

Abstract
CKA, a chemokine receptor antagonist intended for treating inflammatory conditions, produced dose-dependent hepatotoxicity in rats but advanced into the clinic where single doses of CKA up to 600 mg appeared safe in humans. Because existing toxicological platforms used during drug development are not perfectly predictive, a quantitative systems toxicology model investigated the hepatotoxic potential of CKA in humans and rats through in vitro assessments of CKA on mitochondrial respiration, oxidative stress, and bile acid transporters. DILIsym predicted that single doses of CKA caused serum ALT >3xULN in a subset of the simulated rat population, while single doses in a simulated human population did not produce serum ALT elevations. Species differences were largely attributed to differences in liver exposure, but increased sensitivity to inhibition of mitochondrial respiration in the rat also contributed. We conclude that mechanistic modeling can elucidate species differences in the hepatotoxic potential of drug candidates.
Keywords: drug-induced liver injury, systems pharmacology, hepatotoxicity, CKA, quantitative systems toxicology, quantitative systems pharmacology
Drug-induced liver injury (DILI) is one of the primary reasons for the termination of drug candidates during preclinical or clinical development (Ballet, 1997; Kaplowitz, 2005; Schomaker et al., 2013; Tang et al., 2015; Urban et al., 2012). By improving the hepatotoxic preclinical screening for new drugs, drug development may become a more efficient process enabling selection of safer drug candidates. Whilst potential new drugs are tested in many models, existing toxicological platforms are not perfectly predictive. For example, many compounds which are negative in preclinical models are positive in the clinic, and compounds which are positive in preclinical models may not be advanced into the clinic. Computational models can assist in evaluating the hepatotoxic liability of novel therapeutic compounds by translating the compound activity into in vivo findings in multiple species. DILIsym®, a mechanistic, mathematical representation of key mechanisms underlying DILI, can be applied to predict toxicity based on in vitro and/or in vivo data and can provide further insight into the mechanisms responsible for DILI (Bhattacharya et al., 2012; Longo et al., 2016; Shoda et al., 2014; Woodhead et al., 2012). In the current study, we use DILIsym (DILIsym v5A, 2016) to investigate species-related differences in toxicity upon administration of a chemokine receptor antagonist (CKA, 1-[4-chloro-3-trifluoromethyl-benzyl]-5-hydroxy-1-H-indole-2-carboxylic acid) aimed at treating inflammatory conditions (Ulloa et al., 2013).
Chemokine receptors play a key role in the pathogenesis of autoimmune diseases, inflammation, and viral infections (Bodolay et al., 2002; Iwamoto et al., 2008). Drugs that antagonize the chemokine receptor inhibit monocyte recruitment and retention, thereby reducing inflammation (Koch, 2005; Szekanecz et al., 2003). Although many of these compounds have shown initial promise in animal models of inflammation, most have failed to show efficacy in clinical trials (Horuk, 2009; Pease and Horuk, 2009a,b; Ribeiro and Horuk, 2005).
In repeat dose studies in rats, CKA produced hepatoxicity including hepatocyte necrosis, bile duct pathologies, and fibrosis accompanied by elevated liver enzymes and bilirubin. As liver findings were largely reversible and predicted safety margins were deemed good for the intended dose levels in humans, CKA was progressed into the clinic. No significant liver toxicity was observed in healthy volunteers receiving a single oral dose up to 600 mg, while 1/6 subjects receiving the highest dose administered (900 mg) experienced an asymptomatic and transient rise in serum ALT just over 120 U/L. Clinical development of CKA was abandoned primarily due to its short half-life and GI tract toxicity, not due to hepatotoxicity. CKA presents an interesting exemplar compound for investigating the basis for species differences in DILI potential due to differences in the manifestation of hepatotoxic signals in rat and man.
In vitro assays can be used to assess and identify hepatotoxic liabilities of compounds for multiple mechanisms such as reactive oxygen species (ROS) production, alterations in bile acid (BA) homeostasis, and mitochondrial toxicity. It has been reported that CKA causes inhibition of the bile salt export pump (BSEP) (Ulloa et al., 2013), the major efflux transporter that secretes BAs from the hepatocyte into the bile. The only potential contributor to hepatotoxicity seen in rats administered CKA identified so far was BSEP inhibition, but it is well-known that many other mechanisms may contribute to or cause liver injury (Mosedale and Watkins, 2017). In the current study, we assessed the inhibitory effects of CKA on the basolateral efflux transporters (MRP3 and MRP4) that secrete BAs into the blood as well as the sodium-dependent uptake transporter (NTCP) which is responsible for the uptake of BAs from the sinusoids. CKA-mediated oxidative stress and mitochondrial dysfunction were also investigated in whole cell systems.
DILIsym was used to incorporate in vivo exposure predicted by a physiologically based pharmacokinetic (PBPK) submodel with the aforementioned in vitro toxicity data to simulate the in vivo effects of CKA in rats and humans. Responses were analyzed in simulated populations (SimPops™) for both species, which include species-specific, inter-individual variability in parameters relevant to DILI mechanisms. Results from the SimPops were used to assess the primary mechanism underlying CKA-mediated hepatotoxicity and to study possible causes for the observed species difference.
