recently, two main variants of NanA, containing the amino acids KGI or RAV at the catalytic site, were reported to differ in enzyme kinetics and susceptibility to neuraminidase inhibitors (20). It is unknown whether infection with either variant results in a different clinical outcome.
The current study aimed to investigate the effects of soluble NanA on platelet desialylation, platelet activation status, and platelet reactivity and to characterize the pathways involved. Furthermore, the propensity of neuraminidase inhibitors and platelet function inhibitors to prevent these NanA effects on platelets was explored. Finally, we studied whether the KGI and RAV variants of NanA had different effects on platelets and whether these variants were associated with the platelet count in a cohort of individuals with invasive pneumococcal infections.
RESULTS
Soluble NanA from S. pneumoniae desialylates platelets.
To determine whether NanA was released in the medium, supernatants from wild-type (WT) and nanA mutant (ΔnanA) pneumococci were analyzed by SDS-PAGE. The analysis showed a NanA band of the expected size in the WT-derived supernatant, whereas the band was absent in the mutant (see Fig. S1 in the supplemental material).
To study the effect of soluble NanA on the platelet surface sialic acid content, washed platelets were exposed to supernatant with and without NanA, derived from the WT and ΔnanA mutant, respectively. Platelets incubated with NanA resulted in reduced interaction of the sialic acid binding lectins Sambucus nigra lectin (SNA) and Maackia amurensis lectin II (MALII) compared with platelets incubated without NanA, indicating that NanA cleaved sialic acids from platelet surface glycoproteins (Fig. 1A and B; see Fig. S2 in the supplemental material). Binding of SNA lectin on platelets exposed to lower dilutions of ΔnanA-derived supernatant tended to be higher than on those exposed to phosphate-buffered saline (PBS) (Fig. 1A). This may be due to factors in the supernatant that enhance binding of SNA to glycoproteins of nondesialylated platelets. The different binding specificities of MALII, which binds to α-2,3-sialoglycans, and SNA, which binds to α-2,6-sialoglycans, likely explain the differences in binding patterns of the two lectins. Neuraminidase activity in the supernatants was 441.2 μmol/min/liter and 58.9 μmol/min/liter for supernatants with and without NanA, respectively, as measured by the substrate hydrolysis assay. Based on the concentration curve in Fig. 1, we chose to use the 1:400 dilution point in the subsequent experiments. This dilution point is equivalent to 1.10 μmol/min/liter and 0.14 μmol/min/liter for supernatants with and without NanA, respectively.
FIG 1.

Binding of SNA and MALII lectins to NanA-exposed platelets. Binding of SNA (A) and MALII (B) lectins to platelet surface sialic acid was determined after treating washed platelets with serial dilutions (1:80 to 1:10,000) of pneumococcal culture supernatants with (+NanA) and without (−NanA) NanA for 1 h at 37°C. PBS and purified neuraminidase from C. perfringens (NA) were used as negative and positive controls, respectively. The data are presented as mean fluorescent intensity (MFI) plus SD (in arbitrary units [AU]) of the results of two independent experiments with 3 replicates each (n = 6). Differences between the supernatants with and without NanA were analyzed using t tests. **, P < 0.01; *, P < 0.03; ns, not significant. The increase in desialylation with decreases in dilution of supernatants was analyzed using 1-way ANOVA with posttest for the linear trend. For +NanA supernatant, r2 = 0.73 and P < 0.001 for SNA and r2 = 0.44 and P < 0.001 for MALII; for −NanA supernatant, r2 = 0.18 and P = 0.06 for SNA and r2 = 0.65 and P < 0.001 for MALII.
NanA exposure results in platelet hyperreactivity that is largely ADP signaling dependent.
Next, we studied the effects of NanA on platelet activation status and platelet reactivity to ex vivo stimulation. Platelet-rich plasma (PRP) was exposed to NanA for 1 h to assess platelet activation, with subsequent determination of platelet reactivity by addition of increasing concentrations of the platelet agonists ADP, cross-linked collagen-related peptide (CRP-XL), and thrombin receptor activation peptide (TRAP). There was no difference in the expression of the alpha-granule protein P-selectin and binding of fibrinogen following exposure to supernatant with or without NanA. In contrast, following ex vivo stimulation with ADP and a low dose of CRP-XL, platelets exposed to NanA showed higher P-selectin expression and fibrinogen binding than the control without NanA (Fig. 2A and B; see Fig. S3 in the supplemental material). Stimulation with TRAP also induced higher fibrinogen binding, but not P-selectin expression, in NanA-exposed platelets. Similar results for fibrinogen binding were observed using washed platelets, indicating that plasma factors had no effects on the observed outcome (see Fig. S4A in the supplemental material). However, unlike with PRP, fibrinogen binding was also slightly increased in desialylated platelets without secondary stimulation, probably as a result of minimal platelet activation due to the platelet isolation procedure. In order to identify the mechanisms of NanA-induced platelet hyperreactivity involved, we focused on the ADP pathway, because the platelet effects of NanA were more pronounced when ADP or CRP-XL was used as a platelet stimulus than when TRAP was used. The effect of CRP-XL (collagen pathway) is also known to depend on secondary ADP release (21). ADP signaling was inhibited using apyrase, which hydrolyzes soluble ADP, and with ticagrelor, an inhibitor of the ADP receptor P2Y12. Both apyrase and ticagrelor reduced NanA-induced platelet hyperreactivity upon ex vivo stimulation with either CRP-XL or TRAP (Fig. 3), confirming the importance of the ADP signaling pathway.
FIG 2.

Reactivity of NanA-exposed platelets to various platelet agonists. P-selectin (A) and fibrinogen (B) expression on platelets was determined after treating PRP with PBS (negative control), 200 mU purified neuraminidase from C. perfringens (NA), or supernatant with (+NanA) and without (−NanA) NanA for 1 h at 37°C, followed by stimulation with increasing concentrations of ADP, CRP-XL, or TRAP. The data are presented as MFI ± SEM in arbitrary units (n = 10). Differences between +NanA and −NanA supernatants were analyzed using t tests.
FIG 3.

