

ABSTRACT
DSTA4637A, a THIOMAB™ antibody-antibiotic conjugate targeting Staphylococcus aureus, has shown promising bactericidal activity in a mouse model. DSTA4637A consists of a monoclonal anti-S. aureus antibody with an average of two rifalogue antibiotic molecules, dmDNA31, linked to its light chains. The goal of this study was to develop a minimal physiologically-based pharmacokinetic (mPBPK) model to characterize the pharmacokinetic (PK) properties of three analytes of DSTA4637A (i.e., total antibody, antibody-conjugated dmDNA31, and unconjugated dmDNA31) in mice, and to predict pharmacokinetics of DSTA4637A analytes in humans, as well as to provide an initial assessment for potential PK drug-drug interactions (DDI) in clinical trials via cross-species scaling of the mPBPK model. In the proposed model, selected organs, including heart, liver, and kidney, were connected anatomically with plasma and lymph flows. Mouse plasma and tissue concentrations of the three analytes of DSTA4637A were fitted simultaneously to estimate the PK parameters. Cross-species scaling of the model was performed by integrating allometric scaling and human physiological parameters. The final mPBPK model was able to successfully capture PK profiles of three DSTA4637A analytes in mouse plasma and in investigated organs. The model predicted a steady-state peak unbound dmDNA31 concentration lower than 5% of the IC50 of dmDNA31 towards cytochrome P450 following 100 mg/kg weekly intravenous dose, which suggests a low risk of PK DDI in humans for DSTA4637A with co-administered cytochrome P450 substrates. The proposed mPBPK modeling and cross-species scaling approaches provide valuable tools that facilitate the understanding and translation of DSTA4637A disposition from preclinical species to humans.
Keywords: THIOMAB™ antibody antibiotic conjugate, mPBPK, cross-species scaling, drug-drug interactions, Staphylococcus aureus
Introduction
Staphylococcus aureus (S. aureus) is a commensal bacterium and opportunistic pathogen of humans and animals. It is the leading cause of nosocomial infections worldwide with complications such as infective endocarditis, osteomyelitis, fatal necrotizing pneumonia, and sepsis.1 In addition, invasive S. aureus infection is the leading cause of death by an infectious agent in the United States, with a mortality rate of ~ 20%.2 Rapid and wide spread of antibiotic-resistant strains, such as methicillin-resistant S. aureus and recently reported vancomycin and linezolid-resistant S. aureus, have led to increased concern to human health.3 It has been known for more than 50 years that S. aureus applies a “Trojan horse model” to evade host immune responses and antibiotic treatments. S. aureus can invade and survive inside host cells that turn into the “Trojan horses”, which are responsible for the systemic dissemination of the bacteria and recurrence of infection.4,5 Therefore, the need for new therapies that can eliminate intracellular S. aureus in tissues is more pressing than ever.
A novel THIOMAB™ antibody-antibiotic conjugate (TAC), DSTA4637A, composed of an anti-S. aureus THIOMAB™ antibody and a potent antibiotic, 4-dimethylamino piperidino-hydroxybenzoxazino rifamycin (dmDNA31), linked through a protease-cleavable valine-citrulline linker, was developed. DSTA4637A has demonstrated highly efficacious intracellular bactericidal activity against S. aureus both in vitro and in vivo.6 The antibody module specifically targets the β-O-linked N-acetylglucosamine (GlcNAc) sugar modifications on wall teichoic acids of S. aureus and is responsible for opsonization of bacterial cells. Once the opsonized bacteria are taken up into phagolysosomes, proteases such as cathepsins cleave the linker and release active dmDNA31 antibiotic that kills intracellular S. aureus. In addition, DSTA4637A has shown superior potency to vancomycin in a mouse bacteremia model.6 Furthermore, application of the antibody-antibiotic conjugate strategy extends the half-life of dmDNA31 from 3 – 4 hours as a small molecule to ~ 4 days as antibody-conjugated dmDNA31 in mice, which makes less frequent dosing possible.7
