

Mycobacterium tuberculosis causes persistent infection due to its ability to evade host immune responses. M. tuberculosis induces Toll-like receptor 2 (TLR2) signaling, which influences immune responses to M. tuberculosis.
KEYWORDS: Mycobacterium tuberculosis, Toll-like receptor (TLR), lipoprotein, lipoglycan, agonist, immune response
ABSTRACT
Mycobacterium tuberculosis causes persistent infection due to its ability to evade host immune responses. M. tuberculosis induces Toll-like receptor 2 (TLR2) signaling, which influences immune responses to M. tuberculosis. TLR2 agonists expressed by M. tuberculosis include lipoproteins (e.g., LprG), the glycolipid phosphatidylinositol mannoside 6 (PIM6), and the lipoglycan lipomannan (LM). Another M. tuberculosis lipoglycan, mannose-capped lipoarabinomannan (ManLAM), lacks TLR2 agonist activity. In contrast, PILAM, from Mycobacterum smegmatis, does have TLR2 agonist activity. Our understanding of how M. tuberculosis lipoproteins and lipoglycans interact with TLR2 is limited, and binding of these molecules to TLR2 has not been measured directly. Here, we directly measured M. tuberculosis lipoprotein and lipoglycan binding to TLR2 and its partner receptor, TLR1. LprG, LAM, and LM were all found to bind to TLR2 in the absence of TLR1, but not to TLR1 in the absence of TLR2. Trimolecular interactions were revealed by binding of TLR2-LprG or TLR2-PIM6 complexes to TLR1, whereas binding of TLR2 to TLR1 was not detected in the absence of the lipoprotein or glycolipid. ManLAM exhibited low affinity for TLR2 in comparison to PILAM, LM, and LprG, which correlated with reduced ability of ManLAM to induce TLR2-mediated extracellular-signal-regulated kinase (ERK) activation and tumor necrosis factor alpha (TNF-α) secretion in macrophages. We provide the first direct affinity measurement and kinetic analysis of M. tuberculosis lipoprotein and lipoglycan binding to TLR2. Our results demonstrate that binding affinity correlates with the functional ability of agonists to induce TLR2 signaling.
INTRODUCTION
Infection with Mycobacterium tuberculosis can produce both active tuberculosis (TB) and asymptomatic long-term latent M. tuberculosis infection. An estimated 2 billion people are infected with M. tuberculosis, and TB continues to cause millions of deaths each year. The current TB vaccine, Mycobacterium bovis bacillus Calmette-Guerin (BCG), provides only partial protection in childhood and remains ineffective in preventing adult pulmonary TB.
Upon infection, M. tuberculosis is sensed by a number of pattern recognition receptors, including Toll-like receptor 2 (TLR2), which is expressed on the surfaces of macrophages, dendritic cells, and T lymphocytes. TLR2 plays a prominent role in the induction of host immune responses during mycobacterial infection (1–3). The roles of TLR2 in TB are complex; innate immune responses induced by TLR2 can contribute to both host defense and immune evasion (4), and TLR2 also regulates adaptive immune responses (e.g., T lymphocyte responses) (5). TLR2 influences the inflammatory balance of macrophages, which may determine the balance of cytokine secretion (e.g., interleukin 10 [IL-10] versus IL-12) and the extent of Th1 effector T cell function (6).
TLR2 agonists expressed by M. tuberculosis include lipoproteins, lipoglycans, and glycolipids. M. tuberculosis produces close to 100 unique lipoproteins (7), which are predominantly triacylated (8). A number of M. tuberculosis lipoproteins are known to display TLR2 agonist activity, including LprG (9), LprA (10), and LpqH (11). Using a synthetic triacylated lipopeptide (Pam3CSK4), Jin et al. demonstrated ligand-induced formation of TLR2-TLR1 heterodimers and established an X-ray crystallographic structure of Pam3CSK4 bound to TLR2-TLR1 (12), demonstrating that a lipopeptide diacyl structure interacted with TLR2 while the remaining lipopeptide acyl structure interacted with TLR1. Several other studies have demonstrated binding of synthetic lipopeptide to TLR2 (13–15). The TLR2 binding properties of natural lipoproteins expressed by a human pathogen, such as M. tuberculosis, remain to be tested, although TLR2 binding has been reported for the meningococcal porin PorB (16). TLR2 agonist activity has been reported for a larger set of molecules (3, 9, 10, 17–29), but TLR2 binding has not been directly measured for these agonists.
Besides triacylated lipoproteins, a number of structurally diverse M. tuberculosis cell wall lipoglycan and glycolipid molecules induce TLR2 signaling and responses; they include lipoarabinomannan (LAM) and its biosynthetic precursors lipomannan (LM) and phosphatidylinositol mannosides (1, 27–32). LAM molecules from different mycobacterial species differ in the terminal capping of their arabinan domains (33) and have other structural differences. Mannose-capped LAM (ManLAM), which is found in slow-growing mycobacterial pathogens, such as M. tuberculosis and BCG, has an arabinan domain capped by mannosyl residues. Fast-growing Mycobacterium smegmatis expresses PILAM, which has arabinan domains capped with phosphoinositides. While PILAM induces TLR2-mediated responses (1, 31, 34, 35), ManLAM has little or no TLR2 agonist activity in most reported studies (1, 36–39). The ability of ManLAM to physically bind TLR2 has not been assessed directly or in a manner to reveal its potential binding affinity.
M. tuberculosis TLR2 ligands all share acyl structures but otherwise have diverse structures. It is not clear how these structural differences affect the ligand binding and agonist activities of the molecules. This information is necessary to understand the pathophysiological roles of the molecules in TB and to assess potential therapeutic applications of the molecules or versions with structural modifications to adjust their immunomodulatory activities. The data presented here address the relative TLR2 binding and agonist activities of the M. tuberculosis lipoprotein LprG and M. tuberculosis lipoglycans/glycolipids.
