

Humans are constantly exposed to the opportunistic mold Aspergillus fumigatus, and disease caused by this pathogen is often determined by the magnitude of local and systemic immune responses. We have previously shown a protective role for interleukin-22 (IL-22) after acute A. fumigatus exposure.
KEYWORDS: Aspergillus, IL-22, innate-like lymphocyte, lung
ABSTRACT
Humans are constantly exposed to the opportunistic mold Aspergillus fumigatus, and disease caused by this pathogen is often determined by the magnitude of local and systemic immune responses. We have previously shown a protective role for interleukin-22 (IL-22) after acute A. fumigatus exposure. Here, employing IL-22Cre R26ReYFP reporter mice, we identified iNKT cells, γδ T cells, and type 3 innate lymphoid cells (ILC3s) as lung cell sources of IL-22 in response to acute A. fumigatus exposure. As these cells often utilize common γ-chain cytokines for their development or maintenance, we determined the role of IL-7, IL-21, and IL-15 in lung IL-22 induction and A. fumigatus lung clearance. We observed that IL-7, IL-21, and IL-15 were essential for, partially required for, or negatively regulated the production of IL-22, respectively. Deficiency in IL-7 and IL-21, but not IL-15R, resulted in impaired fungal clearance. Surprisingly, however, the absence of IL-7, IL-21, or IL-15R signaling had no effect on neutrophil recruitment. The levels of IL-1α, an essential anti-A. fumigatus proinflammatory cytokine, were increased in the absence of IL-7 and IL-15R but decreased in the absence of IL-21. IL-7 was responsible for maintaining lung iNKT cells and γδ T cells, whereas IL-21 was responsible for maintaining lung iNKT cells and ILC3s. In contrast, IL-15R deficiency had no effect on the absolute numbers of any IL-22 cell source, rather resulting in enhanced per cell production of IL-22 by iNKT cells and γδ T cells. Collectively, these results provide insight into how the IL-22 response in the lung is shaped after acute A. fumigatus exposure.
INTRODUCTION
Exposure to Aspergillus fumigatus may lead to variety of different infections and comorbidities, including development of an aspergilloma, chronic necrotizing aspergillosis, fungal asthma, and an invasive fungal infection (IFI) termed invasive aspergillosis (IA). Incidence rates for IFIs are ∼7% in both solid organ transplants (1) (19% due to IA) and stem cell transplants (2) (43% due to IA). In addition to these individuals with suppressed immune systems, there are several genetic immunodeficiencies in which IA or infection with A. fumigatus is extremely high. The classic example is chronic granulomatous disease (CGD), in which NADPH oxidase deficiency is uniquely associated with the development of IA (3). Individuals with hyper-IgE syndrome have mutations in STAT3, are unable to produce Th17 cells (4), and are severely susceptible to an A. fumigatus lung infection, although most often when cavitary lung lesions are present (5). Additional studies have reported that single-nucleotide polymorphisms (SNPs) in Dectin-1 (6), Toll-like receptor 1 (TLR1)/TLR6 (7), DC-SIGN (6), plasminogen (8), and tumor necrosis factor receptor 1 (TNFR1) (9) are also associated with susceptibility to IA. Although IA is a known infectious complication of the above-mentioned conditions, there is a growing concern for the development of nosocomial IA in the intensive care unit (10), with the underlying disease in these nonneutropenic included patients being high-dose steroids for chronic obstructive pulmonary disease (COPD), cirrhosis/liver failure, and solid cancers (11–14). Finally, simple colonization with or sensitization to A. fumigatus may have dramatic effects on lung function in asthmatics (15–18) and individuals with cystic fibrosis (CF) (19–21) and COPD (22). Therefore, a clearer understanding of protective immune responses in the lung after acute or chronic A. fumigatus exposure may identify therapeutic targets that could improve outcomes in multiple lung diseases (lung transplant, CF, COPD, asthma etc.).
Interleukin-22 (IL-22) is widely acknowledged to promote epithelial antimicrobial responses (23). We have previously reported that mice deficient in Dectin-1 acutely exposed to A. fumigatus had multiple defects in host defense (24). We extended the antifungal contribution of Dectin-1 to the induction of IL-22, as Dectin-1 deficiency resulted in a near total loss of lung IL-22 production after acute A. fumigatus exposure (25). Importantly, genetic deficiency in or neutralization of IL-22 resulted in impaired clearance of A. fumigatus as early as 24 h postchallenge, illustrating a critical role for IL-22 in pathogen elimination during acute infection (25). Although multiple cell sources of IL-22, including CD4+ T cells, CD8+ T cells, γδ T cells, NK cells, iNKT cells, LTi cells, and type 3 innate lymphoid cells (ILC3s), have been identified (reviewed in references 26 and 27), it is not clear which of these are functioning in an innate capacity after acute lung fungal exposure. However, we have previously reported that Dectin-1-dependent IL-22 was possibly produced by a non-CD4+ T cell source in a model of fungal asthma associated with chronic A. fumigatus exposure (28).
The common γ-chain family of cytokines, which includes IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, are most well recognized as essential factors for T cell development and B cell lymphopoiesis (29, 30). In general, IL-2, IL-7, and IL-15 have been shown to preferentially activate STAT5, while IL-4 and IL-21 preferentially activate STAT6 and STAT3, respectively (31). However, common γ-chain cytokines also play important roles in development, maintenance, and homeostasis of innate-like lymphocyte populations. For example, IL-2, IL-7, and IL-9 have recently been linked to ILC2 survival (32–35). Additionally, γδ T cell homeostasis requires IL-7 (36), as do certain iNKT cell populations (37). In the current study, we sought to define the lung cell sources of IL-22 after acute exposure to A. fumigatus and to determine which, if any, common γ-chain cytokines regulate IL-22 production and host defense.
RESULTS
Common γ-chain cytokines play dichotomous roles in lung IL-22 production and A. fumigatus lung clearance.
We have previously reported that IL-22 is essential for fungal clearance after acute A. fumigatus challenge (25). Although originally identified to be produced by CD4+ T cells, IL-22 is now recognized to be produced by many cell types, including γδ T cells, NK cells, iNKT cells, LTi cells, and ILC3s (reviewed in references 26 and 27). T and B lymphocytes, as well as innate-like lymphocytes, express the common γ-chain cytokine receptor, and cytokines in this family are critical for generation, development, or homeostasis of these cell types (36, 38). In addition, we have recently identified a role for the common γ-chain cytokine IL-7 in iNKT- and IL-22-associated immunopathogenesis during A. fumigatus-associated fungal asthma (39). To this end, we examined the impact of common γ-chain cytokines on lung IL-22 induction and clearance of A. fumigatus after acute exposure. In initial studies, we showed that lung IL-22 production is not detected until 24 h after acute A. fumigatus exposure and peaks at 48 h postexposure, followed by a reduction at 72 h (Fig. 1A). With respect to IL-7, mice deficient in this mediator demonstrated significantly higher fungal burdens in the lungs 48 h postexposure (Fig. 1B). An increased fungal burden in Il7−/− mice correlated with a near complete absence of IL-22 production by lung digest cells at 24 h and 48 h postchallenge (Fig. 1C). As neutrophils are central effector cells against A. fumigatus in the lung (40), we assessed their levels in Il7−/− mice. Surprisingly, there was no difference in neutrophil numbers between A. fumigatus-exposed wild-type (WT) and Il7−/− mice (Fig. 1D). As expected based on intact neutrophil recruitment, Il7−/− mice did not succumb to invasive infection (data not shown). Similar to Il7−/− mice, mice deficient in IL-21 also demonstrated an impairment in A. fumigatus lung clearance (Fig. 1E) and a moderate reduction in IL-22 production at 24 and 48 h (Fig. 1F). Once again, neutrophil numbers were not different in Il7−/− mice (Fig. 1G). In contrast to Il7−/− and Il21−/− mice, mice deficient in IL-15R were not more susceptible to acute A. fumigatus exposure (Fig. 1H). Intriguingly, IL-22 was produced at much higher levels at both 24 h and 48 h postchallenge than in similarly exposed WT mice (Fig. 1I). Despite the increase in IL-22, neutrophil levels were not similarly increased (Fig. 1J). Thus, IL-7 and IL-21 are required for optimal IL-22 production and organism clearance after acute A. fumigatus exposure, whereas IL-15R signaling functions as a negative regulator of IL-22. Despite variable IL-22 production in these mice, the level of IL-22 did not correlate with the level of neutrophil recruitment to the lungs.