Plasma pharmacokinetics (PK) of DSTA4637A have been well characterized in both non-infected and S. aureus infected mice by Zhou et al.7 The plasma concentration-time profile of DSTA4637A total antibody (TAb), which measures all drug to antibody ratios (DARs) of DSTA4637A, including fully conjugated, partially deconjugated, and fully deconjugated anti-S. aureus antibodies, and the plasma concentration-time profile of DSTA4637A conjugate, which measures the total concentration of antibody-conjugated dmDNA31 (ac-dmDNA31), showed bi-exponential disposition with rapid distribution and slow elimination phases. Both TAb and ac-dmDNA31 showed linear PK properties in the dose range of 5 – 50 mg/kg in non-infected mice.7 In addition, infection with S. aureus had minimal impact on the linker stability and plasma PK (e.g., clearance and volume of distribution) of TAb and ac-dmDNA31 in the efficacious dose range of 25 – 50 mg/kg.7 However, PK of the three analytes (i.e., TAb, ac-dmDNA31, and unconjugated dmDNA31) in infected organs in mice have not been reported. Particularly the PK of unconjugated dmDNA31 (uc-dmDNA31) was not well characterized due to its extremely low concentration and limited sensitivity of quantitative methods. Assessment of uc-dmDNA31 concentrations in human tissues is even more difficult because of limited access of tissue samples in clinical settings. The uc-dmDNA31 is the active small molecule component of DSTA4637A, and it is responsible for potential drug-drug interactions (DDI). We used the mPBPK from the mouse model to inform us about the uc-dmDNA31 tissue concentrations and potential DDI in humans. The dmDNA31 has been shown to be a relatively weak inhibitor of several human cytochrome P450 (CYP) enzymes, including CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5, in in vitro metabolism studies (Genentech Inc. unpublished data). Thus a priori estimation of uc-dmDNA31 tissue concentrations may provide useful insights about potential dmDNA31-mediated DDI with co-administered CYP substrates in humans.
As described here, we developed a mPBPK model that successfully characterized both plasma and tissue PK of TAb, ac-dmDNA31 and uc-dmDNA31 in S. aureus infected mice. The proposed model also provides a useful approach to predict uc-dmDNA31 concentrations at different dose levels/regimens in S. aureus-infected mice. In addition, this mPBPK modeling approach can be used to predict human plasma PK profile and tissue disposition of uc-dmDNA31 via cross-species scaling, thus providing early risk assessment of potential DDI for the TAC in clinical trials.
Results
DSTA4637A disposition and mPBPK model
Schemas of the proposed mPBPK model for DSTA4637A and sequential deconjugation of dmDNA31 from the TAC molecule are shown in Figure 1. The PK profiles of three DSTA4637A analytes following a single intravenous (IV) dose of 25 or 50 mg/kg DSTA4637A in S. aureus-infected mice are shown in Figures 2, Figures 3 and 4. A typical monoclonal antibody (mAb) bi-exponential concentration-time profile was observed for the disposition of TAb and ac-dmDNA31 in plasma and in tissues, including heart, liver, and kidney. The initial increase in TAb and ac-dmDNA31 concentrations in organs is due to antibody distribution from the central plasma compartment to peripheral tissues. The uc-dmDNA31 showed formation-limited kinetics. In vivo deconjugation of dmDNA31 from TACs (kdc = 0.141 day−1) was 47.5-fold slower than its degradation in mouse plasma (CLp, dmDNA31/Vp, dmDNA31 = 6.70 day−1) (Table 1), and the bi-exponential phases of deconjugation support the proposed sequential release (i.e., two-step release, DAR2 to DAR1 and DAR1 to DAR0) of dmDNA31 from TACs (Figures. 1b and 4). Tissue exposures of TAb were also dose-proportional in the dose range of 25–50 mg/kg in infected mice (Figure 2), which suggests saturated target-binding at the sites of infection, and hence linear PK at doses ≥ 25 mg/kg. Similar relative exposures (% tissue AUC0-14d/plasma AUC0-14d) of TAb and ac-dmDNA31, respectively, were predicted in heart (7.21% and 7.87%), liver (5.44% and 5.81%), and kidney (3.98% and 4.37%) (Figure 5a). The similarity in relative exposures of the two analytes supports the assumption that the distribution of ac-dmDNA31 is mainly driven by the distribution of antibodies. At 3 days post-dose, the observed maximum concentrations of uc-dmDNA31 in liver (184 ng/ml) and kidney (42.5 ng/ml) were much higher than that in plasma (2.45 ng/ml), where uc-dmDNA31 was barely detectable (Figure 4). Interestingly, concentrations of uc-dmDNA31 in heart were not detectable at all in this study.
