

Abstract
Cellular senescence is a tumor-suppressive mechanism which leads to near irreversible proliferative arrest. However, senescent cells can cause tissue dysfunction, in large part because they express a senescence-associated secretory phenotype (SASP) involving secretion of, amongst other factors, proinflammatory cytokines known to compromise neuronal health. Therefore, established neurotoxicants may cause neurotoxicity in vivo, in part by triggering mitotic cells in the brain to undergo senescence and adopt an inflammatory SASP which in turn could cause deleterious effects to surrounding neurons. To begin to address this hypothesis, we examined whether we could screen known neurotoxicants for their ability to cause astrocytes (a mitotic cell type especially important for maintaining neuronal health) to undergo senescence. For this purpose, we utilized inducible pluripotent stem cell-derived human astrocytes and screened an 80 compound neurotoxicant library provided by the Biomolecular Screening Branch of the NIEHS National Toxicology Program. Here we present a screening method based on induction of the senescent marker, senescent-associated beta-galactosidase (SA-β-gal). We describe in detail an automated method for the unbiased quantitation of percentage of SA-β-gal + astrocytes. Although our results suggest that conducting an SA-β-gal senescence screen using human inducible pluripotent stem cell-derived astrocytes may be feasible, they also highlight challenges that likely preclude its adaptation to high-throughput. We also explore the possibility of using primary mouse astrocytes for this purpose and explain why this platform is problematic and very unlikely to yield meaningful results, even in small screens with compound replicates.
Keywords: astrocyte, cellular senescence, iPSC, senescent-associated beta-golactosidase, neurotoxicant screening
Most dividing cell types in peripheral tissues are capable of undergoing cellular senescence, a potent anticancer mechanism that results in near permanent mitotic arrest (Campisi, 2013; Prieur and Peeper, 2008). It occurs in response to stresses that put cells at risk for malignant transformation. Although potentially protective as a tumor-suppressive mechanism, these cells express a senescence-associated secretory phenotype (SASP): the robust secretion of inflammatory cytokines, growth factors and proteases that can contribute to the disruption of tissue structure and function (Coppe et al., 2008).
Cellular senescence of glia has been linked to several neurodegenerative diseases (Baker and Petersen, 2018; Bhat et al., 2012; Chinta et al., 2015; Mombach et al., 2015; Turnquist et al., 2016). Although aging is associated with an increased load of senescent brain cells (Kang et al., 2015; Salminen et al., 2011), it is also possible that chemical exposures could promote the accumulation of senescent cells in the brain. Paraquat (PQ) is a well-established environmental risk factor for Parkinson’s disease (PD) (Pezzoli and Cereda, 2013), that is actively transported across the blood brain barrier (McCormack and Di Monte, 2003). We have recently demonstrated that cultured astrocytes exposed to PQ become senescent and that senescent astrocytes contribute directly to PD neuropathology (Chinta et al., 2018). These data suggest that cellular senescence can be induced in astrocytes in response to an environmental insult, and that senescent induction can in turn contribute to neurodegeneration.
Identifying additional neurotoxicants capable of inducing astrocytic senescence could provide insight into the mechanisms by which they induce neurodegeneration. Additionally, such toxicants may have a pivotal role in the etiology of age related neurodegenerative diseases. For PD in particular, where the majority of cases are idiopathic, with complex interactions between genes and environment, and where environmental exposures have been deemed a culprit—determining if certain compounds cause astrocyte senescence could provide scientific and ultimately therapeutic value. To speak generally to the physiologic relevance of a screen dependent on astrocyte senescence, it is important to distinguish between cellular senescence and quiescence, especially as they relate to astrocyte biology. The major expansion of astrocytic populations occurs in the postnatal period, in both rodent and humans (Bayraktar et al., 2014). Astrocytes can undergo mitosis postnatally, though they do not “generally” do so. This is a reflection of the fact that astrocytes in the healthy adult brain reside in a “quiescent state”, rather than in a “senescent state”. The distinction is that, in the mature brain, a quiescent astrocyte is capable of exiting GO and re-entering the cell cycle but will usually do so only in response to injury or disease—eg, astrocytes proliferate following ischemia forming the infamous glial scars associated with infarcts (Barreto et al., 2011). On the flip side, a senescent astrocyte will have adopted a tumor-suppressive mechanism usually triggered by malignant stresses—including “ageing” and oxidative stress. In this case, the cell will have undergone near irreversible mitotic arrest (Campisi, 2013; Prieur and Peeper, 2008).
This study is primarily meant to establish “proof-of-principle” and to highlight important considerations that should be taken into account when conducting a chemical screen aimed at identifying compounds capable of inducing astrocyte senescence. For this purpose, we utilized an 80 compound neurotoxicant library compiled by the Biomolecular Screening Branch of the NIEHS National Toxicology Program (NTP) (Crofton et al., 2011) in the context of human astrocytes derived from inducible pluripotent stem cells (iPSCs) to screen for induction of the senescent marker, senescence-associated beta-galactosidase (SA-β-gal) (Dimri et al., 1995). We describe a detailed automated means for quantifying percentage of SA-β-gal positive cells, which has up to this point routinely been quantified “by eye” in the senescence field. We point out the challenges related to this screen. Finally, we demonstrate why similarly screening primary mouse astrocytes, for compounds capable of inducing senescence based on SA-β-gal is very unlikely to yield meaningful hits.