MATERIALS AND METHODS
Preclinical experiment
All experiments were conducted in compliance with UK home office licenses issued under the UK Animals (Scientific Procedures) Act 1986 after review by the local ethics committee. A single dose of 50, 200, or 500 mg/kg CKA or vehicle (0.5% w/v hydroxypropyl-methycellulose in 0.1% w/v aqueous polysorbate 80) was administered by oral gavage to fasted male Wistar Hannover rats (Crl: WI(Han), Charles River, Margate, UK; n = 8 except the 500 mg/kg group where n = 4). To determine CKA plasma concentrations, 20 μL microsamples were taken by tail prick 1 h pre-dose and 0.5, 1, 2, 6, 24, and 48 h post-dose into K2EDTA-treated capillary tubes containing 80 μL PBS and plasma was separated by centrifugation (13 000 rpm, 3 min). After protein precipitation with acetonitrile, plasma CKA concentrations were quantified by high performance liquid chromatography with tandem mass spectrometry detection. For clinical chemistry analysis, blood samples from the tail vein (or vena cava for terminal samples) were collected into lithium-heparin tubes 1 h pre-dose and at 0.5, 1, 2, 6, 24, 48 h following CKA; the 500 mg/kg group was last sampled at 24 h. Plasma was separated by centrifugation (1200 × g, 4°C, 10 min) and samples were analyzed on a Roche P Modular analyzer (Roche Diagnostics, Burgess Hill, UK) using a total bilirubin assay, an alanine aminotransferase (ALT/GPT) assay (both from Roche Diagnostics), and a total BA assay (Randox Laboratories Ltd., Crumlin, UK) according to the manufacturers’ instructions.
Clinical trial
A randomized, double blind, placebo-controlled single ascending dose study in healthy volunteers (9 male, 18 female, age: 35–55 years, weight: 52–92 kg, BMI: 19–30 kg/m2) assessed the safety, tolerability, and pharmacokinetics (PK) of CKA. An Independent Ethics Committee (Ärztekammer Berlin, Berlin, Germany) approved the phase 1 study which was performed in accordance with the ethical principles of the Declaration of Helsinki and that are consistent with International Conference on Harmonization/Good Clinical Practice and applicable regulatory requirements and the AstraZeneca Bioethics policy. All participants gave written informed consent.
Using a parallel group design with a rotating placebo group, fasted subjects received CKA doses of 3–900 mg (7 dose levels, 4–6 subjects/dose level) or placebo (2–3 subjects/dose level) as film-coated tablets. The washout period between doses was 2 weeks. For PK analysis, blood samples were taken into lithium-heparin tubes pre-dose and at 0.5, 1, 2, 3, 4, 6, 9, 12, 24, 48 h post-dose. CKA concentrations were determined in plasma obtained after centrifugation and protein precipitation by liquid chromatography and electrospray tandem mass spectrometry. For clinical chemistry analysis (ALT, total bilirubin), blood was sampled into serum gel tubes on day 1 pre-dose and then every 24 h post-dose until day 6.
PBPK submodel development
The disposition of CKA in humans and rats was represented in the PBPK submodel of DILIsym. The structure of the PBPK model is the same for both species and depicted in Figure 1. Distribution of CKA is represented by tissue partition coefficients whose values were optimized using a genetic algorithm in MATLAB (The MathWorks, Natick, MA) to match simulated plasma profiles to data via a least-squares fit. The fraction unbound of CKA in plasma was also optimized in this manner because CKA is highly bound to plasma proteins and measurement errors were assumed. Disposition of CKA metabolites were not modeled due to lack of data. Additional details on the PBPK representation of CKA and drug-specific parameters for each species are in the Supplementary Information.
Figure 1.

CKA chemical structure and physiologically based pharmacokinetic (PBPK) model diagram and results. A, Chemical structure of CKA. B, Schematic diagram representing the PBPK model consisting of blood, gut, liver, muscle, and other tissue compartments. PBPK results for (C) humans and (D) rats. Dark lines represent observed plasma concentrations of CKA. Light lines represent predicted plasma concentrations of CKA based on the PBPK model. Human simulations were run for 96 h, rat simulations for 72 h. The horizontal axis is cropped to provide greater detail during the initial increase in plasma concentration.
In vitro toxicity assessment
To assess CKA effects on mitochondrial function, HepG2 cells were incubated with 0–750 µM CKA for 6 h. Cellular respiration assays (Eakins et al., 2016) were performed to measure the oxygen consumption rate (OCR) using a Seahorse XFe24 Analyzer (Agilent Technologies, Santa Clara, California). Formation of ROS as a measure of oxidative stress was assessed by high content screening using the fluorescent probe dihydroethidium (Woodhead et al., 2017). IC50 values for human MRP3, MRP4 were determined in transporter-overexpressing vesicles derived from HEK293 cells and for human NTCP in transfected CHO cell lines, which were then used to represent inhibition constants in DILIsym (Table 1). The IC50 value for BSEP was taken from literature (Ulloa et al., 2013). Further information on the in vitro experiments and how they were quantified and translated to DILIsym parameter values can be found in the Supplementary Information.