Figure 1.
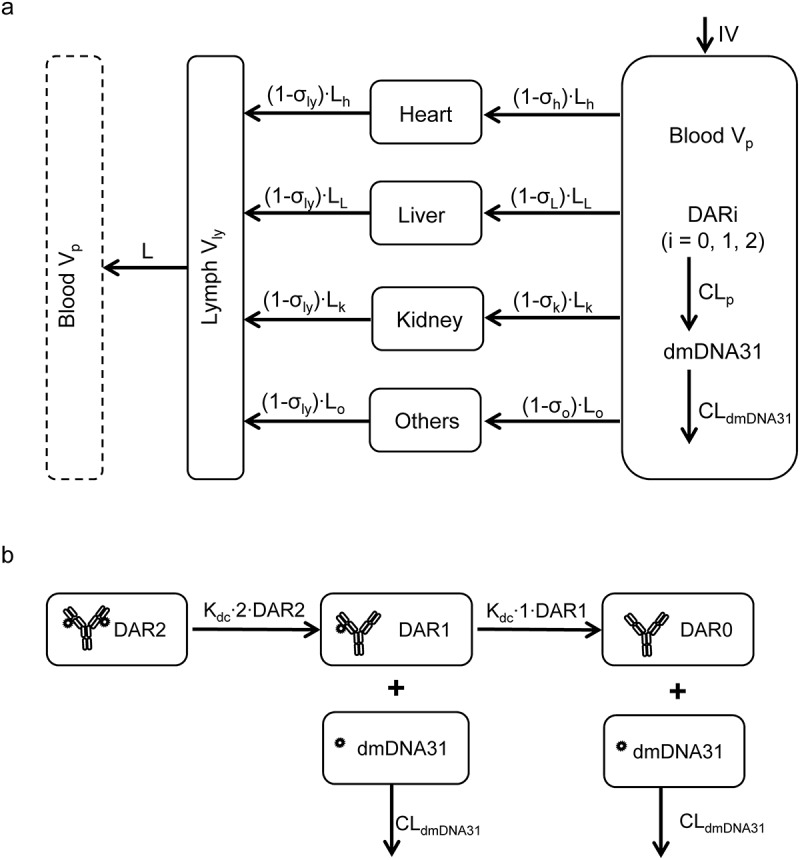
Schematic representation of the mPBPK model for DSTA4637A. (a) The mPBPK model of DSTA4637A disposition. Organs are connected anatomically with plasma and lymph flows. (b) Schematic showing sequential deconjugation of dmDNA31 from DSTA4637A. Models are described by equations in Materials and Methods with symbols defined in Tables 1 and 2.
Figure 2.
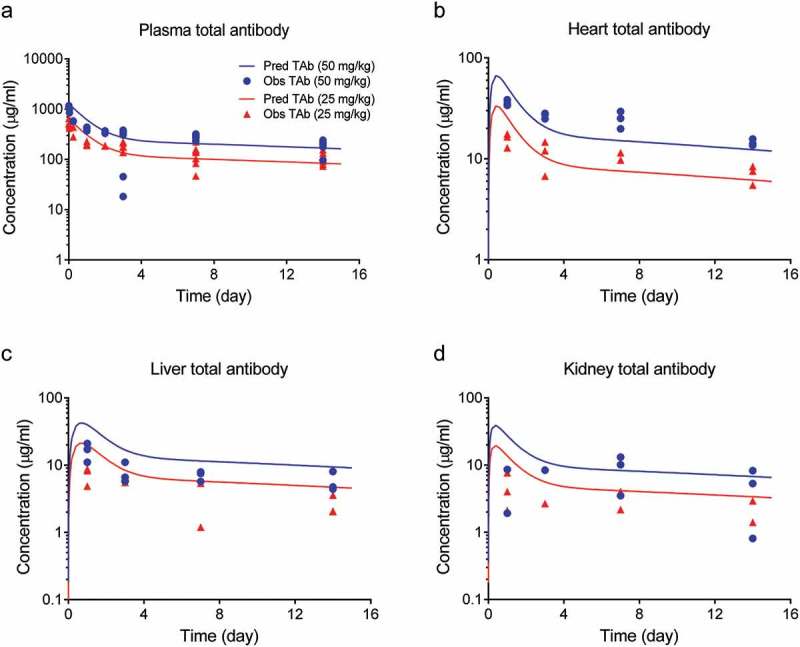
Total antibody concentrations in tissues (a) plasma, (b) heart, (c) liver, and (d) kidney after a single IV dose of 50 mg/kg (blue symbols) or 25 mg/kg (red symbols) of DSTA4637A in S. aureus infected mice. Solid lines represent model fittings, and symbols are observed data.7.
