

Abstract
We have developed an efficient synthetic strategy to assemble C3-symmetric molecules containing propellane moieties as end groups and a benzene ring as a central core. The synthesis of these C3-symmetric molecules involves simple starting materials. Our approach to C3-symmetric compounds relies on a Diels–Alder reaction, cyclotrimerization and ring-closing metathesis as key steps.
Keywords: cyclotrimerization, Diels–Alder, propellane, ring-closing metathesis
Introduction
In 1966 Ginsburg coined the word “propellane” [1–2] and Wiberg reviewed various aspects of medium and small ring propellanes [3–4]. Propellanes consist of tricyclic compounds where three rings are conjoined by a common C–C bond [1,5–6]. Heterocyclic systems contain a heteroatom (e.g., oxygen, nitrogen, and sulfur, etc.) along with carbon atoms. The name of a heterocyclic propellane may be organized by prefixing aza, oxa, etc.
Among various propellanes, nitrogen-containing compounds occupy a special place because they are present as core structural units in bioactive natural products and pharmaceuticals. Some of these propellanes exhibit intresting properties like antibiotic, antifungal, anticancer, platelet-activating factor antagonistic and antibacterial activities. The propellane skeleton is present in many alkaloids such as aknadinine (1), aknadilactam (2), and the known morphinane alkaloid sinococuline (3), which was identified as a bioactive component from S. japonica [7]. In 1963 Brown et al. isolated 1-acetyl-aspidoalbidine (4) from Vallesia dichtoma [8] and subsequently, Djerassi proposed its structure [9]. Another alkaloid (−)-aspidophytine (5) differs from 1-acetylaspidoalbidine (4) only in the degree of unsaturation and the substitution pattern on the aromatic ring (Figure 1).
Figure 1.

Various alkaloids containing propellane frame work.
The design of propellanes demands unique synthetic methods and these include: manganese or palladium-catalyzed transformations [10], the Diels–Alder (DA) reaction [11–12], and rearrangement of spiro-ketones, nucleophilic substitutions of alkenes, and photochemical addition reactions. Multicomponent reactions (MCRs) are also used for the synthesis of hetero-propellanes [13–14]. Recently, heterocyclic propellanes have been reviewed [15–16]. Our group also developed simple synthetic approaches to propellanes via ring-closing metathesis (RCM) as a key step [17–18].
The development of new synthetic strategies to C3-symmetric molecules bearing propellane moieties from commercially available starting materials is worthy of systematic investigation. To this end, our efforts are directed to design star-shaped molecules that involve a wide range of structural variations. To the best of our knowledge there are no synthetic reports available for C3-symmetric molecules bearing propellane moieties. As part of our major program aimed at designing star-shaped C3-symmetric molecules [19–30], here, we conceived new strategies to N-containing star-shaped molecules. Such star-shaped molecules are generally used in organic light-emitting diodes (OLEDs) [31–33], organic photovoltaics (OPVs) [34], organic field-effect transistors (OFETs) [35–36], and other optoelectronic devices. Our approach to C3-symmetric molecules containing propellane moieties involve DA reaction [37], cyclotrimerization [19] and RCM [38–41] as key steps.
Results and Discussion
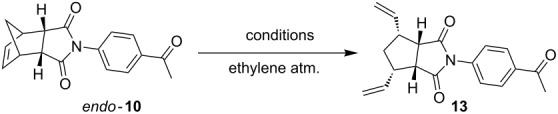
The synthesis of propellane-bearing C3-symmetric derivatives starts with commercially available dicyclopentadiene and maleic anhydride (7). Here, we used a DA reaction of freshly cracked cyclopentadiene (6) and maleic anhydride (7) to obtain the endo-DA adduct 8 [42] in 98% yield. Next, this cycloadduct 8 was treated with commercially available 4-aminoacetophenone (9) in the presence of triethylamine (Et3N) in toluene at 140 °C to obtain the acetophenone derivative 10 in excellent yield (92%) [43]. Later, the acetophenone derivative 10 was subjected to trimerization reaction under ethanol/silicon tetrachloride (EtOH/SiCl4) conditions to deliver the trimerized product 11 (64%). Having the trimerized product 11, we attempted to open the norbornene system due to the fact that not all norbornene rings open up during RCM to generate propellane derivative. After allylation, RCM is not a clean reaction and it gave a mixture of the C3-symmetrical compounds. Therefore it is desirable to open the norbornene double bond before the trimerization sequence. To this end, the trimerized product 11 was treated with Grubbs first generation (G-I) catalyst in CH2Cl2 under ethylene atmosphere but, we were unable to get the ring-opened product 12 (Scheme 1).