To address these questions, we used surface plasmon resonance assays to measure the kinetics and affinities of binding of purified M. tuberculosis molecules to TLR2, and we evaluated the TLR2 agonist activities of these molecules by assessing the induction of extracellular-signal-regulated kinase (ERK) signaling and expression of tumor necrosis factor alpha (TNF-α). The M. tuberculosis molecules that we studied included LprG (a well-studied M. tuberculosis triacylated lipoprotein) and a set of lipoglycans/glycolipids, including phosphatidylinositol mannoside 6 (PIM6), LM, and ManLAM from virulent M. tuberculosis strain H37Rv and PILAM from avirulent M. smegmatis. These studies revealed that lipoprotein and lipoglycan/glycolipid ligands bind to TLR2 and that this binding can lead to the formation of a trimolecular complex of the ligand with TLR2-TLR1. Of the lipoglycans, ManLAM exhibited both lower TLR2 binding and lower TLR2 agonist activity than PILAM. These are the first studies to directly assess binding of any lipoglycan to TLR2 and the first direct studies of binding of an M. tuberculosis lipoprotein to TLR2. The results demonstrate that the diminished TLR2 agonist activity of ManLAM correlates with a low affinity for binding of ManLAM to TLR2.
RESULTS
Mycobacterial lipoproteins and lipoglycans bind directly to isolated TLR2 but not to isolated TLR1.
Triacylated lipoproteins are proposed to promote the formation of a functional TLR2-TLR1 heterodimer and thereby trigger downstream signaling (12, 13), but there have been no reported studies to directly assess binding of M. tuberculosis lipoproteins to TLR2 or TLR1. In addition, TLR2 agonists expressed by M. tuberculosis include lipoglycans, as well as lipoproteins (3, 9, 10, 27–29), yet there have been no studies to directly assess TLR2 binding by any species of lipoglycan. Here, we used surface plasmon resonance (SPR) technology to investigate the binding of a purified M. tuberculosis lipoprotein (LprG) and several mycobacterial lipoglycans (LM, PILAM, and ManLAM) to TLR1 and TLR2. Increasing concentrations of LprG, LM, ManLAM, or PILAM were injected as analytes to measure binding to TLR1 or TLR2 immobilized on a sensor chip. All of the molecules showed no detectable binding to isolated TLR1 (Fig. 1A, D, F, and H), whereas dose-dependent binding to TLR2 was detected for all of the molecules (Fig. 1B, E, G, and I). The presence of acylation on the analyte was critical for TLR2 binding, as nonacylated LprG (NA-LprG) failed to bind to TLR2 (Fig. 1C). We did not assess binding of the glycolipid PIM6 to TLR2 or TLR1 in this assay, as its molecular weight is much lower than those of the lipoglycans, making detection of its binding by SPR problematic, but the lipoglycans share its core glycolipid structure that predicts TLR2 binding, and experiments discussed below (Fig. 2) demonstrated its ability to form trimolecular complexes with TLR2 and TLR1.
FIG 1.

LprG and mycobacterial lipoglycans bind to TLR2 in the absence of TLR1, but not to TLR1 in the absence of TLR2. Binding was evaluated by SPR and measured as resonance units. TLR1 (A, D, F, and H) or TLR2 (B, C, E, G, and I) was immobilized on a CM5 sensor chip. Sensorgrams were obtained by injecting LprG (A and B) or NA-LprG (C) at 625 nM, 2,500 nM, or 5,000 nM or by injecting LM (D and E), ManLAM (F and G), or PILAM (H and I) at 100 nM, 200 nM, 312 nM, or 625 nM. In each graph, the top curve corresponds to the top analyte concentration, and each successive curve below is the next lower analyte concentration. (A, D, F, and H) Only the curves for the two highest concentrations are shown (the other concentrations also revealed no detectable binding). Binding curves were calculated with BIA evaluation 3.1 software with subtraction of nonspecific binding of the analyte to matched sensor chip control cells without immobilized TLR2 or TLR1. The results are representative of three independent experiments.
FIG 2.

Binding of LprG to TLR2 is required to form TLR2-TLR1 complexes. SPR was used to test the abilities of TLR1 and TLR2 to form homodimers or heterodimers in the presence or absence of LprG or PIM6. TLR1 (A, C, D, F, G, and I) or TLR2 (B, E, and H) was immobilized on a CM5 sensor chip. (A to C) TLR1 (A) or TLR2 (B and C) was injected (100 nM) in the absence of a TLR2 agonist. (D to I) Alternatively, LprG (5 μM) or PIM6 (5 μM) was incubated with TLR1 (500 nM) (D and G) or TLR2 (500 nM) (E, F, H, and I) in a total volume of 150 μl HBSN buffer for 2 h at 37°C prior to sample injection. The results are representative of three independent experiments (A to C) or two independent experiments (D to I).
Evaluation of binding kinetics revealed that LprG, LM, ManLAM, and PILAM bind to TLR2, albeit with distinct binding affinities (Table 1). LM, PILAM, and LprG showed high-affinity binding to TLR2 with KD (equilibrium dissociation constant) values in the nanomolar range (Table 1), suggesting physiologically relevant binding. ManLAM bound to TLR2 with much lower affinity, with a KD value of 2.83 × 10−6 M (Table 1). A number of previous studies have reported that ManLAM lacks TLR2 agonist activity (1, 39), but the studies were based on functional outcomes, such as cytokine induction, and direct-binding assays have not been reported. Our binding data suggest that ManLAM is not a robust TLR2 agonist due to its low TLR2 binding affinity, as opposed to an ability to bind without agonist activity.
TABLE 1.
Kinetics of binding of M. tuberculosis lipoproteins and lipoglycans to TLR2a
| Protein | Ka (M−1 s−1) | Kd (1/s) | KD (M) |
|---|---|---|---|
| LprG | 1.04 × 104 | 5.23 × 10−3 | 5.03 × 10−7 |
| LM | 6.91 × 104 | 6.64 × 10−3 | 9.62 × 10−8 |
| ManLAM | 2.77 × 104 | 7.83 × 10−2 | 2.83 × 10−6 |
| PILAM | 1.43 × 104 | 1.84 × 10−3 | 1.29 × 10−7 |
Ka, association rate constant; Kd, dissociation rate constant.
The presence of ligand is required to facilitate TLR2-TLR1 dimerization.
