

ABSTRACT
The tumor suppressor p53 is of crucial importance in the prevention of cellular transformation. In the presence of cellular stress signals, the negative feedback loop between p53 and Mdm2, its main negative regulator, is disrupted, which results in the activation and stabilization of p53. Via a complex interplay between both transcription-dependent and – independent functions of p53, the cell will go through transient cell cycle arrest, cellular senescence or apoptosis. However, it remains difficult to completely fathom the mechanisms behind p53 regulation and its responses, considering the presence of multiple layers involved in fine-tuning them. In order to take the next step forward, novel research tools are urgently needed. We have developed single-domain antibodies, also known as nanobodies, that specifically bind with the N-terminal transactivation domain of wild type p53, but that leave the function of p53 as a transcriptional transactivator intact. When the nanobodies are equipped with a mitochondrial-outer-membrane (MOM)-tag, we can capture p53 at the mitochondria. This nanobody-induced mitochondrial delocalization of p53 is, in specific cases, associated with a decrease in cell viability and with morphological changes in the mitochondria. These findings underpin the potential of nanobodies as bona fide research tools to explore protein function and to unravel their biochemical pathways.
Keywords: cancer, the tumor suppressor p53, cell death, mitochondria, nanobodies, VHH, single-domain-antibody, intrabody
Introduction
Discovered in 1979, tumor suppressor p53, also known as the guardian of the genome, is one of the most scrutinized proteins worldwide.1,2 Despite initial assumptions about p53 acting as an oncogene, it was soon recognized that the protein plays a crucial role in preventing cellular transformation.3 The importance of p53 in cancer biology is accentuated by the fact that mutated p53 is found in 50% or more of all human cancers. Mutated p53 not only loses its wild type functionality, but it can also exert a dominant negative effect over the remaining wild type p53 allele, which eventually will be disabled through loss of heterozygosity (LOH). In addition, certain p53 mutants display gain-of-functions, which promote tumor development.4,5 In other cancers, where wild type p53 is retained, one can often find alterations in genes encoding more upstream or downstream regulators of the p53-pathway.6–10
Under normal circumstances, p53 senses various cellular stresses like DNA damage, the presence of hyperproliferative signals, oxidative stress and hypoxia amongst others. The detection of these stressors results in the disruption of the negative feedback loop between p53 and its main negative regulators, murine double minute 2 (Mdm2) and murine double minute 4 (Mdm4), leading to the stabilization and activation of p53. Cellular fate can vary from transient cell cycle arrest to cellular senescence or apoptosis, and is decided via a complex interplay between both transcription-dependent and – independent functions of p53.11–13 Nevertheless, a recent body of work also brings non-canonical p53 functions into prominence as crucial effectors in tumor suppression.14,15
Despite many efforts in the field, the question regarding which of the p53-dependent processes are most important remains a matter of debate. Obtaining a complete picture regarding p53 regulation and its responses is after all a daunting task, considering the presence of multiple layers in the process of fine-tuning p53 responses. The outcome is not only dependent on the cell type and the sort of cellular stressor, but it is also affected by protein-protein interactions, post-translational modifications, the influence of cross-talking pathways and the differential expression of p53 isoforms.16 Novel and potent research tools to study p53 function and biology could help researchers to take the next step forward. We have developed single-domain antibodies, also known as nanobodies (Nbs) or variable domain of heavy-chain-only antibodies (VHHs), specifically targeted against the transactivation domain of p53 (p53 TAD). This small antibody format is obtained by isolating the variable fragment (VHH) of heavy-chain-only antibodies (HCAbs), which can be found in the sera of Camelidae. Nbs display several interesting biophysical and biochemical properties like a small size (~ 15 kDa), low immunogenicity, high stability under harsh conditions, ease of engineering and cost-effective production. Interestingly, their prolate shape allows them to bind into clefts and cavities thus reaching epitopes which are inaccessible for conventional antibodies.17 Even more, since Nbs retain their functionality in the reducing cytoplasmic environment, they can be applied as an intrabody targeting intracellular proteins. All things considered, Nbs can be applied as a bona fide research tool to explore protein function and to unravel their biochemical pathways.18–21
Here, we introduce various p53 TAD Nbs capable of binding wild type p53 with high affinity without interfering with its function as transcriptional transactivator. Moreover, p53 can be trapped at the mitochondria by equipping the nanobody with a mitochondrial-outer-membrane (MOM)-tag. Interestingly, in specific cases, this mitochondrial delocalization of the p53 protein goes hand in hand with a decrease in cell viability. This observation might be the direct consequence of the transcription-independent functions exerted by p53 near the mitochondria and emphasizes the potential of Nbs as research tools.
Results
In vitro characterization of p53 TAD nanobodies
Phage display resulted in the identification of 15 different Nbs. These Nbs can be subdivided into 11 different groups according to their amino acid sequence. Nbs within a specific group recognize the same epitope and originate from the same B-cell lineage.22 The efficacy of the Nbs was evaluated by means of an indirect enzyme-linked immunosorbent assay (ELISA) and an in vitro pull down assay. The majority of the Nbs were successfully expressed in bacteria. The production of p53 TAD Nb36 was, however, less efficient, whereby either no expression or only limited yields were obtained. Expression of p53 TAD Nb3 was never observed (Figure 1(a,c)). In ELISA experiments, recombinant Nbs are considered positive binders when the ratio of the absorbance for the p53 TAD-coated (positive) and Annexin V-coated (negative) wells minimally exceeds a factor of 3. All Nbs met this criterion, apart from p53 TAD Nb3 and p53 TAD Nb36 for which no expression was observed (Figure 1(b)). Next, we verified whether recombinant hemagglutin (HA)-tagged p53 TAD Nbs were capable of precipitating endogenous p53 from HEK293T cell lysates. All successfully produced Nbs were capable of binding comparable amounts of endogenous p53 (Figure 1(d)). Finally, in order to accurately determine the thermodynamic parameters of the interaction and the binding constant, an isothermal titration calorimetry (ITC) experiment was performed for selected Nbs (e.g., p53 TAD Nb28, p53 TAD Nb43, p53 TAD Nb91). The interaction has a binding stoichiometry of 1:1, is an exothermic process and occurs via an induced-fit mechanism. The equilibrium dissociation constants of the Nbs lie within micromolar and nanomolar range (p53 TAD Nb28 KD = 292 nM ± 21.70 nM, p53 TAD Nb43 KD = 93 nM ± 9.35 nM and p53 TAD Nb91 KD = 2.68 µM ± 0.69 µM) (Figure 2).
Figure 1.
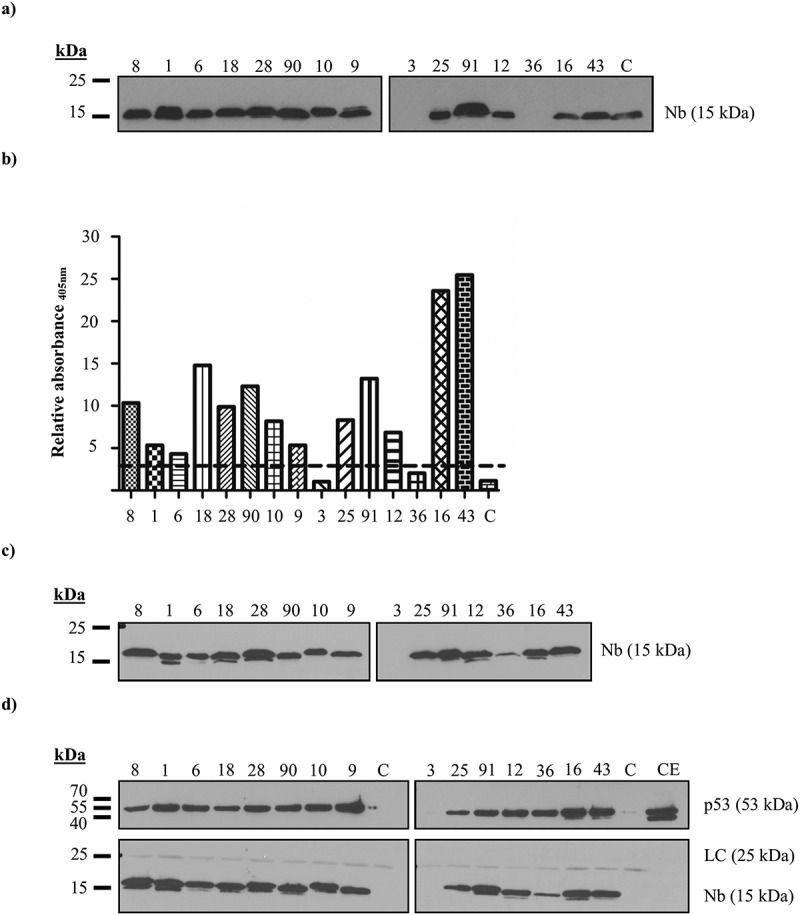
In vitro characterization of p53 TAD nanobodies. (a) Expression levels of the recombinant p53 TAD Nbs in the periplasmic extract. A total amount of 10 µg of nanobody-containing periplasmic extract was loaded onto a 15% SDS gel. An anti-HA antibody was used to visualize the Nbs. High expression yields are observed for 13 out of 15 Nbs, whilst the recombinant production for p53 TAD Nb3 and p53 TAD Nb36 is unsuccessful. (b) Results of ELISA experiment conducted with recombinant p53 TAD Nbs. A 96-well plate, coated with purified p53 TAD (1 µg/ml) or Annexin V (1 µg/ml), was incubated with 20 µl periplasmic extract, containing recombinant p53 TAD Nbs. An irrelevant nanobody was implemented as negative control (C). Nbs were considered as positive binders if the absorbance (at 405 nm) measured for the positive p53 TAD-coated wells exceeds those for the negative Annexin V-coated control wells by a factor of three (represented by a dashed line). All Nbs, with the exception of p53 TAD Nb3 and p53 TAD Nb36 whose recombinant expression were unsuccessful, meet the criterion. (c) Expression levels of the recombinant p53 TAD Nbs in the periplasmic extract. A total amount of 10 µg of nanobody-containing periplasmic extract was loaded onto a 15% SDS gel. An anti-HA antibody was used to visualize the Nbs. High expression yields are observed for 13 out of 15 Nbs, p53 TAD Nb36 displays lower yields, whilst recombinant production of p53 TAD Nb3 is again unsuccessful. (d) In vitro pull-down experiment performed with recombinant HA-tagged p53 TAD Nbs on endogenous p53 originating from crude extract of HEK293T cells. αHA- agarose beads were used for immobilization of the HA-tagged Nbs. A negative control was included where αHA- agarose beads were incubated with crude extract of HEK293T cells in the absence of a HA-tagged nanobody (C). A 40 µg sample of crude extract (CE) was also loaded onto the 15% SDS gel. An anti-HA antibody was used to visualize the Nbs and DO-1 was used to visualize p53. (LC = light chain of IgG antibody).
Figure 2.
