

ABSTRACT
Infections caused by methicillin-resistant Staphylococcus aureus (MRSA) are a growing health threat worldwide. Efforts to identify novel antibodies that target S. aureus cell surface antigens are a promising direction in the development of antibiotics that can halt MRSA infection. We biochemically and structurally characterized three patient-derived MRSA-targeting antibodies that bind to wall teichoic acid (WTA), which is a polyanionic surface glycopolymer. In S. aureus, WTA exists in both α- and β-forms, based on the stereochemistry of attachment of a N-acetylglucosamine residue to the repeating phosphoribitol sugar unit. We identified a panel of antibodies cloned from human patients that specifically recognize the α or β form of WTA, and can bind with high affinity to pathogenic wild-type strains of S. aureus bacteria. To investigate how the β-WTA specific antibodies interact with their target epitope, we determined the X-ray crystal structures of the three β-WTA specific antibodies, 4462, 4497, and 6078 (Protein Data Bank IDs 6DWI, 6DWA, and 6DW2, respectively), bound to a synthetic WTA epitope. These structures reveal that all three of these antibodies, while utilizing distinct antibody complementarity-determining region sequences and conformations to interact with β-WTA, fulfill two recognition principles: binding to the β-GlcNAc pyranose core and triangulation of WTA phosphate residues with polar contacts. These studies reveal the molecular basis for targeting a unique S. aureus cell surface epitope and highlight the power of human patient-based antibody discovery techniques for finding novel pathogen-targeting therapeutics.
Keywords: Wall Teichoic Acid, WTA, Staphylococcus aureus, monoclonal antibodies, antibody structure, antibody-carbohydrate interactions
Introduction
The Gram-positive coccal bacterium Staphylococcus aureus is a common member of the human microbiota, as well as a leading cause of bacterial infections in both hospital and community settings.1,2 While S. aureus infections are usually localized to the skin and respiratory system, life-threatening illness can occur after dissemination of the bacteria to the bloodstream, which can lead to sepsis and infection of the heart (endocarditis), bone (osteomyelitis), and lungs (necrotizing pneumonia).2 While therapy with common β-lactam antibiotics often resolves S. aureus infection, the emergence and spread of methicillin-resistant S. aureus (MRSA) has made treatment increasingly problematic.3 MRSA is resistant to all known β-lactam antibiotics, and reduced susceptibility to last-line antibiotics such as vancomycin, linezolid and daptomycin has also been reported.4
The spread of multi-drug resistance (MDR) in pathogenic bacteria is reducing the number of effective antibiotics for severe human infections,3 yet the discovery of new classes of antibiotics has severely declined in the past few decades.5 The recent increase in MDR infections has revitalized interest in antibody-based approaches to antibiotic discovery,6 with molecules targeting both Gram-positive and Gram-negative pathogens an active area of discovery. The use of animal serum containing antibodies that target bacterial antigens was a common therapeutic approach pre-dating the development of small molecule antibiotics,7 but technological advances in antibody discovery, including B-cell cloning from human patients,8 has invigorated research into monoclonal antibody (mAb)-based antibiotics. Anti-infective biologics with novel mechanisms of action targeting S. aureus alpha toxin, 9,10 protective antigen of Bacillus anthracis,11 toxins A and B from Clostridium difficile,12 and Pseudomonas aeruginosa cell wall components13,14 are currently approved or in clinical development. The presence of cell wall glycopolymers, particularly in Gram-positive bacteria, shields many epitopes that might confer bactericidal activity from antibodies and other host-defense molecules,15 but these cell wall polysaccharides also represent a potential target for mAb therapeutics. An important component of immune defenses against bacteria is a repertoire of germline “natural antibodies” that have affinity for non-protein bacterial epitopes such as carbohydrates.16,17 These antibodies are produced in the absence of T-cell stimulation early in the course of infection, with the best-characterized Gram-positive responses against cell wall epitopes such as poly-N-acetylglucosamine (PNAG) and capsular polysaccharide (CP) teichoic acids from Staphylococcus species18,19 and Streptococcus pneumoniae.20 In addition, CPs are an important component of several bacterial vaccines,21 and opsonizing antibodies targeting polysaccharides have been investigated as therapeutic agents for Burkholderia dolosa22 and S. aureus.8,22
Wall teichoic acids (WTAs) are polyanionic cell-surface glycopolymers attached to the peptidoglycan layer of Gram-positive bacteria.15 WTA varies in structure from species to species, with the S. aureus WTA molecule consisting of a short polysaccharide “linkage unit” connected to a N-acetylmuramic acid residue within the peptidoglycan layer, followed by an extended ribitol poly-phosphate polymer23 (Figure 1). Decorating each ribitol at the 2- and 4-position carbon atoms are an α- or β-linked N-acetylglucosamine (α/β-O-GlcNAc), respectively, and an ester-linked D-alanine (D-alanyl ester). S. aureus WTA is synthesized by a group of eight enzymes found in the Tar (teichoic acid ribitol) operon,24 with functionalization of the ribitol groups performed by the enzymes TarM and TarS (α-GlcNAc and β-GlcNAc)25 and enzymes from the Dlt operon (D-alanyl ester)26 (Figure 1). Importantly, S. aureus WTA has vital roles in colonization and virulence27 with terminal modifications such as D-alanylation and β-O-GlcNAcylation required for resistance to anti-microbial peptides28 and methicillin,29 suggesting that the presence of WTA is essential for productive infection. Moreover, WTA has been shown to be a target of host antibody responses,30,31 and therefore could be a target for antibody-based therapeutic development.
Figure 1.
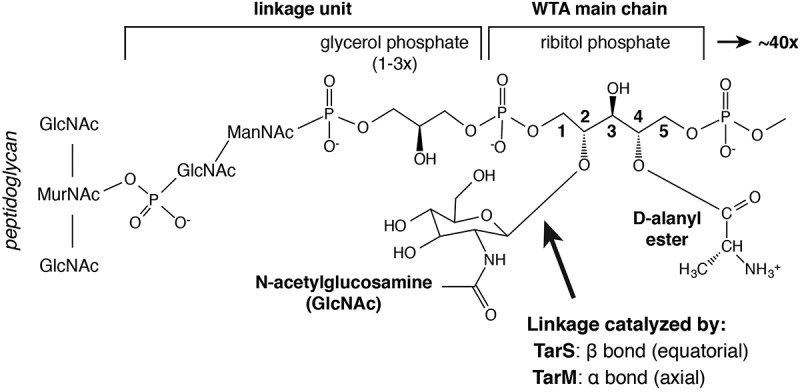
Schematic of S. aureus Wall Teichoic Acid (WTA) molecular structure. WTA polymers are linked to N-acetylmuramic acid (MurNAc) residues in the peptidoglycan backbone. The WTA “linkage unit” is bound to MurNAc via a phosphodiester bond to a N-acetylglucosamine (GlcNAc)–N-acetylmannosamine (ManNAc) disaccharide, which is in turn linked by a second phosphodiester bond to up to three repeating units of glycerol phosphate. The WTA main chain is composed of up to 40 repeats of ribitol phosphate. The linear WTA ribitol sugars are decorated at the 2-position by GlcNAc and at the 4-position by D-Alanine. GlcNAc linkages can be in the equatorial (β, as depicted) or axial (α) anomeric conformation, and are catalyzed by the glycosyltransferases TarS and TarM, respectively.
Conjugating highly potent anti-proliferative compounds to antibodies in order to deliver small drug doses directly to a target cell has been shown to be an effective approach for cancer therapy.32 Intriguingly, recent studies suggest this type of approach may also work for treatment of MDR bacteria. Antibodies specific for S. aureus WTA conjugated to a highly potent rifampicin analog have been shown to have potent bactericidal activity against MRSA in preclinical models of infection.33 Here, we structurally characterized three WTA-reactive antibodies from this antibody-antibiotic conjugate (AAC) program that were cloned from a patient recovering from S. aureus infection. We show that each of the three antibodies is specific for the WTA phosphoribitol repeating unit containing a β-linked GlcNAc at the C2 position. Crystallization of these antibodies with a minimal synthetic unit of WTA shows that all three antibodies utilize structurally distinct mechanisms to interact with WTA, yet follow conserved recognition principles to specifically recognize the β-anomeric form. These structures reveal that the highly abundant WTA polysaccharide on the surface of S. aureus provides a unique epitope that can be targeted by mAbs, and suggest that cell surface glycopolymers may be an underappreciated target for antibodies against MDR bacteria.