Jin et al. found that lipopeptide induced the formation of TLR2-TLR1 heterodimers (12), although some studies have proposed the possibility of TLR2-TLR1 heterodimerization without direct physical binding of a ligand (29). Most studies support recognition of agonists by heterodimers of TLR2 and a coreceptor, either TLR1 or TLR6, although some studies have suggested that TLR2 can recognize agonists independently of TLR1 or TLR6 (40), possibly suggesting the formation of TLR2 homodimers, but such homodimers have not been convincingly demonstrated. We used SPR binding assays to address these questions. We injected TLR1 or TLR2 as an analyte to assess binding to surface-immobilized TLR1 or TLR2. These studies revealed no detectable binding of TLR1 to immobilized TLR1 (Fig. 2A), TLR2 to immobilized TLR2 (Fig. 2B), TLR2 to immobilized TLR1 (Fig. 2C), or TLR1 to immobilized TLR2 under these conditions (data not shown). Thus, TLR1 and TLR2 do not form homodimers or heterodimers in the absence of an additional binding partner.
To investigate the interactions of TLR1 and TLR2 in the presence of a known TLR2 agonist, we preincubated TLR1 or TLR2 with LprG (Fig. 2D to F) or with PIM6 (Fig. 2G to I) for 2 h at 37°C and then injected the resulting material as an analyte to study binding to surface-immobilized TLR1 or TLR2. There was no detectable binding of TLR1 to TLR1 in the presence of LprG (Fig. 2D) or PIM6 (Fig. 2G) and, similarly, no detectable binding of TLR2 to TLR2 in the presence of LprG (Fig. 2E) or PIM6 (Fig. 2H). However, we observed binding of TLR2-LprG to TLR1 (Fig. 2F) and TLR2-PIM6 to TLR1 (Fig. 2I), supporting the conclusion that the presence of a lipoprotein or glycolipid agonist (LprG or PIM6) enabled the formation of a trimolecular complex (TLR2-LprG-TLR1 or TLR2-PIM6-TLR1). We attempted similar experiments to detect binding of TLR2-ManLAM or TLR2-PILAM to TLR1, but we observed high nonspecific signals with these lipoglycans (data not shown), possibly due to formation of nonspecific aggregates under the incubation conditions used to generate the mobile analyte sample, precluding this approach. These results suggest that homodimerization of TLR2 or TLR1 is undetectable even in the presence of an agonist, whereas TLR2-TLR1 heterodimers are induced in the presence of an agonist. Moreover, these conclusions confirm that the model developed by Jin et al. (12) with synthetic Pam3CSK4 lipopeptide also applies to a full-size native lipoprotein and to a glycolipid naturally expressed by M. tuberculosis; these are molecules of potential pathophysiological significance. Finally, binding of TLR2-LprG or TLR2-PIM6 to TLR1 confirmed that surface-immobilized TLR1 was active for specific analyte binding; thus, its lack of binding to the analyte TLR1 (Fig. 1A, D, F, and H and 2A, C, and D) was not due to overall loss of binding activity due to denaturation.
ManLAM is not an effective TLR2 agonist.
We performed studies to test the relationship between TLR2 binding activity and the ability of agonists to induce TLR2 signaling and a cytokine response. M. tuberculosis is known to activate the NF-κB and ERK pathways in a TLR2-dependent manner (6), and purified LprG has been shown to induce ERK activation (41). Murine bone marrow-derived macrophages (BMDMs) were stimulated with LprG or ManLAM for 15 min, washed with cold phosphate-buffered saline (PBS), and lysed in SDS-containing buffer, as previously described (6). Induction of ERK signaling was assessed by phosphorylation of ERK, and NF-κB signaling was assessed by degradation of IκBα (Fig. 3). ManLAM failed to activate NF-κB (as shown by retained expression of intact IκBα) and induced very little ERK phosphorylation. In contrast, LprG induced IκBα degradation and high levels of ERK phosphorylation, as reported previously (6); these effects of LprG were previously shown to be TLR2 dependent (6). Moreover, the low level of ERK phosphorylation induced by ManLAM was TLR2 independent, as it was similarly observed in Tlr2−/− BMDMs. We also compared the abilities of LprG, ManLAM, LM, PIM6, and PILAM at equimolar concentrations to induce TNF-α production by BMDMs and human THP1 cells (Fig. 4). Low concentrations (<15 nM) of LprG, LM, PIM6, and PILAM resulted in robust induction of TNF-α in BMDMs and THP1 cells. In contrast, TNF-α induction by ManLAM occurred only at concentrations above the effective critical micellar concentrations (42), where the acyl chains of micellar LAM are predicted to be less available to bind TLR2 and/or TLR1, although exposed mannan residues could bind other receptors, e.g., dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) or mannose receptor, which have been implicated in ManLAM recognition (43–46). These findings indicate that ManLAM is not an effective TLR2 agonist and has much less ability than established TLR2 agonists to trigger functional cytokine responses by macrophages, consistent with previous publications (1, 30, 36–39, 47). The lack of effective TLR2 agonist activity of ManLAM (Fig. 3 and 4) correlates with its low affinity for binding to TLR2 (Fig. 1 and Table 1), although the difference in agonist activities seen with ManLAM versus LprG may be more profound than expected given the 5.6-fold difference in the KD.
FIG 3.

ManLAM induces only minimal levels of ERK phosphorylation and IκBα degradation. BMDMs from wild-type or Tlr2−/− mice were incubated for 15 min with 30 nM LprG or ManLAM at the indicated concentrations. The cells were lysed and analyzed by Western blotting to detect IκBα degradation and ERK phosphorylation.
FIG 4.