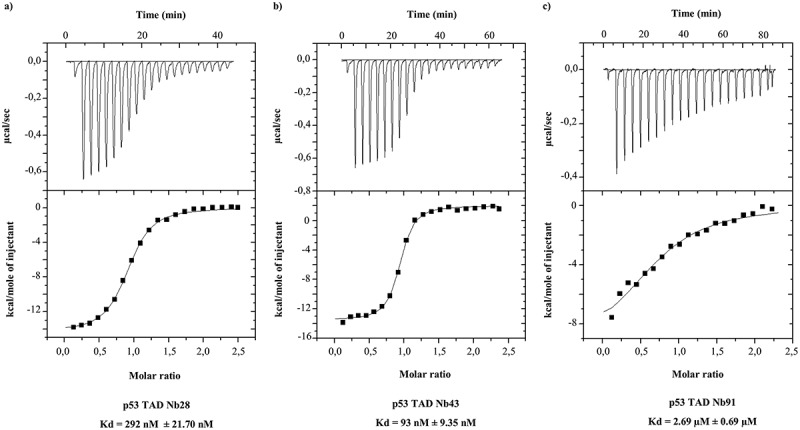
Assessing the thermodynamic parameters of the Nb-p53 TAD interaction. ITC profiles of selected p53 TAD Nbs are shown. Each upper panel displays the amount of heat that is released in function of time in the course of the titration of purified recombinant p53 TAD (sample cell) with recombinant HA-tagged p53 TAD Nbs (injected). The lower panel shows the fitted binding curve of the total amount of heat release per injection as a function of the molar ratio. (a) ITC profile of p53 TAD Nb28. The interaction occurs via an induced-fit mechanism (ΔS = −17.5) and is an exothermic process (ΔH = −1.436 x 104 ± 139.9). The binding affinity is 292 nM (± 21.70 nM) and the reaction occurs via a 1:1 stoichiometry. (b) ITC profile of p53 TAD Nb43. The interaction occurs via an induced-fit mechanism (ΔS = −19) and is an exothermic process (ΔH = −1.55 x 104 ± 129). The binding affinity is 93.46 nM (± 9.35 nM) and the reaction occurs via a 1:1 stoichiometry. (c) ITC profile of p53 TAD Nb91. The interaction occurs via an induced-fit mechanism (ΔS = −6.88) and is an exothermic process (ΔH = −9813 ± 1139). The binding affinity is 2.68 µM (± 0.69 µM) and occurs via a 1:1 stoichiometry.
In vivo characterization of p53 TAD nanobodies
Since p53 is an intracellular protein it was of crucial importance to evaluate the functionality of these Nbs in the intracellular environment. Intracellular stability was tested for 11 of 15 available Nbs. p53 TAD Nb3 and p53 TAD Nb36 were eliminated due to absent/inferior expression yields, whilst p53 TAD Nb90 and p53 TAD Nb6 are members of a group with 5 representatives. In vivo functionality was established via an in vivo pull-down assay for which V5-tagged p53 TAD Nbs were transiently expressed in HEK293T cells. Two negative controls were included: αV5 agarose beads were incubated with the crude lysate of either non-transfected HEK293T cells or with the crude lysate of HEK293T cells that transiently express an irrelevant nanobody (Fsc Nb2),23 targeted against the actin bundling protein fascin (Fsc). The assay indicated that 8 Nbs were capable of binding p53 intracellularly. Intracellular expression of p53 TAD Nb18 and p53 TAD Nb25 could not be detected. p53 TAD Nb91, on the other hand, displays similar expression levels compared to the positive binders, but is not capable of precipitating wild type p53. The rather weak KD (~ 2.69 µM) of this nanobody might be the underlying reason for this observation (Figure 3).
Figure 3.
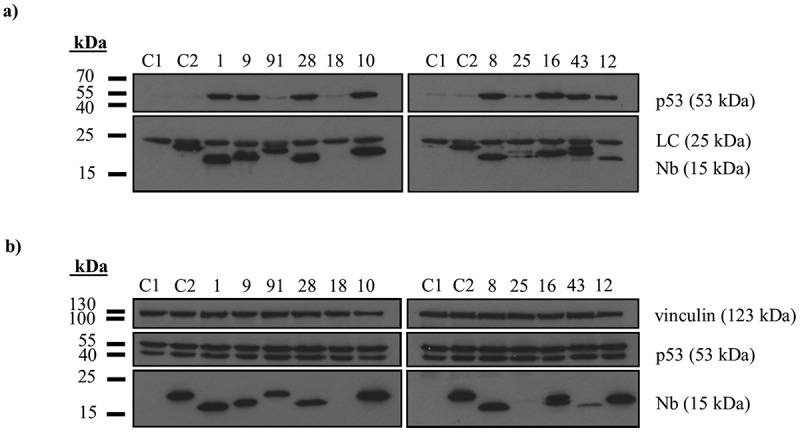
In vivo validation of p53 TAD nanobodies. (a) In vivo pull-down experiment performed with V5-tagged p53 TAD Nbs that were transiently expressed in HEK293T cells. 24 h after transfection, 1 mg of the crude extract was incubated with αV5 agarose beads and it was verified whether or not endogenous p53 co-precipitates with the immobilized Nbs. A negative control was included were αV5 agarose beads were incubated with the crude extract of untransfected cells (C1) or with crude extract of cells that were transfected with V5-tagged Fsc Nb2, a nanobody targeting the actin bundling protein Fsc (C2). Co-precipitation of endogenous p53 was observed for 8 out of 11 p53 TAD Nbs. Nbs incapable of binding endogenous p53 displayed either no intracellular expression (p53 TAD Nb18 and p53 TAD Nb25) or were unable to pull down endogenous p53 despite decent expression levels (p53 TAD Nb91). (b) Expression levels of V5-tagged p53 TAD Nbs in crude extract of HEK293T cells (40 µg). The majority of the Nbs show comparable expression levels, apart from p53 TAD Nb43 (modest expression), p53 TAD Nb18 and p53 TAD Nb25 (absent expression). The crude extracts of both negative controls (C1 and C2) were also checked for nanobody expression. (LC = light chain of IgG antibody).
The transactivation functions of p53 remain undisturbed in the presence of p53 TAD nanobodies
The N-terminal domain of p53 consists of a transactivation domain (p53 TAD1: residues 1–40, p53 TAD2: residues 40–61) and a proline (Pro)-rich region (residues 64–92).24 This domain constitutes a binding site for numerous interacting proteins like transcriptional co-activators, negative regulators and components of the transcription machinery. Disturbance of these interactions might affect the transactivation capacity of wild type p53. Since the epitope recognized by the Nbs is situated within this domain, it is important to assess whether or not the transactivation capacity of p53 remains unaffected.
To this end, a transactivation assay was performed by transiently expressing the Nbs in U2OS pGL13 cells.25 This cell line stably expresses a luciferase reporter gene of which the cDNA is preceded by 13 repeats of the p53 consensus sequence. Nutlin3a, a Mdm2 antagonist known to restore p53 activity, was used as a positive control, whereas Fsc Nb2 was implemented as a negative control. None of the Nbs caused a significant change in luciferase activity compared to Fsc Nb2, which suggests that the transactivation function of p53 remains unaffected (one-way ANOVA, Dunnett’s multiple comparison test) (Figure 4(a)). The same observation was made when transfected U2OS pGL13 cells were additionally treated with 5 µM Nutlin3a (Figure 4(b)). The additional treatment with 5 µM Nutlin3a, however, was responsible for a significant increase in luciferase activity (P < 0.0001, two-way ANOVA). This increase occurred independently of nanobody expression, since a significant interaction between both treatments (i.e., nanobody and Nutlin3a) could not be detected (P = 0.99, two-way ANOVA) (Figure 4(c)). The degree of nanobody expression in U2OS pGL13 cells was verified for each individual experiment, since transfection efficiencies may fluctuate.
Figure 4.
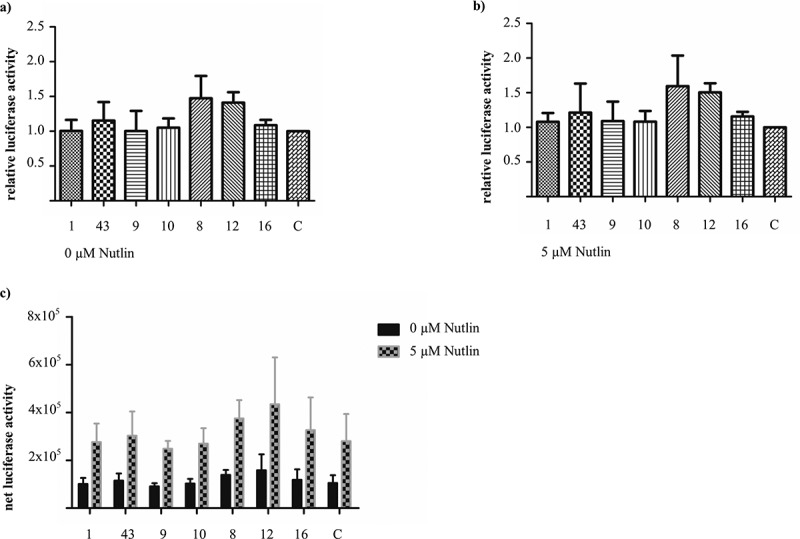
Evaluating p53’s functionality as transactivator in the presence of p53 TAD nanobodies. The U2OS pGL13 cell line expresses a luciferase reporter gene in a stable manner. This reporter gene is preceded by 13 repeats of a p53 response element. Measurement of the luciferase activity in U2OS pGL13 cells, which transiently express p53 TAD Nbs, thus allows the assessment of possible manipulation of p53’s transactivation function by the Nbs. An irrelevant nanobody, Fsc Nb2, was implemented as negative control (C). (a) The Nbs do not significantly affect p53’s transactivation potential compared to the negative control, despite some fluctuations in relative luciferase activity (one-way ANOVA, Dunnett’s multiple comparison test, P-value = 0.55). Luciferase activity was measured 48 h after transfection. (b) Transfected U2OS pGL13 cells received 24 h post-transfection an additional treatment with 5 µM Nutlin3a. Analysis of luciferase activity 24 h post-Nutlin3a treatment, did not reveal significant differences in luciferase activity (one-way ANOVA, Dunnett’s multiple comparison test, P-value = 0.64). (c) Comparison of net luciferase activity measured in transfected U2OS pGL13 cells which received (or did not receive) an additional treatment with 5 µM Nutlin3a. In the presence of 5 µM Nutlin3a a significant increase in luciferase activity can be observed (P-value < 0.0001), whilst the Nbs have no effect (P-value = 0.80). Both treatments do not interact (P-value = 0.99). Data were analyzed via a two-way ANOVA.
Overall, intracellular expression of p53 TAD Nb43 was rather weak, while the expression of the other nanobody constructs varied from modest to strong. This tendency remained unaltered between individually repeated experiments (Supplementary figure 1). The integrity of the transactivation assay was demonstrated by repeating the assay for selected p53 TAD Nbs whereby two additional positive controls were included (i.e., p53 DBD Nb139,20 and HA-tagged p53 mutant (R175H)25) (Supplementary figure 2).