MATERIALS AND METHODS
Chemical library
An 80-compound library (76 unique compounds) was provided to us by the NTP (Supplementary Table 1 and see Crofton et al. for a more detailed description of library). The library contains 37 unique environmental compounds and drugs with reported developmental neurotoxicity (DNT) and/or neurotoxicity (NT) activity, as well as representatives of chemical classes (11 unique flame retardants [FRs] and 17 polycyclic aromatic hydrocarbons [PAHs] “of interest to the NTP, but with limited or unknown NT information”). In addition, the library contains 6 unclassified compounds and 5 negative control compounds for DNT/NT. Stock concentrations were at approximately 20 mM (dimethyl sulfoxide) except for 6 compounds prepared at lower concentrations (0.075–10 mM) due to limited solubility.
Culture of human astrocytes
Human astrocytes derived from iPSCs, purchased from XCell Science, Inc. (Novato California), were cultured at 37°C, 3% O2 and 5% CO2 in XCell’s ‘complete astrocyte medium’ with the addition of penicillin/streptomycin. Cells were not refrozen after initial thawing.
Isolation and culturing of primary mouse astrocytes
Primary cortical astrocytes were prepared as described (Schildge et al., 2013). Briefly, cortices were dissected from postnatal day 2 mice and dissociated, plated and cultured for 2 weeks with removal of microglia by orbital shaking prior to conducting experiments. For experiments described here, cells were not frozen. Cells were culture at 37°C, 3% O2 and 5% CO2 in DMEM medium supplemented with 20% fetal bovine serum and penicillin/streptomycin.
96-well plating and drug treatment
iPSC-derived human astrocytes (or mouse astrocytes) were plated at 10 K cells/well in a 96-well format, where wells were pretreated with matrigel. All day 2 controls were plated on separate plates, so that cell density and % SA-β-gal could be estimated at time of drug treatment. On day 2 postplating, discrete wells were treated with individual compounds diluted in 100 μl of astrocyte medium at a final concentration of 40 μM for 24 h. After, 24 h wells were washed with astrocyte medium 3× and replaced with fresh medium. Medium was replaced 3 days later.
SA-β-gal and diamidino-phenylindole staining of cultured cells
Media was aspirated from cells, and wells were washed 1× with PBS. SA-β-gal staining was carried out using components of a senescence detection kit (BioVision). Cold fixative was added for 5 min. Cells were washed 2 × PBS and incubated in 50 μl of freshly prepared X-gal staining solution (prepared as directed in the kit). Plates were placed in the dark, overnight, in a humidified 37°C incubator. The next day, staining solution was removed and cells were counterstained with diamidino-phenylindole (DAPI) (1:10 000 dilution in PBS for 5 min followed by 3× PBS wash—cells were kept in final solution of PBS). Sealed plates were kept in the dark at 4°C until imaged.
Image acquisition
Corresponding images were acquired (1280 × 1240 pixels) on bright-field (BF) and fluorescence (DAPI) channels, using a 10× objective. Three nonoverlapping images were acquired/well, selected randomly but avoiding the edges of the wells.
Image analysis and quantification of % SA-β-gal positive cells
DAPI images were loaded into ImageJ (open source image analysis software) and converted to 8 bit. The image was processed with “Threshold” (with selection of “dark background”). The image was analyzed with “Analyze particles” (with size set to 100–5000 pixels and circularity set to 0.07–1). Similarly, BF images (SA-β-gal) were loaded and converted to 8 bit. The image was processed with “Threshold” (with selection of “light background”). The image was analyzed with “Analyze particles” (with size set to 100–5000 pixels and circularity set to 0.07–1) (see Supplementary Figure 1 for a walk-through of this process). The % of SA-β-gal (No. SA-β-gal counts/No. DAPI counts) was calculated for each of the 3 images captured/well and the average of the 3 percentages was used to define the % SA-β-gal for that well.
RESULTS
Astrocyte-Senescence Screen Which Controls for Cell Density Results in Identification of False Positives
Our primary objective was to screen the neurotoxicant library in order to determine the feasibility of identifying neurotoxicants, which induce astrocyte senescence. We initially adopted the routine experimental design (principally established for assessing senescence of cultured fibroblasts), whereby senescence is evaluated by comparing control groups at “2 days postplating” versus experimental groups “at 7 days postplating” (Davalos et al., 2013). Senescence experiments are traditionally conducted in this manner because senescent phenotypes such as the SASP take multiple days to manifest and by that time untreated (negative control) groups will have grown to over-confluence, whereas cells subject to a well-established senescence inducer, such as ionizing radiation (IR), will have mitotically arrested shortly after initial exposure. This is problematic because sustained confluence itself is a well-known trigger of cellular senescence (Chow and Rubin, 1996) and thus must be avoided in wells that are purposed to serve as negative controls. This was the impetus behind our decision to control for “cell density” by comparing nonsenescent controls (at 2 days postplating) to senescent cells (7 days postplating). We were able to conduct a preliminary toxicity screen of the 76 unique compounds from the NTP neurotoxicant library (although only 72 compounds were ultimately included in final analyses due to technical issues associated with 4 compounds). Human astrocytes were plated at 10 K cells/well in a 96-well format. On day 2 postplating, discrete wells were treated with individual drugs at 40 μM for 24 h (6 compounds were at lower concentration—see Supplementary Table 1 for chemical names and concentrations therein). At the time of initial drug exposure, 4 untreated negative control wells (“Day-2 NC”) were fixed. SA-β-gal and DAPI-counter staining of these Day-2 NC wells, enabled us to estimate a “mean % SA-β-gal at the time of drug treatment” (Figure 1). Drug-treated wells were fixed at 7 days postplating and % SA-β-gal+ cells/well (here-to-fore referred to simply as “%SA”) was evaluated (Supplementary Figure 1 details our automated method for evaluating %SA).