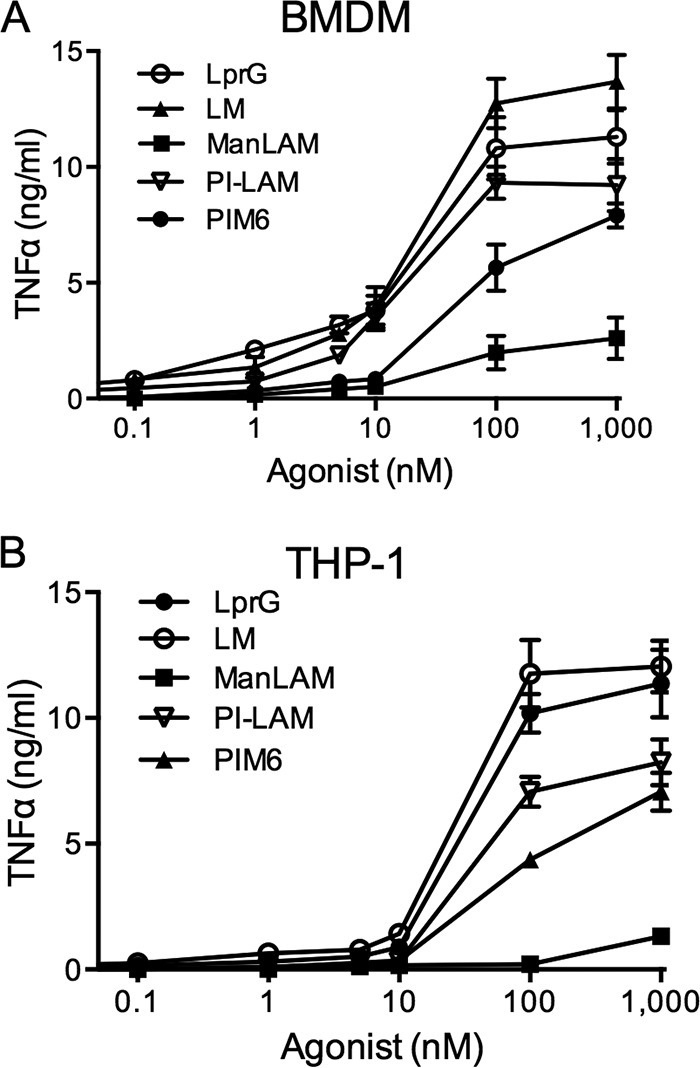
ManLAM induces less TNF-α than lipoproteins or lipoglycans that bind to TLR2 with higher affinity. BMDMs from wild-type mice (A) or THP1 cells (B) were incubated for 24 h with LM, ManLAM, PILAM, PIM6, or LprG at the indicated concentrations (shown in log scale). The supernatants were harvested, and TNF-α production was determined by ELISA. The results are representative of three independent experiments. The data shown represent the means ± standard errors of the mean (SEM) of the results of triplicate assays.
Stimulation of TLR2 signaling by lipoglycans is dependent on TLR1.
Although TLR1 has been shown to bind to TLR2-Pam3CSK4 (12) and has been functionally implicated in the recognition of lipoprotein TLR2 agonists, including the M. tuberculosis lipoprotein LpqH (19-kDa lipoprotein) (19), its role has not been studied in responses to lipoglycan TLR2 agonists. To test the functional role of TLR1 in response to TLR2 agonists, we treated THP1 cells with anti-TLR1, anti-TLR2, both anti-TLR1 and anti-TLR2, or isotype-matched control antibodies for 30 min or left them untreated; incubated the cells with lipoglycan agonists (or LprG for comparison); and assessed induction of TNF-α by enzyme-linked immunosorbent assay (ELISA) (Fig. 5). Incubation with LprG, LM, PIM6, or PILAM resulted in robust induction of TNF-α (Fig. 5A, B, D, and E); as expected, ManLAM induced little or no TNF-α (Fig. 5C). Responses to LM, PIM6, PILAM, and LprG were significantly blocked by treatment with antibody to TLR1 alone, TLR2 alone, or the combination of anti-TLR1 and anti-TLR2 (all relative to appropriate isotype-matched control antibodies). These results establish essential roles of both TLR1 and TLR2 in responses to LM and PILAM, as well as LprG.
FIG 5.

TLR2 and TLR1 are both required for agonist activity of LprG, LM, PILAM, and PIM6. THP1 cells were preincubated for 30 min with or without antibodies (10 μg/ml) as indicated and then incubated for 6 h in the continuous presence or absence of antibodies with LprG (A), LM (B), ManLAM (C), PILAM (D), or PIM6 (E) (all at 125 nM). TNF-α production was determined by ELISA. The results are representative of three independent experiments. The data shown represent the means ± SEM of triplicate assays. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, P > 0.05.
DISCUSSION
M. tuberculosis expresses numerous lipoproteins, lipoglycans, and glycolipids in its complex, hydrophobic cell envelope. These molecules can activate TLR2 signaling in macrophages during infection, leading to downstream protective immune mechanisms, such as TNF-α elaboration, but also causing counterproductive mechanisms leading to bacterial immune evasion, such as expression of IL-10 and downregulation of major histocompatibility complex (MHC) class II (4, 6). Due to their diverse structures, the TLR2 binding properties of the various M. tuberculosis TLR2 agonists remain poorly understood (reviewed in reference 32). Prior to the experiments reported here, direct TLR2 binding studies and quantitative binding data were largely limited to studies of a model lipopeptide, Pam3CSK4 (12), although functional evidence of TLR2 agonist activity exists for a much broader set of molecules (3, 9, 10, 17–29). Information on the binding of agonists by TLR1 is even more limited, with direct binding interactions known for Pam3CSK4 (12) and functional evidence for involvement of TLR1 in responses to a slightly broader set of molecules (13, 19).
Our data demonstrate that LprG, a prototypic M. tuberculosis lipoprotein, binds to TLR2 with high affinity (Fig. 1 and Table 1), and this binding is dependent on the acyl structures of LprG. To our knowledge, this is the first study to directly assess TLR2 binding by an M. tuberculosis lipoprotein with quantitative data on binding kinetics and affinity.
Moreover, our experiments with LM and PILAM extend quantitative understanding of the binding interactions of TLR2 with M. tuberculosis lipoglycans for the first time and provide the first direct assessment of TLR2 binding by any non-lipoprotein/lipopetide agonist. This enabled us to explore why LM and PILAM are TLR2 agonists while the closely related ManLAM is not and to determine whether this difference is based on a lack of ManLAM binding to TLR2 versus the capacity to bind without activating TLR signaling. We observed that LM and PILAM bind TLR2 with high affinity, while ManLAM binds at much lower affinity. ManLAM was not an effective functional activator of TLR2 signaling in macrophages during M. tuberculosis infection, consistent with prior studies (1). While the different functional effects of ManLAM and PILAM have been attributed in part to engagement of additional receptors, e.g., the mannose receptor, which may alter signaling outcomes, our direct-binding data indicate that the low agonist activity of ManLAM correlates with a low affinity for ManLAM binding to TLR2.