Figure 3.
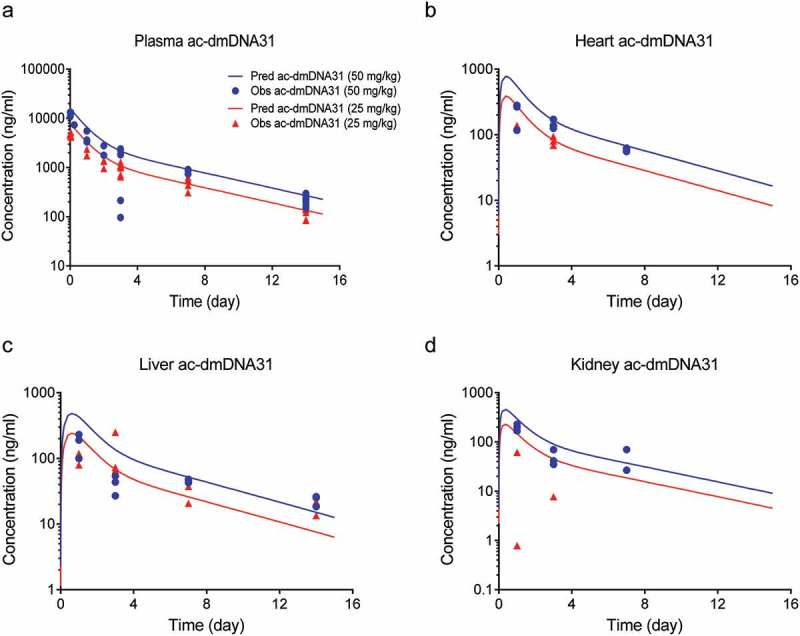
Antibody-conjugated dmDNA31 concentrations in tissues (a) plasma, (b) heart, (c) liver, and (d) kidney after a single IV dose of 50 mg/kg (blue symbols) or 25 mg/kg (red symbols) of DSTA4637A in S. aureus infected mice. Solid lines represent model fittings, and symbols are observed data.7.
Figure 4.
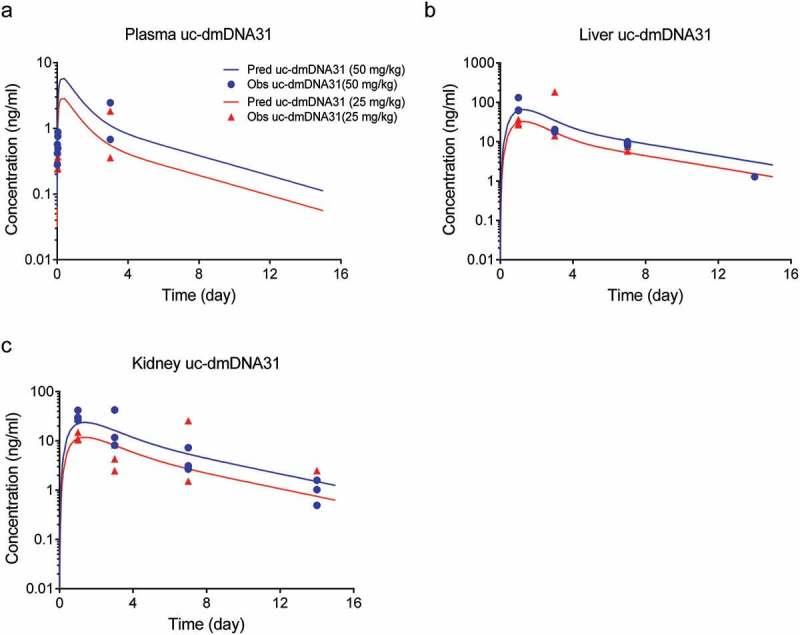
Unconjugated dmDNA31 concentrations in (a) plasma, (b) liver, (c) kidney after a single IV dose of 50 mg/kg (blue symbols) or 25 mg/kg (red symbols) of DSTA4637A in S. aureus infected mice. Solid lines represent model fittings, and symbols are observed data.7.