Results
Characterization of human antibodies specific for S. aureus WTA
Development of the anti-MRSA AAC molecule began with the characterization of a panel of anti-S. aureus antibodies that were cloned from B cells derived from patients recovering from S. aureus infection.33 These mAbs were screened for binding to a panel of clinically relevant S. aureus strains in vitro, and for binding to the highly virulent community-acquired MRSA strain USA300 strain immediately after isolation from infected mouse kidneys. Four of the mAbs (4462, 4497, 6078, and 7578) were found to bind to wild-type S. aureus USA300 when analyzed by flow cytometry (Figure 2). Each of the four mAbs showed a 2 to 3 log increased fluorescence compared to isotype control, indicating high abundance of their cognate epitope. To further characterize the epitope targeted by these antibodies, we generated genetic deletion mutants of two glycosyltransferase enzymes, TarM and TarS. These mutants were created in a protein-A-deficient S. aureus USA300 strain to facilitate analysis of the specific binding of these antibodies to their cognate epitopes. These glycosyltransferases catalyze the addition either α- or β-linked GlcNAc, respectively, to the repeating phospho-ribitol unit.29,34 Using these strains, we tested the ability of the panel of four antibodies to bind S. aureus that displayed WTA containing either α- or β-linked GlcNAc. Flow cytometry analysis showed that three antibodies, 4462, 4497 and 6078, bound the S. aureus USA300 ΔtarM, but not the S. aureus USA300 ΔtarS strain (Figure 2A-C). Conversely, the 7578 antibody only bound the ΔtarS strain (Figure 2D). These data indicate that 4462, 4497, and 6078 are specific for the β-anomeric GlcNAc form of WTA (β-WTA), while 7578 is specific for the α-anomeric form.
Figure 2.
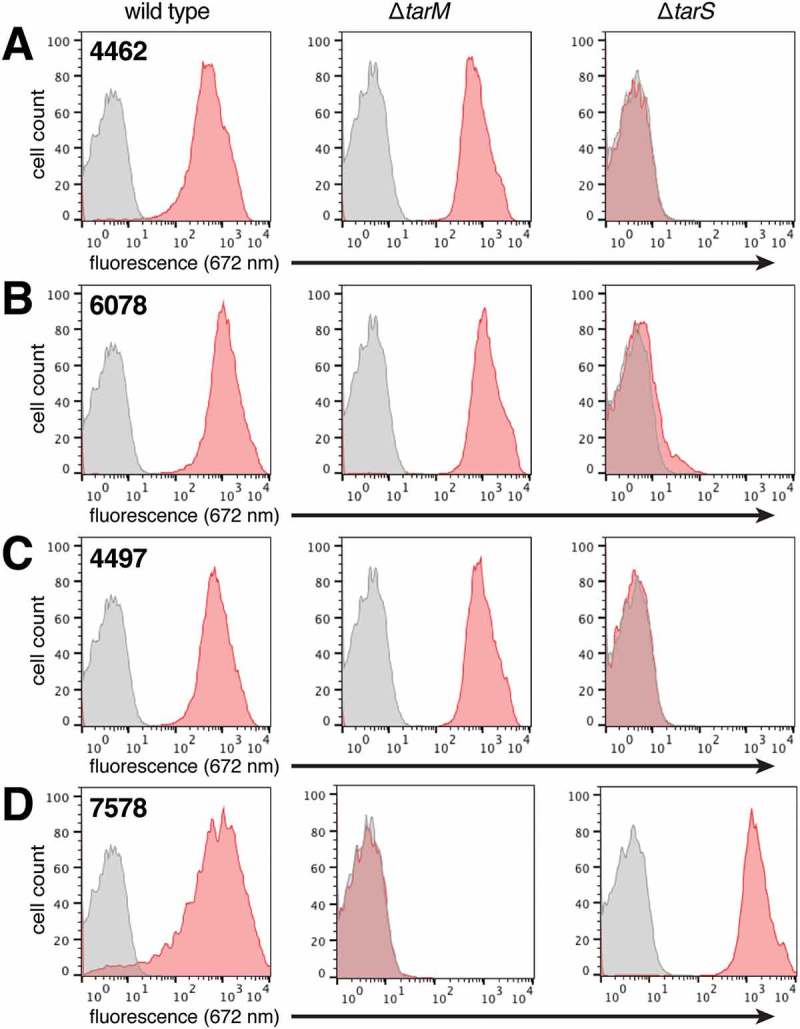
Antibody binding to S. aureus is dependent on WTA glycosyltransferases. Protein A-deficient USA300 S. aureus wild type, ΔtarM, and ΔtarS strains were incubated with anti-WTA (red) or isotype control (grey; human IgG1 anti-cytomegalovirus gD antigen) antibodies, followed by detection with a fluorescently-labeled (DyLight 649) secondary antibody. Stained cells were counted by flow cytometry and data visualized in the displayed histograms. The 4462 (A), 6078 (B), and 4497 (C) antibodies bound to wild type and ΔtarM strains, while 7578 (D) bound to wild type and ΔtarS strains.
To further understand the binding characteristics of these four antibodies, we utilized an 125I-immunoradiometric assay to determine the affinity of the antibodies for USA300 S. aureus bacteria. Saturation binding analysis revealed bivalent affinities in the high picomolar to low nanomolar range for all four antibodies (Figure 3). In addition, this analysis indicated that between 20,000 and 30,000 α- and β-linked binding GlcNAc sites exist on the surface of each bacterium (Figure 3), confirming that GlcNAc-decorated WTA is an abundant epitope at the surface of S. aureus.
Figure 3.
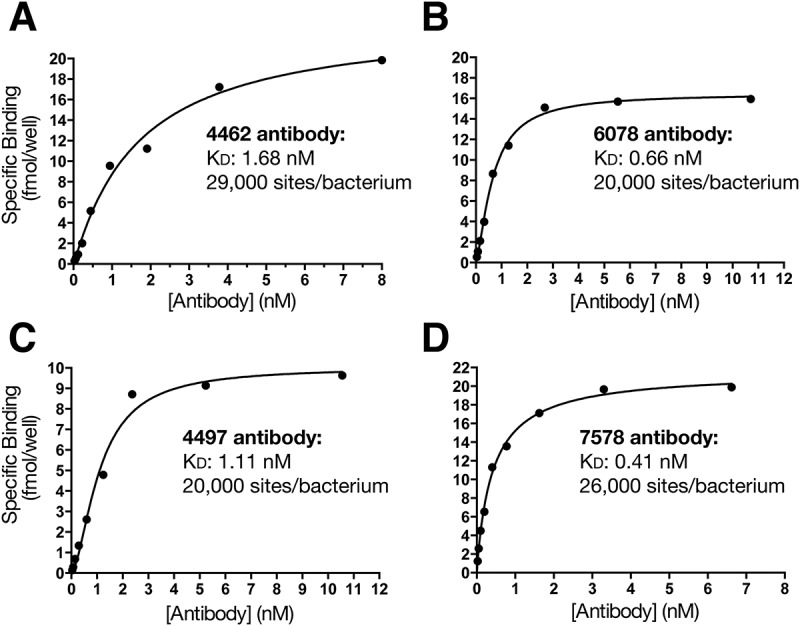
Analysis of anti-WTA antibody binding to S. aureus. Protein A-deficient USA300 S. aureus were incubated with 125I-labelled anti-WTA antibodies and subjected to immunoradiometric assay and saturation binding analysis to determine antibody affinity. Titration curves are shown for 4462 (A), 6078 (B), 4497 (C) and 7578 (D), along with calculated KD values and estimated number of sites/bacterium.