Scheme 1.

Synthesis of the star-shaped norbornene derivative 11 via trimerization.
Later, we considered an alternate route to synthesize compound 12. In this regard, we employed different ruthenium-based catalysts (Figure 2) and reaction conditions to obtain the ring-opening metathesis (ROM) product 13 from norbornene derivate 10. Under these conditions the starting material was not consumed completely. After some experimentation, we found that G-I catalyst (5 mol %) in CH2Cl2 is suitable to generate the ROM product 13 in 56% yield (Table 1).
Figure 2.

Selected list of ruthenium-based catalysts used for ROM.
Table 1.
Different conditions attempted to obtain the ROM product 13.
 | ||||||
| entry | catalyst | mol % | solvent | temp | time (h) | yield (%) |
| 1 | G-I | 5 or 10 | CH2Cl2 | rt | 48 | 56 |
| 2 | G-I | 5 or 10 | CH2Cl2 | reflux | 32 | 48 |
| 4 | G-II | 5 or 10 | toluene | rt | 46 | 24 |
| 5 | G-II | 5 | toluene | reflux | 43 | 20 |
| 6 | Ru-II | 5 or 10 | CH2Cl2 | rt | 48 | 52 |
| 7 | GH-I | 5 or 10 | CH2Cl2 | rt | 40 | 53 |
| 8 | GH-I | 5 or 10 | CH2Cl2 | reflux | 40 | 50 |
Having the ROM product 13 in hand, it was subjected to trimerization in the presence of EtOH/SiCl4 at 0 °C to room temperature to afford the trimerized product 12 in 54% yield. Next, the C3-symmetric product 12 was reacted with allyl bromide in the presence of sodium bis(trimethylsilyl)amide (NaHMDS, 1 M solution in THF) at −75 °C to deliver the RCM precursor 14 in good yield (78%). The hexaallyl derivative 14 was subjected to RCM in the presence of Grubbs second generation (G-II) catalyst in CH2Cl2 under nitrogen to give the propellane moiety bearing C3-symmetric product 15 in good yield (87%). Its structure was established on the basis of NMR spectral data, and its molecular formula was confirmed by HRMS data (Scheme 2).
Scheme 2.

Synthesis of the C3-symmetric molecule 15 bearing propellane moieties via trimerization and RCM.
Along similar lines, we expanded the scope of this strategy. To this end, commercially available anthracene (16) was reacted with maleic anhydride (7) in a screw-capped tube at 150 °C in o-xylene to obtain the DA adduct 17 in 94% yield [44–45]. Later, the DA adduct 17 was treated with 4-aminoacetophenone (9) in the presence of Et3N in toluene at 140 ºC to deliver the acetophenone derivative 18 (91% yield) and it was subjected to trimerization in the presence of EtOH/SiCl4 at 0 °C to rt to obtain the trimerized product 19 in 64% yield. Afterwards, the trimerized product 19 was treated with allyl bromide to accomplish C-allylation in the presence of NaHMDS (1 M solution in THF) at −75 °C to deliver the hexaallyl derivative 20 in 84% yield. Then, RCM in the presence of G-II catalyst in CH2Cl2 under nitrogen atmosphere gave the propellane moieties bearing C3-symmetric product 21 in good yield (91%). Its structure was established with the help of 1H NMR, 13C NMR spectral data and was further supported by HRMS details (Scheme 3).