Mitochondrial delocalization of p53 using p53 TAD nanobodies as a tool
Nbs consist of a single domain and easily allow customized engineering. p53 TAD Nbs were equipped with a mitochondrial-outer-membrane (MOM)-tag in order to delocalize and immobilize p53 at the mitochondrial outer membrane. This MOM-tag is derived from yeast translocase of outer membrane 70 (TOM70) where the basic N-terminus forms a topogenic sequence for protein insertion into the mitochondrial membrane.26 Mitochondrial delocalization of a target protein by Nbs not only confirms their intracellular binding and stability, but this strategy can also be utilized to create a functional protein knock-out by sequestering the target protein into or at specific cellular compartments. In the case of p53, this strategy also might be used to induce cell death, considering the transactivation-independent functions of p53 in the cytoplasm.27–30 First, the capacity of the p53 TAD Nbs to induce a mitochondrial delocalization of either overexpressed or endogenous p53 was verified in U2OS cells and H1299 cells (p53 null) via immunostaining. Next, an 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay was performed to screen for possible effects of the induced delocalization of p53 on cell viability. During these experiments, MOM/V5-tagged Fsc Nb2 was implemented as a negative control together with a condition where cells were mock-transfected.
In the absence of the MOM/V5-tag, p53 TAD Nbs displayed a diffuse staining pattern where they were distributed evenly throughout the cytoplasm and the nucleus. A similar pattern was observed for overexpressed HA-tagged p53 mutant (R175H) in H1299 cells (Supplementary figure 3). Endogenous p53 in U2OS cells was predominantly nuclear enriched (Supplementary figure 4). HA-tagged wild type p53 was ubiquitously distributed throughout U2OS cells, although it variably showed nuclear or cytoplasmic enrichment (Supplementary figure 5). However, once equipped with a MOM/V5-tag, the Nbs adopted a mitochondrial-like staining pattern demonstrating the efficiency by which the MOM/V5-tag redirected the Nbs towards the mitochondria in H1299 cells (Figure 5(a) and Supplementary figure 6a) and in U2OS cells (Figure 6(a) and Supplementary figure 7a). Interestingly, MOM/V5-tagged p53 TAD Nbs were able to capture p53 at the mitochondria (although with variable results for p53 TAD Nb10), resulting in its accumulation at the outer mitochondrial membrane. In contrast, in the presence of MOM/V5-tagged Fsc Nb2 there was no evidence of p53 accumulation near the mitochondria. Remarkably, mitochondrial staining patterns of endogenous p53 were not as frequently observed (Figure 6(b) and Supplementary figure 7b). It thus appeared to be less straightforward to capture endogenous p53 at the mitochondria, compared to the overexpressed p53 constructs (Figure 5(b), 7(a) and Supplementary figures 6b and 8).
Figure 5.
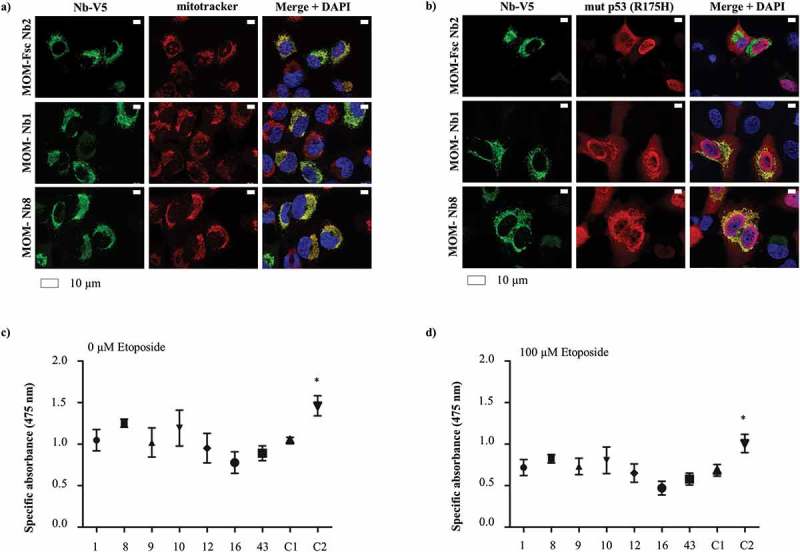
Nanobody-induced mitochondrial delocalization of HA-tagged mutant p53 (R175H) has no impact on cell viability of H1299 cells. H1299 (p53 null) cancer cells transiently express MOM/V5-tagged p53 TAD Nbs and HA-tagged mutant p53 (R175H). The MOM/V5-tagged Fsc Nb2 was used as negative control. (a) Representative epifluorescence images demonstrating the enrichment of MOM/V5-tagged Nbs at the mitochondria, which were labeled via Mitotracker. (b) HA-tagged mutant p53 (R175H) is largely distributed in a mitochondrial-like pattern in the presence of MOM/V5-tagged p53 TAD Nb1 and MOM/V5-tagged p53 TAD Nb8. Similar pattern cannot be observed in the presence of MOM/V5-tagged Fsc Nb2, where p53 displays a more uniform distribution. Visualization of the nuclei was achieved with DAPI, whilst HA-tagged mutant p53 (R175H) and the MOM/V5-tagged Nbs were visualized with an anti-HA antibody and an anti-V5 antibody, respectively. An XTT cell viability assay was performed to evaluate whether mitochondrial delocalization of HA-tagged mutant p53 (R175H) impacts cell viability. The assay was performed in the absence (c) or presence of 100 µM etoposide (d). Two negative controls were implemented, for which cells were either transiently transfected with the MOM/V5-tagged Fsc Nb2 (C1) or were subjected to a mock-transfection (C2). The HA-tagged mutant p53 (R175H) construct was expressed throughout all conditions. The graph represents the mean net absorbance of the formazan dye at 475 nm. None of the MOM/V5-tagged p53 TAD Nbs significantly alters H1299 cell viability compared to MOM/V5-tagged Fsc Nb2. Mock-transfected cells however display a significantly higher cellular proliferation and cell viability than MOM/V5-tagged Fsc Nb2-expressing cells (P < 0.05, one-way ANOVA, Dunnett’s multiple comparison test) (c). Similar results are obtained when cells receive the additional treatment with 100 µM etoposide (d), but globally there is a significant decrease in cell viability (P < 0.0001, two-way ANOVA), which occurs independently from possible effects exerted by the Nbs (P = 0.69, Two-way ANOVA) ((c) and (d)).
Figure 6.
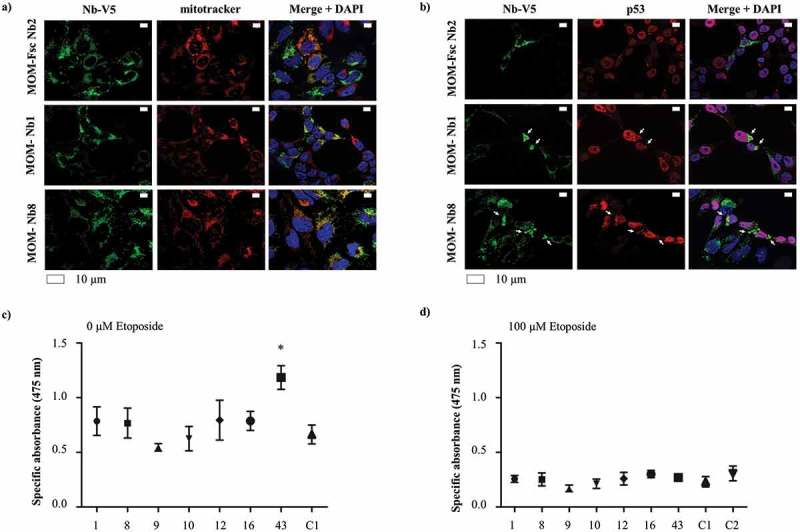
Mitochondrial delocalization of endogenous p53 in U2OS cells without apparent effect on cell viability. U2OS cells transiently express MOM/V5-tagged p53 TAD Nbs. The MOM/V5-tagged Fsc Nb2 was used as negative control. (a) Representative epifluorescence images demonstrating the enrichment of MOM/V5-tagged Nbs at the mitochondria, which were labeled via Mitotracker. (b) Endogenous p53 occasionally adopts a mitochondrial-like pattern in the presence of MOM/V5-tagged p53 TAD Nb1 and MOM/V5-tagged p53 TAD Nb8, albeit with much lower efficiency than observed for overexpressed p53. A similar pattern however cannot be observed in the presence of MOM/V5-tagged Fsc Nb2, where p53 is predominantly located in the nucleus. Visualization of the nuclei was achieved with DAPI, whilst p53 and the MOM/V5-tagged Nbs were visualized with DO-1 and an anti-V5 antibody, respectively. An XTT cell viability assay was performed to evaluate whether mitochondrial delocalization of endogenous p53 impacts cell viability. The assay was performed in the absence (c) or presence of 100 µM etoposide (d). Two negative controls were implemented, for which cells were either transiently transfected with the MOM/V5-tagged Fsc Nb2 (C1) or were subjected to a mock-transfection (C2). The graph represents the mean net absorbance of the formazan dye at 475 nm. A significant change in cell viability is only observed for MOM/V5-tagged p53 TAD Nb43-expressing cells, which display a higher proliferation rate and thus cell viability (P < 0.05, one-way ANOVA Dunnett’s multiple comparison test). (c) When cells are additionally treated with 100 µM etoposide, there is no difference in cell viability between cells that express MOM/V5-tagged p53 TAD Nbs and cells that express the MOM/V5-tagged Fsc Nb2 (one-way ANOVA, Dunnett’s multiple comparison test) (d). The overall viability of U2OS cells significantly decreases in the presence of 100 µM etoposide (P-value = 0.0017, two-way ANOVA). Nanobody treatment also seems to significantly impact cell viability (P-value = 0.0039, two-way ANOVA), however this effect is merely caused by MOM/V5-tagged p53 TAD Nb43. The impact of etoposide treatment is not influenced by the presence of MOM/V5-tagged Nbs (P = 0.0505, two-way ANOVA) ((c) and (d)).
Figure 7.