Design of a minimal WTA epitope
Based on the discovery that our panel of antibodies bound to S. aureus WTA, we sought to generate crystal structures of the antigen-binding fragments (Fabs) in complex with WTA. Importantly, WTA is a large and heterogeneous molecule, and isolation of small WTA fragments amenable to co-crystallization studies was not feasible. To simplify WTA to its basic components for structural interrogation, we generated a synthetic WTA mimic by chemical synthesis.33 This minimal epitope contained the core five carbon ribitol group O-linked to GlcNAc via a β-anomeric (equatorial) bond in the 2-position. This bond arrangement was chosen based on data indicating that all three of the antibodies bind native bacteria displaying the β-O-GlcNAc form of WTA (Figure 2). A phosphate was also introduced at either the 1-position or the 5-position, to mimic the phosphodiester bonds found at both ends of the repeating ribitol phosphate monomer (Figure 4A). It was necessary to test both the 1- and 5-phosphoribitol versions of the WTA mimic because of the molecular asymmetry that results from the location of the GlcNAc at the 2-carbon. These molecules were then used for co-crystallization or ligand soaking experiments with a Fab from each of the β-WTA binding antibodies (Figure 4B-D and Table 1). Importantly, our research showed that α-WTA was not expressed on the surface of all S. aureus isolates, whereas β-WTA was found on all isolates tested.33 Therefore, we only sought to crystallize the β-WTA antibodies 4467, 6078, and 4497 with a synthetic WTA analog, given the potential of these antibodies as therapeutic candidates. Similar α-WTA analogs were not synthesized for crystallization with 7578 due to the lower therapeutic potential of the antibody and difficulty of the required carbohydrate chemistry.
Figure 4.
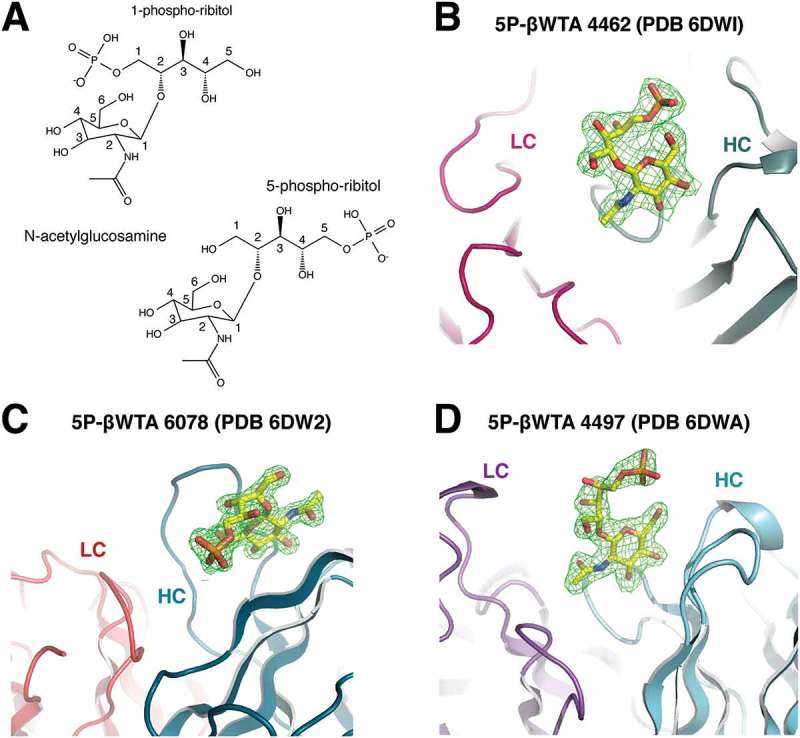
Structure of the synthetic wall teichoic acid epitope. (A) Chemical structures of the synthetic 1-phosphate and 5-phosphate β-WTA epitopes used in crystallization studies. Isolated electron density (contoured at 1σ and shown in magenta) is shown for the 5-phosphate β-WTA epitopes bound to the CDRs of (B) 4462, (C) 6078, and (D) 4497. Light chain (colored shades of red and pink) and heavy chain (colored in shades of cyan) backbones are shown as cartoon representations.
Table 1.
Data collection and refinement statistics for Fab/WTA complexes.
| |
6078/5P-βWTA |
4462/5P-βWTA |
4497/5P-βWTA |
4497 apo |
|---|---|---|---|---|
| PDB ID | ||||
| Data collection | ALS 5.0.2 | ALS 5.0.2 | APS LS-CAT 21-ID-F | SSRL 11–1 |
| Space group | P212121 | P21 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 60.29, 90.25, 105.53 | 122.57, 114.10, 128.30 | 63.68, 114.44, 156.36 | 64.36, 109.67, 156.75 |
| α, β, γ (°) | 90, 90, 90 | 90, 90.46, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–1.70 (1.76–1.70) | 30–2.39 (2.49–2.39) | 50–1.92 (1.99–1.92) | 156.8–2.27 (2.39–2.27) |
| Rsym or Rmerge | 0.054 (0.78) | 0.093 (0.481) | 0.082 (0.757) | 0.038 (0.334) |
| I/σI | 30.4 (2.8) | 13.3 (2.3) | 20.2 (2.2) | 30.6 (4.9) |
| Completeness (%) | 100 (100) | 99.5 (98.7) | 99.5 (98.0) | 99.8 (99.9) |
| Redundancy | 7.2 (7.3) | 3.7 (3.7) | 5.7 (5.4) | 6.5 (6.7) |
| Wilson B-factor | 23.3 | 49.7 | 28.9 | 46.9 |
| Refinement | ||||
| Resolution (Å) | 45.5–1.70 | 29.9–2.39 | 42.6–1.92 | 89.9–2.27 |
| No. reflections (total/test) | 63973/3239 | 138363/6979 | 84098/4243 | 51637/2645 |
| Rwork/Rfree | 18.9/20.8% | 20.3/24.9% | 20.8/23.5% | 18.6/21.7% |
| No. atoms | ||||
| Protein | 3370 | 25321 | 6619 | 6662 |
| WTA | 28 | 112 | 56 | - |
| Malonate | 7 | - | - | - |
| Glycerol | - | 12 | - | 6 |
| Citrate | - | 13 | - | - |
| Acetate | - | - | - | 8 |
| Sodium | 2 | - | - | - |
| Chloride | 6 | - | - | |
| Water | 406 | 738 | 427 | 623 |
| B-factors | ||||
| Protein | 30.3 | 45.4 | 35.8 | 42.3 |
| WTA | 31.7 | 54.3 | 51.0 | - |
| Malonate | 50.0 | - | - | - |
| Glycerol | - | 57.2 | - | 51.9 |
| Citrate | - | 69.0 | - | - |
| Acetate | - | - | - | 56.5 |
| Sodium | 36.1 | - | - | - |
| Chloride | - | 41.8 | - | - |
| Water | 39.9 | 37.2 | 38.4 | 46.6 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.007 | 0.010 |
| Bond angles (°) | 1.17 | 1.23 | 1.10 | 1.17 |
| Ramachandran | ||||
| Most favored | 97.2 | 96.0 | 96.7 | 97.1 |
| Outliers | 0.5 | 0.6 | 0.2 | 0.2 |
*Values in parentheses are for highest-resolution shell.