Figure 1.
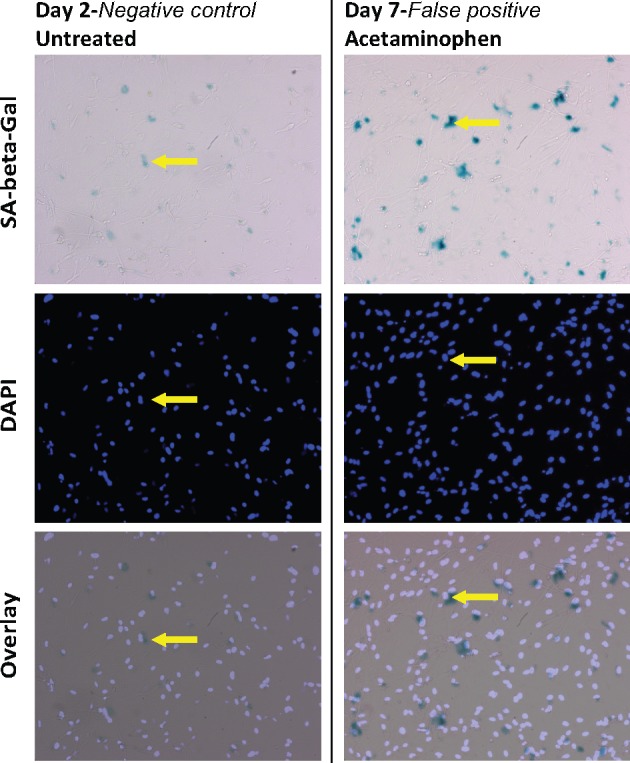
SA-β-gal activity in compound-treated wells compared with activity at 2 days results in the identification of false-positive senescent inducers. Images of cultured human iPSC-derived astrocytes. Untreated control well (left column) fixed on day-2 postplating (the same day that compounds were added to wells). Well treated with 40 μM acetaminophen (included in the library as a negative control for NT) for 24 h and fixed on Day-7 postplating. Staining for SA-β-gal activity (top) and with DAPI (middle) reveals that the number of SA-β-gal+ astrocytes (arrows) has increased substantially over 5 days (postacetaminophen treatment). However, acetaminophen had the second lowest %SA (18.6) of any compound tested, demonstrating that comparing day-2 untreated with toxin-treated at day-7 results in false-positives.
A multitude of chemical screens with diverse endpoint measurements typically define “hits” as compounds that cause an effect > mean + 2× the SD of the negative control, (ie, 95% confidence limit) (Lucanic et al., 2016). However, identifying senescence-inducing compounds based on their having a %SA > mean %SA+ 2× SD of Day-2 NC (6.5% ± 9.5) resulted in an 16.1%SA cutoff and all but a single compound being identified as hits (the lone nonhit, Triphenyl phosphate (10.4%SA), was included in the library due to its use as a FR with unknown neurotoxic potential (Figure 2, right side). This demonstrates erroneous identification of false positives in this screen and suggests that the comparison of treated wells at 7 days versus untreated control wells at 2 days is not a suitable screening method for identifying compounds that induce astrocyte senescence. A routine first pass measure to evaluate the validity of a chosen negative control in a high-throughput screen is to compare the mean value for all compounds, compared with the negative control; this assumes that most compounds are unlikely to have biological activity, and a large difference between the 2 values would not be expected. The fact that %SA day-2 NC was 6.5% and the mean of the treated wells was 33.1% further suggests that the day-2 NC was not an appropriate negative control for the assay. Amongst the likely false-positives were acetaminophen (4-hydroxyacetanilide) which is included in the NTP library as a negative control since meta-analysis supports that it does not act as a neurotoxicant (Crofton et al., 2010). Interestingly, acetaminophen’s %SA (18.6%) was the second lowest % of any of the compounds tested (Figure 1, right side), suggesting that there may be a correlation between a compound’s ability to cause NT and its ability to induce SA-β-gal activation in astrocytes and that quantifying %SA may be yet informative, despite the fact that using the day-2 NC’s mean %SA+ 2× SD as the lower-bound cutoff to define senescence proved to be invalid (Figure 1).
Figure 2.

The use of PQ as a positive control to distinguishes senescent inducers from nonactive compounds. Representative images from which %SA were calculated, reflecting the dynamic range of %SA values. Human astrocytes were stained for SA-β-gal activity (top) and DAPI (middle) to identify SA-β-gal positive cells (eg, arrows). Left most column shows the positive control PQ (%SA = 47.8); second from the left shows the top hit, 3, 3’-Iminodipropionitrile (%SA = 49.6); second from right shows a designated nonhit with a %SA close to the average for all compounds, toluene (%SA = 32.8) and the right most column shows the compound with the lowest %SA, triphenyl phosphate (%SA = 10.4).