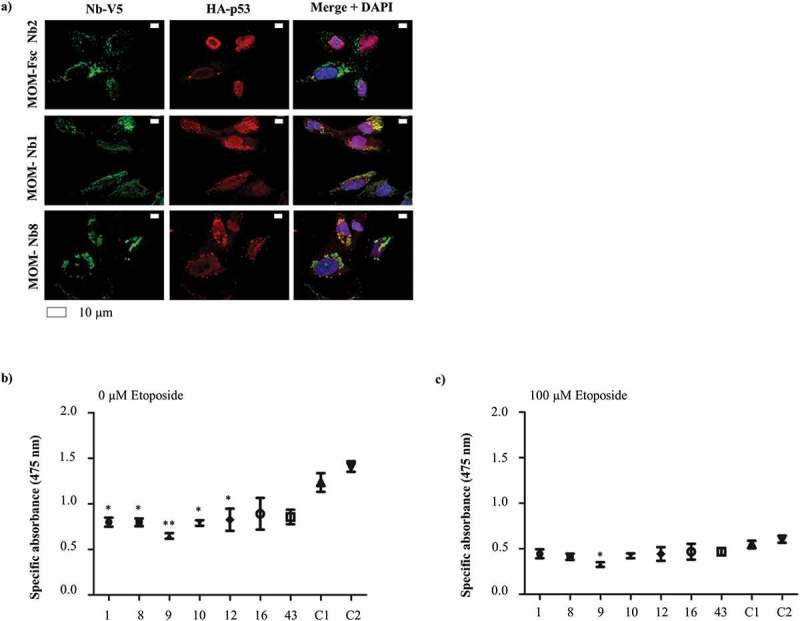
The capture of HA-tagged wild type p53 at the mitochondria by MOM/V5-tagged p53 nanobodies in U2OS cells is connected to a decreased cell viability. U2OS cells transiently express MOM/V5-tagged p53 TAD Nbs and HA-tagged wild type p53 construct. The MOM/V5-tagged Fsc Nb2 was used as negative control. (a) HA-tagged wild type p53 is largely distributed in a mitochondrial-like pattern in the presence of MOM/V5-tagged p53 TAD Nb1 and MOM/V5-tagged p53 TAD Nb8. Similar pattern cannot be observed in the presence of MOM/V5-tagged Fsc Nb2, where p53 is enriched in the nucleus. Visualization of the nuclei was achieved with DAPI, whilst HA-tagged wild type p53 and the MOM/V5-tagged Nbs were visualized with an anti-HA antibody and an anti-V5 antibody, respectively. An XTT cell viability assay was performed to evaluate whether mitochondrial delocalization of HA-tagged wild type p53 impacts cell viability. The assay was performed in the absence (b) or presence of 100 µM etoposide (c). Two negative controls were implemented, for which cells were either transiently transfected with the MOM/V5-tagged Fsc Nb2 (C1) or were subjected to a mock-transfection (C2). The HA-tagged wild type p53 construct was expressed in all conditions. The graph represents the mean net absorbance of the formazan dye at 475 nm. There is a significant reduction in cell viability of U2OS cells in the presence of several MOM/V5-tagged p53 TAD Nbs (P – value < 0.05: p53 TAD Nb1, p53 TAD Nb8, p53 TAD Nb10 and p53 TAD Nb12. P-value < 0.01: p53 TAD Nb9, one-way ANOVA, Dunnett’s multiple comparison test) (b). In the presence of 100 µM etoposide, a significant reduction in cell viability is still observed for p53 TAD Nb9 (P-value < 0.05, one-way ANOVA, Dunnett’s multiple comparison test) (c). Overall, treatment of U2OS cells with 100 µM etoposide causes a significant drop in cell viability (P-value < 0.0001, two-way ANOVA), just as the mere presence of a MOM/V5-tagged p53 TAD nanobody (P-value < 0.0001, two-way ANOVA). There is an interaction between both treatments (P-value = 0.019, two-way ANOVA) ((b) and (c)).
Given the aforementioned transactivation-independent functions exerted by p53, it was valuable to evaluate whether or not the MOM/V5-tagged nanobody-induced mitochondrial delocalization had an effect on cell viability. This was assayed via the XTT cellular proliferation assay kit. Transiently transfected H1299 cells and U2OS cells were plated in triplicate in 96-well microtiter plates. As a positive control, transfected cells were co-treated with 100 µM etoposide, a compound that induces DNA double-strand breaks. Two negative controls were included, where cells were either mock-transfected or were transfected with the MOM/V5-tagged Fsc Nb2-encoding construct. The transfection efficiency was evaluated for each individual experiment via western blotting (Supplementary figures 9 and 10). For statistical analysis a one-way ANOVA with Dunnett’s multiple comparison test was performed where the mean net absorbance (at 475nm) measured for MOM/V5-tagged p53 TAD nanobody-expressing cells was compared with the mean net absorbance measured for MOM/V5-tagged Fsc Nb2-expressing cells.
In H1299 cells (HA-tagged mutant p53 (R175H)) and U2OS cells (endogenous p53), there was no significant decrease in cell viability, neither in the absence nor in the presence of 100 µM etoposide. A significant increase in cellular proliferation, however, was observed for mock-transfected H1299 cells (HA-tagged mutant p53 (R175H)) (P-value < 0.05) and for MOM/V5-tagged p53 TAD Nb43 in U2OS cells (endogenous p53) (P-value < 0.05). In the presence of 100 µM etoposide, this increased proliferation rate was only observed for mock-transfected H1299 cells (P-value < 0.05) (Figures 5 and 6 (c,d)). In contrast, cell viability was significantly reduced when HA-tagged wild type p53 and MOM/V5-tagged p53 TAD Nbs were co-expressed in U2OS cells (p53 TAD Nb1, p53 TAD Nb8, p53 TAD Nb10 and p53 TAD Nb12: P-value < 0.05, p53 TAD Nb9: P-value < 0.01).
When cells received co-treatment with 100 µM etoposide, the significant reduction in cell viability persisted for MOM/V5-tagged p53 TAD Nb9 (P-value < 0.05) (Figure 7(b,c)). For the evaluation of the outcome of etoposide treatment a two-way ANOVA was performed. The treatment of the cells with 100 µM etoposide resulted for each condition in a significant drop of the cell viability (P-value < 0.0001 for H1299 cells (HA-tagged mutant p53 (R175H)) and U2OS cells (HA-tagged wild type p53), P-value < 0.0017 for U2OS cells (endogenous p53)). An interaction between etoposide treatment and nanobody treatment was only observed for U2OS cells (HA-tagged wild type p53) (P-value = 0.019) (Figure 5(d), 6(d) and 7(c)).
The underlying cause of the significantly reduced cell viability of U2OS cells after nanobody-induced delocalization of HA-tagged wild type p53 was more closely investigated. As stated earlier, p53 exerts several transcription-independent functions in the cytoplasm.13 Experiments investigating whether mitochondrial delocalization of HA-tagged wild type p53 resulted in the activation of the intrinsic mitochondrial-mediated apoptotic pathway, were inconclusive. Alternatively, cytoplasmic p53 exerts an inhibitory effect on autophagy by inhibiting the adenosine monophosphate (AMP)-dependent kinase and activating mammalian target of rapamycin (mTOR), although the exact mechanism behind the inhibition of autophagy by cytoplasmic p53 still needs to be elucidated.31 This inhibitory effect of p53 on autophagy might be alleviated through the sequestration of p53 at the mitochondria, resulting in the augmentation of the autophagic flux. The relationship between autophagy and cell death is complex, but high levels of autophagy can promote cell death depending on the stimulus, the cell line and the context.32
We evaluated whether or not the nanobody-induced mitochondrial delocalization of HA-tagged wild type p53 resulted in an increased autophagic flux by monitoring differences in microtubule-associated protein 1A/1B-light chain 3 B-II (LC3B-II) levels. U2OS cells were co-transfected with a MOM/V5-tagged p53 TAD nanobody and HA-tagged wild type p53. As a positive control, cells transfected with HA-tagged wild type p53 were treated with 500 nM rapamycin (mTOR inhibitor). Two negative controls were included, where cells were either co-transfected with MOM/V5-tagged Fsc Nb2 and HA-tagged wild type p53 or were only transfected with HA-tagged wild type p53. LC3B-II levels were compared between samples of the transfected cells which received (or did not receive) an additional treatment with 100 mM NH4Cl, a compound that blocks the proteolytic activity of lysosomes, thereby preventing the turn-over of LC3B-II.33 Increased LC3B-II levels could not be detected for MOM/V5-tagged p53 TAD nanobody-expressing cells compared to MOM/V5-tagged Fsc Nb2-expressing cells (Supplementary figure 11). This implies that nanobody-induced sequestration of HA-tagged wild type p53 has no effect on autophagy in the current setting. The mechanism responsible for the reduced cell viability after nanobody-induced mitochondrial delocalization of HA-tagged wild type p53 thus remains to be clarified.
Discussion
At first, p53 was regarded as an oncogene with transforming capacities, but this perception shifted dramatically and the undeniably crucial role of p53 in the prevention of cancer became apparent. Although p53 was initially considered as undruggable, substantial progress has been made and several compounds are being tested in clinical trials (e.g., PRIMA-1 MET,34 PK7088,35 RG7112,36,37 ALRN-692438). Notwithstanding the progress being made in unraveling p53’s tumor suppressive capacities, several unanswered questions and contradictory findings need to be resolved. For instance, recent findings call the importance of canonical p53 functions like cell cycle arrest, apoptosis and senescence in tumor suppression into question, and propose an important role for novel emerging functions of p53.14 For example, a study was conducted whereby knock-in mice (expressing mutant p53 3KR) still showed tumor suppressive properties despite the complete ablation of the canonical p53 functions. Tumor suppression was associated with the involvement of p53 in metabolic regulation and anti-oxidant function.39 Whether or not these observations are context-dependent or are the mere result of activated back-up mechanisms is not fully clear. In addition, the activity of p53 is not only confined to the nucleus. It has been proven that p53 exerts several transcription-independent functions in the cytoplasm, where it is involved in processes like apoptosis,27 necrosis,28 autophagy and metabolism.30,40 There is still ample room for improvement in deciphering the detailed mechanisms behind these cytoplasmic functions and how these are coordinated with p53’s function as transactivator. Thus, there remains a substantial need for new research tools. We thus developed a set of Nbs targeted against the transactivation domain of p53, which bind p53 with high affinity. The Nbs can be applied as an intrabody, without affecting p53’s function as transactivator. Interestingly, when Nbs are equipped with a MOM-delocalization tag, they can capture p53 at the mitochondrial outer membrane. In specific cases, this delocalization coincides with a decrease in cell viability, which might reflect the activation of a mitochondrial-mediated cell death pathway by p53 or rather results from interference with other cytoplasmic functions of p53.
During the in vitro characterization experiments, it appeared that recombinant production of p53 TAD Nb3 was unsuccessful. This is rather uncommon because phage panning was performed for the retrieval of specific binders in the VHH library rather than direct colony screening. This strategy normally results in the selection of higher affinity binders and Nbs that display better expression levels in bacteria.41 All other Nbs demonstrated high expression yields, apart from p53 TAD Nb36 where expression was rather weak or absent (Figure 1(a,c)). Binding between the successfully produced recombinant Nbs and p53 was confirmed via an indirect ELISA experiment (Figure 1(b)) and an in vitro pull-down experiment (Figure 1(d)).