Scheme 3.

Synthesis of C3-symmetric molecule 21 bearing propellane moieties via trimerization and RCM.
Conclusion
We have demonstrated a simple synthetic methodology to C3-symmetric star-shaped molecules containing propellane moieties at the periphery which may be useful for material science applications. Here, we have prepared DA adducts 8 and 17 from commercially available maleic anhydride (7), which was further utilized for trimerization and RCM sequence. We have successfully synthesized C3-symmetric molecules 15 and 21 bearing propellane moieties by employing RCM in the presence of 2nd generation (G-II) catalyst.
Experimental
General information
Some of these reactions were carried out in screw-capped tubes and other reactions under nitrogen or argon and ethylene atmosphere in oven-dried glassware. Air- and moisture-sensitive reactions were performed in degassed solvents. Transfer of moisture-sensitive materials were carried out using standard syringe−septum techniques. All the commercial grade reagents were used without any purification until otherwise specified. Melting points were recorded on a Veego or Büchi melting point apparatus and are uncorrected. NMR Spectra were generally recorded on Bruker (Avance 400 or Avance III 500) spectrometers operated at 400 or 500 MHz for 1H and 100 or 125.7 MHz for 13C nuclei. NMR Samples were generally made in chloroform-d solvent, and chemical shifts (δ values) are reported in parts per million (ppm). Coupling constants (J values) were reported in hertz (Hz). HRMS measurements were carried out using a Bruker (Maxis Impact) spectrometer. IR spectra were recorded on a Nicolet Impact-400 or Cary 630 FTIR spectrometer.
Synthesis of norbornene-based trimerized product 11
To a solution of norbornene derivative 10 (500 mg, 1.77 mmol) in EtOH (8 mL), silicon tetrachloride (SiCl4, 0.61 mL, 5.36 mmol) was added dropwise at 0 °C and the reaction mixture was stirred for 10–15 min at the same temperature. Later, the reaction mixture was stirred at room temperature for 20 h. After completion of the reaction (TLC monitoring), the reaction mixture was quenched with sat. aq NH4Cl. Thereafter, the reaction mixture was diluted with EtOAc (10 mL) washed with water and brine (2 × 10 mL). Then, the aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by silica gel column chromatography (65% EtOAc/petroleum ether) to afford the trimerized product 11 (321 mg, 64%) as a colourless solid. Rf = 0.54 (6:4 EtOAc/petroleum ether); mp 203–206 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 5.2 Hz, 6H), 7.66 (s, 3H), 7.24 (d, J = 2.4 Hz, 6H), 6.28 (s, 6H), 3.51–3.44 (m, 12H), 1.79 (d, J = 8.8 Hz, 3H), 1.61 (d, J = 8.8 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 177.0, 141.8, 141.3, 134.8, 131.4, 128.2, 127.2, 125.7, 52.4, 46.0, 45.7 ppm; HRMS (ESI, Q-ToF) m/z: [M + H]+ calcd for C51H40N3O6, 790.2912; found, 790.2918; IR (neat)  max: 2918, 1706, 1512, 1371, 1173, 754 cm−1.
max: 2918, 1706, 1512, 1371, 1173, 754 cm−1.