FIG 1.

Common γ-chain cytokines play dichotomous roles in lung IL-22 production and A. fumigatus lung clearance. (A) C57BL/6 wild-type (WT) mice were challenged with live A. fumigatus conidia via intratracheal administration and sacrificed 12, 24, 48, and 72 h postexposure. At each time point, the right lungs were collected and enzymatically digested, and unfractionated lung cells were cultured in triplicate for 24 h. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from three independent experiments (n = 3 mice per time point per experiment) are shown. (B) WT and Il7−/− mice were challenged intratracheally with A. fumigatus conidia, and 48 h after exposure, the lung fungal burden was assessed by real-time PCR analysis of A. fumigatus 18S rRNA levels. Cumulative data from two to three independent experiments (n = 4 to 5 mice per group per experiment) are shown. Data are expressed as the mean level of A. fumigatus 18S rRNA + standard error of the mean (SEM). (C) WT and Il7−/− mice were challenged intratracheally with A. fumigatus conidia, and 24 h and 48 h after exposure, the right lungs were collected and enzymatically digested, and unfractionated lung cells were cultured in triplicate for 24 h. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from three independent experiments (n = 3 to 5 mice per group per experiment) are shown. (D) Total neutrophil population enumerated from whole-lung digests by flow cytometry. Neutrophils were defined as CD45+ CD11b+ Ly6G+. Cumulative data from two independent experiments (n = 3 to 4 mice per group per experiment) are shown. (E) Fungal burden 48 h after challenge in WT and Il21−/− mice. Cumulative data from two to three independent experiments (n = 4 to 5 mice per group per experiment) are shown. Data are expressed as the mean level of A. fumigatus 18S rRNA + SEM. (F) IL-22 production by lung digest cells 48 h after A. fumigatus challenge in WT and Il21−/− mice. Cumulative data from three independent experiments (n = 3 mice per group per experiment) are shown. (G) Total neutrophil numbers in the lung 24 h and 48 h after A. fumigatus challenge in WT and Il21−/− mice. Cumulative data from two independent experiments (n = 3 mice per group per experiment) are shown. (H) Fungal burden 48 h after challenge in WT and Il15r−/− mice. Cumulative data from two independent experiments (n = 4 to 5 mice per group per experiment) are shown. Data are expressed as the mean level of A. fumigatus 18S rRNA + SEM. (I) IL-22 production by lung digest cells 24 h and 48 h after A. fumigatus challenge in WT and Il15r−/− mice. Cumulative data from three independent experiments (n = 3 mice per group per experiment) are shown. (J) Total neutrophil numbers in the lung 48 h after A. fumigatus challenge in WT and Il15r−/− mice. For all graphs, *, **, and *** represent P < 0.05, P < 0.01, and P < 0.0001, respectively. PMN, polymorphonuclear leukocytes.
IL-1α and IL-1β levels are differentially affected by common γ-chain cytokines after acute A. fumigatus exposure.
Renewed interest in the IL-1 family of cytokines has uncovered novel roles for IL-1α and IL-1β in lung defense during acute A. fumigatus exposure (41, 42). Although we have previously reported that IL-1α is dependent on IL-22 after acute A. fumigatus exposure (25), we have recently shown that signaling through the IL-1 receptor is required for optimal IL-22 production after A. fumigatus exposure (43). Despite attenuated IL-22 production, deficiency in IL-7 was associated with elevated levels of IL-1α and IL-1β (Fig. 2A). In contrast, lower IL-22 levels during IL-21 deficiency correlated with lower IL-1α, but not IL-1β, production (Fig. 2B), despite the presence of higher fungal burden. Enhanced IL-22 production in the absence of IL-15 signaling resulted in elevated IL-1α, but not IL-1β (Fig. 2C). Thus, IL-1α and IL-1β production levels are differentially affected by common γ-chain cytokines during acute A. fumigatus exposure.
FIG 2.
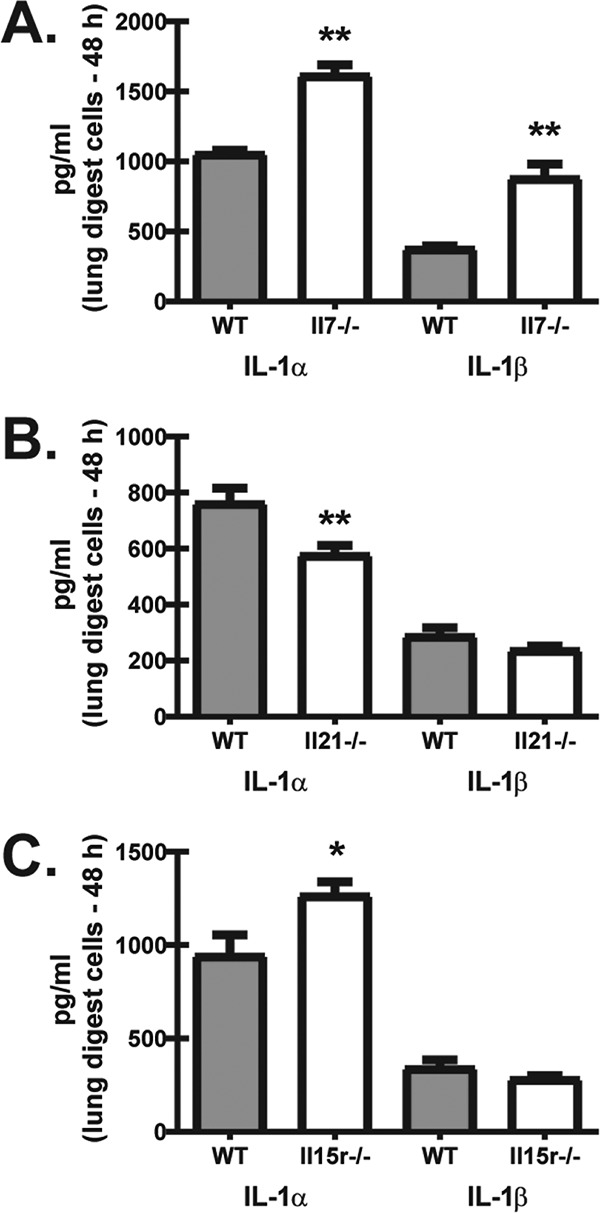
IL-1α and IL-1β levels are differentially affected by common γ-chain cytokines after acute A. fumigatus exposure. Mice were challenged intratracheally with A. fumigatus conidia, and 48 h after exposure, the right lungs were collected and enzymatically digested, and unfractionated lung cells were cultured in triplicate for 24 h. IL-1α and IL-1β levels in Il7−/− mice (A), Il21−/− mice (B), and Il15r−/− mice (C) were quantified in clarified coculture supernatants using a Bio-Plex system. Cumulative data from three independent experiments (n = 3 to 5 mice per group per experiment) are shown. For all graphs, * and ** represent P < 0.05 and P < 0.01, respectively.
Multiple innate and innate-like lymphocytes produce IL-22 after acute A. fumigatus exposure.