Effects of apyrase and ticagrelor on hyperreactivity of NanA-exposed platelets. P-selectin (A and B) and fibrinogen (C and D) expression on platelets was determined after treating PRP with PBS or supernatant containing NanA (+NanA) for 1 h at 37°C, followed by hydrolyzing ADP using 10 μg/ml apyrase (A and C) or blocking ADP receptor using 50 μg/ml ticagrelor (B and D) for 30 min before stimulation with increasing concentrations of CRP-XL or TRAP. The data are presented as MFI ± SEM (in arbitrary units) of the results of two independent experiments with 3 replicates each (n = 6) and were analyzed using t tests. ***, P < 0.001; *, P < 0.05 for +NanA supernatant versus +NanA supernatant with apyrase or ticagrelor.
NanA promotes dense-granule secretion and increases surface expression of GPIIIa.
Platelets contain large quantities of ADP in their dense granules, together with other molecules, such as ATP, calcium, and serotonin. Secretion of ADP from dense granules activates P2Y12, inducing amplification of platelet activation. We show that NanA promotes ADP-induced dense-granule secretion, as suggested by higher expression of the dense-granule marker CD63 on the platelet surface, together with a reduction in intraplatelet mepacrine concentrations (Fig. 4A). Exposure of platelets to NanA without a platelet agonist did not change mepacrine concentrations or CD63 expression, indicating that exposure to NanA alone is insufficient to induce dense-granule secretion.
FIG 4.
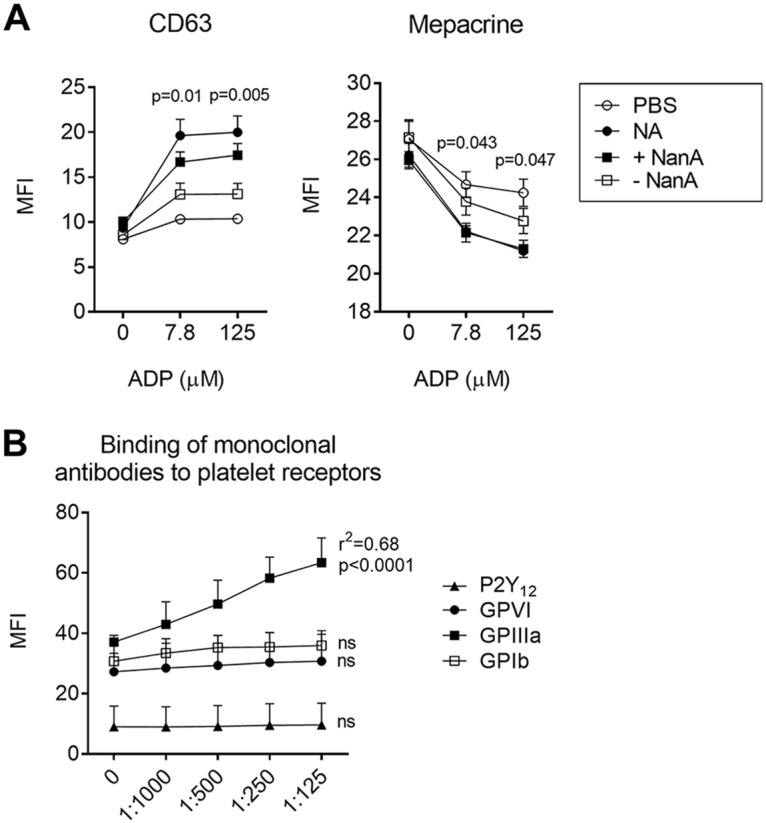
Degranulation of dense granules and binding of monoclonal antibodies to surface receptors in NanA-exposed platelets. (A) CD63 expression and intraplatelet concentrations of mepacrine after treating PRP with PBS (negative control), purified neuraminidase (NA), or supernatants with (+NanA) and without (−NanA) NanA, followed by stimulation with increasing concentrations of ADP. The data are presented as MFI ± SEM (in arbitrary units) of the results of two independent experiments with 3 replicates each (n = 6). Differences between +NanA and –NanA supernatants were analyzed using t tests. (B) Binding of monoclonal antibodies against P2Y12, GPVI, GPIIIa, and GPIbα on platelet surfaces after treating PRP with increasing concentrations of supernatant containing NanA for 1 h at 37°C. The data are presented as MFI plus SD (in arbitrary units) of the results of three independent experiments with 3 replicates each. Increases in expression levels with decreases in dilution of the supernatant were analyzed using 1-way ANOVA with posttest for the linear trend. ns, not significant.
Exposing platelet-rich plasma to increasing concentrations of NanA did not increase binding of antibodies directed against the ADP receptor P2Y12, the collagen receptor GPIV, or the VWF receptor GP1b (Fig. 4B). In contrast, a significant increase in the binding of antibodies against GPIIIa was observed (P < 0.001), probably due to increased exposure of antibody binding sites as a consequence of removal of sialic acid. When associated with GPIIb, GPIIIa constitutes the receptor for fibrinogen and other adhesive proteins, such as VWF.
Neuraminidase inhibitors prevent NanA-induced platelet hyperreactivity.
Next, we explored whether neuraminidase inhibitors could prevent NanA-induced platelet hyperreactivity. Both oseltamivir acid and 2,3-didehydro-2-deoxy-N-acetylneuraminic acid (DANA) prevented NanA-induced platelet hyperreactivity in a dose-dependent manner (Fig. 5).
FIG 5.