Interestingly, LM and PILAM bound to TLR2 with higher affinity than to ManLAM (Table 1), even though these molecules both have PIM core structures, which may provide acyl chain interactions that are central to TLR2-TLR1 binding and induction of TLR2-TLR1 heterodimerization and signaling if the structural and functional model established by Jin et al. based on lipopeptide binding (12) is extrapolated. LM, ManLAM, and PILAM may differ in acylation state, saccharide structures (including terminal mannosylation in ManLAM but not PILAM), and other modifications (acetylation, succinylation, sulfation, and phosphorylation); the specific effects of such structural variations remain to be determined.
Our studies also establish the ability of a pathogen-expressed full-size lipoprotein (LprG) to induce TLR2-TLR1 heterodimerization (or formation of TLR2-LprG-TLR1 complexes), which is known to be required for TLR2 signaling function. While we were unable to detect binding of LprG to TLR1 in the absence of TLR2 or binding of TLR2 to TLR1 in the absence of LprG, we observed binding of TLR2-LprG to TLR1. We also extended these observations beyond lipoprotein agonists by showing that PIM6 binding to TLR2 facilitates TLR2-TLR1 heterodimerization. Although some other TLRs (e.g., TLR4 and TLR9) form homodimers, our data indicate that formation of TLR1-TLR1 or TLR2-TLR2 homodimers does not occur in either the absence or presence of an agonist. It is important to note that binding affinities in the context of TLR2-TLR1 expressed in the membranes of cells may differ from binding as studied by these SPR assays (and other published TLR2-TLR1 binding assays or crystallography of TLR2-TLR1-lipopeptide interactions [12]). This caveat is frequently encountered in studies of membrane receptors and their ligands. Nonetheless, the biological assays of lipoprotein and lipoglycan agonist activities (Fig. 3 to 5) are consistent with the conclusions from the SPR assays. These results suggest that formation of the biologically active heterodimeric signaling receptor may start with an initial binding event between TLR2 and an acylated agonist, followed by binding of that complex to TLR1, but this sequence is not proven and may be affected by the association of both receptors with cell membranes under physiological conditions.
LM and LAM exhibit several types of structural heterogeneity, including varying degrees of acylation, branching, succinylation, sulfation, phosphorylation, and capping of saccharides; these modifications may influence the functional properties of the lipoglycans, including their affinities for TLR2 binding and their TLR2 agonist activities (e.g., their abilities to induce TLR2-dependent ERK activation and expression of TNF-α). Prior studies demonstrated that acylation states of LM determine the ability to activate macrophages through TLR2 signaling and induction of proinflammatory cytokines (34, 48). The mannan chain length of LM and LAM is another factor shown to influence TLR2 agonist activities of M. tuberculosis lipoglycans (49). Torrelles et al. demonstrated that a certain structural subspecies of ManLAM enhanced the production of interferon gamma by CD1-restricted T cells (50). There may be other structural variations that could affect the biological functions of these lipoglycans. The precise characterization of the functional effects of variations in the complex structures of M. tuberculosis lipoglycans and glycolipids remains an important area for future investigation.
We conclude that the binding affinity for TLR2 is directly correlated with the ability of M. tuberculosis lipoproteins and lipoglycans to induce TLR2 signaling and to activate downstream immune functions in macrophages; ManLAM has low affinity for binding to TLR2 and lacks agonist function. These observations extend our understanding of TLR2 interactions with its agonists to molecules naturally expressed by an important human pathogen, including both lipoproteins and lipoglycans/glycolipids, a class of agonists for which TLR2 binding has not previously been assessed.
MATERIALS AND METHODS
Mammalian cell culture.
All animal studies were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University (protocol 2012-0007). Animals were used only to obtain murine BMDMs. C57BL/6J mice (female; 8 to 12 weeks old) were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed under specific-pathogen-free conditions. BMDMs were cultured from suspensions of bone marrow cells from mouse femurs and tibias (6). Cell suspensions were homogenized and filtered through a 70-μm screen. Red blood cells were lysed using ACK lysing buffer (Lonza, Walkersville, MD). The cells were pelleted and cultured in D10F medium composed of Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT) with 10% heat-inactivated fetal bovine serum (Gibco, Carlsbad, CA), 50 μM 2-mercaptoethanol (Bio-Rad, Hercules, CA), 1 mM sodium pyruvate (HyClone), 10 mM HEPES (HyClone), 100 units/ml penicillin, 100 μg/ml streptomycin (HyClone), and 25% LADMAC cell-conditioned medium (51). Bone marrow cultures were incubated at 37°C in a humidified, 5% CO2 atmosphere; the medium was changed on day 5 of culture, and differentiated BMDMs were used on day 7. Human monocytic THP-1 cells (ATCC; TIB-202) were obtained from the ATCC (Manassas, VA) and cultured in complete RPMI medium consisting of RPMI 1640 with 50 mM 2-mercaptoethanol and 10% heat-inactivated fetal bovine serum. To differentiate THP-1 cells into macrophages, the cells were incubated for 2 days in medium containing 20 ng/ml phorbol myristate acetate (PMA) (Sigma; 1-mg/ml stock in dimethyl sulfoxide [DMSO]) with a 2% final concentration of DMSO. The differentiated cells were maintained in complete RPMI.
Reagents.
Fc-tagged TLR–variable lymphocyte receptor hybrid constructs expressing human TLR1 and human TLR2 extracellular domains were a gift from Jie-Oh Lee (Korea Advanced Institute of Science and Technology, Daejeon, South Korea); TLR1 and TLR2 were expressed in Hi5 insect cells (Invitrogen, Carlsbad, CA) and purified as described previously (12). Recombinant M. tuberculosis lipoproteins LprG and NA-LprG were expressed and purified as described previously (9, 10). The M. tuberculosis glycolipid PIM6 and M. tuberculosis lipoglycans LM, ManLAM, and PILAM (ManLAM NR-14848, lots 61983887 and 62976488; LM NR-14850, lot 61977661; PILAM NR-14849, lot 61699475; PIM6 NR-14847, lots 61699473 and 61977659) were obtained from a program supported by the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH (through BEI, Manassas, VA). We performed silver stains on gels to detect potential contaminating protein species and to analyze the purity of the lipoglycan preparations; we detected no contaminants. Furthermore, the LAM, LM, and PIM preparations from BEI had extensive quality control data from laboratories at Colorado State University that were provided by BEI for the lot numbers we used; the data, including gel images, are available for these lot numbers on the BEI website [http://www.beiresources.org]). Analyses included silver staining, Western blotting, and nuclear magnetic resonance (NMR) analyses for LM, ManLAM, and PILAM; PIM6 was analyzed by mass spectrometry. The quality control data from BEI and our own experiments established the purity of these preparations with no detectable protein contamination.