Structures of 4462 and 6078 bound to β-WTA reveal mechanism for β-WTA specificity
To investigate the structure of the 4462 antibody Fab bound to WTA, we purified the Fab from E. coli for crystallization and structural studies. We obtained the co-crystal structure by first growing crystals of the Fab alone, followed by a ligand soaking strategy to place the 5-phosphoribitol β-WTA analog (Table 1). From these crystals, we obtained a 2.4Å resolution structure of the complex in the P21 space group, with eight Fab molecules in the asymmetric unit. Electron density for β-WTA bound to antibody complementarity-determining region (CDR) loops was visible for four of the molecules. The four Fab molecules without β-WTA bound have antibody CDR loops directly involved in crystal contacts, which occlude the β-WTA binding site. Inspection of the 4462 Fab with β-WTA bound revealed that the antibody paratope almost exclusively involves heavy chain residues, with primary molecular interactions coming directly from the CDR H1 and H3 loops (Figure 5A). The 4462 CDR loops present a relatively flat binding interface, with a shallow groove in the center that mediates much of the contact surface between 4462 and the GlcNAc moiety in the WTA analog. This groove is lined on one side by CDR H1 residues Asn32 and Asp33, with the base of the pocket formed by H3 residues Asn95, Ser96, and Gly97 (Figure 5B). The pyranose ring of GlcNAc stacks against the amide side chain of Asn95, while a number of hydrogen bonds are formed between the C3 and C4 pyranose hydroxyl groups and the side chains and main chain amide nitrogen molecules found in Asp33 and Ser96. In addition to these GlcNAc-mediated contacts, the ribitol chain stretches over the top of the CDR H3 loop and positions the C5-linked phosphate group to interact with the Lys94 sidechain. This ionic interaction between Lys94 and the phosphate in WTA mimics an interaction that would take place between 4462 and the WTA phosphodiester, and reveals how the antibody recognizes the β-anomeric, or equatorial, conformation for the GlcNAc-ribitol bond. This interaction is likely key to the specificity of 4462 for β-WTA. Given the positioning of GlcNAc in the CDR H1/H3 pocket, an axial conformation for the ribitol moiety would point to solvent and an attached phosphate would not make contact with the antibody CDR loops (Figure 5B).
Figure 5.
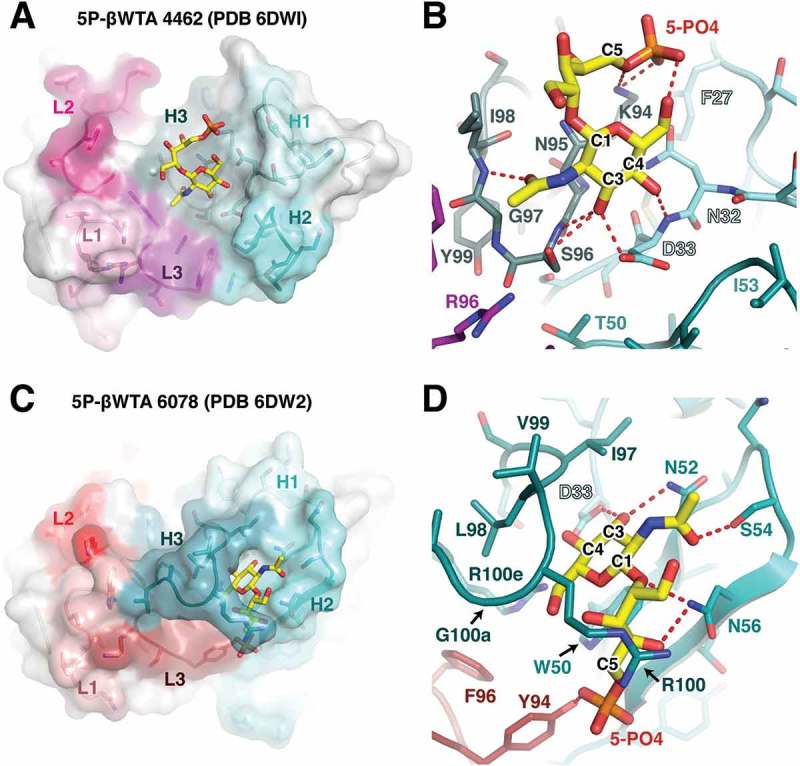
Structural analysis of the 4462 and 6078 antibodies. (A) Overview of the CDR interaction surface of the 4462 Fab bound to 5-phosphate β-WTA. The molecular surface is displayed as partially transparent to show CDR amino acids underneath. Light chain CDRs are labeled and colored in shades of pink, while heavy chain CDRs are labeled and shown in shades of cyan. The bound β-WTA is shown in yellow as a stick representation. (B) Close-up view of the interactions between the 5-phosphate β-WTA and 4462 CDRs. CDR loops are shown as sticks and colored as in (A) with polar contacts shown as red dashed lines. (C) Overview of the CDR interaction surface of the 6078 Fab. Surface is shown as in (A), with light chain CDRs colored in shades of red and heavy chain CDRs shown in shades of teal. (D) Close-up view of the interactions between the 5-phosphate β-WTA and 6078 CDRs. CDR loops are shown as sticks and colored as in (C) with polar contacts shown as red dashed lines.
We also pursued crystallization of the 6078 antibody with the previously described 5-phosphoribitol β-WTA analog. To generate a crystal structure of the complex between 6078 and β-WTA, Fab crystals were grown in the presence of the 5-phosphoribitol β-WTA, which grew in the P212121 space group and diffracted to 1.7Å (Table 1). The structure contained a single Fab molecule bound to β-WTA in the asymmetric unit, and reveals that, like the 4462 antibody, the 6078 antibody paratope primarily consists of heavy chain CDRs (Figure 5C). Yet instead of the GlcNAc residue binding in a central groove, the 6078 antibody has an extended CDR H3 loop that reaches up and surrounds the ligand (Figure 5D). At the base of this pocket is a CDR H2 tryptophan residue (Trp50) that forms a binding site for GlcNAc. The indole side chain of Trp50 stacks against the GlcNAc pyranose ring, generating a favorable carbohydrate-π interaction.35 In addition, the GlcNAc C3 and C4 hydroxyl groups form hydrogen bonds with Asp33 and Asn52 side chains at the base of this pocket, while the carbonyl oxygen from the GlcNAc acetyl group forms a polar contact with Ser54. The 5-phosphoribitol also makes several polar contacts with the antibody, including ribitol interactions with the side chain of Asn56, and phosphate interaction with light chain residue Tyr94. Like 4462, the contact between the CDR L3 Tyr94 and the 5-phosphate would likely be specific for a WTA molecule that contains a β-linked GlcNAc. Intriguingly, there is also an arginine residue in CDR H3 (Arg100) in position to contact a C1-linked phosphate in the context of a native WTA molecule. This structure suggests 6078 may be able to contact both phosphodiester groups, further enhancing specificity and affinity for β-WTA. Despite intensive efforts, we were unable to crystallize the 1-phosphoribitol version of WTA with 6078.
Structural changes in mab 4497 upon binding to β-WTA
As a third comparison to the 4462 and 6078 antibodies, we pursued crystallization of the 4497 antibody to β-WTA, which is also the candidate antibody-antibiotic conjugate in development for treatment of MRSA.33 This Fab was previously crystallized with the 1-phosphoribitol β-WTA analog,33 but also crystallized in the apo form as well as with 5-phosphoribitol β-WTA (Figure 6A). The 4497 Fab crystallized in the P212121 space group, with two equivalent molecules in the asymmetric unit (Table 1). In the apo structure (Figure 6B), the 4497 antibody CDR loops are clearly defined with a large central hydrophobic pocket lined by Trp33 from the CDR H1, as well as Leu32, Tyr91 and Tyr96 contributed by the light chain. This pocket is partially occluded by the rotamer positioning of Asp96 from the CDR H3, which points towards Trp33. Additionally, an unusually long 17-residue CDR L1 projects two basic arginine residues (Arg27d and Arg28) towards the central pocket. This extended loop is encoded by the IgKV4-1*01 germline, but has been mutated at 7 of 17 positions, including addition of the two arginine residues. Notably, Arg27d and Arg28 are splayed apart, and Lys30 pointed away from the central pocket (Figure 6B).
Figure 6.