We have previously reported that IL-22 was primarily produced by a non-CD4+ T cell source during A. fumigatus-associated fungal asthma (28). Antibody-mediated depletion of CD4+ T cells demonstrated no effect on IL-22 production after acute A. fumigatus exposure (Fig. 3A), indicating that these cells play little to no role in IL-22 generation (Fig. 3A). We next sought to identify the non-CD4+ T cell source(s) of IL-22 in the lung by employing IL-22Cre R26ReYFP reporter mice (44), in which yellow fluorescent protein (YFP) expression marks IL-22-producing cells. After exposure of mice to A. fumigatus for 48 h, gating on eYFP+/IL-22+ cells (where eYFP is enhanced YFP) identified a small population of CD1d tetramer-positive (CD1d tetramer+) cells (representative of iNKT cells) (Fig. 3B) and a much larger population of TCR-δ+ cells (representative of γδ T cells) (Fig. 3B). We did not detect eYFP expression in CD11b+ CD11c+ or CD11b+ CD11c+ cells nor in CD8+ or NK1.1+ cells (Fig. 3B). The population of cells not stained by the CD1d tetramer or TCR-δ was analyzed for expression of lineage markers, with a significant portion of cells noted as negative for all lineage markers queried. Upon further analysis, these cells were found to be lineage-negative CD45+ Thy1+ ILC3s (Fig. 3C). To confirm these observations, we examined IL-22 production in mice deficient in these cell types 48 h after A. fumigatus exposure. Surprisingly, despite the identification of iNKT cells as a cell source of IL-22 (Fig. 1A), Cd1d−/− mice did not demonstrate a reduction in IL-22 production 48 h postchallenge (Fig. 3D). However, Cd1d−/− mice did have a significant reduction (∼35%) in IL-22 production at 24 h (Fig. 3D). In contrast, Tcrd−/− mice, deficient in γδ T cells, did not demonstrate a significant decrease in IL-22 production at 24 h post-A. fumigatus exposure but had an ∼80% reduction in IL-22 at 48 h (Fig. 3E). Thus, IL-22 is produced in the lung by multiple innate and innate-like lymphocyte populations in response to fungal exposure, with iNKT cells being an early producer of IL-22 and γδ T cells being a later IL-22 producer.
FIG 3.

Multiple innate and innate-like lymphocytes produce IL-22 during and after acute A. fumigatus exposure. (A) C57BL/6 wild-type mice were administered anti-CD4 depleting antibody GK1.5 or an isotype control 1 day prior to and 1 day after A. fumigatus challenge. At 48 h postchallenge, the right lungs were collected and enzymatically digested, and unfractionated lung cells were cultured in triplicate for 24 h. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from three independent experiments (n = 2 to 3 mice per group per experiment) are shown. (B) Representative plots from IL-22Cre R26ReYFP and WT mice 48 h after A. fumigatus challenge showing live eYFP+ lung digest cells. From the eYFP+ cells, iNKTs were defined as CD1d tetramer+, and γδ T cells were defined as γδ TCR+. CD11b+ CD11c+ and CD11b+ CD11c+ cells were negative for YFP/IL-22, as were CD8+ cells. (C) ILC3s were defined as lineage negative (gate includes CD3, CD4, CD8a, CD11b, CD11c, CD19, Ly6G, NK1.1, FcγR1, αβ TCR, KLRG1, and NKp46), CD1d tetramer−, γδ TCR−, CD4−, CD45+, and Thy1+. (D) C57BL/6 wild-type (WT) and Cd1d−/− mice were challenged intratracheally with A. fumigatus conidia, and 24 and 48 h after exposure, the right lungs were collected and enzymatically digested and unfractionated lung cells were cultured in triplicate for 24 h. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Representative data from one of two experiments (n = 3 mice per group per experiment) are shown. (E) WT and Tcrd−/− mice were challenged and lung cells isolated as described for panel B. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Representative data from one of two experiments (n = 3 mice per group per experiment) are shown. For all graphs, ** and *** represent P < 0.01 and P < 0.0001, respectively.
Common γ-chain cytokines have differential effects on lung IL-22 cell sources after acute A. fumigatus exposure.
In the above studies, lung IL-22 production was found to be completely dependent on IL-7, partially dependent on IL-21, and negatively regulated by IL-15 signaling. Therefore, we determined the effects of these common γ-chain cytokines on the level of IL-22 cell sources (iNKT cells, γδ T cells, and ILC3s) in the lung. The profound reduction in IL-22 production in Il7−/− mice correlated with an ∼3-fold reduction in the absolute numbers of iNKT cells (Fig. 4A) and γδ T cells (Fig. 4B) in the lung. Although trending lower, the absolute numbers of ILC3s were not significantly reduced (Fig. 4C). Unlike the near complete absence of IL-22 in Il7−/− mice, Il21−/− mice exposed to A. fumigatus had an ∼35% reduction in IL-22, which correlated with an ∼2-fold reduction in the absolute numbers of iNKT cells in the lung (Fig. 4D), yet paradoxically an ∼2-fold increase in the numbers of γδ T cells (Fig. 4E). ILC3s were also reduced by ∼2-fold (Fig. 4F) (45). In direct contrast to Il7−/− mice, Il15r−/− mice exposed to A. fumigatus had a dramatically higher production of IL-22 by lung digest cells at 48 h postchallenge. Surprisingly, the absolute numbers of iNKT cells (Fig. 4G), γδ T cells (Fig. 4H), and ILC3s (Fig. 4I) were not significantly different. Collectively, these data demonstrate that IL-7 is crucial for maintaining iNKT cells and γδ T cells in the lung after fungal exposure, whereas IL-21 is required for maintaining optimal numbers of iNKT cells and ILC3s but appears to be a negative regulator of γδ T cells. In contrast, IL-15R signaling serves as a negative regulator of IL-22 production during lung fungal infection, although the mechanism associated with this is not an increased number of IL-22 cell sources.
FIG 4.

Common γ-chain cytokines have differential effects on lung IL-22 cell sources after acute A. fumigatus exposure. WT and Il7−/− mice (A to C), WT and Il21−/− mice (D to F), and WT and Il15r−/− (G to I) mice were challenged intratracheally with A. fumigatus conidia, and 48 h after exposure, the right lungs were collected and enzymatically digested, and IL-22-producing cell populations were enumerated. (A, D, and G) iNKTs were defined as CD19− TCRβ− CD1d tetramer−. (B, D, and H) γδ T cells were defined as CD19− CD4− CD3+ γδ TCR+. (C, F, and I) ILC3s were defined as Lin− CD45+ Thy1+ KLRG1−. Cumulative data from three independent experiments (n = 2 to 3 mice per group per experiment) are shown. For all graphs, * and ** represent P < 0.05 and P < 0.01, respectively.
iNKT cells and γδ T cells produce more IL-22 per cell in the absence of IL-15 signaling after acute A. fumigatus exposure.
We next questioned whether there was a difference in the total number of IL-22-producing cells or a difference in a specific population of IL-22-producing cells in Il15r−/− mice. Intracellular staining for IL-22 in Il15r−/− mice showed no difference in the total number of IL-22-producing cells compared to the number for WT mice (Fig. 5A). Furthermore, we were unable to identify a significant difference in the populations of IL-22+ iNKTs (Fig. 5B), IL-22+ γδ T cells (Fig. 5C), or IL-22+ ILC3s (Fig. 5D) between Il15r−/− and WT mice. As iNKT cells, γδ T cells, and ILC3s were not increased in the lungs of Il15r−/− mice, yet IL-22 production was significantly higher, we determined whether these cell types produced more IL-22 on a per cell basis. For this, lung digest cells were collected 48 h post-A. fumigatus exposure and cultured in the presence of IL-23, followed by permeabilization and staining for intracellular IL-22. This analysis revealed that iNKT cells (Fig. 5E, representative flow; Fig. 5H, geometric mean) and γδ T cells (Fig. 5F, representative flow; Fig. 5I, geometric mean) had higher levels of intracellular IL-22 in Il15r−/− mice than WT mice. In contrast, there was no difference in IL-22 production by ILC3s between WT and Il15r−/− mice (Fig. 5G, representative flow; Fig. 5J, geometric mean). Thus, IL-15 signaling regulates IL-22 production by iNKT cells and γδ T cells, but not ILC3s, during lung fungal infection.