Astrocyte-Senescence Screen Using PQ as a Positive Control
Given the inability to use a Day-2 NC as a threshold to identify compounds capable of inducing astrocyte senescence, we adopted a different approach based on our previous findings that human astrocytes exposed to 200 μM PQ for 24 h undergo senescence. We have previously published extensive evidence, beyond merely examining SA-β-gal, that PQ induces senescence of cultured human astrocytes (Chinta et al., 2018). In our current screen, the average %SA (48.0%) for 4 independent PQ-treated wells would rank it the third highest %SA and a single PQ well had the highest %SA we recorded (59.0%). However, the variability in %SA associated with these 4 PQ wells was high (SD = 9.4%) (Table 1). Despite this variability there was still significant difference for %SA between these PQ-treated wells and 4 untreated negative control (NC) wells at 7 days postplating (mean ± SE: 48.0 ± 5.4% (PQ) and 34.0% ± 2.0 (NC); p < .05), indicating the possibility that a similar screen where at least 4 drug replicates are used could be a feasible means of identifying compounds that significantly induce senescence. However, calculating a Z’-factor in order to estimate a separation band of positive and negative controls (using a 3*SD, and 99.73% confidence limit) indicates that %SA would not likely be an appropriate readout for a high-throughput screening assay, where large library size precludes the testing of compound in replicates (Z’ = −1.7 whereby Z < 0 indicates that the assay “quality” is too low and screening in a high-throughput manner is essentially impossible; see Zhang et al. for details related to calculating Z’.) Despite these important caveats, we prioritized the putative senescence inducers by conducting a “hit analysis” using PQ as a positive control. We defined hits as those compounds with a %SA greater than the mean–SE of the 4 PQ-treated wells (Table 1). This defined senescent inducers as those compounds causing >42.6%SA. This upper-bound threshold may well result in the exclusion of some compounds that are in fact capable of inducing astrocyte senescence. However, given the intrinsic variability of this assay and the relatively high basal %SA in 7-day untreated wells, a more stringent cutoff of mean–SE of positive control is likely necessary than the less stringent but more conventional mean–2*SD of positive control. Adopting this more conservative criterion still resulted in 9 compounds designated as putative inducers of astrocyte senescence (Table 2). Figure 2 shows a sample of representative images from which %SA were calculated, reflecting the dynamic range of %SA values. In high-throughput screens without drug replicates, setting cut-offs and definition of ‘hits’ should merely be considered a means of prioritizing compounds and is somewhat arbitrary; no matter the assay, more stringent cutoffs directs one to the most potent compounds within the chemical library while potentially mitigating the issue of false positives (Zhang et al., 1999). One of the 5 neurotoxicant-negative controls, acetylsalicylic acid, was identified as a hit capable of inducing astrocyte senescence (43%SA). Four drugs occur in duplicate in the library in order to assess “within assay reproducibility” one of which is methyl mercuric (II) chloride. The %SA could not be calculated for methyl mercuric (II) chloride as all cells were killed in both duplicates, however the average SD of the 3 remaining duplicates was approximately 5.8% and the mean of their “percent difference” was 47.5%—further indicative of substantial intraplate variability for this assay—and pointing to the need for compounds to be tested as multi-replicates (Table 1).
Table 1.
Explanation of how astro-senescence hits were defined.
| Senescence Assay Variability | ||
|---|---|---|
| Positive control for senescence | % SA | |
| PQ-1 | 59.1 | |
| PQ-2 | 47.8 | |
| PQ-3 | 36.2 | |
| PQ-4 | 48.9 | |
| Mean PQ wells | 48.0 | |
| SE of PQ wells | 5.4 | |
| Senescence cutoff (Mean–SE) | >42.6 | |
| Replicates | SA-HIT? | |
| Saccharin Sodium Salt hydrate-1 | 29.6 | NO |
| Saccharin Sodium Salt hydrate-2 | 38.3 | NO |
| Triphenyl phosphate-1 | 10.4 | NO |
| Triphenyl phosphate-2 | 28.6 | NO |
| Methyl mercuric (II) chloride-1 | N.D. | NO |
| Methyl mercuric (II) chloride-2 | N.D. | NO |
| Deltamethrin-1 | 37.8 | NO |
| Deltamethrin-2 | 29.8 | NO |
The %SA of individual wells treated with corresponding compounds is listed. The variability about the mean of 4 PQ treated wells defined a Senescence cutoff. Other compounds in duplicate demonstrate variability in measuring this endpoint.
Table 2.
List of compounds that were deemed astro-senescence inducing hits, using a PQ based cutoff.
| Astro-Senescence Inducer | Categorization | % SA | HIT? |
|---|---|---|---|
| 3, 3'-Iminodipropionitrile | DNT/NT | 49.7 | YES |
| Colchicine | DNT/NT | 49.3 | YES |
| Benz(a)anthracene | PAH | 45.0 | YES |
| Thalidomide | DNT/NT | 44.4 | YES |
| Hydroxyurea | DNT/NT | 43.9 | YES |
| Benzo (a) pyrene | PAH | 43.7 | YES |
| Acetylsalicylic acid | NC | 43.3 | YES |
| 6-Hydroxydopaminehydrochloride | DNT/NT | 43.1 | YES |
| 2-Methyoxyethanol | DNT/NT | 42.7 | YES |
| Negative Controls | |||
| Acetylsalicylic acid | NC | 43.3 | YES |
| L-Ascorbic acid | NC | 37.7 | NO |
| D-Glucitol | NC | 35.4 | NO |
| Saccharin Sodium Salt hydrate | NC | 29.7 | NO |
| Acetaminophen | NC | 18.6 | NO |
The %SA of individual wells treated with corresponding compounds is listed. This compiles all the compounds from the library that were categorized as astro-senescence inducers using a PQ %SA based cutoff, as well as compounds originally included in the library as negative controls for neurotoxicity.