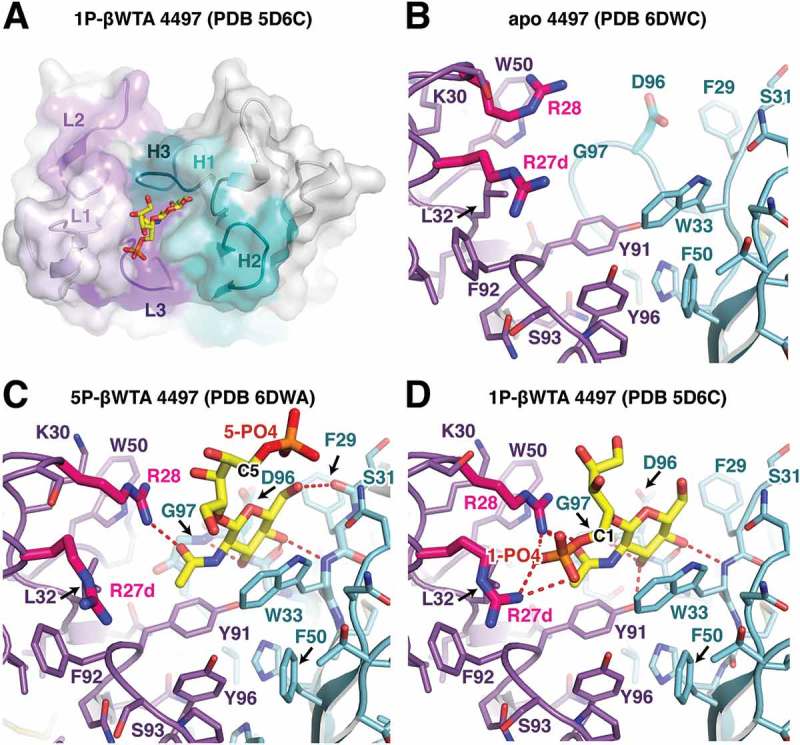
Structural analysis of the 4497 antibody. (A) Overview of the CDR interaction surface of the 4497 Fab in complex with 1-phosphate WTA (PDB ID 5D6C). The molecular surface is displayed as partially transparent to show CDR amino acids underneath. Light chain CDRs are labeled and colored in shades of purple, while heavy chain CDRs are labeled and shown in shades of cyan. The bound 1-phosphate β-WTA is shown in yellow as a stick representation. (B) Close-up view of the CDRs in the apo structure of 4497. CDR loops are shown as sticks and colored as in (A). (C, D) Close-up view of the interactions between the (C) 5-phosphate β-WTA or the (D) 1-phosphate β-WTA (PDB ID 5D6C) and the 4497 CDRs. CDR loops are shown as sticks and colored as in (A) with polar contacts shown as red dashed lines. CDR L1 residues Arg27d and 28, which contact the 1-phosphate, are shown in magenta to highlight changes in conformation between the apo- and WTA-bound structures.
Upon binding to the 5-phosphoribitol β-WTA analog, the GlcNAc residue drops in directly between the heavy and light chain CDRs, with the pyranose hydroxyl groups making backbone contacts with heavy chain residues Ser31, Trp33, Gly95, and Gly97 (Figure 6C). Similar to the 6078 antibody, the GlcNAc pyranose ring is stacked against a tryptophan residue, in this case heavy chain CDR1 residue Trp33. GlcNAc binding also induces a number of structural changes in the 4497 CDR loops. This includes rotation of residue Asp96, which switches rotamers to make room for the GlcNAc moiety, and a shift in light chain residue Arg28, which moves downward into the pocket to interact with GlcNAc. In addition, Lys30 swings towards the sugar-binding pocket to form a π-cation interaction with Trp50. Interestingly, no contact is made between the antibody and the phosphate attached to the ribitol C5 position. In contrast, the structure of the 1-phosphoribitol β-WTA bound to 449733 showed that the phosphate group attached to the ribitol C1 position is a key element of the WTA interaction (Figure 6D). This structure shows that the CDR L1 arginine sidechains (Arg27d and 28) form a tweezers-like motif to engage the 1-phosphate in salt bridge contacts from two sides. Arg28 remains in a similar conformation as seen in the 5-phosphoribitol β-WTA structure, with the guanidine nitrogen engaging the GlcNAc carbonyl oxygen atom and two phosphate oxygen atoms. The lack of movement in the Arg28 sidechain between the 1- and 5-phosphoribitol structures suggests the Arg28 sidechain conformation is driven by hydrogen bond formation with the GlcNAc acetyl group (Figure 6C and D). Like the 4462 and 6078 antibodies, the 4497 CDRs engage both the GlcNAc sugar ring as well as a ribitol phosphate group in order to specifically recognize the stereochemistry of β-WTA. In addition, the formation of favorable intra-chain interactions, such as light chain Lys30-Trp50 π-cation interaction and the heavy chain Arg94-Asp96 salt bridge, likely stabilizes antibody paratopic residues in a manner favorable for WTA binding.
Sequence characterization of β-WTA specific antibodies
Polysaccharide antigens are typically described as “T-cell independent” antigens, given their inability to interact with class I or II MHC molecules and recruit T-cell help via T-cell receptor (TCR) engagement and germinal center formation.17 Paradoxically, these polysaccharide-reactive antibodies are often somatically hypermutated, which suggests that somatic hypermutation in B cells and subsequent production of affinity-matured antibodies does not always require TCR engagement.36,37 We set out to characterize the sequence of these three β-WTA-reactive antibodies to better understand their evolution from germline sequences (Figure 7). All three antibodies are classified as the human IgG1 isotype and utilize unique IgK light chain alleles. The kappa germline alleles for 4462, 4497, and 6078 are IgKV3-15*01, IgKV4-1*01, and IgKV1-5*03, respectively. For the heavy chain, 4462, 4497, and 6078 germline alleles are IgHV3-23*01, IgHV3-74*01, and IgHV1-8*01. To help understand how somatic hypermutation may have altered the mature antibody sequence, we compared the cloned antibody sequences to the germline sequence. All three antibodies are somatically mutated (Figure 7), with all heavy and light chains having 10–30% mutation rates. Given that the three antibodies have undergone significant somatic hypermutation, we were interested in further understanding how these antibodies developed. In particular, we were interested in whether these antibodies arose randomly by clonal selection, or whether there was evidence that the germline sequences might provide a paratope favorable for binding GlcNAc or other pyranose-based sugars. To begin, we identified all residues in 4462, 4497, and 6078 that contact β-WTA in the crystal structures (Figure 7). These contact residues were then compared to their corresponding V-region germline residues to determine whether they are conserved or arose by mutation.
Figure 7.
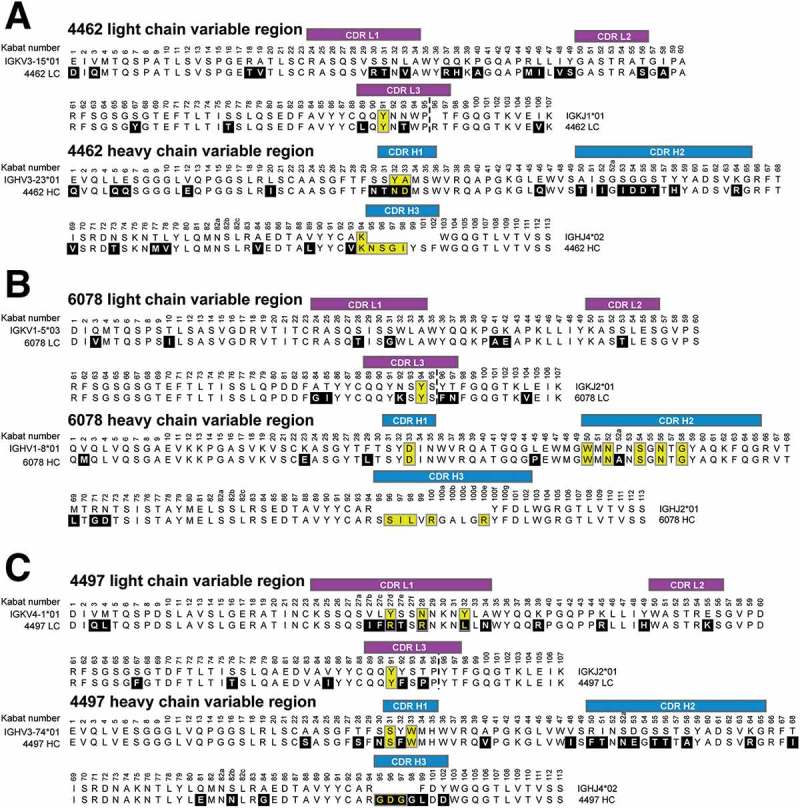
Comparison of anti-β-WTA antibodies to their germline sequences. Alignment of the (A) 4462, (B) 6078, and (C) 4497 light and heavy chain Fab sequences to their respective germline sequences. Antibody light chain CDRs are labeled in purple, and heavy chain CDRs labeled in blue. Amino acids changed from the germline are boxed in black with white lettering. CDR amino acids that contact the WTA epitope (4.5Å cutoff) are boxed in yellow. Germlines matches for antibodies 4462, 6078 and 4497 were determined using the IMGT/V-QUEST server (http://www.imgt.org).