Synthesis of ring open metathesis (ROM) product 13
The solution of compound 10 (500 mg, 1.76 mmol) in dry CH2Cl2 (25 mL) was degasified by ethylene and G-I (5 mol %) was added to the reaction mixture at rt. Further, the reaction mixture was stirred for 48 h under ethylene atmosphere at rt. After completion of the reaction (TLC monitoring), the solvent was removed under reduced pressure. Later, the crude product was purified by silica gel column chromatography (30% EtOAc/petroleum ether) to obtain the ROM product 13 as a colourless solid (310 mg, 56%); Rf = 0.68 (4:6 EtOAc/petroleum ether); mp 143–145 °C; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 6.12–6.03 (m, 2H), 5.20–5.15 (m, 4H), 3.43 (q, J = 2.0 Hz, 2H), 3.08–3.00 (m, 2H), 2.60 (s, 3H), 2.08–2.02 (m, 1H), 1.57 (t, J = 6.4 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 197.2, 175.4, 136.7, 136.2, 136.1, 129.2, 126.5, 116.3, 49.1, 46.4, 35.4, 26.8 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C19H19NO3·Na, 332.1257; found, 332.1254; IR (neat)  max: 2325, 1671, 1263, 746 cm−1.
max: 2325, 1671, 1263, 746 cm−1.
Synthesis of trimerized compound 12
Based on the earlier procedure of trimerization, compound 13 (500 mg, 1.61 mmol) was treated with SiCl4 (0.55 mL, 4.84 mmol) in the presence of EtOH (8 mL) for 16 h to afford trimerized product 12 after silica gel column chromatography (60% EtOAc/petroleum ether) as a colourless solid (254 mg, 54%); mp 152–154 °C; Rf = 0.55 (5:5 EtOAc/petroleum ether); 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.0 Hz, 6H), 7.70 (s, 3H), 7.37 (d, J = 8.4 Hz, 6H), 6.15–6.07 (m, 6H), 5.19 (q, J = 8.4 Hz, 12H), 3.43 (dd, J1 = 2.0 Hz, J2 = 2.0 Hz, 6H), 3.07–2.99 (m, 6H), 2.04 (q, J = 6.8 Hz, 3H), 1.58 (q, J = 13.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 175.7, 141.7, 141.1, 136.4, 131.4, 128.1, 126.9, 125.6, 116.1, 49.1, 46.3, 35.5 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C57H51N3O6·Na, 896.3670; found; 896.3678; IR (neat)  max: 2342, 1709, 1512, 1183, 919, 736 cm−1.
max: 2342, 1709, 1512, 1183, 919, 736 cm−1.
Synthesis of trimerized product 19
Based on the earlier procedure of trimerization, compound 18 (500 mg, 1.27 mmol) was treated with SiCl4 (0.43 mL, 3.83 mmol) in the presence of EtOH (8 mL) for 20 h to give the trimerized product 19 after silica gel (100–200 mesh) column chromatography (50% EtOAc/petroleum ether) as a colourless solid (324 mg, 64%); Rf = 0.61 (4:6 EtOAc/petroleum ether); mp 224–226 °C; 1H NMR (500 MHz, CDCl3) δ 7.56 (s, 3H), 7.51 (d, J = 8.0 Hz, 6H), 7.43 (d, J = 3.0 Hz, 6H), 7.37 (d, J = 3.0 Hz, 6H), 7.24–7.22 (m, 12H), 6.62 (d, J = 8.5 Hz, 6H), 4.92 (s, 6H), 3.41 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3) δ 176.3, 141.5, 141.4, 138.9, 128.2, 127.4, 127.1, 127.0, 125.3, 124.5, 47.2, 46.1 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C78H51N3O6·Na, 1148.3670; found, 1148.3672; IR (neat)  max: 2318, 1266, 745, 707 cm−1.
max: 2318, 1266, 745, 707 cm−1.