Table 1.
Toxicity Parameters for Human and Rat
| Human | Rat | |
|---|---|---|
| Bile acid transporter inhibition constant (µM) | ||
| BSEP | 94a | 129.7b |
| MRP3 | 11.2 | 11.2c |
| MRP4 | 12.3 | 12.3c |
| NTCP | 19.5 | 19.5c |
| Mitochondrial toxicity constant (mM) | ||
| ETC inhibition constant | 14.2 | 1.42 |
| ROS production constant (mL/mol/h) | ||
| ROS production constant | 7278 | 9705 |
Transporter inhibition constants for CKA pertaining to bile acid and bilirubin were used as Ki values in simulating CKA-mediated hepatotoxicity. The electron transport chain (ETC) inhibition constant and reactive oxygen species (ROS) production constant parameters were optimized to data using MITOsym and DILIsym, respectively. IC50 values, non-competitive inhibition assumed for efflux transporters (BSEP, MRP3, MRP4), competitive inhibition assumed for uptake transporter (NTCP). For transporters where data were lacking, the same IC50 value was assumed across species, as indicated.
Data from Astra Zeneca (unpublished).
From Ulloa et al (2013).
Assumed to be the same as human.
Human and rat simulated populations (SimPops) and toxicity predictions
Simulated human (n = 285) and rat (n = 294) populations have been created within DILIsym by varying 34 parameters in the BA, ROS, and mitochondrial submodels in addition to system-specific parameters such as body weight. The selected parameters were chosen based on a genetic algorithm in MATLAB and the resulting simulated populations have accurately predicted the incidence of hepatotoxicity associated with various drugs (Woodhead et al., 2014; Yang et al., 2014) Parameters that varied in both the rat and human SimPops are listed in Supplementary Table 2. To replicate the preclinical and clinical studies, simulations ran for 96 and 72 h following single doses of 300, 600, and 900 mg in humans and 50, 200, and 500 mg/kg in rats, respectively.
RESULTS
Biomarker Observations in Preclinical and Clinical Studies
Serum ALT activity is one of the primary biomarkers of liver injury and was used to determine the hepatotoxic risk of CKA in rats and humans. At the 200 mg/kg dose level, 25% (2 out of 8) of the rats experienced ALT elevations >3× upper limit of normal (ULN, ULN = 30 U/L). Additionally, 75% (3 out of 4) of rats receiving single doses of 500 mg/kg had severe increases in serum ALT >3×ULN.
After 300 mg (n = 5) and 600 mg (n = 4) doses of CKA administered to humans, 0% of patients had increases in ALT. At the highest dose administered (900 mg), 1 out of 6 patients (16.7%) experienced an asymptomatic and transient increase in ALT that just surpassed the 3×ULN threshold while the remaining patients did not experience increases in ALT.
Quantification of In Vitro Toxicity Data in DILIsym
In the mitochondrial respiration assay, CKA slightly increased OCR at lower concentrations but significantly decreased OCR at ≥500 µM, indicating that CKA causes mitochondrial uncoupling and ETC inhibition (ETCi). In a concentration-dependent manner, CKA increased ROS-induced oxidative stress and inhibited human BA transporters: BSEP, MRP3, MRP4, and NTCP. IC50 values were directly used in DILIsym. Figure 2 shows observed and simulated changes in in vitro mitochondrial function and oxidative stress in response to CKA. The estimated parameters for BA transporter inhibition, ETCi, and ROS production are shown in Table 1.
Figure 2.

Oxygen consumption rate (OCR) and oxidative stress measurements in response to CKA treatment. Observed and simulated percentage changes in (A) OCR and (B) oxidative stress in CKA-treated cells compared with vehicle control. In vitro respiration data were obtained from HepG2 cells incubated with CKA and measured using the Seahorse XF instrument. Simulated OCR responses were generated using MITOsym with the electron transport chain (ETC) inhibition parameter value optimized to the measured data. Oxidative stress data were obtained from HepG2 cells treated with CKA and measured using fluorescent imaging. Simulated oxidative stress responses were generated using DILIsym with the ROS production constant value optimized to the measured data.
Physiologically Based Pharmacokinetics (PBPK) Model
The DILIsym PBPK submodel was used to describe the disposition of CKA in humans and rats. Simulated CKA plasma concentration profiles were within 2 SD of the mean observed concentrations at each time point in both humans following a single oral dose of 600 or 900 mg and rats following a single oral dose of 200 or 500 mg/kg. Measured and simulated plasma profiles are shown in Figure 1.
CKA Hepatotoxicity in Simulated Rat Populations
The rat SimPops predicted dose-dependent elevations in serum ALT >3×ULN in 0.0–36.4% of the population receiving 50–500 mg/kg doses (Table 2). Simulations reasonably recapitulated dose-dependent CKA hepatotoxicity and range of observed peak ALT values in rats. The rat baseline ALT level was 30 U/L, increased from the DILIsym default value of 21 U/L to reflect observations in the preclinical study. Mean simulated maximum ALT levels were 35 and 131 U/L at the 200 and 500 mg/kg doses, respectively, somewhat underpredicting the mean ALT values observed in the preclinical rat studies (83 and 205 U/L). Simulated ranges of peak ALT levels for 200 and 500 mg/kg doses were 30–138 and 30–1348 U/L, respectively, spanning the observed ranges in the preclinical study (67–102 and 70–335 U/L). Dose-response results for maximum serum ALT values are shown in Figure 3.