FIG 5.

iNKT cells and γδ T cells produce more IL-22 per cell in the absence of IL-15 signaling after acute A. fumigatus exposure. (A to D) WT and Il15r−/− mouse whole-lung digests were stimulated with IL-23 (4 ng/ml) for 4 h before surface staining, permeabilization, and intracellular staining for IL-22. Populations were defined as described in Materials and Methods. (A) Total IL-22-producing cells enumerated 48 h postexposure by flow cytometry in whole-lung digests from WT and Il15r−/− mice. Cumulative data from two independent experiments (n = 2 to 3 mice per group per experiment) are shown. (B) Total IL-22-producing iNKT cells enumerated 48 h postexposure by flow cytometry in whole-lung digests from WT and Il15r−/− mice. Cumulative data from two independent experiments (n = 2 to 3 mice per group per experiment) are shown. (C) Total IL-22-producing γδ T cells enumerated 48 h postexposure by flow cytometry in whole-lung digests from WT and Il15r−/− mice. Cumulative data from two independent experiments (n = 2 to 3 mice per group per experiment) are shown. (D) Total IL-22-producing ILC3s enumerated 48 h postexposure by flow cytometry in whole-lung digests from WT and Il15r−/− mice. Cumulative data from two independent experiments (n = 2 to 3 mice per group per experiment) are shown. (E to J) WT and Il15r−/− mice were exposed to live A. fumigatus conidia via intratracheal administration and sacrificed 48 h postexposure. Cells were enumerated and then stimulated with IL-23 (4 ng/ml) for 4 h before surface staining, permeabilization, and intracellular staining for IL-22. (E to G) Histograms from concatenated samples representing each innate IL-22 cell population. Data are representative of two independent experiments. Gray, WT; black, Il15r−/−. (H to J) Mean fluorescence intensity (MFI) quantified for iNKTs, γδ T cells, and ILC3s in WT and Il15r−/− mice. Cumulative data of two independent experiments (n = 3 mice per group per experiment) are shown. For all graphs, ** and *** represent P < 0.01 and P < 0.0001, respectively.
DISCUSSION
Therapeutics such as granulocyte-macrophage colony-stimulating factor (GM-CSF; sargramostim [Leukine]) and gamma interferon (IFN-γ; Actimmune) are FDA-approved treatments for immunosuppression associated with bone marrow transplantation and for combatting infectious complications associated with chronic granulomatous disease (CGD) (46, 47). Development of these therapies could not have been possible without a better understanding of innate immune mechanisms that are protective against invasive fungal infections. We have previously reported that IL-22 deficiency or neutralization leads to impaired lung clearance of A. fumigatus (25). Lack of IL-22 resulted in impaired inflammatory cytokine and chemokine production and impaired antifungal activity of lung lavage fluid (25). The purpose of the current study was to identify the cell sources of IL-22 and to clarify mediators that may positively or negatively affect these IL-22 cell sources.
To date, CD4 T cells, CD8 T cells, iNKT cells, γδ T cells, and ILC3s have been identified as cell sources of IL-22 (reviewed in reference 48). In addition, neutrophils have been identified as an IL-22 cell source in the gut during experimental colitis (49). Finally, IL-23R reporter mice identified a CD11b+ CD11c+ cell population that produced IL-22 during experimental autoimmune encephalomyelitis (50). Collectively, these studies document the diversity of IL-22 cell sources at multiple sites. We have previously shown that IL-22 protein levels in lung digest cell cultures replicate those observed in whole-lung homogenates (25). Data presented here now show that IL-22 is not detected in or produced by lung digest cell cultures until 24 h post-A. fumigatus challenge, which increases at 48 h and begins to decline by 72 h. Based on this observation, we hypothesized that an innate-like lymphocyte or a myeloid cell population was responsible for IL-22 production. γδ T cells have previously been reported to produce IL-17A during A. fumigatus exposure, and mice deficient in these cells were susceptible to infection (51). However, this study failed to determine whether γδ T cells also produced IL-22. iNKT cells are thought to be a primary source of IFN-γ during A. fumigatus exposure, although small populations may produce IL-17A (52), a phenotype that is associated with reduced fungal clearance in iNKT cell-deficient mice. Here again, IL-22 levels were not investigated. In human peripheral blood mononuclear cells (PBMCs) stimulated with A. fumigatus, the primary source of IL-22 has been reported to be CD4+ T cells, with these cells producing IL-22 alone or double-producing IL-22 and either IL-17A or IFN-γ (53). This study however did not further examine other non-CD4+ cell populations, despite showing that ∼10% of IL-22 was produced by a non-CD4+, non-CD8+, non-CD56+ population. In the current study, we employed IL-22Cre R26ReYFP fate reporter mice, which have previously demonstrated that the majority of IL-22+ cells in the naive lung are γδ T cells (∼65%), followed by CD4+ cells (∼20%) and ILC3s (∼10%) (44). At 48 h post-A. fumigatus exposure, we observed three YFP+ cell populations that we subsequently identified as iNKT cells, γδ T cells, and ILC3s. We did not observe CD11b+ or CD11c+ cells that were also YFP+. An intriguing finding from our work is the observation that iNKT cells appear to be the “first line” of IL-22 production, as mice deficient in iNKT cells had a blunted IL-22 response 24 h after challenge but intact IL-22 48 h after challenge. In contrast, mice deficient in γδ T cells had the opposite phenotype, in that these cells were more critical for IL-22 production 48 h after exposure. Although we are unaware of mice with a specific deletion of ILC3s, we speculate that these cells produce IL-22 at both time points.
IL-7 is a well-studied common γ-chain cytokine with a known role in the development, homeostasis, or expansion of many cell types that also produce IL-22 (36, 37, 54–56). Previous studies have shown that IL-7 is required for the development of γδ T cells in multiple peripheral and mucosal tissues (57). In contrast, although numbers of iNKT cells in peripheral lymphoid tissues are reduced in Il7ra−/− mice, they are not completely absent, implicating a role for IL-7 in iNKT cell expansion rather than in their development (58). Likewise, a recent study has shown that ILC3s in the small intestine lamina propria are lower in number in Il7ra−/− mice yet are competent to produce IL-22 during Citrobacter rodentium infection (59). Moreover, our data indicate that IL-7 was required for optimal levels of iNKT cells and γδ T cells in the lung during acute fungal exposure, with each population reduced by 3-fold in Il7−/− mice. In contrast to C. rodentium infection (59), however, IL-7 was absolutely required for IL-22 production, as lung cells from Il7−/− mice produced less than 10% of that observed for WT mice. These results are in line with reports documenting IL-7 involvement in IL-17A production in iNKT (58) and γδ T cell in vitro cultures (60).
IL-21 is recognized as a critical mediator driving effector and memory CD8 T cell responses (61) and T follicular helper cell differentiation (62) and as an autocrine signal for CD4 T cells producing IL-17 and IL-22 (63). Regarding iNKT cells, IL-21 may be produced by, as well as activate, iNKT cells (64). Support for IL-21 in iNKT cell maintenance comes from studies in humans showing that a loss-of-function mutation in Il21r results in blunted iNKT cell numbers in peripheral blood (65). Our data also support a role for the IL-21/IL-21R axis in maintaining iNKT cell levels in the lung, as these cells were reduced by a little more than half after acute fungal exposure. γδ T cells may also be a source of IL-21 in response to IL-1β and IL-23 stimulation (66). However, mice deficient in IL-21R have an increase in IL-17A-producing γδ T cells during influenza lung infection (67). We extend this observation here by demonstrating that Il21−/− mice also have increased numbers of γδ T cells in the lung during acute fungal exposure. To our knowledge, however, we are the first to demonstrate that optimal ILC3 levels in the lung during infection require IL-21. The finding that IL-22 is only partially reduced during IL-21 deficiency is putatively due to higher γδ T cell numbers (some of which are producing IL-22) in the presence of the lower numbers of iNKT cells and ILC3s.