Screening for Neurotoxicants That Induce Astrocytic Death
As described above, we investigated the possibility of using an increase in the percent of cells expressing SA-β-gal (relative to some standard) as an indicator that a compound is likely inducing astrocyte senescence. However, we recognize that this readout may be difficult to interpret given that some compounds would be expected to kill astrocytes. Indeed, it is expected that this screen would pull-out compounds which preferentially kill nonsenescent cells, since such effects would increase %SA, even if the absolute number of SA+ cells remained unchanged. Therefore, it may be advisable in a senescence screen to do a first pass filtering in order to exclude compounds which significantly kill astrocytes in order to eliminate some of this complexity. We therefore performed a simple cytotoxicity assay in parallel to our senescence assay. DAPI-counts of the day-2 untreated control wells enabled us to estimate a “mean cell density at the time of drug treatment” (Figure 3). We defined “astrocytic cytotoxicity” based on the number of cells in toxicant-treated wells being <84% of that of the mean of untreated day-2 controls, which equated to the mean minus 2×SD of the 4 day-2 control wells (Table 3). In other words, this endpoint defines cytotoxicity based on the fact that there were significantly (95% limit) fewer cells in a well than we predict would have been there 5 days earlier. This is obviously a conservative definition of cytotoxicity as normally cell density would increase substantially over this 5-day period as cells continue to divide; we estimate that on average cell density increases by >30% over this period—average of all toxin treated wells equals 136% relative to day-2 controls. There are 2 reasons we would advise adopting this conservative approach in a senescence screen: first, if a net loss of cells is observed compared with density at the time of drug treatment, it assures that what is being observed is cell death rather than decreased rates of proliferation (the expected effect for compounds that induce senescence); second, robustly filtering out all cytotoxic compounds including those with more mild effects on cell viability is not advisable given what we already know to be generally true for senescent-inducing agents, including PQ: the concentration range that effects cell viability partially overlaps with the range that induces senescence (Park et al., 2013). Using this measure, we identified 7 unique putative toxicants that caused loss of astrocytes (Table 4). Figure 3 shows a sampling of images from which cell densities were assessed relative to day-2 NC. None of the 6 chemicals included in the library as negative controls for neurotoxicants (Acetaminophen, D-glucitol, acetylsalicylic acid, L-ascorbic acid, and saccharin sodium salt) caused astrocyte cell death. The average SD of intraplate duplicates was approximately 7.3% (the mean of their percent difference was 8.2%), indicative of assay variability; though paired duplicates were consistent in terms of either both being above or below the 84% cutoff. Of note, methyl mercuric (II) chloride resulted in complete absence of DAPI-stained astrocytes in both duplicated wells (Figure 1; Table 4), indicative of its potent toxicity and validating the screen’s potential to at the very least consistently identify the most toxic compounds. Filtering for cytotoxicity would have excluded 2 of our senescent-inducing compounds (colchicine and 2-methyoxyethanol). Importantly, none of the 4 positive control PQ wells would have been excluded based on such filtering. On the other hand, using more sensitive “viability” measures such as an MTT assay certainly would exclude compounds such as PQ with pronounced effects on cellular metabolism. Indeed, Pei et al. previously probed the NTP neurotoxicant library for compounds that are toxic to iPSC-derived astrocytes as well as neural stem cells and neurons using the MTT assay and identified 15 toxins (10 μM–24 h) or 38 toxins (100 μM–24 h) that had significant effects on astrocyte ‘viability’. Their study corroborates 4 out of the 7 astro-toxic hits we identified in our current screen (Table 4), suggesting that methyl mercuric (II) chloride; valinomycin; colchicine and 2-methyoxyethanol “robustly” induce astrocyte cell death—although curiously one of their methyl mercuric (II) chloride duplicates showed no effect in the MTT assay even at 100 μM (Pei et al., 2016).
Figure 3.

Compounds with effects on the viability of astrocytes can be identified. Representative images of human astrocytes stained for DAPI (top) and analysis (nuclei counts—bottom) to quantify relative cell density compared with day-2 untreated controls (left most column). Images include a sample of raw data that reflects the dynamic range of compounds’ effects on cell viability. Second from left shows a nontoxic compound, acetaminophen (150.3%), second from right shows a highly astrotoxic compound, methyl mercuric (II) chloride (0%) and the right column shows the compound with the lowest relative density, that was not designated astrotoxic, benzo(a) pyrene (90.5%).
Table 3.