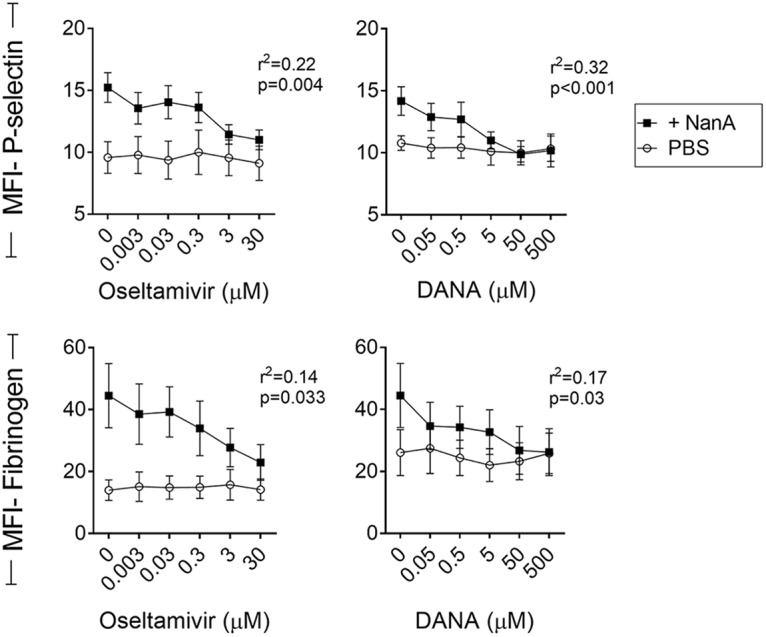
Inhibition of NanA by oseltamivir acid and DANA. P-selectin and fibrinogen expression on platelets was determined after treating PRP with PBS or supernatant containing NanA in the presence of increasing concentrations of oseltamivir acid or DANA for 1 h at 37°C and then stimulating with 125 μM ADP. The data show MFI ± SEM (in arbitrary units) of the results of two independent experiments with 3 replicates each (n = 6). Decreases in P-selectin and fibrinogen expression levels with increased concentrations of oseltamivir acid or DANA in +NanA samples were analyzed using 1-way ANOVA with posttest for the linear trend.
NanA induces platelet interaction with leukocytes.
Sialic acid is negatively charged, which gives a cell a high electronegative surface charge. Removal of sialic acid from the platelet surface reduces the overall electronegative charge, and this may increase intercellular interactions. Analysis of whole blood exposed to NanA showed increased aggregates of platelets with monocytes, neutrophils, and T cells compared with the control without NanA (Fig. 6). Addition of oseltamivir showed a reduction in platelet-leukocyte interaction, although the differences were not statistically significant (see Fig. S4C in the supplemental material).
FIG 6.

Interaction of NanA-exposed platelets with leukocytes. Levels of platelet interaction with monocytes, neutrophils, and T cells after treating whole blood with PBS as a negative control, 200 mU purified neuraminidase from C. perfrigens (NA), or supernatants with (+NanA) and without (−NanA) NanA for 1 h at 37°C. The data are presented as MFI ± SEM (in arbitrary units) of the results of two independent experiments with 3 replicates each (n = 6) analyzed using t tests.
Variation in the catalytic site of NanA had no effect on platelet hyperreactivity.
Despite the highly conserved activity of NanA, a recent study revealed the evolutionary diversity of the enzyme (20). Two semiconserved epitopes were identified in the catalytic site of NanA, KGI (Lys-Gly-Ile) and RAV (Arg-Ala-Val), which differed in their Michaelis constants (Kms), the substrate concentration at 1/2 the maximum velocity. The Km of the RAV variant (NanA-RAV) was found to be significantly lower than that of the KGI variant (NanA-KGI), suggesting that the RAV variant has higher activity. In a cohort of 281 patients with S. pneumoniae bacteremia, 239 had the KGI variant and 42 had the RAV variant. Platelet counts measured at hospital admission did not differ between the two groups (Fig. 7A). Unfortunately, nadir platelet counts were available for a few patients in the KGI group only and were therefore not used in the analysis. The types of disease manifestation and severities of disease were also comparable between the groups (data not shown). A subset of S. pneumoniae isolates from patients in either group were cultured, and the supernatants were used to study the effect of NanA-KGI and NanA-RAV on platelet desialylation. There were no differences in binding of SNA and MALII lectins to platelet sialic acid between platelets exposed to supernatants of the KGI or RAV variant (Fig. 7B and C). In addition, there was no difference in neuraminidase activity between the two NanA variants as measured by a substrate-based assay (Fig. 7D). These observations suggest there is no difference in the enzymatic activity of NanA variants and no difference in clinical outcomes after infection with S. pneumoniae isolates producing either variant.
FIG 7.
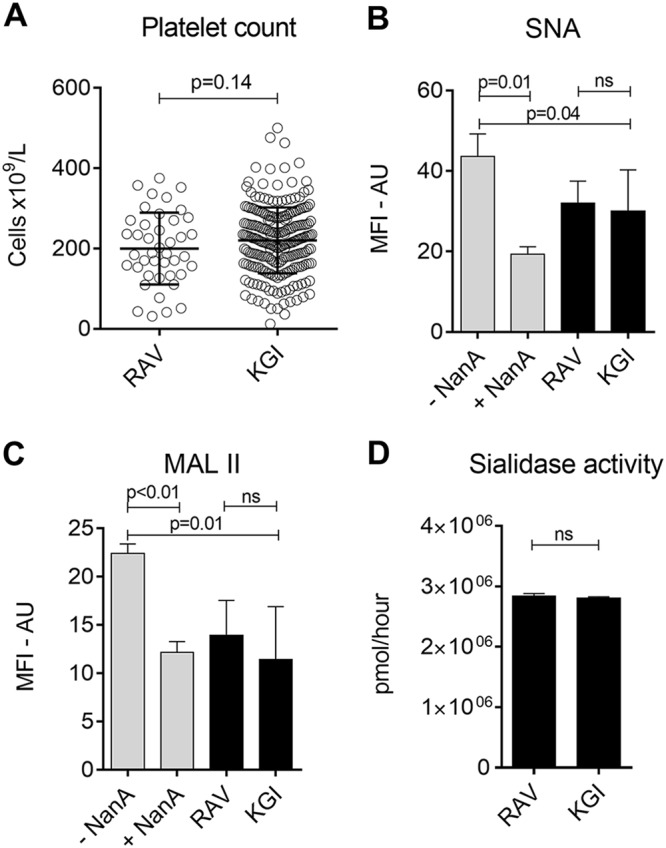
Differential effects of NanA-RAV and NanA-KGI on platelet function. (A) Platelet counts of patients infected with S. pneumoniae containing the KGI or RAV NanA variant at admission (KGI, n = 239; RAV, n = 42). (B and C) Binding of SNA and MALII lectins to surface sialic acid after treating platelets with supernatants derived from cultures of isolates producing either NanA-RAV or NanA-KGI. +NanA and −NanA supernatants were used as positive and negative controls, respectively. The RAV and KGI groups consisted of four isolates each tested in three platelet donors. (D) Enzymatic activity of the RAV versus the KGI NanA variants in hydrolyzing sialic acid substrate [2-(4-methylumbelliferyl)α-d-N-acetylneraminic acid]. The data are presented as means with SD and were analyzed using t tests. ns, not significant.