Table 2.
Summary of CKA-Mediated Hepatotoxicity in Rat and Human SimPops and Preclinical and Clinical Observations
| Species | Rat | Rat | Human | Human | Human |
|---|---|---|---|---|---|
| Simulationsa | |||||
| Dose | 200 mg/kg | 500 mg/kg | 300 mg | 600 mg | 900 mg |
| Population size | n = 294 | n = 294 | n = 285 | n = 285 | n = 285 |
| ALT > 3× ULN (%)b | 2.4 | 36.4 | 0 | 0 | 0 |
| ALT > 5× ULN (%)b | 0 | 20.1 | 0 | 0 | 0 |
| ALT > 10× ULN (%)b | 0 | 7.8 | 0 | 0 | 0 |
| Preclinical/clinical trials | |||||
| Dose | 200 mg/kg | 500 mg/kg | 300 mg | 600 mg | 900 mg |
| Population size | n = 8 | n = 4 | n = 5 | n = 4 | n = 6 |
| ALT > 3× ULN (%)b | 25 | 75 | 0 | 0 | 16.7 |
| ALT > 5× ULN (%)b | 0 | 50 | 0 | 0 | 0 |
| ALT > 10× ULN (%)b | 0 | 25 | 0 | 0 | 0 |
Human simulations were run for 96 h, rat simulations for 72 h.
Upper limit of normal (ULN) was 30 U/L in rat simulations and preclinical trials. ULN was 40 U/L in human simulations and clinical trials.
Figure 3.
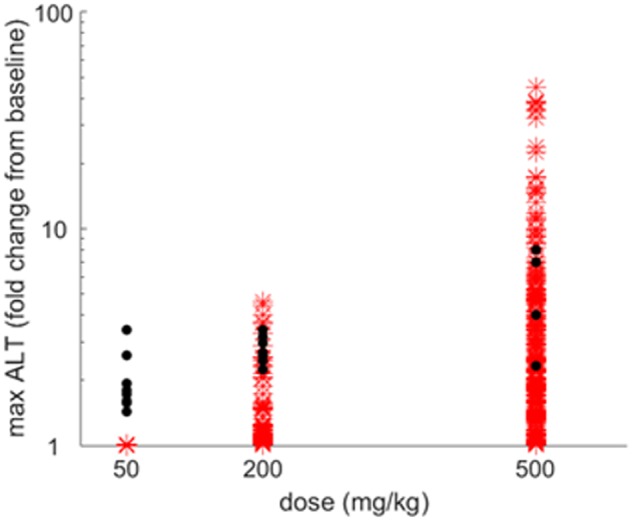
Simulated and observed peak alanine transaminase (ALT) levels in rats administered 50, 200, or 500 mg/kg CKA. Maximum ALT fold change from baseline for increasing doses of CKA. Dots represent data from preclinical studies and asterisks denote simulation results for rat SimPops (n = 294). Results are quantitatively summarized in Table 2.
Next, two approaches were taken to determine which toxicity mechanism(s) accounted for the toxicity observed in rats.
First, a strictly qualitative approach was performed through a combinatorial analysis. All toxicity mechanisms were initially deactivated and then one mechanism at a time was activated to study the effect each solo mechanism had on ALT elevations. Next, all possible combinations of two mechanisms were activated to determine if the interplay between multiple mechanisms affected the results (Figure 4). From this qualitative analysis, mitochondrial toxicity via ETCi was implicated as the key driver in observed rat hepatotoxicity with minor contributions from BA transporter inhibition.
Figure 4.

Qualitative sensitivity analysis based on toxicity mechanisms in rat SimPops. A single dose of 500 mg/kg CKA was simulated in rat SimPops with a single mechanism (top row) or combination of two DILI mechanisms (bottom row). (A) bile acid (BA) transporter inhibition only, (B) reactive oxygen species (ROS) generation only, (C) mitochondrial electron transport chain (ETC) inhibition only, (D) ETC inhibition + ROS generation, (E) ETC inhibition + BA transporter inhibition, (F) BA transporter inhibition + ROS generation. Simulations with a single DILI mechanism (top row) suggest that ETC inhibition is the main contributor to CKA-mediated rat hepatotoxicity. When two DILI mechanisms are combined, ETC inhibition and BA transporter inhibition together produce a greater peak plasma alanine transaminase (ALT), suggesting BA transporter inhibition as the secondary contributor to hepatotoxicity. Qualitative results are supported by multiple regression analysis results shown in Supplementary Table 2.