Synthesis of hexaallyl derivative 14
To the solution of compound 12 (200 mg, 0.22 mmol) in anhydrous THF (15 mL) was added NaHMDS (2 mL of 1 M solution in THF, 1.93 mmol) at −75 °C and the reaction mixture was stirred for 30 min under nitrogen atmosphere. Then allyl bromide (0.11 mL, 1.60 mmol) was added to the reaction mixture and stirred for 2 h at −75 °C. Later, the reaction mixture was stirred to room temperature for 10 h. After completion of the reaction (TLC monitoring), the reaction mixture was quenched with 1 M aq HCl solution, and the aqueous layer was extracted by EtOAc (3 × 10 mL). Then the organic fraction was washed with brine solution, dried over Na2SO4 and concentrated. The crude residue was purified by silica gel column chromatography (10% EtOAc/petroleum ether) to afford hexa-allyl derivative 14 as a colourless solid (199 mg, 78%). Rf = 0.60 (3:7 EtOAc/petroleum ether); mp 204–206 °C; 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 5.5 Hz, 6H), 7.69 (s, 3H), 7.33 (d, J = 8.5 Hz, 6H), 6.08–5.96 (m, 12H), 5.28–5.13 (m, 24H), 2.77–2.66 (m, 18H), 2.04–2.00 (m, 3H), 1.65 (q, J = 12.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 178.2, 141.9, 141.3, 136.5, 132.8, 131.5, 128.1, 127.1, 125.7, 120.3, 117.1, 59.9, 51.2, 36.6, 35.1 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C75H75N3O6·Na, 1136.5548; found, 1136.5544; IR (neat)  max: 2345, 1671, 1263, 746 cm−1.
max: 2345, 1671, 1263, 746 cm−1.
Synthesis of hexaallyl product 20
Based on the earlier procedure of allylation, compound 19 (336 mg, 0.29 mmol) was treated with NaHMDS (2.3 mL of 1 M solution in THF, 2.39 mmol) and allyl bromide (0.14 mL, 1.93 mmol) for 17 h to deliver hexaallyl product 20 after silica gel column chromatography (20% EtOAc/petroleum ether) as a colourless solid (345 mg, 84%); Rf = 0.83 (2:8 EtOAc/petroleum ether); mp 195–197 °C; 1H NMR (400 MHz, CDCl3) δ 7.57 (s, 3H), 7.51 (d, J = 8.8 Hz, 6H), 7.40 (q, J = 3.2 Hz, 6H), 7.32 (q, J = 3.2 Hz, 6H), 7.24–7.20 (m, 12H), 6.58 (d, J = 8.0 Hz, 6H), 6.33–6.23 (m, 6H), 5.20 (dd, J1 = 11.6 Hz, J2 = 17.2 Hz, 12H), 4.68 (s, 6H), 2.45 (dd, J1 = 5.6 Hz, J2 = 5.6 Hz, 6H), 2.16 (q, J = 8.8 Hz, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 178.5, 141.7, 141.4, 139.9, 139.4, 133.6, 131.0, 128.1, 127.2, 127.1, 126.7, 126.5, 125.6, 125.3, 119.4, 55.7, 51.6, 37.3 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C96H75N3O6·Na, 1388.5548; found, 1389.5585; IR (neat)  max: 2925, 2335, 1706, 1461, 1376, 1273, 741 cm–1.
max: 2925, 2335, 1706, 1461, 1376, 1273, 741 cm–1.
General procedure for ring-closing metathesis (RCM)
The solution of hexaallyl derivatives 14 or 20 in dry CH2Cl2 (20 mL) was degassed by nitrogen and G-II (10 mol %) was added to the reaction mixture. Further, the reaction mixture was stirred for 6 h under nitrogen atmosphere at room temperature. After completion of the reaction (TLC monitoring), the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography (EtOAc/petroleum ether) to afford the propellane bearing C3-symmetric products 15 or 21.
Synthesis of RCM derivative 15
Colourless solid, 87% (121 mg, starting with 150 mg of hexaallyl compound 14); Rf = 0.60 (3:7 EtOAc/petroleum ether); mp 272–275 °C; 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 2.0 Hz, 6H), 7.69 (s, 3H), 7.35 (d, J = 8.4 Hz, 6H), 6.08–5.99 (m, 12H), 5.18–5.14 (m, 12H), 2.76 (dd, J1 = 4.0 Hz, J2 = 3.2 Hz, 6H), 2.67–2.61 (m, 6H), 2.23 (dd, J1 = 2.0 Hz, J2 = 2.0 Hz, 6H), 2.04–1.98 (m, 3H), 1.58 (q, J = 12.8, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 178.2, 141.8, 141.2, 136.2, 131.6, 128.5, 128.1, 126.9, 125.7, 116.4, 58.7, 53.3, 35.7, 30.9 ppm; HRMS (ESI, Q-ToF) m/z: [M + Na]+ calcd for C69H63N3O6·Na, 1052.4609; found, 1052.4617; IR (neat)  max: 2305, 1651, 1363, 844 cm−1.
max: 2305, 1651, 1363, 844 cm−1.