DISCUSSION
In this study, we demonstrate that pneumococcal NanA induces removal of sialic acid from the platelet surface, resulting in platelet hyperreactivity. These effects were dependent on ADP signaling and could be prevented by ADP receptor antagonists, such as ticagrelor, as well as neuraminidase inhibitors, such as oseltamivir acid.
Sialic acid on the platelet surface was determined using the lectins SNA and MALII, which both bind N-acetylneuraminic acid found on platelet glycoproteins. Pneumococci also harbor other neuraminidases than NanA, e.g., NanB and NanC, explaining why supernatant from ΔnanA mutants also induced some sialic acid depletion. While NanA can cleave α-2,3-, α-2,6-, and α-2,8-linked sialic acids, NanB exhibits a preference for α-2,3-linked sialic acids (17). In our experiments, only the NanA gene was mutated. Traces of NanB and NanC could still be present in the supernatant, cleaving some of the α-2,3 links that can be detected by MALII, but not SNA. This accounts for the low binding of MALII, but not SNA lectin, to platelets exposed to the supernatant of the mutant strain. However, NanA is the strongest neuraminidase and virulence factor for sialic acid removal. NanB, on the other hand, is involved only in metabolic activities (17, 18). In our experiments, we carefully selected the dilution point of the supernatant where the effect of NanB was minimal, diminishing any potential effects of the contaminant on the observed outcomes of platelet reactivity.
Our findings show that NanA-induced sialic acid removal does not directly lead to platelet activation but has an indirect effect, as it leads to increased sensitivity for platelet activation by other platelet agonists. These findings are in line with a 1975 report by Greenberg and colleagues, who found increased agonist-induced platelet aggregation after removal of sialic acid by neuraminidase (22). However, they conducted a noncontrolled experiment with the ADP-hydrolyzing agent apyrase, leading to inconclusive results on the role of ADP. Our study clearly identified the importance of ADP signaling in NanA-induced platelet hyperreactivity and showed that both ADP receptor antagonists and neuraminidase inhibitors may prevent these effects. Although oseltamivir acid is designed for influenza virus neuraminidase, it can effectively inhibit S. pneumoniae neuraminidase due to close similarities in the active sites of the two proteins (19).
Different mechanisms may explain why NanA-desialylated platelets are hyperreactive, as is also shown in Fig. S5 in the supplemental material. First, the plasma membrane Orai1 channels contain α-2,6-linked sialic acids (23), which can be cleaved off by NanA. Orai1 mediates calcium influx into the cell, and its desialylation has been associated with increased calcium uptake (23), a process known to enhance platelet activation (24). Second, desialylation may lead to a reduction in the negative charge of the cell membrane, which may enhance transport of positively charged calcium ions over the cell membrane. Third, binding of desialylated VWF (asialo-VWF) to platelets is known to induce degranulation of platelet dense granules (25), an effect that can be blocked by apyrase and ticagrelor (26).
Neuraminidase could act on platelets, as well as on sialylated plasma proteins, such as fibrinogen, in the PRP. However, several studies have reported that the sialic acid content of fibrinogen does not affect its interactions with platelets (27, 28). The observed increase in fibrinogen binding in our assay was therefore unlikely to have been influenced by desialylated fibrinogen molecules. Although aggregation of NanA-exposed platelets was not measured, it is expected, as platelet aggregation correlates with fibrinogen binding to platelets.
Platelet activation is an important process in inflammation and host defense (29–31); however, hyperreactive platelets may also increase the risk for cardiovascular events. The importance of cardiovascular complications in pneumococcal infections is increasingly recognized (9, 32). ADP receptor antagonists are among the most commonly used drugs in cardiovascular medicine, and neuraminidase inhibitors are frequently prescribed for patients with pneumonia during the influenza season; therefore, it is important to better understand the effects of NanA-mediated desialylation of platelets. From studies in murine models, it is known that neuraminidase inhibitors may ameliorate sepsis by dampening inflammation (33). Grewal et al. found that clearance of desialylated platelets protects the host from streptococcus-associated coagulopathy (16). Interestingly, loss of sialic acid is a well-known platelet clearance mechanism, and in past years, different authors have reported that oseltamivir acid may increase platelet counts under conditions such as immune thrombocytopenia (15, 34), sepsis (35), and suspected influenza (36).
Platelet-monocyte clusters, which we observed in vitro, are also observed during S. pneumoniae infections (4) and are known to mediate the development of cardiovascular events (37). We observed increased platelet-leukocyte aggregates in whole blood exposed to NanA; however, desialylation of other sialylated blood components (38), apart from platelets, may have promoted the observed intercellular aggregation. Platelets engage with leukocytes mainly through the surface P-selectin receptor expressed on activated platelets. Platelet-leukocyte interactions therefore increase with increased platelet activation. Removal of sialic acid from cell surfaces reduces the cell surface electronegative charge, allowing more cell-cell interactions to occur. Our results show that desialylated platelets are hyperreactive to stimulation. We hypothesize that under pathological conditions where the vasculature is rich in mediators with the potential to activate platelets, platelet-leukocyte interactions are also enhanced.
NanA may influence the binding of agonists to other platelet receptors; however, only anti-GPIIIa binding increased significantly when platelets were exposed to NanA, while no increased binding of anti-P2Y12, anti-GPIV, or anti-GP1b antibodies was found. Fibrinogen binding to platelets occurs only after the formation of GPIIb/GPIIIa complexes, which is driven by activating agents such as ADP (39), which explains the increase in availability of GPIIIa receptor without fibrinogen binding in unstimulated samples. This observation is contrary to that of Grewal and colleagues (16), who did not observe a change in binding of anti-CD61 (GPIIIa) in desialylated platelets. Our results are in line with previous studies that indicated that glycosylation of P2Y12 receptor (40) or GPVI (41) affects their function and ligand binding (only GPVI) but not their surface expression. Conversely, amplification of platelet hyperreactivity through ADP signaling may be blunted by neuraminidase, as Zhong et al. (40) further showed, using CHO cells, that nonglycosylated P2Y12 receptors are defective in the P2Y12-mediated inhibition of adenylyl cyclase activity.