In general, antibodies are interesting tools to investigate the cellular function of proteins, because of their high affinity and specificity. However, not all antibody formats are sufficiently stable and robust in the reducing cytoplasmic environment. For example, full-length antibodies contain several inter- and intramolecular disulfide bridges, which cannot be formed in the cytoplasm resulting in their partial unfolding and loss-of-function.42 Nbs, however, display a remarkably stable behavior and can resist chemical and thermal denaturation. This makes them suitable candidates for use as an intrabody.17 The intracellular stability of our nanobody set was tested via an in vivo pull down experiment (Figure 3). The majority of the Nbs experienced no hinderance from the reducing environment and were capable of binding endogenous p53 when expressed intracellularly. p53 TAD Nb91 was, in spite of its satisfactory intracellular expression, not able to pull down p53. Its rather low affinity (~ 2.68 ± 0.69 µM) probably accounts for this observation (Figure 2(c)). Curiously, intracellular expression could not be detected for p53 TAD Nb18 and p53 TAD Nb25, resulting in a negative outcome in the in vivo pull down (Figure 3). Possibly, these two Nbs are an exception to the rule and display a low intracellular stability resulting in their unfolding.
The nanobody set was raised specifically against the N-terminal transactivation domain of p53. This domain forms a promiscuous binding site for components of the transcriptional machinery, like the general transcription factors TFIID and TFIIH,43,44 for its negative regulators Mdm2 and Mdm4,45,46 and for the transcriptional co-activators p300/CREB-binding protein (CBP).47 The Nbs had no effect on the transactivation functions of p53, and thus probably do not interfere with crucial protein-protein interactions occurring at this domain (Figure 4). Interestingly, only a few selected hydrophobic amino acids (F19, L22, W23, L25 and L26 (p53 TAD1); W53 and F54 (p53 TAD2)) in the transactivation domains of p53 are crucial for its functionality (reviewed by Raj N et al.).48 This observation was demonstrated via the creation of knock-in mice expressing different p53 transcriptional activation mutants, where the mere substitution of four amino acids (p53 L25Q, W26S, F53Q, F54S) resulted in the creation of a transactivation-dead p53 mutant.49 These dramatic effects are the result of the disruption of critical interactions between p53 and crucial cofactors. For example, F19, W23 and L26 mediate the interaction between p53, Mdm2 and Mdm4,46,50
F19, L22 and W23 coördinate the interaction between p53 and the TFIID complex,51 and F19, L22, L25, W53 and F54 are important residues for the interaction between p53 and p300/CBP.52,53 Considering this, and the fact that the transactivation functions of p53 are left intact in the presence of the Nbs, it is reasonable to state that the p53 TAD Nbs do not bind near these crucial amino acids. It seems most likely that interaction occurs either at the opposite side of the binding region for cofactors or near the Pro-rich domain of p53. It is also possible, albeit less likely, that the Nbs selectively bind one transactivation domain, since there is functional redundancy between both transactivation domains.
As previously mentioned, the activity of p53 is not confined to the nucleus as p53 also exerts transcription-independent functions in the cytoplasm. Of these, its capacity to induce the intrinsic mitochondrial-mediated apoptotic pathway is the best characterized.54 It has been demonstrated that Fsc and survivin Nbs, equipped with a MOM-tag, are able to trap their target at the mitochondria and, by doing so, limit their free diffusion in cells, creating a functional knock-out.18,23 We equipped the p53 TAD Nbs with a MOM/V5-tag and applied the delocalization strategy to p53. Mitochondrial delocalization of p53 by the p53 TAD Nbs was observed for each condition, albeit with varying success in the case of p53 TAD Nb10. This observation once more confirms the intracellular binding capacity of p53 TAD Nbs. The mitochondrial delocalization of HA-tagged wild type p53 significantly reduced cell viability of U2OS cells (Figure 7(b)). In apoptotic cells, the mitochondria form dense perinuclear clusters rather than showing a uniform distribution throughout the cytoplasm.55
Mitochondrial fragmentation can also be observed during necrotic cell death and mitophagy.56 When HA-tagged p53 co-localizes with the MOM/V5-tagged p53 TAD Nbs, which reside at the mitochondrial outer membrane, one can observe these dense perinuclear clusters (Figure 7(a) and Supplementary figure 8). A similar pattern is observed for delocalized endogenous wild type p53 (Figure 6(b) and Supplementary figure 7b). Here, however, no significant decrease in cell viability was detected (Figure 6(c)). This can be explained by the lower efficiency by which endogenous wild type p53 was captured at the mitochondria. Interestingly, these dense perinuclear clusters cannot be observed for the delocalized HA-tagged mutant p53 (R175H) (Figure 5(b) and Supplementary figure 6b), compatible with the absence of an effect on cell viability (Figure 5(c)). This is consistent with several studies showing that the R175H p53 mutant does not exhibit apoptotic activity.27,57,58 The R175H p53 mutant is largely unfolded at body temperature, since the substitution disrupts a salt bridge with D184 and causes a distortion of the zinc coordination sphere.4 Therefore, this mutant is incapable of binding the anti-apoptotic B-cell lymphoma 2 (Bcl2)-family members (Bcl2 and B-cell lymphoma-extra large (Bcl-xL)), and cannot neutralize their anti-apoptotic effect on Bcl2-associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak), thus preventing outer mitochondrial membrane permeabilization.58 Indeed, several biophysical characterization studies show that the DNA-binding domain of p53 is crucial for its interaction with Bcl-xL, Bcl2 and Bak.58–60 Furthermore, it has been shown that both mutant and wild type p53 interact with pro-caspase 3 near the mitochondria, but in contrast to wild type p53, mutant p53 impairs the activation of pro-caspase 3 by upstream caspases, thus preventing apoptosis.61 The true reason behind the reduced cell viability caused by nanobody-induced mitochondrial delocalization of HA-tagged wild type p53 in U2OS cells, however, has not yet been clarified. There are no indications that cell death is caused by an increased autophagic flux, since augmented LC3B-II levels could not be detected (Supplementary figure 11). Nanobody-induced sequestration of p53 at the mitochondria thus apparently does not alleviate the inhibitory effects of cytoplasmic p53 on autophagy.31 Experiments whereby apoptosis was assessed in a more direct manner were inconclusive, although the morphological alterations observed for the mitochondria suggest that a mitochondrial-mediated cell death pathway is activated. There are, however, multiple cell death pathways that can be activated by cytoplasmic p53. Apart from direct activation of the intrinsic mitochondrial-mediated apoptotic pathway, p53 is also directly involved in the activation of necrosis by interacting with cyclophilin D in the mitochondrial matrix.28 Alternatively, caspase-independent cell death after mitochondrial outer membrane permeabilization has been observed in a wide variety of cell types.62 In MCF-7 cells, accumulation of p53 at the mitochondria led to the activation of Bak, which subsequently initiated cell death in a caspase-independent way.29
Altogether, we have demonstrated the utility of p53 TAD Nbs as a research tool, with a potential in unraveling cytoplasmic functions of p53. The delocalization strategy, for example, can be of use to further look into these functions. For instance, the potency of mutant p53 to regulate mitochondrial apoptosis has not been completely clarified. In vitro studies show that some p53 mutants are still capable of inducing Bak oligomerization that results in the release of cytochrome c. In vivo analysis, however, showed that Bak activation by mutant p53 alone is not sufficient for induction of apoptosis and that other proteins and post-translational modifications play a crucial role.57 We have shown that p53 TAD Nbs are capable of inducing mitochondrial delocalization of both wild type p53 and mutant p53 whereby indications of cell death are only present for the former. We thus believe that the Nbs can assist in the identification of protein interactions crucial for the induction of cell death via post-fixation labeling.63 In this way, which interactors are either absent or prevent the induction of mitochondrial-mediated cell death by mutant p53 can be clarified.
Insight into the relevance of nuclear or cytoplasmic functions of p53 in tumor suppression can also be obtained by equipping the Nbs with a nuclear localization signal (NLS)-tag or a nuclear export signal (NES)-tag. In this way, free diffusion of the protein can be limited to either the nucleus or the cytoplasm.18,63 This is a gentler and more specific way of manipulating the p53 pathway, since it has been shown that small compounds like pifithrin-α also affect other pathways.64 Alternatively, Nbs are valuable tools for spatiotemporal antigen tracking of intracellular proteins.65–67 One could use p53 TAD Nbs to track the cellular movements of both wild type p53 and p53 mutants in response to several cellular stresses, which might help to define how the nuclear and cytoplasmic routes of p53 are coordinated.
Nbs have also proven to be valuable for mass spectrometry applications.68,69 The use of p53 TAD Nbs in combination with mass spectrometry can be powerful to detect novel protein-protein interactions, especially when combining the nanobody footprinting technique and the BioID proximity-labeling strategy since they allow the detection of weak and transient interactions that might have been missed in previous research.63,70,71 Nanobody footprinting is a technique wherein Nbs, recognizing a different epitope on the same antigen, are used to precipitate the protein of interest and its associated proteins from crude extracts. Thereafter, the identity of the interacting proteins is recovered via mass spectrometry but, rather than screening for the presence of proteins, one screens for proteins that are missing in either of the two conditions. In this way, only authentic interactions remain, and nonspecific binders are excluded. Nbs can also be used as a crystallization chaperone for the structural investigation of proteins.22 Several biophysical studies on the interaction between p53 and Bcl2/Bcl-xL were conducted with truncated or mutated versions of these proteins because these studies require stable and soluble proteins.54 In the presence of p53 TAD Nbs, the protein complex between full-length p53 and Bcl2/Bcl-xL might be stabilized, which can result in a more representative image of the in vivo situation. The same strategy can be applied for the visualization of the weak p53-Bax interaction.72
In summary, p53 TAD Nbs are bonafide research tools that can be of use in versatile applications allowing researchers to further decipher the mysteries of the complex and intricate p53 pathway.
Materials and methods
Antibodies and reagents
Mouse monoclonal αHA agarose (clone HA-7, A2095), mouse αV5 agarose affinity gel (clone V5-10, A7345), mouse monoclonal anti-vinculin (clone hVIN-1, V9131), rabbit polyclonal αV5 (V8137), mouse monoclonal anti-p53 (DO-1, P6874), DAPI (D8417), etoposide (E1383), nutlin-3a (N6287) and casein (C7078) were purchased from Sigma-Aldrich. Mouse monoclonal αV5 (R960-25), Alexa Fluor 488/594 goat anti-rabbit or mouse (A11034, A11032) and Clean-Blot IP detection reagent (HRP) (21230) were purchased from Thermo Fisher Scientific. Mouse monoclonal anti-HA (clone 12CA5, 11583816001) and XTT cell proliferation kit II (11465015001) were purchased from Roche. Mito Tracker Orange CMTMRos (M7510) was purchased from Molecular Probes. Goat polyclonal anti-beta actin (ab8229) and rabbit, anti-goat IgG H&L (HRP) (ab6741) were purchased from Abcam. ECL anti-mouse or rabbit IgG horseradish peroxidase-linked substrates (NA931, NA9340) were purchased from GE Healthcare. JetPrime transfection reagent (114–15) was purchased from Polyplus Transfection. Goat anti-mouse IgG heavy and light chain antibody AP (A90-116AP) was purchased from Bethyl Laboratories. Mouse monoclonal anti-HA (901501) was purchased from Biolegend. Rapamycin (S1039) was purchased from Selleck Chemicals. Rabbit polyclonal αLC3B-II (2755S) was purchased from Cell Signaling Technologies. For additional information regarding the lot number the original immunogen used for antibody production and the used dilutions for the experiments, see supplementary data (Supplementary table 2).