In an alternative approach, we quantitatively analyzed the correlation between parameters varied in the rat SimPops and the predicted ALT elevations by performing a multiple regression analysis. The strongest correlated parameters represent mechanisms responsible for CKA-mediated ALT elevations. Parameters varied in the rat SimPops were normalized prior to performing the multiple regression analysis to account for differing orders of magnitude. This approach also identified mitochondrial toxicity as the major mechanism contributing to observed CKA hepatotoxicity in rats.
Mitochondrial toxicity via ETCi was the only mechanism capable of increasing serum ALT by itself in simulations. The multiple linear regression analysis confirmed these results by determining that the parameters most strongly correlated with ALT elevations were those found in the mitochondrial toxicity submodel in DILIsym. The Supplementary Information contains further details on these analyses.
CKA Hepatotoxicity in Simulated Human Populations
In contrast to the simulated rat population, no individual in the human SimPops demonstrated ALT elevations >3×ULN at 300–900 mg doses (Table 2).
Investigating Differences Between Preclinical and Clinical Findings
Next, the basis for the differences between simulated rats and humans was explored. Possible sources may stem from species differences in hepatic drug exposure and toxicity mechanism parameters (such as ETCi) or from differences in sensitivity between species. Using DILIsym, it is possible to explore potential causes by adjusting the model to reflect these hypotheses.
Simulated human liver AUC of CKA at its maximum clinical dose of 900 mg was 0.03 mg*h/mL, which was 100-fold smaller than simulated rat liver AUC of CKA at the toxic dose of 200 mg/kg. To explore CKA doses and exposures that might cause hepatotoxicity in humans, higher dose levels of CKA (up to ∼250-fold greater dose than the maximum clinical dose) were simulated in the human population (Table 3). At 223 g, human liver AUC was similar to that in rats at the hepatotoxic dose of 500 mg/kg, and 57.1% of the simulated humans developed ALT elevations >3×ULN. On the other hand, at the dose of 100 g, human liver AUC was similar to that in rats at the hepatotoxic dose of 200 mg/kg, but none of the simulated humans developed ALT elevations >3×ULN. These results suggest that while the low hepatic exposure in humans compared with rats at tested preclinical/clinical doses is the underlying main reason for species differences in CKA-mediated hepatotoxicity, other factors such as DILI mechanism might have also contributed to species differences in DILI sensitivity.
Table 3.
Results Investigating Species Differences in CKA-Mediated Toxicity
| Species | Single Dose | Liver Cmax (mg/mL) | Liver AUC0–72 (mg*h/mL) | ALT > 3× ULNb |
|---|---|---|---|---|
| Rata | 200 mg/kg | 0.40 | 3.00 | 7/294 (2.4%) |
| (n = 294) | 500 mg/kg | 1.03 | 7.51 | 107/294 (36.4%) |
| Humana | 0.9 g | 0.04 | 0.03 | 0/285 (0%) |
| (n = 285) | 10 g | 0.41 | 0.27 | 0/285 (0%) |
| 100 g | 4.52 | 2.88 | 0/285 (0%) | |
| 223 g | 11.37 | 7.50 | 148/285 (57.1%) |
Human simulations were run for 96 h, rat simulations for 72 h.
Upper limit of normal (ULN) was 30 U/L in rat simulations, 40 U/L in human simulations.
In the subsequent analysis, species differences in susceptibility to ETCi were explored because ETCi was the major mechanism underlying ALT elevations in the simulated rat populations. As noted in Table 1, the ETCi parameter value for rats is more potent than for humans; this is based on the translation factor from MITOsym to DILIsym, which was derived from differences in the respiratory characteristics between primary human hepatocytes, HepG2 cells, and primary rat hepatocytes in response to rotenone, a well-known ETC inhibitor (Yang et al., 2015; Longo et al., 2016, Supplementary Table 4). By maintaining the same toxicity mechanism parameters for ROS generation and BA inhibition and setting the human ETCi parameter value to that of the rat (ETCi = 1.42 mM), results from the ensuing human population were used to investigate this hypothesis. Even when 900 mg CKA, the highest clinical dose, was simulated in the human population with the rat ETCi parameter, no ALT elevations were predicted. However, when 500 mg/kg CKA was simulated in the rat population with the less potent human ETCi parameter (ETCi = 14.2 mM), ALT elevations disappeared. From these two simulations, the potency of ETCi was capable of contributing to species differences in ALT elevations when hepatic CKA exposure was high (e.g., in rats). The cumulative results indicate that differences observed in preclinical and clinical findings are likely attributed to differences in CKA liver exposure at tested preclinical/clinical doses, while increased sensitivity to inhibition of mitochondrial respiration in the rat also contributed.