SPR binding assays.
SPR binding experiments were performed as described previously (52) with a BIAcore 3000 instrument, CM5 (carboxymethylated) sensor chips, and HBSN running buffer (10 mM HEPES and 150 mM NaCl, pH 7.4). N-hydroxysuccinimide (NHS), ethylene diamine (EDC), and ethanolamine were obtained from GE Healthcare Biosciences (Pittsburgh, PA). TLR1 or TLR2 was immobilized on CM5 sensor chips by amine coupling (53). The chip cells were equilibrated with HBSN. Seventy milliliters of a 1:1 mixture of 0.1 M NHS and 0.1 M EDC was injected to activate carboxyl groups on the CM5 matrix, and the TLR (30 μg/ml in 10 mM acetate buffer, pH 4.5) was injected. Residual NHS esters were deactivated with 70 ml of 1 M ethanolamine. Control flow cells on the chips were activated and deactivated in the same manner but without TLR addition to yield blank surfaces. Analytes, including LprG, NA-LprG, LM, ManLAM, PILAM, and PIM6, were diluted in HBSN and injected over chip-immobilized TLR1 or TLR2 at a flow rate of 30 μl/min. At the end of the analyte injection, the complexes were allowed to dissociate for 10 min in HBSN running buffer. The chip surfaces were regenerated by 2 or 3 injections of 20 ml 50% ethylene glycol, followed by an equilibration delay of 10 min with HBSN at a flow rate of 5 ml/min before it was used for another analyte injection. Controls were obtained by injection of the analyte solution into a cell with a blank chip surface. The final amount of bound analyte, expressed in resonance units (RU), was calculated by subtracting the RU of the blank cell from the RU of the TLR-conjugated cell. Sensorgrams were analyzed using BIA evaluation 3.1 software (GE Healthcare).
Western blots.
Macrophages (106 per condition) were treated with ManLAM (31.25 nM, 62.5 nM, or 125 nM) or 30 nM LprG (6) for 15 min, washed with cold PBS, and lysed using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and protein phosphatase inhibitor cocktail (Pierce, Rockford, IL). Protein lysates were boiled in reducing sample buffer (Bio-Rad), loaded on SDS-PAGE gels (Bio-Rad), and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were blocked in 5% dry milk in PBS with 0.1% Tween 20, incubated with primary antibodies overnight at 4°C, and then washed three times in PBS with 0.1% Tween 20. The membranes were then treated with secondary antibodies for 1 h at room temperature, washed in PBS with 0.1% Tween 20 three times, and treated with chemiluminescence reagent (Pierce). The membranes were exposed to autoradiography film (Amersham GE Healthcare, Pittsburg, PA) and developed on an automated film processor (Konica Minolta, Wayne, NJ). The antibodies included anti-phospho-ERK1/2 (clone D13.14.4E), anti-ERK1/2 (clone 137F5), anti-β-actin (clone D6A8), horseradish peroxidase-linked goat anti-rabbit IgG (catalog number 7074), and horseradish peroxidase-linked horse anti-mouse IgG (catalog number 7076) (all from Cell Signaling Technology, Danvers, MA) and anti-IκBα (clone T.937.7) from Thermo Scientific/Pierce Biotechnology (Rockford, IL).
ELISA.
Macrophages (105 per condition) were treated with LprG, LM, ManLAM, PILAM, or PIM6 (0.1 nM to 1,000 nM) for 24 h. The plates were centrifuged at 1,500 × g, and the supernatants were collected and stored at −80°C. TNF-α concentrations were quantified by ELISA with kits from R&D Systems (Minneapolis, MN; DY208) and a model 680 microplate reader (Bio-Rad).
Receptor-blocking assay.
Antibodies were from eBioscience (San Diego, CA). PMA-activated THP1 cells (105 per condition) were incubated for 30 min with or without anti-TLR2 (clone TL2.1) and/or anti-TLR1 (clone GD2.F4) or similar combinations of isotype-matched control antibodies at a 10-μg/ml final concentration at 37°C in 5% CO2. The cells were incubated for 6 h in the continuous presence of antibodies with TLR2 agonist (LprG, ManLAM, LM, PILAM, or PIM6) at 125 nM. The supernatants were collected and stored at −80°C. TNF-α production was quantified by ELISA.
Statistical analysis.
Statistical analyses were performed using GraphPad (La Jolla, CA) Prism 5.01 software. Fisher's exact test or Student's two-tailed t test was used to analyze the statistical significance of differences between groups. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Jie-Oh Lee for kindly providing constructs for expression of TLR2 and TLR1 and Satya Yadav and the Cleveland Clinic Molecular Biotechnology Core for technical assistance with SPR assays.
This work was supported in whole or part by National Institutes of Health grants R01AI069085, R01AI034343, R01AI027243, T32GM007250, and T32AI089474.
We declare that we have no conflicts of interest related to the contents of this article.
S.S., W.H.B., and C.V.H. designed the study. S.S., E.T.R., and M.G.D. performed the experiments. S.S., E.T.R., M.G.D., C.V.H., and W.H.B. analyzed the results. M.G.D. developed the constructs for protein purification, and S.S. and M.G.D. purified TLR2, TLR1, and LprG proteins for use in experiments. S.S., E.T.R., W.H.B., and C.V.H. wrote the paper. We all reviewed the results and approved the final version of the manuscript.