Synthesis of RCM derivative 21
Colourless solid, 91% (258 mg, starting with 300 mg of hexaallyl product 20); Rf = 0.75 (3:7 EtOAc/petroleum ether); mp 264–267 °C; 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 3H), 7.49 (d, J = 6.8 Hz, 6H), 7.43 (d, J = 2.4 Hz, 6H), 7.34 (d, J = 2.4 Hz, 6H), 7.25–7.23 (m, 12H), 6.57 (d, J = 7.6 Hz, 6H), 5.83 (s, 6H), 4.47 (s, 6H), 2.90 (d, J = 14.4 Hz, 6H), 1.80 (d, J = 14.8 Hz, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 180.0, 141.6, 141.4, 140.4, 140.2, 131.2, 128.0, 127.9, 127.2, 127.1, 127.0, 126.5, 125.6, 125.2, 57.4, 51.5, 30.2 ppm; HRMS (ESI, Q-ToF) m/z: [M + K]+ calcd for C90H63N3O6·K, 1320.4348; found, 1320.4344; IR (neat)  max: 2328, 1708, 1383, 837, 690 cm−1.
max: 2328, 1708, 1383, 837, 690 cm−1.
Supporting Information
Copies of 1H, 13C NMR and HRMS spectra of new compounds.
Acknowledgments
We thank the Department of Science and Technology (DST), New Delhi, India, for financial support and IIT Bombay, for recording spectral data. S.K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship (SR/S2/JCB-33/2010), Praj industries, Pune for Pramod Chaudhari Chair Professorship (Green Chemistry) and CSIR (02(0272)/16/EMR-II). S.T. thanks the IIT Bombay for the award of a research fellowship. V.R.A. thanks the CSIR-New Delhi and IIT Bombay for the award of a research fellowship.
This article is part of the thematic issue "Progress in metathesis chemistry III".
References
- 1.Altman J, Babad E, Itzchaki J, Ginsburg D. Tetrahedron. 1966;22:279–304. doi: 10.1016/S0040-4020(01)82189-X. [DOI] [Google Scholar]
- 2.Ginsburg D. Acc Chem Res. 1969;2:121–128. doi: 10.1021/ar50016a005. [DOI] [Google Scholar]
- 3.Wiberg K B. Acc Chem Res. 1984;17:379–386. doi: 10.1021/ar00107a001. [DOI] [Google Scholar]
- 4.Wiberg K B. Acc Chem Res. 1996;29:229–234. doi: 10.1021/ar950207a. [DOI] [Google Scholar]
- 5.Weber R W, Cook J M. Can J Chem. 1978;56:189–192. doi: 10.1139/v78-030. [DOI] [Google Scholar]
- 6.Schneider L M, Schmiedel V M, Pecchioli T, Lentz D, Merten C, Christmann M. Org Lett. 2017;19:2310–2313. doi: 10.1021/acs.orglett.7b00836. [DOI] [PubMed] [Google Scholar]
- 7.Carroll A R, Arumugan T, Redburn J, Ngo A, Guymer G P, Forster P I, Quinn R J. J Nat Prod. 2010;73:988–991. doi: 10.1021/np100009j. [DOI] [PubMed] [Google Scholar]
- 8.Brown K, Jr, Budzikiewicz H, Djerassi C. Tetrahedron Lett. 1963;4:1731–1736. doi: 10.1016/S0040-4039(01)90904-9. [DOI] [Google Scholar]
- 9.Walser A, Djerassi C. Helv Chim Acta. 1965;48:391–404. doi: 10.1002/hlca.19650480220. [DOI] [Google Scholar]