DISCUSSION
CKA, an anti-inflammatory class drug candidate, was associated with dose-related increases in liver enzymes during preclinical studies but appeared to have minimal liver signals in clinical trials. Based only on in vitro experimental data, CKA appears to increase ROS production, inhibit ETC activity, and inhibit BA transporters (Ulloa et al., 2013). Although the inhibition of BA transporters led to a slight increase in plasma BAs in preclinical studies, the direct link to toxicity cannot be made without further investigation. To study the effect of these mechanisms, the current study used multiple, integrated DILIsym submodels representing drug distribution, mitochondrial toxicity, oxidative stress, BA physiology and pathophysiology, hepatocyte life cycle, and liver injury biomarkers to simulate the response to CKA in human and rat populations. Population variability was incorporated into the simulations through the use of prefabricated SimPops which included variability in parameters related to DILI mechanisms (Supplementary Table 2) (Woodhead et al., 2014; Yang et al., 2014). The mechanistic model could distinguish between species and predicted that toxicity should be observed in the rat population but not in humans, in agreement with observations from preclinical and clinical trials. Although the incidence rates of ALT >3×ULN were underpredicted compared with both preclinical and clinical data, this could be due to several reasons such as the small number of animals treated in the preclinical studies (n = 4–8) and patients in clinical studies (n = 6 at 900 mg), the lack of possible metabolite effects included in the simulations, or differences between the studied and simulated populations. Although a single patient experienced ALT elevations that just surpassed the 3× threshold, ALT elevations resolved quickly and CKA was generally thought to be safe in humans.
Inhibition of ETC activity and the subsequent reduction in mitochondrial energy production appears to be the primary mechanism of CKA-induced hepatotoxicity in rats based on a qualitative sensitivity analysis and a quantitative multiple regression analysis. Additionally, CKA-mediated BA transporter inhibition contributes to elevations in liver enzyme levels in rats while the increase in ROS production appears to have a minimal effect (Figure 4). Specifically, CKA-mediated ALT elevations were associated with decreased basal ETC flux values in the rat SimPops (Supplementary Table 2 and Supplementary Figure 1). Decreased reserve mitochondrial ETC function and decreased Vmax for canalicular bulk BA transport were also linked to simulated rat ALT elevations. The basal ETC flux parameter represents the functional amount of ETC activity, but the respiratory reserve parameter denotes the additional ETC activity that can compensate for when mitochondrial function is compromised (Longo et al., 2016). For individuals appearing more susceptible to CKA-mediated injury, the decrease in either of these quantities accounts for the inability of an individual to compensate for reductions in ATP production caused by CKA-mediated ETCi. A decrease in Vmax for canalicular BA transport corresponds to a reduced rate for bulk BA transport through canalicular transporters such as BSEP (Woodhead et al., 2014), which CKA inhibits at high concentrations.
Based on DILIsym predictions, the higher liver exposure in rats and the more potent inhibition of rat ETC enzymes are predicted to be predominant causes for the observed species difference in serum ALT elevations (Table 3). In this scenario, humans must be administered very large doses of CKA to result in significant ALT increases. At the same time, the coupled effect of increased liver exposure of CKA and the more potent inhibition of ETC activity account for the incidence of serum ALT elevations observed in rats. No serum ALT elevations were predicted when the rat value for ETCi was replaced with the value obtained in human hepatocytes. While this may suggest that a species difference in ETCi is responsible for the observed ALT elevations found in rats, species differences in liver exposure also contributed. Although not applicable in this study, DILIsym can be used in a similar manner to investigate potential causes of species differences based on other toxicity mechanisms, i.e., BA inhibition and ROS generation.
Intracellular drug concentrations are important in accurately predicting toxicity and drug interactions when whole-cell based assay systems are employed (Groothuis et al., 2015). Because the intracellular drug concentration is influenced by a number of factors such as the physiochemical properties of the drug, cell physiology, active transport mechanisms, and culture conditions, the nominal media concentration may not reflect the true intracellular concentration and may not translate directly to an in vivo extracellular to intracellular ratio. However, there is no agreement yet on the best approach to estimate intracellular concentrations in whole cell-based assays, and they are not often measured due to the requirement of LC/MS/MS analysis which increases time and cost. In the current study, mitochondrial toxicity and ROS assays were performed in whole-cell based assay systems; since the intracellular concentration of CKA was not measured experimentally, a model-based method was employed to estimate the intracellular concentration of CKA based on known physicochemical and pharmacokinetic properties of CKA (Supplementary Information). Sensitivity analyses revealed that CKA-mediated ALT elevations in the rat SimPops were sensitive to the method used to estimate intracellular concentration from the culture media concentration in the mitochondrial assays (Supplementary Figure 2). The estimation method influences the optimization of the ETCi parameter, which is used to represent the effect that CKA has on mitochondrial function. The simulated incidence of elevated serum ALT >3×ULN increased from 36.4% to 100% when the ETCi parameter was optimized with the nominal media concentrations or intracellular concentrations estimated using Kp (the total liver-to-total plasma partition coefficient) rather than the intracellular concentrations estimated using Kp,u (the total liver-to-unbound plasma partition coefficient) (the default assumption used in the current study because Seahorse media did not include FBS). In contrast, the simulated incidence of CKA-mediated hepatotoxicity was insensitive to the estimation technique used for intracellular concentration with regards to ROS assay. Although the estimation techniques produced different ROS production constants, serum ALT levels were unaffected by the change in this parameter (results not shown). This is likely due to ROS generation not having a strong impact on CKA-mediated injury. Overall, the use of PBPK model predicted Kp,u (our default assumption) reasonably recapitulated observed CKA-mediated hepatotoxicity, whereas use of nominal concentration or Kp significantly over-predicted in vivo hepatotoxicity, highlighting the importance of accurate estimation of intracellular concentrations from in vitro toxicity assays for this type of quantitative mechanistic modeling.