Table 1.
Estimated pharmacokinetic parameters for the mPBPK model of DSTA4637A.
| Parameter (unit) | Definition | Estimates (CV %) | 95% confidence interval |
|---|---|---|---|
| σh | Vascular reflection coefficient of heart | 0.934 (0.683) | 0.922 – 0.947 |
| σl | Vascular reflection coefficient of liver | 0.957 (0.399) | 0.950 – 0.965 |
| σk | Vascular reflection coefficient of kidney | 0.964 (0.404) | 0.956 – 0.972 |
| σo | Vascular reflection coefficient of lumped tissues | 0.910 (0.804) | 0.896 – 0.924 |
| kdc (1/day) | Deconjugation rate constant | 0.141 (5.19) | 0.126 – 0.155 |
| CLp (ml/day/kg) | TAC (DAR 0 – 2) plasma proteolytic clearance | 5.26 (fixed)1 | |
| Vp,uc-dmDNA31 (ml/kg) | uc-dmDNA31 plasma volume of distribution | 1.91 × 103 (17.1) | 1.27 × 103 – 2.56 × 103 |
| Vl,uc-dmDNA31 (ml/kg) | uc-dmDNA31 liver volume of distribution | 12.8 (92.0) | 0 – 35.9 |
| Vk,uc-dmDNA31 (ml/kg) | uc-dmDNA31 kidney volume of distribution | 13.5 (43.8) | 1.86 – 25.1 |
| CLp,uc-dmDNA31 (ml/day/kg) | uc-dmDNA31 plasma clearance | 1.28 × 104 (19.6) | 7.86 × 103 – 1.77 × 104 |
| CLl,uc-dmDNA31 (ml/day/kg) | uc-dmDNA31 liver clearance | 38.9 (15.8) | 26.8 – 51.0 |
| CLk,uc-dmDNA31 (ml/day/kg) | uc-dmDNA31 kidney clearance | 18.1 (15.6) | 12.6 – 23.7 |
1Estimated CLp value did not change significantly using different analysis methods, including non-compartmental analysis, two-compartment model, and mPBPK model, and hence it was fixed to the average estimated value.
Figure 5.