Explanation of how astro-toxic hits were defined
| Toxicity Assay Variability | ||
|---|---|---|
| Cell density at Day2 | % Mean Day 2 | |
| Day2-1 | 104.5 | |
| Day2-2 | 97.5 | |
| Day2-3 | 107.6 | |
| Day2-4 | 90.4 | |
| MEAN Day 2-Control wells | 100 | |
| SD of Day 2-Control wells | 7.7 | |
| Toxicity Cutoff (Mean Day 2–2× SD) | < 84.6 | |
| Replicates | Astro-toxic-HIT? | |
| Saccharin Sodium Salt hydrate-1 | 129.3 | NO |
| Saccharin Sodium Salt hydrate-2 | 136.3 | NO |
| Triphenyl phosphate-1 | 128.7 | NO |
| Triphenyl phosphate-2 | 111.5 | NO |
| Deltamethrin-1 | 138.2 | NO |
| Deltamethrin-2 | 158.0 | NO |
| Methyl mercuric (II) chloride-1 | 0 | YES |
| Methyl mercuric (II) chloride-2 | 0 | YES |
The cell density at Day 7 for individual wells treated with corresponding compounds is listed. This density is represented relative to the density at day 2 (the time point at which compounds were first applied). The variability about the mean of the 4 Day 2 wells defined a toxicity cutoff. Other compounds in duplicate demonstrate variability in measuring this endpoint.
Table 4.
List of compounds that were deemed astro-toxic, using ‘cell density at time of compound application’ as a baseline.
| Astro-Toxic Compound | Categorization | Cell Density relative to 2 Day | HIT? | MTT-Based Astro-toxic by Pei et al |
|---|---|---|---|---|
| Methyl mercuric (II) chloride | DNT/NT | 0.0 | YES | YES (100 μM) |
| Bis(tributyltin)oxide | DNT/NT | 0.0 | YES | NO |
| Valinomycin | Other | 21.7 | YES | YES (100 μM) |
| Colchicine | DNT/NT | 47.8 | YES | YES (100 μM) |
| 2-Methyoxyethanol | DNT/NT | 74.5 | YES | YES (10 and 100 μM) |
| Benzo(b)fluoranthene | PAH | 79.6 | YES | NO |
| Berberine chloride | Other | 80.9 | YES | NO |
| Negative Controls | ||||
| Acetaminophen | NC | 150.3 | NO | YES (100 μM) |
| L-Ascorbic acid | NC | 143.9 | NO | YES (10 and 100 μM) |
| D-Glucitol | NC | 138.2 | NO | YES (100 μM) |
| Acetylsalicylic acid | NC | 148.4 | NO | YES (100 μM) |
| Saccharin Sodium Salt hydrate | NC | 136.3 | NO | NO |
The cell density of individual wells treated with corresponding compounds is listed. This compiles all the compounds from the library that were categorized as astro-toxic, as well as compounds originally included in the library as negative controls for neurotoxicity.
Potential for Primary Mouse Astrocytes to Serve as a Platform for Screening Toxicants
In addition to conducting our %SA screen on iPSC-derived human astrocytes, we also performed control experiments to assess the potential for cultured primary mouse astrocytes isolated from postnatal mouse brain to serve as a screening platform where potentially PQ’s %SA could be similarly used as a threshold for identifying hits. However, mouse astrocytic cultures were found to express high basal levels of SA-β-gal (approximately 70% SA) and, perhaps as a result, demonstrated extreme cytotoxic sensitivity to PQ, such that >30 μM-induced substantial cell death (Figure 4). This is compared with human astrocytes where after 7 days postseeding %SA was observed to be approximately 30% and where moderate cell death was not observed until PQ reached 400 μM (data not shown_).
Figure 4.

Primary mouse astrocytes have high basal SA-β-gal activity and demonstrate sensitivity to PQ. Representative images of primary mouse astrocytes stained for SA-β-gal activity. Astrocytes were treated with PQ (0, 15, 31, and 62.5 μM) for 24 h and fixed 5 days later. Astrocytes show high levels of SA-β-gal activity (left most panel) and show sensitivity to PQ, such that no cells remain when astrocytes were treated with 62.5 μM PQ for 24 h (right most panel).
DISCUSSION
Although astrocytes derived from human iPSCs could potentially serve as a suitable platform for conducting a screen to identify compounds capable of inducing astrocyte senescence, our study here suggests that conducting such screens based on SA-β-gal may be challenging. As we discovered, these cells’ SA-β-gal activity increases as they are cultured (even at 3% oxygen) precluding the possibility for using an increase above “%SA right before toxin treatment” as a means to identify senescent inducing hits. Instead, we propose that a method based on a defining a hit as inducing a “%SA within the levels induced by the positive control—PQ”, may be a more suitable approach. This is of course contingent on PQ being a reliable positive control for astrocyte senescence induction. The evidence for PQ inducing astrocyte senescence can be summarized as follows: decreased expression of the mitotic marker—Ki67; decreased BrdU incorporation; increased SA-β-gal activity; increased expression of p16; decreased expression of laminB1 within nuclear laminae and decreased nuclear expression of HMGB1; increased number of 53BP1 foci—indicative of DNA damage and important in initiation of a SASP and increased expression and secretion of various SASP factors (Chinta et al., 2018). Defining hits provides an important “prioritization cutoff” (ie, which of these “hits” are likely to pass more rigorous validation studies). Our reasoning for choosing PQ at 200 μM as a “positive control” was not because it is allows for a less or more stringent definition of senescence compared with some other reference points, it is rather due to our extensive published data validating that this exposure condition induces astrocyte senescence. Based on results from this study, we suggest use of this “PQ filtering mask” to increase the % of hits that are likely to ultimately “pass validation” in follow-up experiments.