- 10.Asahi K, Nishino H. Tetrahedron. 2008;64:1620–1634. doi: 10.1016/j.tet.2007.12.017. [DOI] [Google Scholar]
- 11.Diels O, Alder K. Chem Ber. 1929;62:2081–2087. doi: 10.1002/cber.19290620829. [DOI] [Google Scholar]
- 12.Diels O, Alder K. Justus Liebigs Ann Chem. 1931;486:191–202. doi: 10.1002/jlac.19314860110. [DOI] [Google Scholar]
- 13.Yamamoto N, Fujii H, Nemoto T, Nakajima R, Momen S, Izumimoto N, Hasebe K, Mochizuki H, Nagase H. Bioorg Med Chem Lett. 2011;21:4104–4107. doi: 10.1016/j.bmcl.2011.04.147. [DOI] [PubMed] [Google Scholar]
- 14.Alizadeh A, Bayat F, Sadeghi V. Lett Org Chem. 2015;12:153–158. doi: 10.2174/1570178612666150108003359. [DOI] [Google Scholar]
- 15.Pihko A J, Koskinen A M. Tetrahedron. 2005;61:8769–8807. doi: 10.1016/j.tet.2005.06.013. [DOI] [Google Scholar]
- 16.Yavari I, Khajeh-Khezri A. Mol Diversity. 2017;21:849–854. doi: 10.1007/s11030-017-9761-8. [DOI] [PubMed] [Google Scholar]
- 17.Kotha S, Chinnam A K, Tiwari A. Beilstein J Org Chem. 2013;9:2709–2714. doi: 10.3762/bjoc.9.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotha S, Manivannan E, Ganesh T, Sreenivasachary N, Deb A. Synlett. 1999:1618–1620. doi: 10.1055/s-1999-2896. [DOI] [Google Scholar]
- 19.Kotha S, Chakraborty K, Brahmachary E. Synlett. 1999:1621–1623. doi: 10.1055/s-1999-2895. [DOI] [Google Scholar]
- 20.Dash B P, Satapathy R, Maguire J A, Hosmane N S. Org Lett. 2008;10:2247–2250. doi: 10.1021/ol8005248. [DOI] [PubMed] [Google Scholar]
- 21.Dash B P, Satapathy R, Gaillard E R, Maguire J A, Hosmane N S. J Am Chem Soc. 2010;132:6578–6587. doi: 10.1021/ja101845m. [DOI] [PubMed] [Google Scholar]
- 22.Kashiki T, Kohara M, Osaka I, Miyazaki E, Takimiya K. J Org Chem. 2011;76:4061–4070. doi: 10.1021/jo2005044. [DOI] [PubMed] [Google Scholar]
- 23.Mbyas Saroukou M S, Skalski T, Skene W G, Lubell W D. Tetrahedron. 2014;70:450–458. doi: 10.1016/j.tet.2013.11.043. [DOI] [Google Scholar]
- 24.Dash J, Trawny D, Rabe J P, Reissig H-U. Synlett. 2015;26:1486–1489. doi: 10.1055/s-0034-1380716. [DOI] [Google Scholar]
- 25.Preis E, Dong W, Brunklaus G, Scherf U. J Mater Chem. 2015;3:1582–1587. doi: 10.1039/C4TC02664K. [DOI] [Google Scholar]
- 26.Shah S R, Thakore R R, Vyas T A, Sridhar B. Synlett. 2016;27:294–300. doi: 10.1055/s-0035-1560576. [DOI] [Google Scholar]
- 27.Kotha S, Todeti S, Gopal M B, Datta A. ACS Omega. 2017;2:6291–6297. doi: 10.1021/acsomega.7b00941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotha S, Todeti S, Das T, Datta A. ChemistrySelect. 2018;3:136–141. doi: 10.1002/slct.201702675. [DOI] [Google Scholar]