REFERENCES
- 1.Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. 1999. Human Toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol 163:3920–3927. [PubMed] [Google Scholar]
- 2.Underhill DM, Ozinsky A, Smith KD, Aderem A. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A 96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, Brennan PJ, Bloom BR, Godowski PJ, Modlin RL. 1999. Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science 285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 4.Harding CV, Boom WH. 2010. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol 8:296–307. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reba SM, Li Q, Onwuzulike S, Ding X, Karim AF, Hernandez Y, Fulton SA, Harding CV, Lancioni CL, Nagy N, Rodriguez ME, Wearsch PA, Rojas RE. 2014. TLR2 engagement on CD4 T cells enhances effector functions and protective responses to Mycobacterium tuberculosis. Eur J Immunol 44:1410–1421. doi: 10.1002/eji.201344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richardson ET, Shukla S, Sweet DR, Wearsch PA, Tsichlis PN, Boom WH, Harding CV. 2015. Toll-like receptor 2-dependent extracellular signal-regulated kinase signaling in Mycobacterium tuberculosis-infected macrophages drives anti-inflammatory responses and inhibits Th1 polarization of responding T cells. Infect Immun 83:2242–2254. doi: 10.1128/IAI.00135-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutcliffe IC, Harrington DJ. 2004. Lipoproteins of Mycobacterium tuberculosis: an abundant and functionally diverse class of cell envelope components. FEMS Microbiol Rev 28:645–659. doi: 10.1016/j.femsre.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Tschumi A, Nai C, Auchli Y, Hunziker P, Gehrig P, Keller P, Grau T, Sander P. 2009. Identification of apolipoprotein N-acyltransferase (Lnt) in mycobacteria. J Biol Chem 284:27146–27156. doi: 10.1074/jbc.M109.022715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drage MG, Tsai HC, Pecora ND, Cheng TY, Arida AR, Shukla S, Rojas RE, Seshadri C, Moody DB, Boom WH, Sacchettini JC, Harding CV. 2010. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol 17:1088–1095. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. 2006. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol 177:422–429. doi: 10.4049/jimmunol.177.1.422. [DOI] [PubMed] [Google Scholar]
- 11.Pai RK, Pennini ME, Tobian AA, Canaday DH, Boom WH, Harding CV. 2004. Prolonged Toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect Immun 72:6603–6614. doi: 10.1128/IAI.72.11.6603-6614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. 2007. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Ranoa DR, Kelley SL, Tapping RI. 2013. Human lipopolysaccharide-binding protein (LBP) and CD14 independently deliver triacylated lipoproteins to Toll-like receptor 1 (TLR1) and TLR2 and enhance formation of the ternary signaling complex. J Biol Chem 288:9729–9741. doi: 10.1074/jbc.M113.453266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasselon T, Detmers PA, Charron D, Haziot A. 2004. TLR2 recognizes a bacterial lipopeptide through direct binding. J Immunol 173:7401–7405. doi: 10.4049/jimmunol.173.12.7401. [DOI] [PubMed] [Google Scholar]
- 15.Kajava AV, Vasselon T. 2010. A network of hydrogen bonds on the surface of TLR2 controls ligand positioning and cell signaling. J Biol Chem 285:6227–6234. doi: 10.1074/jbc.M109.063669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massari P, Visintin A, Gunawardana J, Halmen KA, King CA, Golenbock DT, Wetzler LM. 2006. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J Immunol 176:2373–2380. doi: 10.4049/jimmunol.176.4.2373. [DOI] [PubMed] [Google Scholar]
- 17.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 18.Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, Weis JJ. 1999. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by Toll-like receptor 2. J Immunol 163:2382–2386. [PubMed] [Google Scholar]
- 19.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 20.Latz E, Franko J, Golenbock DT, Schreiber JR. 2004. Haemophilus influenzae type b-outer membrane protein complex glycoconjugate vaccine induces cytokine production by engaging human Toll-like receptor 2 (TLR2) and requires the presence of TLR2 for optimal immunogenicity. J Immunol 172:2431–2438. doi: 10.4049/jimmunol.172.4.2431. [DOI] [PubMed] [Google Scholar]
- 21.Singleton TE, Massari P, Wetzler LM. 2005. Neisserial porin-induced dendritic cell activation is MyD88 and TLR2 dependent. J Immunol 174:3545–3550. doi: 10.4049/jimmunol.174.6.3545. [DOI] [PubMed] [Google Scholar]
- 22.Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. 2001. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19 kD lipoprotein of Mycobacterium tuberculosis. J Immunol 167:910–918. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 23.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. 2004. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol 173:2660–2668. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 24.Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. 2003. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a potential mechanism for immune evasion. J Immunol 171:175–184. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 25.Drage MG, Pecora ND, Hise AG, Febbraio M, Silverstein RL, Golenbock DT, Boom WH, Harding CV. 2009. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell Immunol 258:29–37. doi: 10.1016/j.cellimm.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lancioni CL, Li Q, Thomas JJ, Ding X, Thiel B, Drage MG, Pecora ND, Ziady AG, Shank S, Harding CV, Boom WH, Rojas RE. 2011. Mycobacterium tuberculosis lipoproteins directly regulate human memory CD4(+) T cell activation via Toll-like receptors 1 and 2. Infect Immun 79:663–673. doi: 10.1128/IAI.00806-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, Fenton MJ. 2001. Different Toll-like receptor agonists induce distinct macrophage responses. J Leukoc Biol 69:1036–1044. [PubMed] [Google Scholar]
- 28.Gilleron M, Quesniaux VF, Puzo G. 2003. Acylation state of the phosphatidylinositol hexamannosides from Mycobacterium bovis bacillus Calmette Guerin and mycobacterium tuberculosis H37Rv and its implication in Toll-like receptor response. J Biol Chem 278:29880–29889. doi: 10.1074/jbc.M303446200. [DOI] [PubMed] [Google Scholar]