SA-β-gal activation is not required for senescence as evidence when the gene encoding β-gal is experimentally knocked down (Lee et al., 2006), although the marker is consistently activated in senescent cells precluding the likelihood of false negatives (Dimri et al., 1995). However, its activity can be elevated in nonsenescent cells (Huang and Rivera-Perez, 2014; Yang and Hu, 2005), raising the concern of false positives. Similar to molecular pathways that control apoptosis, depending on the particular cellular stress and the specific cell type, it has become more and more evident that no single maker, including SA-β-gal, will be sufficient on its own in identifying “all (and only)” senescent cells in “all” contexts (Hernandez-Segura et al., 2017). No matter the chosen readout, any toxin that purports to increase senescence will require follow-up validation using additional senescent cell markers.
With that in mind, SA-β-gal staining does provide substantial protocol streamlining and cost benefits over (immunofluorescence) IF-based screens. For technical reasons, SA-β-gal staining is not compatible with IF staining, precluding dual “staining” screens (despite attempts to troubleshoot this issue). We believe that a more efficient approach is to use SA-β-gal activity levels to help prioritize top hits and then use additional expressional analysis experiments (as we did with PQ), to help validate putative senescence-inducing compounds. Biran et al. recently published a method for combining flow cytometry with high-content image analysis, allowing for quantification (still based on SA-β-gal staining) of senescent cells. The real advance of this methodology as laid out by the authors over previous techniques, is the potential for superior assessment of cellular senescence in vivo (eg, “which cell types become senescent with age and other questions of that nature”). However, the technique could be amended to assess senescence of cultured astrocytes. As these authors concur, combinatorial staining of SA-β-gal and other senescent markers using conventional staining techniques is problematic. Their approach does allow for simultaneous assessment of SA-β-gal and other senescent markers on an individual cell basis, potentially allowing for a more accurate assessment of senescent cells. Theirs represents a promising method that could hold potential for validating candidates, particularly for ultimately validating candidates in vivo (Biran et al., 2017).
The very nature of assays which count % positive cells is not ideal as a screening readout since the 100% “ceiling” means such assays may have a relatively limited dynamic range (Zhang et al., 1999). There are 2 empirical factors which we specifically measured in human astrocytes which further compound this issue: first, the high basal %SA of astrocytes and second, the high variability associated with this readout. What adjustments could be made to decrease basal %SA in order to improve the quality of the assay? Astrocytes are a highly heterogeneous population in vivo (Allaman et al., 2011) so it is possible that a different source of iPSC-derived astrocytes may have lower basal SA-β-gal activity. Additionally, it may be possible to look at time points earlier than 5 days posttoxin treatment, eg, at a time point where PQ treatment has already resulted in an increase in %SA but where basal activity has not yet ramped-up. Baring such optimizations, in order to identify putative senescent inducers it will be necessary to carry out the screen minimally in quadruplicate and to define hits as being those which are not significantly different from a positive control (ie, PQ, also in quadruplicate), and those which have a %SA significantly greater than a day matched untreated control (ie, Day-7 NC, also in quadruplicate).
We additionally concluded from our results that the primary mouse astrocyte platform was not ideal for conducting a senescent screen due to the fact that the concentration range at which toxicants would induce quantifiable increased senescence, but not cell death, is likely extremely narrow—obviously not ideal conditions for conducting a wide-panel screen where one is attempting to tease apart toxicants’ effects on senescence from general cytotoxicity. If the correlation between basal SA-β-gal expression and toxin sensitivity generally holds true for all cell types, this would mean that any cell type which shows high basal activity of SA-β-gal in culture is not likely to serve as an ideal platform for a senescence screen due to the fact that senescent inducers will likely cause such cells to die.
We recognize that the concentration of compounds we have tested here is relatively high for a toxin screen. However, it is worth pointing out that in cases where the toxin turns out to be a general effector of senescence, preoccupation with the inability of the toxin to cross the blood brain barrier may be a moot, as cerebrovascular structures formed from senescent endothelial cells and pericytes have been shown to be compromised (Yamazaki et al., 2016). Our approach here was empirically driven by our findings that 100–250 μM PQ-induced substantial senescence without severely compromising astrocyte viability (Chinta et al., 2018). However, with an appreciation that many of the neurotoxins in the library have been demonstrated to be toxic at or below this range (Crofton et al., 2010) we more than halved our starting test concentration to 40 μM. This is still a much higher exposure level than people are likely to encounter even in “hazardous” environments; however, this raises the interesting question of how one can model exposure to toxins hypothesized to cause premature cellular aging that ultimately contributes to age-related neurodegeneration. How can environmental toxins be tested, in acute experimental screens, to reveal those which when present over a human life-time, may cause a low grade stress that contributes to age-related diseases? With this in mind, testing with higher concentrations for shorter periods of times and specifically monitoring for proxies of aging seems a worthwhile first step for highlighting chemicals with the potential to exacerbate aging and its numerous associated diseases.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by an NIEHS (R21 ES025641) and an Ellison Senior Scholar in Aging Award (J.K.A.). G.W. was funded by an NRSA Institutional Postdoctoral Training Grant (T32).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the NTP library provider, Mamta Behl and Ray Tice (of the NIEHS Neurotoxicology Program) and Shankar Chinta for his technical and scientific contributions toward this project.