- 29.Kotha S, Todeti S, Das T, Datta A. Tetrahedron Lett. 2018;59:1023–1027. doi: 10.1016/j.tetlet.2018.01.084. [DOI] [Google Scholar]
- 30.Thallapally P K, Chakraborty K, Carrell H L, Kotha S, Desiraju G R. Tetrahedron. 2000;56:6721–6728. doi: 10.1016/S0040-4020(00)00493-2. [DOI] [Google Scholar]
- 31.Justin Thomas K R, Lin J T, Tao Y-T, Ko C-W. Chem Mater. 2002;14:1354–1361. doi: 10.1021/cm010976q. [DOI] [Google Scholar]
- 32.Shirota Y. J Mater Chem. 2000;10:1–25. doi: 10.1039/A908130E. [DOI] [Google Scholar]
- 33.Kimura M, Kuwano S, Sawaki Y, Fujikawa H, Noda K, Taga Y, Takagi K. J Mater Chem. 2005;15:2393–2398. doi: 10.1039/b502268a. [DOI] [Google Scholar]
- 34.Yu G, Gao J, Hummelen J C, Wudl F, Heeger A J. Science. 1995;270:1789–1791. doi: 10.1126/science.270.5243.1789. [DOI] [Google Scholar]
- 35.Hoang M H, Cho M J, Kim D C, Kim K H, Shin J W, Cho M Y, Joo J-s, Choi D H. Org Electron. 2009;10:607–617. doi: 10.1016/j.orgel.2009.02.021. [DOI] [Google Scholar]
- 36.Ponomarenko S A, Kirchmeyer S, Elschner A, Huisman B-H, Karbach A, Drechsler D. Adv Funct Mater. 2003;13:591–596. doi: 10.1002/adfm.200304363. [DOI] [Google Scholar]
- 37.Fringuelli F, Taticchi A. Dienes in the Diels–Alder Reaction. New York: Wiley; 1990. [Google Scholar]
- 38.Shafi S, Kędziorek M, Grela K. Synlett. 2011:124–128. doi: 10.1055/s-0030-1259083. [DOI] [Google Scholar]
- 39.Grubbs R H, O’Leary D J. Handbook of Metathesis, Application in Organic Synthesis. 2nd ed. Weinheim, Germany: Wiley-VCH; 2015. [Google Scholar]
- 40.Kotha S, Aswar V R. Org Lett. 2016;18:1808–1811. doi: 10.1021/acs.orglett.6b00537. [DOI] [PubMed] [Google Scholar]
- 41.Kotha S, Shah V R, Mandal K. Adv Synth Catal. 2007;349:1159–1172. doi: 10.1002/adsc.200600469. [DOI] [Google Scholar]
- 42.Kiriazis A, af Gennäs G B, Talman V, Ekokoski E, Ruotsalainen T, Kylänlahti I, Rüffer T, Wissel G, Xhaard H, Lang H, et al. Tetrahedron. 2011;67:8665–8670. doi: 10.1016/j.tet.2011.09.044. [DOI] [Google Scholar]
- 43.Kocyigit U M, Budak Y, Gürdere M B, Tekin Ş, Köprülü T K, Ertürk F, Özcan K, Gülçin İ, Ceylan M. Bioorg Chem. 2017;70:118–125. doi: 10.1016/j.bioorg.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Marsh B J, Adams H, Barker M D, Kutama I U, Jones S. Org Lett. 2014;16:3780–3783. doi: 10.1021/ol5016702. [DOI] [PubMed] [Google Scholar]
- 45.Obermayer D, Znidar D, Glotz G, Stadler A, Dallinger D, Kappe C O. J Org Chem. 2016;81:11788–11801. doi: 10.1021/acs.joc.6b02242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H, 13C NMR and HRMS spectra of new compounds.