First, we analyzed heavy chain residues in the 4462 antibody that contact the GlcNAc core sugar (Figure 7A). Two CDR H1 residues involved in GlcNAc interaction (Asn32, and Asp33) are changed from the germline sequence, while CDR L3 Tyr91 is a germline residue but is buried and is likely a structurally important residue for heavy/light chain pairing. Much of the interaction with WTA is also mediated by CDR H3 residues (Lys94, Asn95, Ser96, Gly97, and Ile98) that, except for Lys94, are part of insertions introduced by VDJ recombination in this antibody (Figures 7A and 5B). This analysis suggests that 4462 did not arise from a germline precursor that has affinity for β-WTA. Instead, it seems likely that the initial affinity for β-WTA was likely derived from the H3 loop, followed by CDR H1 mutations that may have improved interactions with the GlcNAc residue.
Next, we analyzed 6078 residues that contact GlcNAc and found that all amino acids involved in binding outside the CDR H3 are germline-encoded (Figure 7B). This includes CDR L3 Tyr94, CDR H1 Asp33, and CDR H2 Trp50, Asn52, Ser54, and Asn56 (Figures 7B and 5D). We then analyzed the 4497 contacts and found that CDR L3 Tyr91 along with CDR H1 Ser31 and Trp33 are unchanged from the germline sequence, while the CDR L1 residues that contact the 1-phosphate (Arg27d, Arg28, and Leu32) are mutated from germline (Figures 7C and 6D). Intriguingly, the tryptophan residues in both 6078 and 4497 (CDR H2 Trp50 and H1 Trp33, respectively) that mediate carbohydrate-π stacking interactions with the GlcNAc pyranose core are germline-encoded, while other germline residues (CDR H1 Asp33 and H2 Asn52 in 6078, and CDR L3 Tyr91 in 4497) interact with the sugar hydroxyl groups. Both of these antibodies also contain CDR H3 residues that contribute to binding, but, given the CDR H3 residues vary depending on the D and J segments utilized, it is difficult to speculate on the contribution of the CDR H3 loop. Regardless, our analysis indicates that structurally significant GlcNAc-contacting residues in 6078 and 4497 are encoded by the heavy and light chain V region germlines.
Discussion
The use of cell wall carbohydrates to elicit strong antibacterial immune responses has been a cornerstone of vaccine development for decades.21 Yet the molecular characterization of anti-polysaccharide mAbs for therapeutic purposes has been much more limited, particularly from a structural perspective. Indeed, there are less than twenty crystal structures of antibodies in complex with cell wall polysaccharides in the Protein Data Bank (PDB) database,38 with most of these antibodies targeting O-chain and core epitopes from Gram-negative lipopolysaccharides. To our knowledge, our structures are the first atomic descriptions of antibodies in complex with an epitope from the Gram-positive restricted teichoic acid family of polysaccharides. Wall teichoic acid is a major component of the cell wall structure of S. aureus,39 and along with PNAG22 and the glycosylated SD repeats (SDRs) of adhesive factors such as ClfA,8 is a key exposed epitope on the surface of the bacteria. Importantly, we showed by immunoradiometric assay that each S. aureus cell exposes between 20,000 and 30,000 binding sites for each of our anti-WTA antibodies (Figure 3), highlighting the abundance of the WTA epitope at the S. aureus surface. Antibodies that bind S. aureus, and more specifically the S. aureus-specific GlcNAc modifications of wall teichoic acid and SDR-containing proteins, have been shown to comprise a significant percentage of the human serum IgG content, even from healthy normal donors. 8,33 Given S. aureus is a known human commensal that often colonizes the nostrils and skin, the anti-WTA antibody repertoire likely represents an important component of the human immune system to control S. aureus colonization and infection.40
All three of the antibodies described in this study developed from unique germline IgK and IgH variable sequences, and have undergone significant somatic hypermutation from the initial sequence, resulting in structurally diverse mechanisms for WTA recognition (Figure 4). Yet structural analysis shows that all three antibodies employ similar recognition principles to specifically interact with the β-anomeric form of WTA. For recognition of the N-acetylglucosamine residue, each antibody utilizes heavy chain residues as a platform to stack against the pyranose core, along with C3 and C4 hydroxyl hydrogen bonds to lock the GlcNAc residue in the specific antibody-bound conformation. With the GlcNAc held in position, peripheral basic residues can then triangulate the phosphodiester linkages that form the backbone of WTA. Importantly, the GlcNAc–ribitol linkage in WTA can take the axial (α) or equatorial (β) form, creating two distinct anomeric conformations for phosphoribitol in relation to the GlcNAc. Therefore, triangulation of the phosphate is key to imparting binding specificity towards the β-form of WTA.
The formation of a carbohydrate-π interaction between a sugar ring face and a tryptophan residue combined with multiple hydrogen bonds with sugar hydroxyl groups has been previously recognized as a common interaction “motif” for antibody recognition of carbohydrate epitopes.41 What is noteworthy about our structural analysis is that this antibody–carbohydrate interaction motif is germline-encoded for both the 4497 and 6078 antibodies. Key GlcNAc contact residues in 4497 and 6078, including the pyranose core-binding tryptophan, are preserved from the germline sequence (Figure 7). This raises the interesting possibility that the germline VH genes from which 4497 and 6078 developed may contain a paratope with baseline affinity for pyranose saccharides that serves as a handle for affinity maturation towards specific bacterial cell wall structures. A similar paradigm was identified for antibodies that recognize inner core sugars of LPS on the surface of pathogenic Chlamydia strains,42 reinforcing the idea that a subset of germline VH gene segments may contain structural elements favorable for development of anti-polysaccharide antibodies.
The three anti-WTA antibodies described here have undergone significant somatic hypermutation from the germline sequence, which, based on our structural analysis, has likely improved the affinity for WTA. We were unable, however, to quantitatively interrogate binding of WTA to these antibodies using biophysical methods such as surface plasmon resonance or calorimetry, given the complexity of the native WTA molecule and the difficulty of generating the synthetic epitope. Regardless, affinity analysis using wild-type S. aureus bacteria (Figure 3) as well as co-crystallization of the Fabs with the synthetic WTA epitope indicate a remarkably high affinity of the intact IgG for the native WTA molecule. In contrast to our understanding of B-cell development in response to protein antigens, how polysaccharide-based T-cell independent antigens stimulate somatic hypermutation to produce affinity-matured Ig subtypes is not completely understood, but has been proposed to occur after stimulation of innate immune receptors (Toll-like receptors) and CD40-like co-stimulatory molecules at the mucosal interface.36,37 Our work suggests that these intriguing antibody subtypes can develop against multiple types of bacterial cell wall polysaccharides, and highlights the power of modern antibody discovery techniques for finding antibodies that target these unique epitopes on bacteria of emerging therapeutic importance.
Materials and methods:
Bacterial strains
All bacterial experiments were done with S. aureus strain USA300 NRS384 obtained from the Network on Antibiotic Resistance in Staphylococcus aureus (NARSA) Program (isolate NRS384) supported under NIAID/NIH Contract No. HHSN272200700055C (https://www.beiresources.org). Protein A-deficient S. aureus USA300 mutant strains lacking tarM or tarS were described previously. 8,33 The protein A-deficient S. aureus strain was used to prevent protein A-mediated antibody binding and enable analysis of WTA-specific antibody binding.
Antibody cloning and fluorescent labeling
Recombinant full-length human IgG1 antibodies were cloned from peripheral plasma and memory B cells from patients recovered from S. aureus infection as previously described.8 Briefly, the cognate antibody heavy and light chain pairs were cloned using the SymplexTM technology,43 followed by transfection of mammalian cells to enable IgG expression.44 Antibodies against WTA were selected based on primary screens for binding to cell wall preparations of S. aureus USA300 or Newman, followed by secondary screens for binding to whole bacteria from a variety of clinically relevant S. aureus strains and to S. aureus immediately after isolation from infected mouse kidneys. Isolation and use of these antibodies were approved by the regional ethical review board.