Homeostasis of peripheral iNKT cells was previously found to require IL-15 (68), although IL-15 is dispensable for the development of IL-17A-producing iNKT cells in the spleen (58). IFN-γ-producing, but not IL-17A-producing, splenic γδ T cells require IL-15 (58). However, data suggest that IL-17A-producing γδ T cells in lymph nodes are restricted by IL-15R signaling (45), although some reports suggest a role for IL-15 in γδ T cell development in the lymph nodes (36) and skin (69). IL-15 was recently demonstrated to sustain IL-7R-independent small intestine lamina propria ILC3 development (59). However, IL-15 may function in ILC survival in general, as reports suggest a role for IL-15 in intestinal ILC1 and ILC2 differentiation (70). Overall, these observations suggest that the role of IL-15 is diverse based on the type of cell and its cytokine-producing phenotype. In our study, the absence of IL-15 signaling (i.e., IL-15R deficiency) did not affect the numbers of iNKT cells, γδ T cells, and ILC3s in the lung after fungal exposure, suggesting that in a mucosal tissue (lung) during an infectious process (fungal exposure), IL-15 is not essential for the presence of these cell types. Rather, the lack of IL-15 signaling in iNKT cells and γδ T cells resulted in higher IL-22 production. In contrast to findings for the small intestine lamina propria during C. rodentium infection (59), IL-15 signaling did not positively or negatively affect IL-22 production by ILC3s.
The receptor for IL-1 was originally shown to have little to no role in A. fumigatus host defense (71); however, renewed interest in the IL-1 family of cytokines has uncovered novel roles for IL-1α and IL-1β in early neutrophil recruitment and macrophage antifungal responses (41). More recently, IL-1α was required for the elimination of highly virulent, highly germinating A. fumigatus strains (42). We have previously demonstrated that IL-1α is dependent on IL-22 after A. fumigatus lung exposure (25). However, we have also recently reported that IL-22 production is highly dependent on IL-1R signaling (43, 72). Therefore, IL-1α and IL-1β exist as both upstream promoters of IL-22 production and downstream effectors of IL-22-mediated antifungal defense. Our data here demonstrate that IL-1α and IL-1β were increased in the absence of the common γ-chain cytokine IL-7, which we hypothesize is a result of increased fungal burden in these mice rather than a direct effect of IL-7 on their production. Nevertheless, as neutrophil recruitment was intact in Il7−/− mice, this suggests that susceptibility to A. fumigatus is a specific result of impaired IL-22-induced soluble antifungal activity. It is not clear why IL-1α was lower in the absence of IL-21, as fungal burden was also increased in Il21−/− mice, and in theory, so might IL-1α. However, IL-1α was recently reported to be primarily produced by radiosensitive myeloid cells, including macrophages (42). As macrophages express the receptor for IL-21 (73), it is possible that the lack of this common γ-chain cytokine directly affects the production of IL-1α by this cell type. Future studies will probe the relationship between IL-21 and IL-1α generation. IL-1α was observed to be increased in the absence of IL-15R, and we speculate that this is a result of the increased IL-22 response.
In summary, we have shown that IL-22 is primarily produced by three innate-like lymphocyte subsets, iNKT cells, γδ T cells, and ILC3s, in the lung after acute fungal exposure. The common γ-chain cytokine IL-7 was indispensable for IL-22 production, whereas the common γ-chain cytokine IL-21 was only partially required for IL-22 production. Signaling through the receptor for the common γ-chain cytokine IL-15 had a profound negative effect on IL-22 production. Collectively, these results provide insight into the parameters of IL-22 induction in the lung during fungal exposure.
MATERIALS AND METHODS
Mice.
WT BL/6 mice, 6 to 8 weeks of age, were obtained from The Jackson Laboratory (Bangor, ME) or Taconic (Hudson, NY). Il7−/− mice were a kind gift from Chris Klug (UAB). Il21−/− mice were provided in collaboration with Allan Zajac (UAB). Cd1d−/−, Tcrd−/−, Il15r−/− B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J and C57BL/6-Il22tm1.1(icre)Stck/J mice (IL-22 reporter mice, which were developed using a sequence encoding Cre recombinase cloned into the Il22 locus and crossed with reporter mice expressing enhanced yellow fluorescence protein [eYFP] under the control of the endogenous Rosa26 promoter [44]) were obtained from The Jackson Laboratory. All animals were housed in a specific-pathogen-free, Association for Assessment and Accreditation of Laboratory Animal Care-certified facility and handled according to Public Health Service Office of Laboratory Animal Welfare policies after review by the UAB Institutional Animal Care and Use Committee.
Preparation of A. fumigatus, in vivo challenge, and lung fungal burden assessment.
A. fumigatus isolate 13073 (ATCC, Manassas, VA) was maintained on potato dextrose agar for 5 to 7 days at 37°C. Conidia were harvested by washing the culture flask with 50 ml of sterile phosphate-buffered saline supplemented with 0.1% Tween 20. The conidia were then passed through a sterile 40-μm nylon membrane to remove hyphal fragments and enumerated on a hemacytometer. For challenge, mice were lightly anesthetized with isoflurane and administered 7 × 107 A. fumigatus conidia in a volume of 50 μl intratracheally as previously described (24, 25). Briefly, mice are held in a vertical, upright position, and the tongue is withdrawn from the mouth using forceps. A pipette is used to deliver the 50-μl inoculum to the caudal oropharynx in which normal breathing results in fluid aspiration into the lungs (74). For lung fungal burden analysis, the left lungs were collected at 48 h postexposure and homogenized in 1 ml of phosphate-buffered saline (PBS). Total RNA was extracted from 0.1 ml of unclarified lung homogenate using the MasterPure yeast RNA purification kit (Epicentre Biotechnologies, Madison, WI), which includes a DNase treatment step to eliminate genomic DNA as previously reported (75). Total RNA was also extracted from serial 1:10 dilutions of live A. fumigatus conidia (101 to 109) and DNase treated to form a standard curve. The lung A. fumigatus burden was analyzed with a real-time PCR measurement of the A. fumigatus 18S rRNA (76) and quantified using a standard curve of A. fumigatus conidia, as previously described (75). As a validation of the real-time PCR method, heat-killed A. fumigatus conidia did not yield a signal by real-time PCR and were unable to grow on potato dextrose agar plates (75). In addition, no amplification controls (i.e., no reverse transcriptase included in the cDNA reaction) yielded a signal of <0.001% by real-time PCR, indicating that the DNase treatment step efficiently eliminated contaminating A. fumigatus DNA (as DNA is not predicative of organism viability [77]).
Lung cell isolation, flow cytometry, and analysis of IL-22.