The current study has several limitations. First, the major metabolite of CKA was not tracked because pharmacokinetic and in vitro experimental data were not available. Activity of any unaccounted metabolite will have the potential to influence the aforementioned toxicity mechanisms and increase CKA’s hepatotoxic effect (Yang et al., 2017). It is possible that the model prediction might change if the metabolite’s effects were available and included. Determining metabolite effects remains a challenge during early drug development because pure metabolites are not often available in sufficient quantities for testing. Refining the model as new information becomes available during drug development will increase its predictability. Second, the intracellular concentration of CKA in the in vitro cell-based assays was not directly measured. In the absence of experimental data, a PBPK model-based approach was employed to estimate the intracellular concentrations in the mitotoxicity assay, assuming negligible protein binding in the assay media. In general, PBPK models assuming flow-limited distribution and well-stirred tissue kinetics have been shown to provide accurate predictions of tissue exposure for passively distributed compounds (Chu et al., 2013). Along similar lines, plasma and liver concentrations are closely correlated for passively distributed compounds such as CKA (Li et al., 2016). Thus, the CKA PBPK models which were optimized to plasma profiles provide strong predictions for liver concentrations and the resulting partition coefficients for liver (Supplementary Table 3). Using this approach, the hepatotoxic potential of CKA in rats and humans was adequately predicted in the current study. However, this does not necessarily hold for transporter substrates where translation of in vitro data to in vivo prediction is still a challenging area (Chu et al., 2013). For these reasons, among others, it is recommended to directly measure intracellular concentrations for the most accurate toxicity predictions.
Toxicity signals are commonly seen in preclinical studies, such as with CKA in rats, but it is not always clear if they are relevant to clinical situations. The current paradigm relies on introducing wide exposure margins to account for lack of translation across species. Here, we have shown that QST modeling using DILIsym software can account for both exposure differences and differences in off-target potency across species, displaying the novelty of QST modeling as a tool to refine preclinical-to-clinical extrapolations of hepatic safety. It was demonstrated that species differences in ETCi and exposure could account for the hepatotoxicity signal observed in rats and the relative liver safety observed with CKA treatment in humans. In similar cases where risk profiles may be deemed unacceptable for progression, the ability of DILIsym to allow for exploration of mechanisms driving toxicity provides key feedback to chemistry that could be incorporated to find new chemical species that would potentially avoid further toxicity risk.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge Kevin Hickling, Mathew Smith, Lorraine Jones, Steven Silvester, Nicola Derbyshire, Debbie Mason, and Peter Hall (Drug Safety and Metabolism, AstraZeneca) for their excellent technical assistance in supporting the preclinical single dose rat study. C. Battista, K. Yang, B.A. Howell, and S.Q. Siler are employees of DILIsym Services, Inc., a company that licenses the DILIsym software for commercial use and serves as the coordinating member of the DILI-sim Initiative. P.B. Watkins directs the DILI-sim Initiative. C. Battista, K. Yang, B.A. Howell, S.Q. Siler, and P.B. Watkins have a financial stake in DILIsym Services, Inc. J.T. Mettetal and S.H. Stahl are employees of AstraZeneca.
FUNDING
This work was supported by the DILI-sim Initiative. For more information on the DILI-sim Initiative, see http://www.DILIsym.com.
This work was supported in part by an appointment (C. Battista) to the Research Participation Program at the Center for Drug Evaluation and Research, U.S. Food and Drug Administration, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and Food and Drug Administration.