Antibody staining for flow cytometry
S. aureus, cultured at 37°C for 20 h on tryptic soy agar plates with 5% defibrinated sheep blood, were suspended at 109 CFU/mL of HB buffer (Hank’s balanced salt solution containing 0.1% bovine serum albumin and 10 mM Hepes, pH 7.4) and incubated with 200 μg/mL of rabbit IgG (Sigma Aldrich) for 1 h to block non-specific IgG binding sites. Test IgG1 antibodies (anti-β-WTA, anti-α-WTA or isotype control-anti CMV-gD; 1 μg/mL) were added directly for 20 min. After washing twice in HB buffer, bacteria were incubated with DyLight 649 conjugated Affinipure F(ab’)2 Fragment Donkey Anti-human IgG (H + L) (Jackson ImmunoResearch Laboratories, cat. 709–496-149), for 20 min. Bacteria were washed twice with HB buffer and once in phosphate-buffered saline (PBS), fixed in PBS 2% paraformaldehyde, and analyzed by flow cytometry (BD FACSAria).
Affinity analysis and epitope counting
Affinity analysis and epitope counting was performed by saturation binding analysis using radio-iodinated anti-WTA antibodies. The 4462, 4497, 6078, and 7578 antibodies were iodinated using the Iodogen method (ThermoFisher). Protein A-deficient S. aureus USA300 bacteria, cultured on tryptic soy agar with 5% defibrinated blood at 37°C for 20 h, were washed with PBS, fixed with 2% paraformaldehyde in PBS, washed again and resuspended at 8 × 105 CFU/mL in PBS.
Saturation binding assays were carried out in 96-well plates. To initiate the radioligand binding, serially-diluted 125I antibody was added to each well and the plate incubated for 120 min with gentle agitation at room temperature. The experiment was stopped by transferring the solutions to buffer-soaked Millipore filtration units and vacuum filtration. Filters were then washed several times with ice-cold PBS. Filters were dried under air, punched out and placed individually into plastic vials. The vials were scintillation counted using a PerkinElmer 1470 gamma counter.
To analyze the data, for each antibody concentration, non-specific binding was subtracted from total binding to give specific binding. Bound CPM values were converted to fmoles/well from the specific activity of the radiotracer. Data was plotted and fit using the non-linear curve fitting routines in Prism (Graphpad Software) to obtain the KD and Bmax for each antibody. Bmax values were calculated as both fmole/well and sites per cell, the latter obtained from the average cell number in each well.
Antibody purification and crystallization
For crystallization of the 6078/WTA complex, the Fab was expressed in E. coli and purified via Protein G and SP sepharose cation exchange chromatography. After purification the 6078 Fab was concentrated to 15 mg/mL in MES buffer (20 mM MES, pH 5.5, 250 mM NaCl) and mixed with a 2:1 mol/mol ratio of the 5P-βWTA analog (diluted in water) for sparse matrix crystallization screening. An initial hit was found in 2.4 M sodium malonate, which was further optimized to provide diffraction quality crystals. Ultimately, data was collected on a crystal grown by the vapor diffusion method in a sitting drop containing 0.5 ul protein and 0.5 uL 1.9 M sodium malonate. 6078 crystals were cryo-protected in mother liquor, flash frozen in liquid nitrogen, and stored for data collection at 100K.
For the 4462/WTA complex, the Fab was again expressed in E. coli and purified via Protein G and SP sepharose cation exchange chromatography. After purification the 4462 Fab was concentrated to 20 mg/mL in MES buffer (20 mM MES, pH 5.5, 250 mM NaCl) for sparse matrix crystallization screening. The apo 4462 Fab crystallized in 0.1 M citric acid, pH 3.5 containing 25% PEG3350 as a precipitant. Apo 4462 crystals were then soaked with mother liquor containing 0.5 mM of 5P-βWTA analog for two days. 4462 crystals were cryoprotected in 35% PEG3350, flash frozen in liquid nitrogen, and stored for data collection at 100K.
For 4497 Fab crystallization, the Fab was also expressed in E. coli and purified via Protein G and SP sepharose cation exchange chromatography. After purification the 4497 Fab was concentrated to 30 mg/mL in MES buffer (20 mM MES, pH 5.5, 250 mM NaCl) for sparse matrix crystallization screening. For crystallization with the 5P-βWTA analog, the Fab was mixed with a 2:1 mol/mol ratio of the WTA analog. An initial hit was found in a condition from the JCSG Core III, which contained 0.16 M calcium acetate, 0.08 M sodium cacodylate, pH 6.5, 14.4% PEG8000, and 20% glycerol, which after optimization ultimately yielded diffraction quality crystals. 4497 crystals were cryo-protected in mother liquor, flash frozen in liquid nitrogen, and stored for data collection at 100K.
Data collection and structure determination
For the 6078/5P-βWTA and 4462/5P-βWTA complexes, data was collected at the Advanced Light Source (ALS) beamline 5.0.2 at a wavelength of 1.0Å. The 4497/5P-βWTA data was collected at the Advanced Photon Source (APS) beamline LS-CAT 21-ID-F at a wavelength of 0.979Å. The apo 4497 data was collected at the Stanford Synchotron Radiation Laboratory (SSRL) beamline 11–1 at a wavelength of 0.979Å. All data was collected under cryo-cooled conditions (100K). Data was reduced using HKL2000/SCALEPACK45 or XDS/XSCALE.46 Each structure was solved by sequential molecular replacement searches with PHASER47 using Fab constant and variable regions (Protein Data Bank accession 4I77) as individual search models. Iterative rounds of model adjustment with COOT48 were followed by simulated annealing, coordinate, and b-factor refinement with PHENIX49 and BUSTER (Global Phasing Ltd) were performed to build the final models. Full data processing, refinement, and model quality statistics can be found in Table 1. Structural data has been submitted to the RCSB and can be found under PDB codes 6DW2 (6078/5P-βWTA), 6DWA (4497/5P-βWTA), 6DWC (4497 apo), and 6DWI (4462/5P-βWTA). All figures were generated using PYMOL (Schrodinger).
Acknowledgments
The authors would like to thank Peter S. Andersen, Magnus Strandh and Klaus Koefoed (Symphogen A/S, Lyngby, Denmark) for their contributions to the generation and initial analysis of the anti-WTA antibodies. The authors would also like to thank Elizabeth Luis for her help in facilitating the epitope counting experiments.