Mice were anesthetized with intraperitoneal ketamine/xylazine and sacrificed by exsanguination 12, 24, or 48 h postinfection. Both lungs were collected and minced in IMDM medium (Sigma, St. Louis, MO) supplemented with 1% pen-strep-glut (Mediatech, Herndon, VA), 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), and 0.4 mg/ml polymyxin B (Thermo Fisher), followed by incubation for 50 min with tissue culture-grade type IV collagenase (1 mg/ml; Sigma, St. Louis, MO) in a 37°C orbital shaker at 100 rpm. The cell suspension was filtered through a sterile 70-μm nylon filter, and red blood cells were lysed with ACK buffer and finally filtered through sterile 40-μm nylon (Lonza, Walkersville, MD) to create lung cell preparations. For assessing IL-22 production, cells were enumerated on a hemacytometer and plated at 1 × 106 cells in a volume of 0.2 ml. Supernatants were collected after 24 h, clarified by centrifugation, and stored at −80°C. IL-22 levels were quantified in supernatants by ELISA (R&D Systems) (24). For intracellular IL-22 staining, lung digest cells were stimulated with 4 ng/ml IL-23 (R&D Systems) for 4 h, and brefeldin A (eBioscience) was then added per the manufacturer's instruction for an additional 3 h. For surface and intracellular staining, cells were incubated with Mouse BD Fc Block (clone 2.4G2; BD Pharmingen), followed by viability assessment (LIVE/DEAD fixable aqua; Life Technologies). Cell populations were identified with a combination of the following markers: anti-CD45R allophycocyanin (APC)–Cy7 (cloneRA3-6B2; BioLegend), anti-CD4 fluorescein isothiocyanate (FITC) (clone GK1.5; BioLegend), anti-CD3 phycoerythrin (PE) (clone17A2; BD Pharmingen), anti-TCRβ PE (clone H57-597; eBioscience), anti-γδ TCR APC (clone eBioGL3; eBioscience), PBS 57 loaded CD1d tetramer APC (NIH tetramer core), anti-CD19 biotin (clone eBio1D3; eBioscience), anti-CD3 biotin (clone 17A2; eBioscience), anti-CD4 biotin (clone GK1.5; eBioscience), anti-CD8a biotin (clone 53-6.7; eBioscience), anti-γδ TCR biotin (clone UC7-13DS; eBioscience), anti-CD11b biotin (M1/70; eBioscience), anti-CD11c biotin (clone N418; eBioscience), anti-Ly6G biotin (clone RB6-8C5; eBioscience), anti-FcεR1 biotin (clone MAR1; eBioscience), anti-NK1.1 biotin (clone PK136; eBioscience), anti-KLRG1 biotin (clone2F1; eBioscience), anti-CD127 PE (clone A7R34; eBioscience), anti-Thy1.2 PE–Cy7 (clone 53-2.1; BD Pharmingen), anti-CD45 BV785 (clone 30-F11; BioLegend), anti-RORγt BV421 (cloneQ31-378; BD Horizon), BV421 streptavidin, and/or BV650 streptavidin. After surface staining for appropriate cell markers, cells were fixed and permeabilized via the Foxp3/transcription factor staining buffer set (eBioscience) and stained for IL-22 PE (clone 1H8PWSR; eBioscience). All staining was carried out at 4°C. Data were acquired using a BD LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar). Data acquisition and analysis were supported by the UAB Rheumatic Disease Core Center NIH P30 AR048311.
Statistics.
Data were analyzed using GraphPad Prism version 5.0 statistical software (GraphPad Software, San Diego, CA). Comparisons between groups when data were normally distributed were made with the two-tailed unpaired Student t test. Significance was accepted at a P value of <0.05.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants HL096702, HL122426, and HL136211.
REFERENCES
- 1.Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, Anaissie EJ, Brumble LM, Herwaldt L, Ito J, Kontoyiannis DP, Lyon GM, Marr KA, Morrison VA, Park BJ, Patterson TF, Perl TM, Oster RA, Schuster MG, Walker R, Walsh TJ, Wannemuehler KA, Chiller TM. 2010. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis 50:1101–1111. doi: 10.1086/651262. [DOI] [PubMed] [Google Scholar]
- 2.Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, Ito J, Andes DR, Baddley JW, Brown JM, Brumble LM, Freifeld AG, Hadley S, Herwaldt LA, Kauffman CA, Knapp K, Lyon GM, Morrison VA, Papanicolaou G, Patterson TF, Perl TM, Schuster MG, Walker R, Wannemuehler KA, Wingard JR, Chiller TM, Pappas PG. 2010. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001-2006: overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin Infect Dis 50:1091–1100. doi: 10.1086/651263. [DOI] [PubMed] [Google Scholar]
- 3.Forehand JR, Johnston RB. 1994. Chronic granulomatous disease: newly defined molecular abnormalities explain disease variability and normal phagocyte physiology. Curr Opin Pediatr 6:668–675. doi: 10.1097/00008480-199412000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding M, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC. 2008. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V, Puck JM, Holland SM. 2007. Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol 119:1234–1240. doi: 10.1016/j.jaci.2006.12.666. [DOI] [PubMed] [Google Scholar]
- 6.Sainz J, Lupianez CB, Segura-Catena J, Vazquez L, Rios R, Oyonarte S, Hemminki K, Forsti A, Jurado M. 2012. Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS One 7:e32273. doi: 10.1371/journal.pone.0032273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kesh S, Mensah NY, Peterlongo P, Jaffe D, Hsu K, van den Brink M, O'reilly R, Pamer E, Satagopan J, Papanicolaou GA. 2005. TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann N Y Acad Sci 1062:95–103. doi: 10.1196/annals.1358.012. [DOI] [PubMed] [Google Scholar]
- 8.Zaas AK, Liao G, Chien JW, Weinberg C, Shore D, Giles SS, Marr KA, Usuka J, Burch LH, Perera L, Perfect JR, Peltz G, Schwartz DA. 2008. Plasminogen alleles influence susceptibility to invasive aspergillosis. PLoS Genet 4:e1000101. doi: 10.1371/journal.pgen.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sainz J, Salas-Alvarado I, Lopez-Fernandez E, Olmedo C, Comino A, Garcia F, Blanco A, Gomez-Lopera S, Oyonarte S, Bueno P, Jurado M. 2010. TNFR1 mRNA expression level and TNFR1 gene polymorphisms are predictive markers for susceptibility to develop invasive pulmonary aspergillosis. Int J Immunopathol Pharmacol 23:423–436. doi: 10.1177/039463201002300205. [DOI] [PubMed] [Google Scholar]
- 10.Meersseman W, Lagrou K, Maertens J, Wilmer A, Hermans G, Vanderschueren S, Spriet I, Verbeken E, Van Wijngaerden E. 2008. Galactomannan in brochoalveolar lavage fluid—a tool for diagnosing aspergillosis in intensive care unit patients. Am J Respir Crit Care Med 177:27–34. doi: 10.1164/rccm.200704-606OC. [DOI] [PubMed] [Google Scholar]
- 11.Trof RJ, Beishuizen A, Debets-Ossenkopp YJ, Girbes AR, Groeneveld AB. 2007. Management of invasive pulmonary aspergillosis in non-neutropenic critically ill patients. Intensive Care Med 33:1694–1703. doi: 10.1007/s00134-007-0791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandewoude KH, Blot SI, Depuydt P, Benoit D, Temmerman W, Colardyn F, Vogelaers D. 2006. Clinical relevance of Aspergillus isolation from respiratory tract samples in critically ill patients. Crit Care 10:R31. doi: 10.1186/cc4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garnacho-Montero J, Amaya-Villar R, Ortiz-Leyba C, Leon C, Alvarez-Lerma F, Nolla-Salas J, Iruretagoyena JR, Barcenilla F. 2005. Isolation of Aspergillus spp. from the respiratory tract in critically ill patients: risk factors, clinical presentation and outcome. Crit Care 9:R191–R199. doi: 10.1186/cc3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barberan J, Sanz F, Hernandez JL, Merlos S, Malmierca E, Garcia-Perez FJ, Sanchez-Haya E, Segarra M, Garcia de la Llana F, Granizo JJ, Gimenez MJ, Aguilar L. 2012. Clinical features of invasive pulmonary aspergillosis vs. colonization in COPD patients distributed by gold stage. J Infect 65:447–452. doi: 10.1016/j.jinf.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Zureik M, Neukirch C, Leynaert B, Liard R, Bousquet J, Neukirch F. 2002. Sensitisation to airborne moulds and severity of asthma: cross sectional study from European Community respiratory health survey. BMJ 325:411–414. doi: 10.1136/bmj.325.7361.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Driscoll BR, Powell G, Chew F, Niven RM, Miles JF, Vyas A, Denning DW. 2009. Comparison of skin prick tests with specific serum immunoglobulin E in the diagnosis of fungal sensitization in patients with severe asthma. Clin Exp Allergy 39:1677–1683. doi: 10.1111/j.1365-2222.2009.03339.x. [DOI] [PubMed] [Google Scholar]