Generation of p53 TAD nanobodies
Nbs targeted against the transactivation domain of p53 (p53 TAD, AA 1–102) were generated in collaboration with the VIB Nanobody Service Facility as previously described.18 All animal work was performed by the VIB Nanobody Service Facility and has Public Health Service-approved animal welfare assurance from the Office of Laboratory Animal Welfare (F16-00131 (A5593-01)). In short, immunization of an alpaca occurred via subcutaneous injection of 150 µg of purified human p53 TAD on days 0, 7, 14, 21, 28 and 35. At day 39, the anticoagulated blood was collected and lymphocyte preparation was conducted. Next, a VHH library was constructed which was subjected to phage display in order to identify antigen-specific Nbs. The constructed VHH library contained 3 × 108 independent transformants, of which 80% harbored the vector with the right insert size. Next, 4 consecutive rounds of panning were performed on the solid-phase coated antigen. After the 3rd and 4th round of panning, the phage population was enriched for antigen-specific phages. Subsequently, 95 colonies were randomly selected from both the 3rd as the 4th panning round and were analyzed by ELISA for the presence of antigen-specific Nbs in the periplasmic extracts. This screening revealed 23 positive colonies in round 3 of panning and 30 positive colonies in round 4. After sequencing, these 53 colonies appeared to represent 15 different Nbs which can be classified into 11 different groups.
cDNA cloning
The expression plasmid pMECS was used to subclone the nanobody into the pcDNA3.1 V5/His6 and pcDNA3.1 MOM/V5 vector. The cloning experiments were carried out using the Cold FusionTM cloning kit (System Biosciences). The following primers were employed to subclone the Nbs into the pcDNA3.1 V5/His6 vector: 5ʹ TTG GTA CCG AGC TCG GCC ACC ATG CAG GTG CAG CTG CAG GAG 3ʹ (forward primer) and 5ʹ TAG ACT CGA GCG GCC GCT GGA GAC GGT GAC CTG 3ʹ (reverse primer). Subcloning of the Nbs into the pcDNA3.1 MOM/V5 vector was executed using following primers: 5ʹ TC GAT TCT ACG CGT ACC GGT GCC CAG GTG CAG CTG CAG GAG 3ʹ (forward primer) and 5ʹ GGT GAT GAT GAC CGG CTA GCT GGA GAC GGT GACC TG 3ʹ (reverse primer).
Cell culture and transfection
HEK293T, U2OS pGL13, U2OS and H1299 cell lines were cultivated in DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum. The cells were grown at 37°C in a humidified incubator at 10% CO2 (HEK293T, U2OS pGL13 and U2OS cells) or 5% CO2 (H1299 cells). The JetPrime transfection reagent (Polyplus Transfection) was used to transiently express the Nbs in the aforementioned cell lines. Transfection was performed according to the manufacturers’ instructions.
Investigation of in vitro binding characteristics of p53 TAD nanobodies
ELISA
Expression and purification of recombinant p53 TAD nanobodies and untagged p53 TAD
The p53 TAD Nbs, cloned in the pMECS vector, are equipped with a PelB signal sequence at their N-terminus. This signal sequence targets the nanobody to the periplasmic space of E. coli, which allows their retrieval from the periplasm. Heat-shock competent E. coli cells (WK6 strain, created by Dr. Gholamreza Hassanzadeh-Ghassabeh (Nanobody Service Facility, VIB)) were transformed with p53 TAD nanobody constructs (pMECS) and were grown overnight at 37°C on a lysogeny broth (LB)-agar plate containing 100 µg/ml ampicillin and 1% glucose. Individual colonies were picked, and the tip was dipped into one well of a 96-well plate to which 100 µl tryptone yeast (TY) (2x) media, supplemented with 10% glycerol, 1% glucose and 100 µg/ml ampicillin, was added. The plates were incubated overnight at 37°C. Subsequently, 1 ml of TY (2x) media was added to a 96 deep-well plate and each well was inoculated with 20 µl of the corresponding pre-culture. The plates were incubated at 37°C in a thermal shaker until an optical density (OD)600 of 1.00 was reached. Once the required optical density was obtained, nanobody expression was induced by adding 1 mM isopropyl β-D-thiogalactoside (IPTG). Cultures were incubated for another 3–5 hours in a thermal shaker at 37°C, and then centrifuged for 20 min at 3,000 x g. The supernatant was discarded, and plates were incubated upside down overnight at −80°C. Next, 100 µl phosphate-buffered saline (PBS; -Ca2+/Mg2+) was added to the culture plates, followed by 30 min incubation at room temperature (RT) on a shaker. Finally, plates were centrifuged for 20 min at 3,000 x g, retrieving the periplasmic extract.
Recombinant production of untagged p53 TAD (AA 1–102, pTYB12 vector) was performed as previously described.73 Briefly, freshly transformed E. coli cells (BL21 strain, C6000-03, Invitrogen) were grown at 37°C until an OD600 of 2.00 was reached, and at that point the expression of p53 TAD was induced by adding 0.5 mM IPTG. Thereafter, cultures were incubated overnight at 20°C. The cells were pelleted by centrifugation (11,000 x g for 20 min at 4°C), and the bacterial pellet was resuspended in chitin column buffer (20 mM Tris, 500 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.00). After French press and sonication, the suspension was centrifuged (29,000 x g for 30 min at 4°C), obtaining the bacterial protein lysate. Affinity purification, using a chitin resin (NEB), was performed according to the manufacturers’ instructions, and led to the purification of p53 TAD. Ion exchange chromatography (MONO Q HR 10/10 column, GE Healthcare) was performed for further purification of p53 TAD. The same protocol was used for the recombinant production of Annexin V.
ELISA
A 96-well plate (Maxisorp, Nunc) was coated with 1 µg/ml p53 TAD in coating buffer (0.1 M NaHCO3, pH 8.20) or with 1 µg/ml Annexin V (negative control) and plates were incubated overnight at 4°C. In order to prevent non-specific protein binding, a blocking step was performed during which plates were incubated with 0.1% casein in PBS (-Ca2+/Mg2+) for 1 h at RT. Subsequently, 20 µl periplasmic extract was added to both p53 TAD-coated and Annexin V-coated wells and the extract was further diluted with PBS (-Ca2+/Mg2+) until a total volume of 100 µl/well was reached. After 1 h incubation time at RT, plates were successively incubated with an anti-HA antibody (1 h at RT) and a goat-anti-mouse alkaline phosphatase (AP) antibody (1 h at RT). Finally, 100 µl of developing solution was added to the plates and absorbance was measured at 405 nm via a spectrophotometer. The developing solution consisted of 2 mg/ml AP substrate dissolved in AP blot buffer (100 mM Tris, 50 mM MgCl2.6 H2O, 100 mM NaCl, pH 9.50). In between each step, the plates were washed several times with 0.1% Tween in PBS (-Ca2+/Mg2+). A nanobody is considered as a possible binder if a 3-fold difference in absorbance was observed between the p53 TAD-coated (positive) and Annexin V-coated (negative) wells.
In vitro pull down
Expression and purification of recombinant nanobodies
The recombinant production of p53 TAD Nbs (pMECS vector) used for pull down experiments was performed as previously described.74 Briefly, E. coli cells (WK6 strain) transformed with the pMECS-nanobody plasmids were grown at 37°C until an OD600 of 0.60–0.80 was reached. Nanobody expression was induced by adding 1 mM IPTG, followed by overnight incubation at 28°C. Bacterial cultures were pelleted by centrifugation (11,000 x g for 20 min at 4°C). The periplasmic extract, containing the recombinant Nbs, was obtained through the administration of an osmotic shock using Tris-EDTA-sucrose (TES) buffer (0.2 M Tris, 0.5 mM EDTA, 0.5 M sucrose, pH 8.00). The concentration of the periplasmic extract was determined via the Bradford protein assay (Bio-Rad Laboratories) according to the manufacturers’ prescription.
In vitro pull down
The in vitro pull down was performed as previously described.18 In brief, cell lysis of HEK293T cells was performed using an ice-cold Tris-lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethane sulfonyl fluoride (PMSF) and 200 µg/ml protease inhibitor cocktail mix). Hereafter, 1 mg of crude HEK293T extract was incubated with 10 µg periplasmic extract and 10 µl settled αHA agarose beads. To exclude non-specific binding of p53 with the beads, a negative control was implemented where 1 mg of crude extract was incubated with αHA agarose beads in the absence of a nanobody. The samples were rotated for 2 h at 4°C, allowing the immobilization of the recombinant HA-tagged Nbs onto αHA-agarose beads. Beads were washed with Tris-lysis buffer and boiled for 5 min in Laemmli SDS sample buffer to elute the proteins. Proteins were separated according to their molecular weight via SDS-PAGE, which was followed by western blot analysis.
Isothermal titration calorimetry
Expression and purification of recombinant nanobodies
Expression of recombinant p53 TAD Nbs was performed as described earlier (i.e., in vitro pull-down experiment), but rather than preparing a periplasmic extract containing the Nbs, immobilized metal affinity chromatography (IMAC) was used for further purification of the Nbs which express a His6-tag at their C-terminus. Bacterial pellets are resuspended in lysis buffer (50 mM Tris-HCl, 0.1 mM β-mercaptoethanol, 100 mM KCl, 1 mM PMSF, 200 µg/ml protease inhibitor cocktail mix, 1% NP-40 and 0.2 mg/ml lysozyme, pH 8.25) and the suspension is rotated for 40 min at RT to achieve cell lysis. Next, the mixture is sonicated (Vibracell, Sonics and Materials) and centrifuged (29,000 x g for 20 min at 4°C) to acquire the bacterial lysate. Thereafter, the bacterial lysate was incubated with Ni2+ -chelating beads (ProBond Nickel-chelating resin, Invitrogen) and the mixture was rotated for 2 h at 4°C. After a number of washing steps (20 mM Tris, 500 mM KCl, 20 mM imidazole, 0.1 mM beta-mercaptoethanol, 10% glycerol, 1 mM PMSF and 200 µg/ml protease inhibitor cocktail mix, pH 8.25), Nbs were eluted from the beads (20 mM Tris, 100 mM KCl, 500 mM imidazole, 0.1 mM beta-mercaptoethanol, 10% glycerol, 1 mM PMSF and 200 µg/ml protease inhibitor cocktail mix, pH 8.25). A final purification step was performed by size-exclusion chromatography using a Superdex 75 PG 16/60 column (GE Healthcare).
ITC
The thermodynamic parameters of the interaction between p53 TAD Nbs and the p53 TAD was determined for a few selected Nbs (p53 TAD Nb28, p53 TAD Nb43 and p53 TAD Nb91) via isothermal titration calorimetry using a Microcal VPITC MicroCalorimeter (Malvern instruments Ltd). Both purified p53 TAD Nbs as purified untagged p53 TAD were dialyzed against 20 mM Tris, 150 mM NaCl, pH 7.5. Binding affinity was measured at 30°C and the experiment was performed as described earlier.74 In short, 9–10 µM p53 TAD was titrated with 90–100 µM p53 TAD Nbs. Data were analyzed with the Microcal Origin software and were fitted to a ‘One Set of Sites’ model.