Abbreviation
- AAC
antibody-antibiotic conjugate
- CDR
complementarity-determining region
- CP
capsular polysaccharide
- Fab
Fragment antigen-binding
- GlcNAc
N-acetylglucosamine
- mAbs
monoclonal antibodies; MDR: multi-drug resistant
- MHC
major histocompatibility complex
- MRSA
Methicillin resistant Staphylococcus aureus
- PNAG
poly-N-acetylglucosamine
- SDR
Serine-Aspartate repeats
- TCR
T- cell receptor
- TI
T-cell independent
- WTA
Wall Teichoic Acid
References
- 1.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, RN Jones, Beach M. SENTRY Partcipants Group . Survey of infections due to staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin Infect Dis. 2001;32(Suppl 2):S114–32. doi: 10.1086/318467. [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med. 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 48:1–12. doi: 10.1086/597588. [DOI] [PubMed] [Google Scholar]
- 4.Nannini E, Murray BE, Arias CA. 2010. Resistance or decreased susceptibility to glycopeptides, daptomycin, and linezolid in methicillin-resistant Staphylococcus aureus. Curr Opin Pharmacol. 10:516–521. doi: 10.1016/j.coph.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev. 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. 2013. Platforms for antibiotic discovery. Nat Rev Drug Discov. 12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 7.Casadevall A, Scharff MD. Serum therapy revisited: animal models of infection and development of passive antibody therapy. Antimicrob Agents Chemother. 38;1994:1695–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hazenbos WLW, Kajihara KK, Vandlen R, Morisaki JH, Lehar SM, Kwakkenbos MJ, Beaumont T, Bakker AQ, Phung Q, Swem LR, et al. 2013. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog. 9:e1003653. doi: 10.1371/journal.ppat.1003653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oganesyan V, Peng L, Damschroder MM, Cheng L, Sadowska A, Tkaczyk C, Sellman BR, Wu H, Dall’Acqua WF. 2014. Mechanisms of neutralization of a human anti-α-toxin antibody. J Biol Chem. 289:29874–29880. doi: 10.1074/jbc.M114.601328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tkaczyk C, Hua L, Varkey R, Shi Y, Dettinger L, Woods R, Barnes A, MacGill RS, Wilson S, Chowdhury P, et al. 2012. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin Vaccine Immunol. 19:377–385. doi: 10.1128/CVI.05589-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Migone T-S, Subramanian GM, Zhong J, Healey LM, Corey A, Devalaraja M, Lo L, Ullrich S, Zimmerman J, Chen A, et al. 2009. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med. 361:135–144. doi: 10.1056/NEJMoa0810603. [DOI] [PubMed] [Google Scholar]
- 12.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Leney M, Sloan S, et al. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 13.DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, Bonnell J, Fleming R, Bezabeh B, Dimasi N, et al. 2014. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med. 6:262ra155–5. doi: 10.1126/scitranslmed.3009655. [DOI] [PubMed] [Google Scholar]
- 14.Secher T, Fas S, Fauconnier L, Mathieu M, Rutschi O, Ryffel B, Rudolf M. 2013. The anti-Pseudomonas aeruginosa antibody Panobacumab is efficacious on acute pneumonia in neutropenic mice and has additive effects with meropenem. PLoS ONE. 8:e73396. doi: 10.1371/journal.pone.0073396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidenmaier C, Peschel A. 2008. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Micro. 6:276–287. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
- 16.Grönwall C, Vas J, Silverman GJ. 2012. Protective roles of natural IgM Antibodies. Front Immunol. 3:66. doi: 10.3389/fimmu.2012.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kearney JF, Patel P, Stefanov EK, King RG. Natural antibody repertoires: development and functional role in inhibiting allergic airway disease. Annu Rev Immunol. 2015;33:475–504. [DOI] [PubMed] [Google Scholar]
- 18.Maira-Litràn T, Kropec A, Goldmann D, Pier GB. 2004. Biologic properties and vaccine potential of the staphylococcal poly-N-acetyl glucosamine surface polysaccharide. Vaccine. 22:872–879. doi: 10.1016/j.vaccine.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Skurnik D, Kropec A, Roux D, Theilacker C, Huebner J, Pier GB. 2012. Natural antibodies in normal human serum inhibit Staphylococcus aureus capsular polysaccharide vaccine efficacy. Clin Infect Dis. 55:1188–1197. doi: 10.1093/cid/cis624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.AlonsoDeVelasco E, Verheul AF, Verhoef J, Snippe H. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol Rev. 59;1995:591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hütter J, Lepenies B. Carbohydrate-based vaccines: an overview. Methods Mol Biol. 1331;2015:1–10. [DOI] [PubMed] [Google Scholar]
- 22.Skurnik D, Davis MR, Benedetti D, Moravec KL, Cywes-Bentley C, Roux D, Traficante DC, Walsh RL, Maira-Litràn T, Cassidy SK, et al. Targeting pan-resistant bacteria with antibodies to a broadly conserved surface polysaccharide expressed during infection. J Infect Dis. 2012;205:1709–1718. doi: 10.1093/infdis/jis213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swoboda JG, Campbell J, Meredith TC, Walker S. 2010. Wall teichoic acid function, biosynthesis, and inhibition. Chem Eur J of Chem Bio. 11:35–45. doi: 10.1002/cbic.200900557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian Z, Yin Y, Zhang Y, Lu L, Li Y, Jiang Y. 2006. Genomic characterization of ribitol teichoic acid synthesis in Staphylococcus aureus: genes, genomic organization and gene duplication. BMC Genomics. 7:74. doi: 10.1186/1471-2164-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown S, Y-H Z, Walker S. 2008. A revised pathway proposed for Staphylococcus aureus wall teichoic acid biosynthesis based on in vitro reconstitution of the intracellular steps. Chem Biol. 15:12–21. doi: 10.1016/j.chembiol.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neuhaus FC, Heaton MP, Debabov DV, Zhang Q. 1996. The dlt operon in the biosynthesis of D-alanyl-lipoteichoic acid in Lactobacillus casei. Microb Drug Resist. 2:77–84. doi: 10.1089/mdr.1996.2.77. [DOI] [PubMed] [Google Scholar]
- 27.Ewc S, Brown ED. 2014. Taking aim at wall teichoic acid synthesis: new biology and new leads for antibiotics. J Antibiot. 67:43–51. doi: 10.1038/ja.2013.100. [DOI] [PubMed] [Google Scholar]
- 28.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 29.Brown S, Xia G, Luhachack LG, Campbell J, Meredith TC, Chen C, Winstel V, Gekeler C, Irazoqui JE, Peschel A, et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc Natl Acad Sci USA. 2012; doi: 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathenson SG, Strominger JL. Enzymatic synthesis and immunochemistry of N-acetylglucosaminylribitol linkages in the teichoic acids of Staphylococcus aureus stains. J Biol Chem. 237;1962:3839–3841. [PubMed] [Google Scholar]
- 31.Torii M. 1964. Separation of teichoic acid of staphylococcus aureus into two immunologically distinct specific polysaccharides with alpha- and -n-acetylglucosaminyl linkages respectively: antigenicity of teichoic acids in man. J Exp Med. 120:13–29. doi: 10.1084/jem.120.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sievers EL, Senter PD. 2013. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 33.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Morisaki JH, et al. Novel antibody-antibiotic conjugate eliminates intracellular Saureus. Nature. 2015;527:323–328. doi: 10.1038/nature15724. [DOI] [PubMed] [Google Scholar]
- 34.Xia G, Maier L, Sanchez-Carballo P, Li M, Otto M, Holst O, Peschel A. 2010. Glycosylation of wall teichoic acid in Staphylococcus aureus by TarM. J Biol Chem. 285:13405–13415. doi: 10.1074/jbc.M109.096172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laughrey ZR, Kiehna SE, Riemen AJ, Waters ML. 2008. Carbohydrate-pi interactions: what are they worth? J Am Chem Soc. 130:14625–14633. doi: 10.1021/ja803960x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weill J-C, Weller S, Reynaud C-A. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–285. [DOI] [PubMed] [Google Scholar]
- 37.Cerutti A, Puga I, Cols M. 2011. Innate control of B cell responses. Trends Immunol. 32:202–211. doi: 10.1016/j.it.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haji-Ghassemi O, Blackler RJ, Martin Young N, Evans SV. 2015. Antibody recognition of carbohydrate epitopes†. Glycobiology. 25:920–952. doi: 10.1093/glycob/cwu134. [DOI] [PubMed] [Google Scholar]
- 39.Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev. 67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spellberg B, Daum R. 2012. Development of a vaccine against Staphylococcus aureus. Semin Immunopathol. 34:335–348. doi: 10.1007/s00281-011-0293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson IA, Stanfield RL. 1995. A Trojan horse with a sweet tooth. Nat Struct Biol. 2:433–436. doi: 10.1038/nsb0695-433. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen HP, Seto NOL, MacKenzie CR, Brade L, Kosma P, Brade H, Evans SV. 2003. Germline antibody recognition of distinct carbohydrate epitopes. Nat Struct Biol. 10:1019–1025. doi: 10.1038/nsb1014. [DOI] [PubMed] [Google Scholar]
- 43.Meijer P-J, Andersen PS, Haahr Hansen M, Steinaa L, Jensen A, Lantto J, Oleksiewicz MB, Tengbjerg K, Poulsen TR, Coljee VW, et al. 2006. Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J Mol Biol. 358:764–772. doi: 10.1016/j.jmb.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 44.Meijer P-J, Nielsen LS, Lantto J, Jensen A. Human antibody repertoires. Methods Mol Biol. 525;2009:261–277–xiv. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data collected in Oscillation Mode. Methods Enzymology. 1997;276:1–20. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch W XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potterton E, Briggs P, Turkenburg M, Dodson E. 2003. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 59:1131–1137. doi: 10.1107/S0907444903008126. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]