- 17.Fairs A, Agbetile J, Hargadon B, Bourne M, Monteiro WR, Brightling CE, Bradding P, Green RH, Mutalithas K, Desai D, Pavord ID, Wardlaw AJ, Pashley CH. 2010. IgE sensitization to Aspergillus fumigatus is associated with reduced lung function in asthma. Am J Respir Crit Care Med 182:1362–1368. doi: 10.1164/rccm.201001-0087OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maurya V, Gugnani HC, Sarma PU, Madan T, Shah A. 2005. Sensitization to Aspergillus antigens and occurrence of allergic bronchopulmonary aspergillosis in patients with asthma. Chest 127:1252–1259. doi: 10.1378/chest.127.4.1252. [DOI] [PubMed] [Google Scholar]
- 19.Valenza G, Tappe D, Turnwald D, Frosch M, Konig C, Hebestreit H, Abele-Horn M. 2008. Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J Cyst Fibros 7:123–127. doi: 10.1016/j.jcf.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Burgel PR, Baixench MT, Amsellem M, Audureau E, Chapron J, Kanaan R, Honore I, Dupouy-Camet J, Dusser D, Klaassen CH, Meis JF, Hubert D, Paugam A. 2012. High prevalence of azole-resistant Aspergillus fumigatus in adults with cystic fibrosis exposed to itraconazole. Antimicrob Agents Chemother 56:869–874. doi: 10.1128/AAC.05077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraemer R, Delosea N, Ballinari P, Gallati S, Crameri R. 2006. Effect of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. Am J Respir Crit Care Med 174:1211–1220. doi: 10.1164/rccm.200603-423OC. [DOI] [PubMed] [Google Scholar]
- 22.Bafadhel M, McKenna S, Agbetile J, Fairs A, Desai D, Mistry V, Morley JP, Pancholi M, Pavord ID, Wardlaw AJ, Pashley CH, Brightling CE. 2014. Aspergillus fumigatus during stable state and exacerbations of COPD. Eur Respir J 43:64–71. doi: 10.1183/09031936.00162912. [DOI] [PubMed] [Google Scholar]
- 23.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, De Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 24.Werner J, Metz AE, Horn D, Faro-Trindade I, Schoeb TR, Hewitt MM, Schwiebert LM, Brown GD, Steele C. 2009. Requisite role for the Dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 182:4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, Chan YR, Ouyang W, Brown GD, Weaver CT, Steele C. 2012. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun 80:410–417. doi: 10.1128/IAI.05939-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werner JL, Gessner MA, Lilly LM, Nelson MP, Metz AE, Horn D, Dunaway CW, Deshane J, Chaplin DD, Weaver CT, Brown GD, Steele C. 2011. Neutrophils produce IL-17A in a Dectin-1- and IL-23-dependent manner during invasive fungal infection. Infect Immun 79:3966–3977. doi: 10.1128/IAI.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zenewicz LA, Flavell RA. 2011. Recent advances in IL-22 biology. Int Immunol 23:159–163. doi: 10.1093/intimm/dxr001. [DOI] [PubMed] [Google Scholar]
- 28.Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, Brown GD, Steele C. 2012. The beta-glucan receptor Dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol 189:3653–3660. doi: 10.4049/jimmunol.1201797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, Li WQ, Aiello FB, Mazzucchelli R, Asefa B, Khaled AR, Durum SK. 2005. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev 16:513–533. doi: 10.1016/j.cytogfr.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Hara T, Shitara S, Imai K, Miyachi H, Kitano S, Yao H, Tani-ichi S, Ikuta K. 2012. Identification of IL-7-producing cells in primary and secondary lymphoid organs using IL-7-GFP knock-in mice. J Immunol 189:1577–1584. doi: 10.4049/jimmunol.1200586. [DOI] [PubMed] [Google Scholar]
- 31.Shuai K, Liu B. 2003. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 32.Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, Tan SY, Forbes-Blom E, Tong PL, Koller Y, Shklovskaya E, Iwashima M, McCoy KD, Le Gros G, Fazekas de St Groth B, Weninger W. 2015. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol 136:1653–1663. doi: 10.1016/j.jaci.2015.03.043. [DOI] [PubMed] [Google Scholar]
- 33.Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, Radtke F, Hardman CS, Hwang YY, Fallon PG, McKenzie AN. 2012. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol 13:229–236. doi: 10.1038/ni.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, Hendriks RW. 2012. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol 42:1106–1116. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 35.Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B. 2013. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med 210:2951–2965. doi: 10.1084/jem.20130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baccala R, Witherden D, Gonzalez-Quintial R, Dummer W, Surh CD, Havran WL, Theofilopoulos AN. 2005. Gamma delta T cell homeostasis is controlled by IL-7 and IL-15 together with subset-specific factors. J Immunol 174:4606–4612. doi: 10.4049/jimmunol.174.8.4606. [DOI] [PubMed] [Google Scholar]
- 37.Liang B, Hara T, Wagatsuma K, Zhang J, Maki K, Miyachi H, Kitano S, Yabe-Nishimura C, Tani-Ichi S, Ikuta K. 2012. Role of hepatocyte-derived IL-7 in maintenance of intrahepatic NKT cells and T cells and development of B cells in fetal liver. J Immunol 189:4444–4450. doi: 10.4049/jimmunol.1201181. [DOI] [PubMed] [Google Scholar]
- 38.Rochman Y, Spolski R, Leonard WJ. 2009. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol 9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reeder KM, Dunaway CW, Blackburn JP, Yu Z, Matalon S, Hastie AT, Ampleford EJ, Meyers DA, Steele C. 2018. The common gamma-chain cytokine IL-7 promotes immunopathogenesis during fungal asthma. Mucosal Immunol doi: 10.1038/s41385-018-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnes PD, Marr KA. 2007. Risks, diagnosis and outcomes of invasive fungal infections in haematopoietic stem cell transplant recipients. Br J Haematol 139:519–531. doi: 10.1111/j.1365-2141.2007.06812.x. [DOI] [PubMed] [Google Scholar]
- 41.Caffrey AK, Lehmann MM, Zickovich JM, Espinosa V, Shepardson KM, Watschke CP, Hilmer KM, Thammahong A, Barker BM, Rivera A, Cramer RA, Obar JJ. 2015. IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog 11:e1004625. doi: 10.1371/journal.ppat.1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caffrey-Carr AK, Kowalski CH, Beattie SR, Blaseg NA, Upshaw CR, Thammahong A, Lust HE, Tang YW, Hohl TM, Cramer RA, Obar JJ. 2017. IL-1alpha is critical for resistance against highly virulent Aspergillus fumigatus isolates. Infect Immun 85:e00661-17. doi: 10.1128/IAI.00661-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garth JM, Reeder KM, Godwin MS, Mackel JJ, Dunaway CW, Blackburn JP, Steele C. 2017. IL-33 signaling regulates innate IL-17A and IL-22 production via suppression of prostaglandin E2 during lung fungal infection. J Immunol 199:2140–2148. doi: 10.4049/jimmunol.1602186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahlfors H, Morrison PJ, Duarte JH, Li Y, Biro J, Tolaini M, Di Meglio P, Potocnik AJ, Stockinger B. 2014. IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J Immunol 193:4602–4613. doi: 10.4049/jimmunol.1401244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colpitts SL, Puddington L, Lefrancois L. 2015. IL-15 receptor alpha signaling constrains the development of IL-17-producing gammadelta T cells. Proc Natl Acad Sci U S A 112:9692–9697. doi: 10.1073/pnas.1420741112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richard S, Schuster MW. 2002. Stem cell transplantation and hematopoietic growth factors. Curr Hematol Rep 1:103–109. [PubMed] [Google Scholar]