REFERENCES
- Ballet F. (1997). Hepatotoxicity in drug development: detection, significance and solutions. J. Hepatol 26, 26–36. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S., Shoda L. K., Zhang Q., Woods C. G., Howell B. A., Siler S. Q., Woodhead J. L., Yang Y., McMullen P., Watkins P. B., Andersen M. E., et al. (2012). Modeling drug- and chemical-induced hepatotoxicity with systems biology approaches. Front Physiol 3, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodolay E., Koch A. E., Kim J., Szegedi G., Szekanecz Z. (2002). Angiogenesis and chemokines in rheumatoid arthritis and other systemic inflammatory rheumatic diseases. J Cell Mol Med 6, 357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X., Korzekwa K., Elsby R., Fenner K., Galetin A., Lai Y., Matsson P., Moss A., Nagar S., Rosania G. R., et al. (2013). Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther 94, 126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakins J., Bauch C., Woodhouse H., Park B., Bevan S., Dilworth C., Walker P. (2016). A combined in vitro approach to improve the prediction of mitochondrial toxicants. Toxicol In Vitro 34, 161–170. [DOI] [PubMed] [Google Scholar]
- Groothuis F. A., Heringa M. B., Nicol B., Hermens J. L. M., Blaauboer B. J., Kramer N. I. (2015). Dose metric considerations in in vitro assays to improve quantitative in vitro-in vivo dose extrapolations. Toxicology 332, 30–40. [DOI] [PubMed] [Google Scholar]
- Horuk R. (2009). Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8, 23–33. [DOI] [PubMed] [Google Scholar]
- Iwamoto T., Okamoto H., Toyama Y., Momohara S. (2008). Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients: chemokines in the joints of patients. FEBS J 275, 4448–4455. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. (2005). Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov 4, 489–499. [DOI] [PubMed] [Google Scholar]
- Koch A. E. (2005). Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum 52, 710–721. [DOI] [PubMed] [Google Scholar]
- Li R., Maurer T. S., Sweeney K., Barton H. A. (2016). Does the systemic plasma profile inform the liver profile? Analysis using a physiologically based pharmacokinetic model and individual compounds. AAPS J 18, 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo D. M, Yang Y., Watkins P. B., Howell B. A., Siler S. Q. (2016). Elucidating differences in the hepatotoxic potential of tolcapone and entacapone with DILIsym ®, a mechanistic model of drug-induced liver injury: modeling tolcapone and entacapone with DILIsym ®. CPT Pharmacomet Syst Pharmacol 5, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosedale M., Watkins P. (2017). Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 101, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J. E., Horuk R. (2009a). Chemokine receptor antagonists: part 1. Expert Opin Ther Pat 19, 39–58. [DOI] [PubMed] [Google Scholar]
- Pease J. E., Horuk R. (2009b). Chemokine receptor antagonists: part 2. Expert Opin Ther Pat 19, 199–221. [DOI] [PubMed] [Google Scholar]
- Ribeiro S., Horuk R. (2005). The clinical potential of chemokine receptor antagonists. Pharmacol Ther 107, 44–58. [DOI] [PubMed] [Google Scholar]
- Schomaker S., Warner R., Bock J., Johnson K., Potter D., Van Winkle J., Aubrecht J. (2013). Assessment of emerging biomarkers of liver injury in human subjects. Toxicol Sci 132, 276–283. [DOI] [PubMed] [Google Scholar]
- Shoda L. K., Woodhead J. L., Siler S. Q., Watkins P. B., Howell B. A. (2014). Linking physiology to toxicity using DILIsym(®), a mechanistic mathematical model of drug-induced liver injury. Biopharm Drug Dispos 35, 33–49. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z., Kim J., Koch A. E. (2003) Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol 15, 15–21. [DOI] [PubMed] [Google Scholar]
- Tang D. M., Koh C., Twaddell W. S., von Rosenvinge E. C., Han H. (2015). Acute hepatocellular drug-induced liver injury from bupropion and doxycycline. ACG Case Rep J 3, 66–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa J. L., Stahl S., Yates J., Woodhouse N., Kenna J. G., Jones H. B., Waterton J. C., Hockings P. D. (2013). Assessment of gadoxetate DCE-MRI as a biomarker of hepatobiliary transporter inhibition. NMR Biomed 26, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban T. J., Goldstein D. B., Watkins P. B. (2012). Genetic basis of susceptibility to drug-induced liver injury: what have we learned and where do we go from here? Pharmacogenomics 13, 735–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead J. L., Howell B. A., Yang Y., Harrill A. H., Clewell H. J., Andersen M. E., Siler S. Q., Watkins P. B. (2012). An analysis of N-acetylcysteine treatment for acetaminophen overdose using a systems model of drug-induced liver injury. J Pharmacol Exp Ther 342, 529–540. [DOI] [PubMed] [Google Scholar]
- Woodhead J. L., Brock W. J., Roth S. E., Shoaf S. E., Brouwer K. L. R., Church R., Grammatopoulos T. N., Stiles L., Siler S. Q., Howell B. A., et al. (2017). Application of a mechanistic model to evaluate putative mechanisms of tolvaptan drug-induced liver injury and identify patient susceptibility factors. Toxicol Sci 155, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead J. L., Yang K., Brouwer K. L. R., Siler S. Q., Stahl S. H., Ambroso J. L., Baker D., Watkins P. B., Howell B. A. (2014). Mechanistic modeling reveals the critical knowledge gaps in bile acid-mediated DILI. CPT Pharmacomet Syst Pharmacol 3, e123.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead J. L., Yang K., Siler S. Q., Watkins P. B., Brouwer K. L. R., Barton H. A., Howell B. A. (2014). Exploring BSEP inhibition-mediated toxicity with a mechanistic model of drug-induced liver injury. Front Pharmacol 5, 240.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Battista C., Woodhead J. L., Stahl S. H., Mettetal J. T., Watkins P. B., Siler S. Q., Howell B. A. (2017). Systems pharmacology modeling of drug – induced hyperbilirubinemia: Differentiating hepatotoxicity and inhibition of enzymes/transporters. Clin Pharmacol Ther 101, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Woodhead J. L., Watkins P. B., Howell B. A., Brouwer K. L. R. (2014). Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin Pharmacol Ther 96, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Nadanaciva S., Will Y., Woodhead J. L., Howell B. A., Watkins P. B., Siler S. Q. (2015). MITOsym®: a mechanistic, mathematical model of hepatocellular respiration and bioenergetics. Pharm Res 32, 1975–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.