Assessing in vivo properties of p53 TAD nanobodies
In vivo pull-down assay
JetPrime transfection reagent was used to transiently express p53 TAD Nbs (subcloned into the pcDNA3.1 vector) in HEK293T cells. The Nbs express a V5-tag at their C-terminal end and thus can be immobilized onto αV5-agarose beads. Cell lysis, measurement of the concentration of the crude extracts and pull down were performed as described earlier (i.e., in vitro pull down), but for the pull-down experiment 1 mg of each individual crude extract was incubated with 10 µl settled αV5-agarose beads. An irrelevant Nb, targeted against Fsc, was implemented as a negative control.
Transactivation assay
p53 TAD Nbs (subcloned into a pcDNA3.1 vector) were transiently expressed in U2OS pGL13 cells, which were seeded into a T25 culture flask, using the JetPrime transfection reagent. Cells were trypsinized 24 hours after transfection and were re-seeded into a 96-well format (10,000 cells/well). Transfected cells received (or did not receive) an additional treatment with 5 µM Nutlin3a. The next day, luciferase activity was measured by a Topcount luminometer (Canberra-Packard). First, cells were lysed in 50 µl luciferase lysis buffer (25 mM Tris-phosphate, 2 mM dithiotreitol (DTT), 2 mM trans-1,2-diaminocyclo-hexane -N, N, N′, N′-tetra acetic acid (CDTA), 10% glycerol, 1% Triton X-100 in 500 ml dH20, pH 7.80). After 10 min incubation at RT, 35 µl luciferase substrate buffer (40 mM Tricine, 2.14 mM (MgCO3)4Mg(OH)2.5H2O, 5.34 mM MgSO4, 66.6mM dithiotreitol (DTT), 0.2 mM EDTA, 509 µM Coenzyme A, 734 µM ATP, 940 µM D-Luciferin in 500 ml dH2O) was added to each well and the read-out was measured immediately.
Mitochondrial delocalization strategy: immunofluorescence and microscopy
p53 TAD Nbs were subcloned into the pcDNA3.1 MOM/V5 vector, resulting in the presence of a mitochondrial outer membrane (MOM)-tag and a V5-tag at their N-terminal end. These constructs were transiently expressed in U2OS cells and H1299 cells, whether or not in the presence of HA-tagged wild type p53 or HA-tagged mutant p53 (R175H) (subcloned into pcDNA3.1 vector), employing the JetPrime transfection reagent. Experiments were performed as described earlier.18 In short, cells were seeded onto collagen-coated coverslips and were transfected 24 h after cell-seeding. Another 24 h later, immunostaining was performed. Mitotracker Orange (M-7510, Thermo Fisher Scientific) was used for visualization of mitochondria, polyclonal rabbit anti-V5 (V8127, Sigma) was used for visualization of the nanobody, monoclonal mouse DO-1 (P6874, Sigma) or monoclonal mouse anti-HA (12CA5, Roche) was used for visualization of p53 and 4ʹ,6-diamidino-2-phenylindole (DAPI) (D8417, Sigma) was used for staining of the nucleus. Coverslips were mounted on microscopic slides using 1% n-propyl gallate in glycerol as mounting medium and were sealed with nail polish. Analysis of the cells was performed using a Zeiss Axiovert 200M fluorescence microscope with Apotome module (Zeiss x63, 1.4-NA Oil Plan Apochromat Objective, Carl Zeiss) and Axiovision 4.5 software (Zeiss).
XTT assay
U2OS cells and H1299 cells were seeded into a T75 culture flask. The next day, cells were transfected with MOM/V5-tagged p53 TAD nanobody plasmids and/or HA-tagged wild type p53 or HA-tagged mutant p53 (R175H) plasmids using the JetPrime transfection reagent. Cells were re-seeded 24 h after transfection into a 96-well format, where 100 µl of each cell suspension was added per well (triplicate samples). The dilution of the cell suspension differed depending on the executed experiment: 2.5*105 cells/ml (endogenous p53, U2OS cells), 5*105 cells/ml (HA-tagged wild type p53, U2OS cells) and 1.25*105 cells/ml (HA-tagged mutant p53 (R175H), H1299 cells). Blank background control wells, containing only cell medium, were also provided. Next, the cells received (or did not receive) an additional treatment with 100 µM etoposide for a duration of 20 h. Finally, 50 µl of activated XTT-solution was added to the cells and control wells and the absorbance of the samples was measured 6 h (U2OS) or 4 h (H1299) later at 475 nm and 660 nm. The specific absorbance was obtained by subtracting the average value for the blank background control wells and the average value for the non-specific readings (A660) from the values measured at 475 nm.
Autophagy: assessment of changes in LC3B-II levels
U2OS cells were seeded into 6-well plates. The next day, cells were transfected with MOM/V5-tagged p53 TAD nanobody plasmids and HA-tagged wild type p53 using the JetPrime transfection reagent. A negative control was implemented whereby cells were co-transfected with MOM/V5-tagged Fsc Nb2 and HA-tagged wild type p53 or were only transfected with HA-tagged wild type p53. As a positive control, an additional condition was provided whereby cells, transfected with HA-tagged wild type p53, were treated with 500 nM rapamycin for a duration of 4 h. This was performed in duplicate, whereby transfected cells received (or did not receive) an additional treatment with 100 mM NH4Cl for a duration of 4 h. Cell lysis was performed 24 h after transient transfection using RIPA lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 200 µg/ml protease inhibitor cocktail mix, pH 7.6). Thereafter, a 40 µg sample of crude extract was loaded onto a 15% SDS-gel and proteins were separated according to their molecular weight, followed by western blot analysis.
Funding Statement
This work was supported by the Agentschap voor Innovatie door Wetenschap en Technologie [IWT-141238]; Fonds Wetenschappelijk Onderzoek [G.0559.16N]; Ghent University (BOF-GOA) [BOF13/GOA/010].
Acknowledgments
The authors thank Dr. Gholamreza Hassanzadeh-Ghassabeh (Nanobody Service Facility, Vlaams Instituut voor Biotechnologie (VIB), Brussels, Belgium) for the generation and isolation of p53 TAD-specific Nbs and the creation of the WK6 E. coli strain. The H1299 cell line and U2OS pGL13 cell line were a kind gift from Prof. Dr Joost Schymkowitz and Prof. Dr Frederic Rousseau (VIB-KULeuven). The U2OS cell line was a kind gift from Rene Bernards (Netherlands Cancer Institute, Amsterdam). The HEK293T cell line was a kind gift from Prof. Dr Sven Eyckerman (VIB-UGent center for medical biotechnology).
Disclosure statement
In accordance with Taylor & Francis policy and my ethical obligation as a researcher, I am reporting that Prof. Dr Jan Gettemans has a financial and/or business interests in Gulliver Biomed BVBA, a company that may be affected by the research reported in the enclosed paper. I have disclosed those interests fully to Taylor & Francis.
Supplemental data
Supplemental data for this article can be accessed here.
Abbreviations
- AMP-dependent kinase
adenosine monophosphate-dependent kinase
- AP
alkaline phosphatase
- Bak
Bcl-2 homologous antagonist/killer
- Bax
Bcl-2 associated X
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- CBP
CREB-binding protein
- CDTA
trans-1,2-diaminocyclo-hexane -N, N, N′, N′-tetra acetic acid
- CE
crude extract
- DAPI
4ʹ,6-diamidino-2-phenylindole
- DTT
dithiotreitol
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- Fsc
fascin
- HA-tag
hemagglutinin tag
- HCAbs
heavy-chain-only antibodies
- IMAC
immobilized metal affinity chromatography
- IPTG
isopropyl β-D-thiogalactoside
- ITC
isothermal titration calorimetry
- LB
lysogeny broth
- LC
light chain IgG antibody
- LC3
microtubule-associated protein 1A/1B-light chain 3
- LOH
loss of heterozygosity
- Mdm2
murine double minute 2
- Mdm4
murine double minute 4
- MOM
mitochondrial outer membrane
- mTOR
mammalian target of Rapamycin
- Nbs
nanobodies
- NES
nuclear export signal
- NLS
nuclear localization signal
- OD
optical density
- p53 TAD
transactivation domain of p53
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
- Pro-rich
proline-rich
- RT
room temperature
- TES buffer
tris-EDTA-sucrose buffer
- TFIID
transcription factor II D
- TFIIH
transcription factor II H
- TOM70
translocase of outer membrane 70
- TY media
tryptone yeast media
- VHHs
variable domain of heavy-chain-only antibodies
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
References
- 1.Lane DP, Crawford LV.. T antigen is bound to a host protein in SV40-transformed cells. Nature. 278;1979:261–263. [DOI] [PubMed] [Google Scholar]
- 2.Linzer DI, Levine AJ.. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 17;1979:43–52. [DOI] [PubMed] [Google Scholar]
- 3.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 57;1989:1083–1093. [DOI] [PubMed] [Google Scholar]
- 4.Joerger AC, Fersht AR. The p53 Pathway: origins, Inactivation in Cancer, and Emerging Therapeutic Approaches In: Kornberg RD, ed. Annual Review of Biochemistry. Vol. 85, Palo Alto, CA:Annual Reviews; 2016. p. 375–404. doi: 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- 5.Muller PAJ, Vousden KH. 2013. p53 mutations in cancer. Nat Cell Biol. 15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 6.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–130. [DOI] [PubMed] [Google Scholar]
- 7.Levine AJ. 2009. The common mechanisms of transformation by the small DNA tumor viruses: the inactivation of tumor suppressor gene products: p53. Virology. 384:285–293. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 8.Manfredi JJ. 2010. The Mdm2-p53 relationship evolves: mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 24:1580–1589. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proceedings of the National Academy of Sciences of the United States of America 2001; 98:11598–11603. doi: 10.1073/pnas.181181198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikolaev AY, Li MY, Puskas N, Qin J, Gu W. Parc: A cytoplasmic anchor for p53. Cell. 112;2003:29–40. [DOI] [PubMed] [Google Scholar]
- 11.Hu W, Feng Z, Levine AJ. 2012. The Regulation of Multiple p53 stress responses is mediated through MDM2. Genes & Cancer. 3:199–208. doi: 10.1177/1947601912454734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comel A, Sorrentino G, Capaci V, Del Sal G. 2014. The cytoplasmic side of p53 ‘ s oncosuppressive activities. FEBS Lett. 588:2600–2609. doi: 10.1016/j.febslet.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 14.Hager KM, Gu W. 2014. Understanding the non-canonical pathways involved in p53-mediated tumor suppression. Carcinogenesis. 35:740–746. doi: 10.1093/carcin/bgt487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Reports. 3:1339–1345. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Meek DW. 2015. Regulation of the p53 response and its relationship to cancer. Biochem J. 469:325–346. doi: 10.1042/BJ20150517. [DOI] [PubMed] [Google Scholar]