Xu et al. showed with a molecular simulation model and with purified NanA variants containing KGI and RAV motifs in the catalytic domain that NanA-RAV has higher enzymatic activity than the NanA-KGI variant (20). We could not confirm this result, as evidence for a difference in neither clinical outcomes nor in vitro platelet activation assays between the two variants was found. Our findings suggest, therefore, that exposure to either variant may have similar clinical consequences. Although we were not able to demonstrate removal of asialoglycoproteins from the circulation of pneumonia patients in our cohort, Grewal et al. have shown, using a mouse model, that intravenous administration of pneumococcal sialidase leads to platelet desialylation and subsequent removal from the circulation (16). These observations suggest that a similar effect could occur in human patients with streptococcus bacteremia.
Siglecs are sialic acid binding receptors on cell surfaces involved in the transduction of inhibitory signals. Siglec-7 is expressed on platelet surfaces following platelet activation, where engagement of purinergic receptors, as well as integrin αIIbβ3, is crucial (42). Siglec-7 binds to α-2,3, α-2,6, and α-2,8 linkages, which can all be cleaved by NanA. Engagement of Siglec-7 with its ligand leads to platelet apoptosis by the intrinsic pathway and an extramitochondrial pathway but has no effect on platelet activation, aggregation, or secretion (42, 43). Although platelet desialylation, particularly of the GP1b receptor, has been associated with platelet apoptosis, the role of siglec-7 was not discussed (15). Since siglec-7 induces apoptosis after engagement with its ligand, gangliosides, it would be expected that removal of platelet sialic acid would affect this apoptotic pathway. Whether platelet desialylation by NanA interferes with siglec-inhibitory signals requires further investigation. Given the existing information, it is unlikely that interference of neuraminidase with siglec-7 ligands would affect platelet activation.
One limitation of our study is that we did not purify NanA from the culture supernatants used. However, we controlled for possible effects of other pneumococcus-derived products in the supernatant by using a control NanA mutant strain with the same biological properties as the NanA-producing strain except for NanA secretion. We are therefore confident that the effects we observed are caused by NanA and not by other streptococcal exoglycosidases, such as BgaA (a β-galactosidase) and StrH (a β-acetylglucosaminidase), which are primarily membrane bound (44).
Collectively, our findings show that exposure of platelets to soluble NanA induces removal of sialic acid from the platelet surface, which in turn leads to platelet hyperreactivity and increased interaction with leukocytes. Neuraminidase inhibitors may prevent these effects on platelets caused by NanA, but the clinical consequences are currently unknown.
MATERIALS AND METHODS
Ethical statement.
Clinical data were obtained from a cohort of adult patients with a first episode of bacteremic pneumococcal pneumonia admitted to two Dutch hospitals, namely, the Canisius-Wilhelmina Ziekenhuis in Nijmegen and Maasziekenhuis Pantein in Boxmeer, between 2000 and 2011. This cohort study was approved by the local Medical Ethics Committees of both participating hospitals. Analyses of the current study were within the goals of the cohort study, and no additional institutional approval was required. All data and samples used were anonymous.
Construction of a ΔnanA mutant.
A directed deletion of nanA was made in the pneumococcal TIGR4 strain by allelic replacement of the target gene with an antibiotic resistance marker, as described previously (45). Briefly, overlap extension PCR was used to insert the spectinomycin resistance cassette of the pR412 plasmid between the two 500-bp flanking sequences adjacent to the target gene. The resulting PCR products were introduced by competent stimulating peptide 2 (CSP-2)-induced transformation into TIGR4. Directed mutants were obtained by selective plating and were checked for correct integration of the antibiotic resistance cassette into the target gene by PCR using control primers located inside the gene. Subsequently, the WT TIGR4 strain was transformed with chromosomal DNA isolated from the mutants to prevent the accumulation of inadvertent mutations elsewhere on the chromosome. In addition, loss of the target gene was confirmed by quantitative PCR (qPCR). The primers (Biolegio, Nijmegen, The Netherlands) used in this study are listed in Table S1 in the supplemental material.
Alignment of pneumococcal NanA variants.
A total of 281 pneumococcal isolates were obtained from an unbiased cohort of patients with invasive pneumococcal disease in Nijmegen, The Netherlands, between 2001 and 2011. All the patients gave written consent to participate. The study was approved by the Local Medical Ethics Committees of the participating hospitals. Details of clinical data on patient characteristics, comorbidities, severity of disease, and clinical outcome were described by Cremers et al. (46). Genomic DNA preparation and whole-genome sequencing were described by Cremers et al. (47). The NanA sequences were aligned using Multiple-Sequence Comparison by Log Expectation (MUSCLE) (48) software.
Development of culture supernatants and NanA protein quantification.
Single bacterial colonies of WT and ΔnanA TIGR4 were cultured in casein tryptone (CAT) medium supplemented with 0.015 M K2HPO4, 5 μl catalase (Sigma), and 0.02 g N-acetyl-d-mannosamine (ManNAc) and harvested at an optical density (OD) of 0.2. After removal of remnant bacterial and culture components, the supernatants were concentrated using Microcon 30-kDa centrifugation filter devices (Merck Millipore).
The concentrated supernatants were incubated at 100°C for 5 min in sample buffer (60 mM Tris-HCl, pH 6.8, 2% SDS, 2% β-mercaptoethanol, trace bromophenol blue), analyzed on Tris-glycine SDS-PAGE gels in a Protean II xi cell electrophoresis system (Bio-Rad), and visualized by Coomassie staining. Densitometry analysis was performed with ImageJ (49). Average spot intensities of NanA (∼80 kDa) and bovine serum albumin (BSA) (∼55 kDa), used as a loading control (marker), were determined. The absence of the ∼80-kDa band in the NanA mutant confirmed the presence of a protein of this size in the gel. Average spot intensity of NanA was corrected with the average spot intensity of BSA and depicted as the relative corrected spot intensity compared to WT TIGR4. Enzymatic activity of NanA in the supernatant was confirmed by a fluorometric method using 0.45 mM 2-(4-methylumbelliferyl)α-d-N-acetylneuraminic acid as the substrate. Thirty microliters of sample was mixed with equal volumes of substrate and reaction buffer, pH 7.0, and incubated at 37°C for 2 h, followed by 400 μl of stop solution (containing glycine and Trizol). After centrifugation, the supernatant was transferred onto a clean reaction plate, and the fluorescence was measured. Total enzymatic activity was calculated in moles of substrate hydrolyzed per unit per time (hours).