- 29.Tapping RI, Tobias PS. 2003. Mycobacterial lipoarabinomannan mediates physical interactions between TLR1 and TLR2 to induce signaling. J Endotoxin Res 9:264–268. doi: 10.1179/096805103225001477. [DOI] [PubMed] [Google Scholar]
- 30.Quesniaux VJ, Nicolle DM, Torres D, Kremer L, Guerardel Y, Nigou J, Puzo G, Erard F, Ryffel B. 2004. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J Immunol 172:4425–4434. doi: 10.4049/jimmunol.172.7.4425. [DOI] [PubMed] [Google Scholar]
- 31.Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. 1999. The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for Toll-like receptors. J Immunol 163:6748–6755. [PubMed] [Google Scholar]
- 32.Zahringer U, Lindner B, Inamura S, Heine H, Alexander C. 2008. TLR2—promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213:205–224. [DOI] [PubMed] [Google Scholar]
- 33.Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 83:91–97. doi: 10.1016/S1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 34.Gilleron M, Nigou J, Nicolle D, Quesniaux V, Puzo G. 2006. The acylation state of mycobacterial lipomannans modulates innate immunity response through Toll-like receptor 2. Chem Biol 13:39–47. doi: 10.1016/j.chembiol.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 35.Vignal C, Guerardel Y, Kremer L, Masson M, Legrand D, Mazurier J, Elass E. 2003. Lipomannans, but not lipoarabinomannans, purified from Mycobacterium chelonae and Mycobacterium kansasii induce TNF-alpha and IL-8 secretion by a CD14-Toll-like receptor 2-dependent mechanism. J Immunol 171:2014–2023. doi: 10.4049/jimmunol.171.4.2014. [DOI] [PubMed] [Google Scholar]
- 36.Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, Inoue H, Tanaka M, Yoneyama M, Oh-Hora M, Akashi K, Yamasaki S. 2014. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity 41:402–413. doi: 10.1016/j.immuni.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Mazurek J, Ignatowicz L, Kallenius G, Svenson SB, Pawlowski A, Hamasur B. 2012. Divergent effects of mycobacterial cell wall glycolipids on maturation and function of human monocyte-derived dendritic cells. PLoS One 7:e42515. doi: 10.1371/journal.pone.0042515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puissegur MP, Lay G, Gilleron M, Botella L, Nigou J, Marrakchi H, Mari B, Duteyrat JL, Guerardel Y, Kremer L, Barbry P, Puzo G, Altare F. 2007. Mycobacterial lipomannan induces granuloma macrophage fusion via a TLR2-dependent, ADAM9- and beta1 integrin-mediated pathway. J Immunol 178:3161–3169. doi: 10.4049/jimmunol.178.5.3161. [DOI] [PubMed] [Google Scholar]
- 39.Dao DN, Kremer L, Guerardel Y, Molano A, Jacobs WR Jr, Porcelli SA, Briken V. 2004. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun 72:2067–2074. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ. 2006. TLR1- and TLR6-independent recognition of bacterial lipoeptides. J Biol Chem 281:9049–9057. doi: 10.1074/jbc.M512525200. [DOI] [PubMed] [Google Scholar]
- 41.Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. 2006. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 176:4323–4330. doi: 10.4049/jimmunol.176.7.4323. [DOI] [PubMed] [Google Scholar]
- 42.Riviere M, Moisand A, Lopez A, Puzo G. 2004. Highly ordered supra-molecular organization of the mycobacterial lipoarabinomannans in solution. Evidence of a relationship between supra-molecular organization and biological activity. J Mol Biol 344:907–918. [DOI] [PubMed] [Google Scholar]
- 43.Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y. 2003. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med 197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda N, Nigou J, Herrmann JL, Jackson M, Amara A, Lagrange PH, Puzo G, Gicquel B, Neyrolles O. 2003. The cell surface receptor DC-SIGN discriminates between Mycobacterium species through selective recognition of the mannose caps on lipoarabinomannan. J Biol Chem 278:5513–5516. doi: 10.1074/jbc.C200586200. [DOI] [PubMed] [Google Scholar]
- 45.Torrelles JB, Azad AK, Schlesinger LS. 2006. Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J Immunol 177:1805–1816. doi: 10.4049/jimmunol.177.3.1805. [DOI] [PubMed] [Google Scholar]
- 46.Nigou J, Zelle-Rieser C, Gilleron M, Thurnher M, Puzo G. 2001. Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J Immunol 166:7477–7485. doi: 10.4049/jimmunol.166.12.7477. [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee D, Roberts AD, Lowell K, Brennan PJ, Orme IM. 1992. Structural basis of capacity of lipoarabinomannan to induce secretion of tumor necrosis factor. Infect Immun 60:1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doz E, Rose S, Nigou J, Gilleron M, Puzo G, Erard F, Ryffel B, Quesniaux VF. 2007. Acylation determines the Toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J Biol Chem 282:26014–26025. doi: 10.1074/jbc.M702690200. [DOI] [PubMed] [Google Scholar]
- 49.Nigou J, Vasselon T, Ray A, Constant P, Gilleron M, Besra GS, Sutcliffe I, Tiraby G, Puzo G. 2008. Mannan chain length controls lipoglycans signaling via and binding to TLR2. J Immunol 180:6696–6702. doi: 10.4049/jimmunol.180.10.6696. [DOI] [PubMed] [Google Scholar]
- 50.Torrelles JB, Sieling PA, Zhang N, Keen MA, McNeil MR, Belisle JT, Modlin RL, Brennan PJ, Chatterjee D. 2012. Isolation of a distinct Mycobacterium tuberculosis mannose-capped lipoarabinomannan isoform responsible for recognition by CD1b-restricted T cells. Glycobiology 22:1118–1127. doi: 10.1093/glycob/cws078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sklar MD, Tereba A, Chen BD, Walker WS. 1985. Transformation of mouse bone marrow cells by transfection with a human oncogene related to c-myc is associated with the endogenous production of macrophage colony stimulating factor 1. J Cell Physiol 125:403–412. doi: 10.1002/jcp.1041250307. [DOI] [PubMed] [Google Scholar]
- 52.Shukla S, Richardson ET, Athman JJ, Shi L, Wearsch PA, McDonald D, Banaei N, Boom WH, Jackson M, Harding CV. 2014. Mycobacterium tuberculosis lipoprotein LprG binds lipoarabinomannan and determines its cell envelope localization to control phagolysosomal fusion. PLoS Pathog 10:e1004471. doi: 10.1371/journal.ppat.1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnsson B, Lofas S, Lindquist G. 1991. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem 198:268–277. doi: 10.1016/0003-2697(91)90424-R. [DOI] [PubMed] [Google Scholar]