- 17.Muyldermans S. Nanobodies: natural single-domain antibodies In: Kornberg RD, ed. Annual review of biochemistry. Vol. 82 Palo Alto, CA: Annual Reviews; 2013. p. 775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 18.Beghein E, Van Audenhove I, Zwaenepoel O, Verhelle A, De Ganck A, Gettemans J. A new survivin tracer tracks, delocalizes and captures endogenous survivin at different subcellular locations and in distinct organelles. Sci Rep. 2016;6:Art. 31177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertier L, Boucherie C, Zwaenepoel O, Vanloo B, Van Troys M, Van Audenhove I, Gettemans J. 2017. Inhibitory cortactin nanobodies delineate the role of NTA- and SH3-domain-specific functions during invadopodium formation and cancer cell invasion. Faseb J. 31:2460–2476. doi: 10.1096/fj.201600810RR. [DOI] [PubMed] [Google Scholar]
- 20.Bethuyne J, De Gieter S, Zwaenepoel O, Garcia-Pino A, Durinck K, Verhelle A, Hassanzadeh-Ghassabeh G, Speleman F, Loris R, Gettemans J. 2014. A nanobody modulates the p53 transcriptional program without perturbing its functional architecture. Nucleic Acids Res. 42:12928–12938. doi: 10.1093/nar/gku962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Audenhove I, Denert M, Boucherie C, Pieters L, Cornelissen M, Gettemans J. 2016. Fascin Rigidity and L-plastin Flexibility Cooperate in Cancer Cell Invadopodia and Filopodia. J Biol Chem. 291:9148–9160. doi: 10.1074/jbc.M115.706937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, Muyldermans S, Hol WG, Kobilka BK, Steyaert J. 2014. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 9:674–693. doi: 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Audenhove I, Boucherie C, Pieters L, Zwaenepoel O, Vanloo B, Martens E, Verbrugge C, Hassanzadeh-Ghassabeh G, Vandekerckhove J, Cornelissen M, et al. Stratifying fascin and cortactin function in invadopodium formation using inhibitory nanobodies and targeted subcellular delocalization. FASEB J. 2014;28:1805–1818. doi: 10.1096/fj.13-242537. [DOI] [PubMed] [Google Scholar]
- 24.Joerger AC, Fersht AR. 2008. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 26.McBride HM, Millar DG, Li JM, Shore GC. A signal-anchor sequence selective for the mitochondrial outer membrane. J Cell Biol. 119;1992:1451–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Molecular Cell. 11;2003:577–590. [DOI] [PubMed] [Google Scholar]
- 28.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. 2012. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang JJ, Guo WH, Zhou H, Luo N, Nie CL, Zhao XY, Yuan Z, Liu XY, Wei YQ. 2015. Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget. 6:17192–17205. doi: 10.18632/oncotarget.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu CL, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mrakovcic M, Frohlich LF. p53-Mediated molecular control of autophagy in tumor cells. Biomolecules. 2018;8:Art. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fitzwalter BE, Thorburn A. 2015. Recent insights into cell death and autophagy. FEBS J. 282:4279–4288. doi: 10.1111/febs.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orhon I, Reggiori F. Assays to Monitor Autophagy Progression in Cell Cultures. Cells. 2017;6:Art. UNSP 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. 2016. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol. 6:21. doi: 10.3389/fonc.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu XR, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, Fersht AR. 2013. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41:6034–6044. doi: 10.1093/nar/gkt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clinical Cancer Res: an Official J American Assoc Cancer Res. 2016;22:868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia MX, Tardell C, Garrido R, Lee E, Kolinsky K, et al. MDM2 Small-Molecule Antagonist RG7112 Activates p53 Signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 38.Meric-Bernstam F, Saleh MN, Infante JR, Goel S, Falchook GS, Shapiro G, Chung KY, Conry RM, Hong DS, Wang JS-Z, et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J Clin Oncol. 2017;35:2505. [Google Scholar]
- 39.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang P, Du WJ, Wang XW, Mancuso A, Gao XA, Wu MA, Yang XL. 2011. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 13:310–U278. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 74;2001:277–302. [DOI] [PubMed] [Google Scholar]
- 42.Helma J, Cardoso MC, Muyldermans S, Leonhardt H. 2015. Nanobodies and recombinant binders in cell biology. J Cell Biol. 209:633–644. doi: 10.1083/jcb.201409074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Lello P, Jenkins LM, Jones TN, Nguyen BD, Hara T, Yamaguchi H, Dikeakos JD, Appella E, Legault P, Omichinski JG. 2006. Structure of the Tfb1/p53 complex: insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol Cell. 22:731–740. doi: 10.1016/j.molcel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier JL, Triezenberg SJ, Reinberg D, Flores O, Ingles CJ, et al. BINDING OF BASAL TRANSCRIPTION FACTOR TFIIH TO THE ACIDIC ACTIVATION DOMAINS OF VP16 AND P53. Mol Cell Biol. 1994;14:7013–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chi SW, Lee SH, Kim DH, Ahn MJ, Kim JS, Woo JY, Torizawa T, Kainosho M, Han KH. 2005. Structural details on mdm2-p53 interaction. J Biol Chem. 280:38795–38802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- 46.Popowicz GM, Czarna A, Holak TA. 2008. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 7:2441–2443. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 47.Scolnick DM, Chehab NH, Stavridi ES, Lien MC, Caruso L, Moran E, Berger SL, Halazonetis TD. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 57;1997:3693–3696. [PubMed] [Google Scholar]
- 48.Raj N, Attardi LD. The transactivation domains of the p53 protein. Cold Spring Harb Perspect Med. 2017;7:Art. a026047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brady Colleen A, Jiang D, Mello Stephano S, Johnson Thomas M, Jarvis Lesley A, Kozak Margaret M, Broz Daniela K, Basak S, Park Eunice J, McLaughlin Margaret E, et al. Distinct p53 transcriptional programs dictate acute DNA-Damage responses and tumor suppression. Cell. 145;2011:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 274;1996:948–953. [DOI] [PubMed] [Google Scholar]
- 51.Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proceedings of the National Academy of Sciences of the United States of America 1995; 92:5154–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller Jenkins LM, Feng H, Durell SR, Tagad HD, Mazur SJ, Tropea JE, Bai Y, Appella E. 2015. Characterization of the p300 Taz2-p53 TAD2 complex and comparison with the p300 Taz2-p53 TAD1 complex. Biochemistry. 54:2001–2010. doi: 10.1021/acs.biochem.5b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proceedings of the National Academy of Sciences of the United States of America 2007; 104:7009–7014. doi: 10.1073/pnas.0702010104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Speidel D. 2010. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 20:14–24. doi: 10.1016/j.tcb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Bottone MG, Santin G, Aredia F, Bernocchi G, Pellicciari C, Scovassi AI. 2013. Morphological features of organelles during apoptosis: an Overview. Cells. 2:294–305. doi: 10.3390/cells2020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elgass K, Pakay J, Ryan MT, Palmer CS. 2013. Recent advances into the understanding of mitochondrial fission. Biochim Et Biophys Acta-Molecular Cell Res. 1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Pietsch EC, Perchiniak E, Canutescu AA, Wang GL, Dunbrack RL, Murphy ME. 2008. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J Biol Chem. 283:21294–21304. doi: 10.1074/jbc.M710539200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, Pan H, Kessler H, Pancoska P, Moll UM. 2006. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 59.Follis AV, Llambi F, Ou L, Baran K, Green DR, Kriwacki RW. 2014. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat Struct Mol Biol. 21:535–543. doi: 10.1038/nsmb.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sot B, Freund SM, Fersht AR. 2007. Comparative biophysical characterization of p53 with the pro-apoptotic BAK and the anti-apoptotic BCL-xL. J Biol Chem. 282:29193–29200. doi: 10.1074/jbc.M705544200. [DOI] [PubMed] [Google Scholar]
- 61.Frank AK, Pietsch EC, Dumont P, Tao J, Murphy ME. 2011. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol Ther. 11:740–745. doi: 10.4161/cbt.11.8.14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tait SW, Green DR. 2008. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 27:6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Audenhove I, Van Impe K, Ruano-Gallego D, De Clercq S, De Muynck K, Vanloo B, Verstraete H, Fernandez LA, Gettemans J. 2013. Mapping cytoskeletal protein function in cells by means of nanobodies. Cytoskeleton (Hoboken, NJ). 70:604–622. doi: 10.1002/cm.21122. [DOI] [PubMed] [Google Scholar]
- 64.Komarova EA, Neznanov N, Komarov PG, Chernov MV, Wang K, Gudkov AV. 2003. p53 inhibitor pifithrin alpha can suppress heat shock and glucocorticoid signaling pathways. J Biol Chem. 278:15465–15468. doi: 10.1074/jbc.C300011200. [DOI] [PubMed] [Google Scholar]
- 65.Jullien D, Vignard J, Fedor Y, Bery N, Olichon A, Crozatier M, Erard M, Cassard H, Ducommun B, Salles B, et al. Chromatibody, a novel non-invasive molecular tool to explore and manipulate chromatin in living cells. J Cell Sci. 2016;129:2673–2683. doi: 10.1242/jcs.183103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maier J, Traenkle B, Rothbauer U. Real-time analysis of epithelial-mesenchymal transition using fluorescent single-domain antibodies. Sci Rep. 2015;5:Art. 13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Panza P, Maier J, Schmees C, Rothbauer U, Sollner C. 2015. Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development. 142:1879–1884. doi: 10.1242/dev.118943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pleiner T, Bates M, Trakhanov S, Lee CT, Schliep JE, Chug H, Bohning M, Stark H, Urlaub H, Gorlich D. 2015. Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. eLife. 4:e11349. doi: 10.7554/eLife.06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Y, Pellarin R, Fridy PC, Fernandez-Martinez J, Thompson MK, Li Y, Wang QJ, Sali A, Rout MP, Chait BT. 2015. A strategy for dissecting the architectures of native macromolecular assemblies. Nat Methods. 12:1135–1138. doi: 10.1038/nmeth.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beghein E, Gettemans J. 2017. Nanobody technology: A versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front Immunol. 8:771. doi: 10.3389/fimmu.2017.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 73.Van Overbeke W, Verhelle A, Everaert I, Zwaenepoel O, Vandekerckhove J, Cuvelier C, Derave W, Gettemans J. 2014. Chaperone nanobodies protect gelsolin against MT1-MMP degradation and alleviate amyloid burden in the gelsolin amyloidosis mouse model. Mol Ther. 22:1768–1778. doi: 10.1038/mt.2014.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Den Abbeele A, De Clercq S, De Ganck A, De Corte V, Van Loo B, Soror SH, Srinivasan V, Steyaert J, Vandekerckhove J, Gettemans J. 2010. A llama-derived gelsolin single-domain antibody blocks gelsolin-G-actin interaction. Cell Mol Life Sci. 67:1519–1535. doi: 10.1007/s00018-010-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.