Platelet sialic acid assays.
Washed platelets were isolated from 3.2% sodium-citrate-anticoagulated whole blood (Becton Dickinson), as previously described (50), and resuspended in HEPES Tyrode buffer at 4 × 108 platelets/ml. The platelets were exposed to serial dilutions of culture supernatant for 1 h at 37°C and washed twice with PBST (PBS plus 0.05% Tween 20) supplemented with 0.01% prostaglandin. PBS and 200 μM purified NA from Clostridium perfringens (Sigma-Aldrich) were used as negative and positive controls, respectively. Purified neuraminidase was used to demonstrate that observed effects with supernatants containing NanA were similar to those induced by other neuraminidases. Cells were then stained with the platelet identification marker CD61 (Beckman Coulter) and sialic acid binding lectins SNA and MALII (both from Vector Laboratories) for 30 min at room temperature (RT). The cells were washed twice, fixed using 0.2% paraformaldehyde, and analyzed using a Cytomics FC500 flow cytometer and CXP acquisition software (Beckman Coulter).
Platelet function assays and platelet-leukocyte complexes.
Platelet function was determined using a flow cytometry assay, as previously described (51). Whole blood or PRP was incubated with 200 mU purified neuraminidase and supernatants with and without NanA for 1 h at 37°C, followed by stimulation with increasing concentrations of ADP, CRP-XL, or TRAP. The cells were labeled with antibodies directed against CD61, P-selectin (BioLegend) as a marker of platelet degranulation, and fibrinogen (Dako) as a marker of integrin αIIbβ3 activation. The ADP-stimulatory pathway was blocked by hydrolyzing ADP using 10 μg/ml of apyrase (Sigma-Aldrich) or by blocking the ADP receptor P2Y12 with 50 μg/ml ticagrelor (Bio Connect B.V.) for 30 min before addition of stimuli. These concentrations of apyrase and ticagrelor reduced about 40% of secondary stimulation with 7.8 μM ADP for both P-selectin and fibrinogen binding (see Fig. S4B in the supplemental material). This observation indicates that apyrase and ticagrelor provide adequate inhibition of the CRP- and TRAP-induced ADP positive-feedback loop, as activated platelets are expected to release smaller amounts of ADP than the 7.8 μM used in our experiments. Quantification of platelet dense granules was performed by incubating NanA-exposed PRP with 50 μM mepacrine for 10 min, followed by flow cytometry analysis to determine the intraplatelet mepacrine content. Furthermore, dense-granule degranulation was analyzed by measuring the expression of CD63 (BioLegend) on the platelet membrane. Platelet-leukocyte interactions were determined by gating CD45-positive cells that were also positive for both platelet (anti-CD42b; BD Biosciences) and monocyte (CD14), neutrophil (CD56), or T-cell (CD3) markers (Beckman Coulter).
Surface expression of platelet glycoproteins.
To study whether desialylation of platelets affects the surface expression of glycoproteins, PRP was treated with increasing concentrations of WT supernatant for 1 h at 37°C. Cells were labeled with antibodies against the VWF receptor GPIbα (anti-CD42b; BD Biosciences), the fibrinogen receptor GPIIIa (anti-CD61; Beckman Coulter), the collagen receptor GPVI (anti-GPVI; BD Biosciences), and the ADP receptor P2Y12 (Fabgennix). The effects of neuraminidase inhibitors on platelet function were assessed by treating PRP with supernatant containing NanA in the presence of increasing concentrations of oseltamivir acid (Bioconnect B.V.) and DANA (Sigma-Aldrich), followed by stimulation with 125 μM ADP.
Statistical analysis.
Statistical differences were analyzed using Student's t test or one-way analysis of variance (ANOVA), with posttests as indicated. Data are presented as means with standard deviations (SD) or means with standard errors of the mean (SEM) of independent measurements. Statistical analysis was done using GraphPad Prism 6 software. Flow cytometry analysis was done using Beckman Coulter Kaluza software version 1.2.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marc Eleveld, Erika van der Maten, Astrid van Rens, and Rahajeng Tunjungputri for laboratory assistance.
This project was funded by a grant from the Netherlands Fellowship Programme awarded to V.K.
Author contributions: M.I.D.J., Q.D.M., and A.J.V.D.V. conceived the study; V.K., J.D.L., and C.E.V.D.G.-D.J. performed the experiments; V.K., J.D.L., and A.J.C. analyzed the data; C.B., G.J.A., P.G.D.G., B.T.M., and D.L. provided technical advice and support; V.K., M.I.D.J., Q.D.M., J.D.L., and A.J.V.D.V. wrote the manuscript. We all contributed significantly to the final version of the manuscript.
We declare that we have no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00213-18.