REFERENCES
- Allaman I., Belanger M., Magistretti P. J. (2011). Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 34, 76–87. [DOI] [PubMed] [Google Scholar]
- Baker D. J., Petersen R. C. (2018). Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Invest. 128, 1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto G. E., Sun X., Xu L., Giffard R. G. (2011). Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One 6, e27881.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar O. A., Fuentealba L. C., Alvarez-Buylla A., Rowitch D. H. (2014). Astrocyte development and heterogeneity. Cold Spring Harb. Perspect. Biol. 7, a020362.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R., Crowe E. P., Bitto A., Moh M., Katsetos C. D., Garcia F. U., Johnson F. B., Trojanowski J. Q., Sell C., Torres C. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7, e45069.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biran A., Zada L., Abou Karam P., Vadai E., Roitman L., Ovadya Y., Porat Z., Krizhanovsky V. (2017). Quantitative identification of senescent cells in aging and disease. Aging Cell 16, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta S. J., Woods G., Demaria M., Rane A., Zou Y., McQuade A., Rajagopalan S., Limbad C., Madden D. T., Campisi J., et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta S. J., Woods G., Rane A., Demaria M., Campisi J., Andersen J. K. (2015). Cellular senescence and the aging brain. Exp. Gerontol. 68, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow M., Rubin H. (1996). Evidence for cellular aging in long-term confluent cultures: Heritable impairment of proliferation, accumulation of age pigments and their loss in neoplastic transformation. Mech. Ageing Dev. 89, 165–183. [DOI] [PubMed] [Google Scholar]
- Coppe J. P., Patil C. K., Rodier F., Sun Y., Munoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., Campisi J. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton K. M., Mundy W. R., Lein P. J., Bal-Price A., Coecke S., Seiler A. E., Knaut H., Buzanska L., Goldberg A. (2010). Developmental neurotoxicity testing: Recommendations for developing alternative methods for the screening and prioritization of chemicals. Altex 28, 9–15. [PubMed] [Google Scholar]
- Davalos A. R., Kawahara M., Malhotra G. K., Schaum N., Huang J., Ved U., Beausejour C. M., Coppe J. P., Rodier F., Campisi J. (2013). p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A., de Jong T. V., Melov S., Guryev V., Campisi J., Demaria M. (2017). Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T., Rivera-Perez J. A. (2014). Senescence-associated beta-galactosidase activity marks the visceral endoderm of mouse embryos but is not indicative of senescence. Genesis 52, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C., Xu Q., Martin T. D., Li M. Z., Demaria M., Aron L., Lu T., Yankner B. A., Campisi J., Elledge S. J. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. Y., Han J. A., Im J. S., Morrone A., Johung K., Goodwin E. C., Kleijer W. J., DiMaio D., Hwang E. S. (2006). Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195. [DOI] [PubMed] [Google Scholar]
- Lucanic M., Garrett T., Yu I., Calahorro F., Asadi Shahmirzadi A., Miller A., Gill M. S., Hughes R. E., Holden-Dye L., Lithgow G. J. (2016). Chemical activation of a food deprivation signal extends lifespan. Aging Cell 15, 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack A. L., Di Monte D. A. (2003). Effects of L-dopa and other amino acids against paraquat-induced nigrostriatal degeneration. J. Neurochem. 85, 82–86. [DOI] [PubMed] [Google Scholar]
- Mombach J. C., Vendrusculo B., Bugs C. A. (2015). A Model for p38MAPK-Induced Astrocyte Senescence. PLoS One 10, e0125217.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Jo Y. H., Cho C. H., Choe W., Kang I., Baik H. H., Yoon K. S. (2013). ATM-deficient human fibroblast cells are resistant to low levels of DNA double-strand break induced apoptosis and subsequently undergo drug-induced premature senescence. Biochem. Biophys. Res. Commun. 430, 429–435. [DOI] [PubMed] [Google Scholar]
- Pei Y., Peng J., Behl M., Sipes N. S., Shockley K. R., Rao M. S., Tice R. R., Zeng X. (2016). Comparative neurotoxicity screening in human iPSC-derived neural stem cells, neurons and astrocytes. Brain Res. 1638, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzoli G., Cereda E. (2013). Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80, 2035–2041. [DOI] [PubMed] [Google Scholar]
- Prieur A., Peeper D. S. (2008). Cellular senescence in vivo: A barrier to tumorigenesis. Curr. Opin. Cell Biol. 20, 150–155. [DOI] [PubMed] [Google Scholar]
- Salminen A., Ojala J., Kaarniranta K., Haapasalo A., Hiltunen M., Soininen H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11. [DOI] [PubMed] [Google Scholar]
- Schildge S., Bohrer C., Beck K., Schachtrup C. (2013). Isolation and culture of mouse cortical astrocytes. J Vis Exp. 19, pii: 50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist C., Horikawa I., Foran E., Major E. O., Vojtesek B., Lane D. P., Lu X., Harris B. T., Harris C. C. (2016). p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 23, 1515–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y., Baker D. J., Tachibana M., Liu C. C., van Deursen J. M., Brott T. G., Bu G., Kanekiyo T. (2016). Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 47, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N. C., Hu M. L. (2005). The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 40, 813–819. [DOI] [PubMed] [Google Scholar]
- Zhang J. H., Chung T. D., Oldenburg K. R. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.