- 47.Marciano BE, Wesley R, De Carlo ES, Anderson VL, Barnhart LA, Darnell D, Malech HL, Gallin JI, Holland SM. 2004. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis 39:692–699. doi: 10.1086/422993. [DOI] [PubMed] [Google Scholar]
- 48.Lim C, Savan R. 2014. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev 25:257–271. doi: 10.1016/j.cytogfr.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. 2013. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A 110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, Oukka M. 2009. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol 182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romani L, Fallarino F, De Luca A, Montagnoli C, D'Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P. 2008. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 52.Cohen NR, Tatituri RV, Rivera A, Watts GF, Kim EY, Chiba A, Fuchs BB, Mylonakis E, Besra GS, Levitz SM, Brigl M, Brenner MB. 2011. Innate recognition of cell wall beta-glucans drives invariant natural killer T cell responses against fungi. Cell Host Microbe 10:437–450. doi: 10.1016/j.chom.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gresnigt MS, Becker KL, Smeekens SP, Jacobs CW, Joosten LA, van der Meer JW, Netea MG, van de Veerdonk FL. 2013. Aspergillus fumigatus-induced IL-22 is not restricted to a specific Th cell subset and is dependent on complement receptor 3. J Immunol 190:5629–5639. doi: 10.4049/jimmunol.1202601. [DOI] [PubMed] [Google Scholar]
- 54.Vonarbourg C, Diefenbach A. 2012. Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Semin Immunol 24:165–174. doi: 10.1016/j.smim.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Mebius RE. 2003. Organogenesis of lymphoid tissues. Nat Rev Immunol 3:292–303. doi: 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 56.Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, Acha-Orbea H, Finke D. 2007. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity 26:643–654. doi: 10.1016/j.immuni.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Maki K, Sunaga S, Komagata Y, Kodaira Y, Mabuchi A, Karasuyama H, Yokomuro K, Miyazaki JI, Ikuta K. 1996. Interleukin 7 receptor-deficient mice lack gamma delta T cells. Proc Natl Acad Sci U S A 93:7172–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webster KE, Kim HO, Kyparissoudis K, Corpuz TM, Pinget GV, Uldrich AP, Brink R, Belz GT, Cho JH, Godfrey DI, Sprent J. 2014. IL-17-producing NKT cells depend exclusively on IL-7 for homeostasis and survival. Mucosal Immunol 7:1058–1067. doi: 10.1038/mi.2013.122. [DOI] [PubMed] [Google Scholar]
- 59.Robinette ML, Bando JK, Song W, Ulland TK, Gilfillan S, Colonna M. 2017. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat Commun 8:14601. doi: 10.1038/ncomms14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michel ML, Pang DJ, Haque SF, Potocnik AJ, Pennington DJ, Hayday AC. 2012. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing gammadelta cells. Proc Natl Acad Sci U S A 109:17549–17554. doi: 10.1073/pnas.1204327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tian Y, Cox MA, Kahan SM, Ingram JT, Bakshi RK, Zajac AJ. 2016. A context-dependent role for IL-21 in modulating the differentiation, distribution, and abundance of effector and memory CD8 T cell subsets. J Immunol 196:2153–2166. doi: 10.4049/jimmunol.1401236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jogdand GM, Mohanty S, Devadas S. 2016. Regulators of Tfh cell differentiation. Front Immunol 7:520. doi: 10.3389/fimmu.2016.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. 2007. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, Godfrey DI. 2007. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol 178:2827–2834. doi: 10.4049/jimmunol.178.5.2827. [DOI] [PubMed] [Google Scholar]
- 65.Wilson RP, Ives ML, Rao G, Lau A, Payne K, Kobayashi M, Arkwright PD, Peake J, Wong M, Adelstein S, Smart JM, French MA, Fulcher DA, Picard C, Bustamante J, Boisson-Dupuis S, Gray P, Stepensky P, Warnatz K, Freeman AF, Rossjohn J, McCluskey J, Holland SM, Casanova JL, Uzel G, Ma CS, Tangye SG, Deenick EK. 2015. STAT3 is a critical cell-intrinsic regulator of human unconventional T cell numbers and function. J Exp Med 212:855–864. doi: 10.1084/jem.20141992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. 2009. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 67.Moser EK, Sun J, Kim TS, Braciale TJ. 2015. IL-21R signaling suppresses IL-17+ gamma delta T cell responses and production of IL-17 related cytokines in the lung at steady state and after influenza A virus infection. PLoS One 10:e0120169. doi: 10.1371/journal.pone.0120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ranson T, Vosshenrich CA, Corcuff E, Richard O, Laloux V, Lehuen A, Di Santo JP. 2003. IL-15 availability conditions homeostasis of peripheral natural killer T cells. Proc Natl Acad Sci U S A 100:2663–2668. doi: 10.1073/pnas.0535482100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Creus A, Van Beneden K, Stevenaert F, Debacker V, Plum J, Leclercq G. 2002. Developmental and functional defects of thymic and epidermal V gamma 3 cells in IL-15-deficient and IFN regulatory factor-1-deficient mice. J Immunol 168:6486–6493. doi: 10.4049/jimmunol.168.12.6486. [DOI] [PubMed] [Google Scholar]
- 70.Van Acker A, Gronke K, Biswas A, Martens L, Saeys Y, Filtjens J, Taveirne S, Van Ammel E, Kerre T, Matthys P, Taghon T, Vandekerckhove B, Plum J, Dunay IR, Diefenbach A, Leclercq G. 2017. A murine intestinal intraepithelial NKp46-negative innate lymphoid cell population characterized by group 1 properties. Cell Rep 19:1431–1443. doi: 10.1016/j.celrep.2017.04.068. [DOI] [PubMed] [Google Scholar]
- 71.Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L. 2004. The contribution of Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- 72.Lilly LM, Scopel M, Nelson MP, Burg AR, Dunaway CW, Steele C. 2014. Eosinophil deficiency compromises lung defense against Aspergillus fumigatus. Infect Immun 82:1315–1325. doi: 10.1128/IAI.01172-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vallieres F, Girard D. 2013. IL-21 enhances phagocytosis in mononuclear phagocyte cells: identification of spleen tyrosine kinase as a novel molecular target of IL-21. J Immunol 190:2904–2912. doi: 10.4049/jimmunol.1201941. [DOI] [PubMed] [Google Scholar]
- 74.Nemzek JA, Ebong SJ, Kim J, Bolgos GL, Remick DG. 2002. Keratinocyte growth factor pretreatment is associated with decreased macrophage inflammatory protein-2alpha concentrations and reduced neutrophil recruitment in acid aspiration lung injury. Shock 18:501–506. doi: 10.1097/00024382-200212000-00003. [DOI] [PubMed] [Google Scholar]
- 75.Mattila PE, Metz AE, Rapaka RR, Bauer LD, Steele C. 2008. Dectin-1 Fc targeting of Aspergillus fumigatus beta-glucans augments innate defense against invasive pulmonary aspergillosis. Antimicrob Agents Chemother 52:1171–1172. doi: 10.1128/AAC.01274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bowman JC, Abruzzo GK, Anderson JW, Flattery AM, Gill CJ, Pikounis VB, Schmatz DM, Liberator PA, Douglas CM. 2001. Quantitative PCR assay to measure Aspergillus fumigatus burden in a murine model of disseminated aspergillosis: demonstration of efficacy of caspofungin acetate. Antimicrob Agents Chemother 45:3474–3481. doi: 10.1128/AAC.45.12.3474-3481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hummel M, Spiess B, Roder J, von Komorowski G, Durken M, Kentouche K, Laws HJ, Morz H, Hehlmann R, Buchheidt D. 2009. Detection of Aspergillus DNA by a nested PCR assay is able to improve the diagnosis of invasive aspergillosis in paediatric patients. J Med Microbiol 58:1291–1297. doi: 10.1099/jmm.0.007393-0. [DOI] [PubMed] [Google Scholar]