REFERENCES
- 1.Kadioglu A, Andrew PW. 2004. The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol 25:143–149. doi: 10.1016/j.it.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, Kerrigan SW, Watson SP. 2014. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood 123:3166–3174. doi: 10.1182/blood-2013-11-540526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, Jenkinson HF, Kerrigan SW. 2010. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemost 8:2757–2765. doi: 10.1111/j.1538-7836.2010.04093.x. [DOI] [PubMed] [Google Scholar]
- 4.de Stoppelaar SF, Claushuis TAM, Schaap MCL, Hou B, van der Poll T, Nieuwland R, van 't Veer C. 2016. Toll-like receptor signalling is not involved in platelet response to Streptococcus pneumoniae in vitro or in vivo. PLoS One 11:e0156977. doi: 10.1371/journal.pone.0156977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brogly N, Devos P, Boussekey N, Georges H, Chiche A, Leroy O. 2007. Impact of thrombocytopenia on outcome of patients admitted to ICU for severe community-acquired pneumonia. J Infect 55:136–140. doi: 10.1016/j.jinf.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. 2012. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol 34:5–30. doi: 10.1007/s00281-011-0286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semple JW, Italiano JE, Freedman J. 2011. Platelets and the immune continuum. Nat Rev Immunol 11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 8.Schrottmaier WC, Kral JB, Badrnya S, Assinger A. 2015. Aspirin and P2Y12 inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb Haemost 114:478–489. doi: 10.1160/TH14-11-0943. [DOI] [PubMed] [Google Scholar]
- 9.Rae N, Finch S, Chalmers JD. 2016. Cardiovascular disease as a complication of community-acquired pneumonia. Curr Opin Pulm Med 22:212–218. doi: 10.1097/MCP.0000000000000261. [DOI] [PubMed] [Google Scholar]
- 10.Trappetti C, Kadioglu A, Carter M, Hayre J, Iannelli F, Pozzi G, Andrew PW, Oggioni M. 2009. Sialic acid: a preventable signal for pneumococcal biofilm formation, colonization, and invasion of the host. J Infect Dis 199:1497–1505. doi: 10.1086/598483. [DOI] [PubMed] [Google Scholar]
- 11.Brittan JL, Buckeridge TJ, Finn A, Kadioglu A, Jenkinson HF. 2012. Pneumococcal neuraminidase A: an essential upper airway colonization factor for Streptococcus pneumoniae. Mol Oral Microbiol 27:270–283. doi: 10.1111/j.2041-1014.2012.00658.x. [DOI] [PubMed] [Google Scholar]
- 12.Crook M. 1991. Sialic acid: its importance to platelet function in health and disease. Platelets 2:1–10. doi: 10.3109/09537109109005496. [DOI] [PubMed] [Google Scholar]
- 13.Madoff MA, Ebbe S, Baldini M. 1964. Sialic acid of human blood platelets. J Clin Invest 43:870–877. doi: 10.1172/JCI104972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, Hoffmeister KM. 2015. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med 21:47–54. doi: 10.1038/nm.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X-H, Wang Q-M, Zhang J-M, Feng F-E, Wang F-R, Chen H, Han W, Xu L, Liu K, Huang X. 2015. Desialylation is associated with apoptosis and phagocytosis of platelets in patients with prolonged isolated thrombocytopenia after allo-HSCT. J Hematol Oncol 8:116. doi: 10.1186/s13045-015-0216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grewal PK, Aziz PV, Uchiyama S, Rubio GR, Lardone RD, Le D, Varki NM, Nizet V, Marth JD. 2013. Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc Natl Acad Sci U S A 110:20218–20223. doi: 10.1073/pnas.1313905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu G, Kiefel MJ, Wilson JC, Andrew PW, Oggioni MR, Taylor GL. 2011. Three Streptococcus pneumoniae sialidases: three different products. J Am Chem Soc 133:1718–1721. doi: 10.1021/ja110733q. [DOI] [PubMed] [Google Scholar]
- 18.Gualdi L, Hayre JK, Gerlini A, Bidossi A, Colomba L, Trappetti C, Pozzi G, Docquier J, Andrew P, Ricci S, Oggioni MR. 2012. Regulation of neuraminidase expression in Streptococcus pneumoniae. BMC Microbiol 12:200. doi: 10.1186/1471-2180-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gut H, Xu G, Taylor GL, Walsh MA. 2011. Structural basis for Streptococcus pneumoniae NanA inhibition by influenza antivirals zanamivir and oseltamivir carboxylate. J Mol Biol 409:496–503. doi: 10.1016/j.jmb.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 20.Xu Z, von Grafenstein S, Walther E, Fuchs JE, Liedl KR, Sauerbrei A, Schmidtke M. 2016. Sequence diversity of NanA manifests in distinct enzyme kinetics and inhibitor susceptibility. Sci Rep 6:25169. doi: 10.1038/srep25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nieswandt B, Bergmeier W, Eckly A, Schulte V, Ohlmann P, Cazenave J-P, Zirngibl H, Offermanns S, Gachet C. 2001. Evidence for cross-talk between glycoprotein VI and Gi-coupled receptors during collagen-induced platelet aggregation. Blood 97:3829–3835. doi: 10.1182/blood.V97.12.3829. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg J, Packham MA, Cazenave JP, Reimers HJ, Mustard JF. 1975. Effects on platelet function of removal of platelet sialic acid by neuraminidase. Lab Invest 32:476–484. [PubMed] [Google Scholar]
- 23.Dörr K, Kilch T, Kappel S, Alansary D, Schwär G, Niemeyer BA, Peinelt C. 2016. Cell type-specific glycosylation of Orai1 modulates store-operated Ca 2+ entry. Sci Signal 9:ra25. doi: 10.1126/scisignal.aaa9913. [DOI] [PubMed] [Google Scholar]
- 24.Varga-Szabo D, Braun A, Nieswandt B. 2011. STIM and Orai in platelet function. Cell Calcium 50:270–278. doi: 10.1016/j.ceca.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 25.De Marco L, Girolami A, Russell S, Ruggeri ZM. 1985. Interaction of asialo von Willebrand factor with glycoprotein Ib induces fibrinogen binding to the glycoprotein IIb/IIIa complex and mediates platelet aggregation. J Clin Invest 75:1198–1203. doi: 10.1172/JCI111816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyú D, Glenn JR, White AE, Fox SC, van Giezen H, Nylander S, Heptinstall S. 2011. Mode of action of P2Y12 antagonists as inhibitors of platelet function. Thromb Haemost 105:96–106. [DOI] [PubMed] [Google Scholar]
- 27.Harfenist E, Packham M, Mustard J. 1984. Effects of variant gamma chains and sialic acid content of fibrinogen upon its interactions with ADP-stimulated human and rabbit platelets. Blood 64:1163–1168. [PubMed] [Google Scholar]
- 28.Park K, Gerndt SJ, Cooper SL. 1986. The effect of fibrinogen sialic acid residues on ex vivo platelet deposition on biomaterials. Thromb Res 43:293–302. doi: 10.1016/0049-3848(86)90149-0. [DOI] [PubMed] [Google Scholar]
- 29.Jenne CN, Kubes P. 2015. Platelets in inflammation and infection. Platelets 26:286–292. doi: 10.3109/09537104.2015.1010441. [DOI] [PubMed] [Google Scholar]
- 30.Kral JB, Schrottmaier WC, Salzmann M, Assinger A. 2016. Platelet interaction with innate immune cells. Transfus Med Hemother 43:78–88. doi: 10.1159/000444807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas MR, Storey RF. 2015. The role of platelets in inflammation. Thromb Haemost 114:449–458. doi: 10.1160/TH14-12-1067. [DOI] [PubMed] [Google Scholar]
- 32.Cangemi R, Casciaro M, Rossi E, Calvieri C, Bucci T, Calabrese CM, Taliani G, Falcone M, Palange P, Bertazzoni G, Farcomeni A, Grieco S, Pignatelli P, Violi F, Albanese F, Biliotti E, Carnevale R, Catasca E, Celestini A, Esvan R, Fazi L, Marinelli P, Mordenti M, Napoleone L, Palumbo Pastori Perri Proietti M, Capparuccia MR, Russo A, Russo R, Sarallo V, Salvatori G, Scarpellini MG, Ullo I. 2014. Platelet activation is associated with myocardial infarction in patients with pneumonia. J Am Coll Cardiol 64:1917–1925. doi: 10.1016/j.jacc.2014.07.985. [DOI] [PubMed] [Google Scholar]
- 33.Chen G-Y, Chen X, King S, Cavassani KA, Cheng J, Zheng X, Yu H, Qu J, Fang D, Wu W, Bai X-F, Liu J-Q, Woodiga SA, Chen C, Sun L, Hogaboam CM, Kunkel SL, Zheng P, Liu Y. 2011. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol 29:428–435. doi: 10.1038/nbt.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, Vadasz B, Carrim N, Grozovsky R, Ruan M, Zhu L, Zeng Q, Tao L, Zhai Z-M, Peng J, Hou M, Leytin V, Freedman J, Hoffmeister KM, Ni H. 2015. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun 6:7737. doi: 10.1038/ncomms8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson JC, Kawasaki N. 2011. Sialidase inhibitors DAMPen sepsis. Nat Biotechnol 29:406–407. doi: 10.1038/nbt.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen AJG, Peng J, Zhao H-G, Hou M, Ni H. 2015. Sialidase inhibition to increase platelet counts: a new treatment option for thrombocytopenia. Am J Hematol 90:E94–E95. doi: 10.1002/ajh.23953. [DOI] [PubMed] [Google Scholar]
- 37.Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, Marchese P, Frelinger AL, Goldberg RJ, Michelson AD. 2001. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol 38:1002–1006. doi: 10.1016/S0735-1097(01)01485-1. [DOI] [PubMed] [Google Scholar]
- 38.Varki A. 2008. Sialic acids in human health and disease. Trends Mol Med 14:351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett JS, Vilaire G. 1979. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J Clin Invest 64:1393–1401. doi: 10.1172/JCI109597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong X, Kriz R, Seehra J, Kumar R. 2004. N-linked glycosylation of platelet P 2 Y 12 ADP receptor is essential for signal transduction but not for ligand binding or cell surface expression. FEBS Lett 562:111–117. doi: 10.1016/S0014-5793(04)00191-7. [DOI] [PubMed] [Google Scholar]
- 41.Kunicki TJ, Cheli Y, Moroi M, Furihata K. 2005. The influence of N-linked glycosylation on the function of platelet glycoprotein VI. Blood 106:2744–2749. doi: 10.1182/blood-2005-04-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen KA, Hamzeh-Cognasse H, Palle S, Anselme-Bertrand I, Arthaud C-A, Chavarin P, Pozzetto B, Garraud O, Cognasse F. 2014. Role of Siglec-7 in apoptosis in human platelets. PLoS One 9:e106239. doi: 10.1371/journal.pone.0106239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, Garraud O. 2015. The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol 6:83. doi: 10.3389/fimmu.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King SJ, Hippe KR, Weiser JN. 2006. Deglycosylation of human glycoconjugates by the sequential activities of exoglycosidases expressed by Streptococcus pneumoniae. Mol Microbiol 59:961–974. doi: 10.1111/j.1365-2958.2005.04984.x. [DOI] [PubMed] [Google Scholar]
- 45.Burghout P, Bootsma HJ, Kloosterman TG, Bijlsma JJE, de Jongh CE, Kuipers OP, Kuipers OPM. 2007. Search for genes essential for pneumococcal transformation: the RADA DNA repair protein plays a role in genomic recombination of donor DNA. J Bacteriol 189:6540–6550. doi: 10.1128/JB.00573-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cremers AJH, Meis JF, Walraven G, Van Der Gaast-De Jongh CE, Ferwerda G, Hermans PWM. 2014. Effects of 7-valent pneumococcal conjugate 1 vaccine on the severity of adult 2 bacteremic pneumococcal pneumonia. Vaccine 32:3989–3994. doi: 10.1016/j.vaccine.2014.04.089. [DOI] [PubMed] [Google Scholar]
- 47.Cremers AJH, Mobegi FM, De Jonge MI, Van Hijum SAFT, Meis JF, Hermans PWM, Ferwerda G, Bentley SD, Zomer AL. 2015. The post-vaccine microevolution of invasive Streptococcus pneumoniae. Sci Rep 5:14952. doi: 10.1038/srep14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tunjungputri RN, van der Ven AJ, Riksen N, Rongen G, Tacke S, van den Berg TNA, Fijnheer R, Gomes ME, Dinarello CA, van de Veerdonk FL, Gasem M, Netea MG, Joosten LAB, De Groot PG, de Mast Q. 2015. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2- and TLR4-mediated inflammatory responses. Thromb Haemost 113:1035–1045. doi: 10.1160/TH14-07-0579. [DOI] [PubMed] [Google Scholar]
- 51.Tunjungputri RN, van de Heijden W, Urbanus RT, de Groot PG, van der Ven A, de Mast Q. 2017. Higher platelet reactivity and platelet-monocyte complex formation in Gram-positive sepsis compared to Gram-negative sepsis. Platelets 28:595–601. doi: 10.1080/09537104.2016.1252837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.