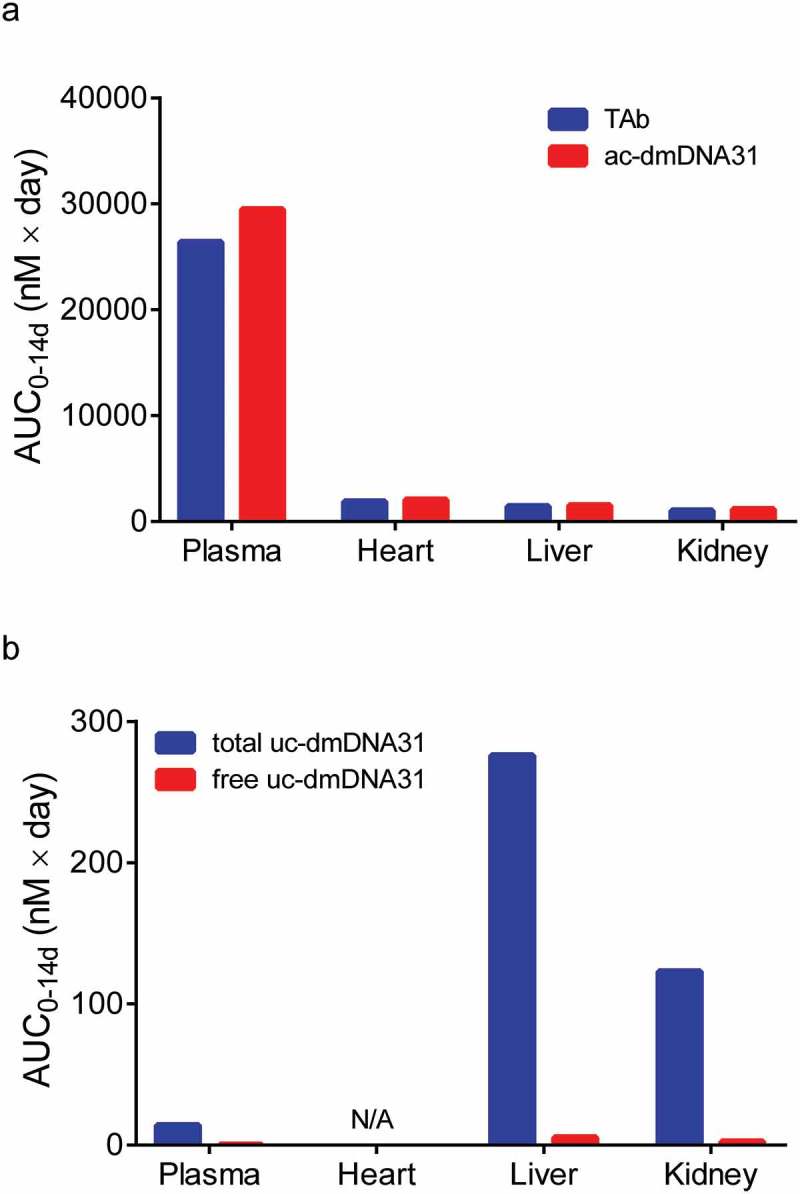
Model predicted AUC0-14d values of (a) TAb and ac-dmDNA31, and (b) total uc-dmDNA31 and free uc-dmDNA31 in tissues after a single IV dose of 50 mg/kg DSTA4637A in S. aureus infected mice. N/A = prediction not available.
Development of the mPBPK model for TAC disposition was based on the PK study of DSTA4637A in S. aureus infected mice.7 Concentrations of TAb, ac-dmDNA31, and uc-dmDNA31 were simultaneously fitted to estimate model parameters (i.e., naïve pooled method). The final mPBPK model was successful in capturing bi-exponential PK profiles for all DSTA4637A analytes (TAb, ac-dmDNA31, and uc-dmDNA31) in plasma, heart, liver, and kidney (Figure 2, 3, and 4). Although the majority of TAb and ac-dmDNA31 were present in plasma, uc-dmDNA31 accumulated in liver and kidney, with 19.4- and 8.62-fold higher model predicted AUC0-14d than that in plasma. In contrast, uc-dmDNA31 was undetectable in heart, albeit ac-dmDNA31 concentrations were comparable to the values in liver and kidney (Figure 5). This higher accumulation of uc-dmDNA31 in kidney, but not in plasma and heart, in infected mice correlated with a relatively high number of bacteria in kidney compared to the values in plasma and heart, but whether infection with S. aureus could lead to the dmDNA31 accumulation in tissues at therapeutic dose levels need to be confirmed in future studies.
The proposed mPBPK model was able to estimate PK parameters with relatively good accuracy and precision. CV% of the estimates varied from 0.399% to 92.0% (Table 1). In heart, liver, and kidney, the estimated σ were similar (0.934, 0.957 0.964, respectively). The clearance of uc-dmDNA31 in plasma (CLp, dmDNA31) was estimated to be 1.28 × 104 ml/day/kg, which is close to the measured value 1.01 × 104 ml/day/kg that was obtained from a separate study in mice with intravenously injected carbon-14-labeled dmDNA31 (Genentech Inc. unpublished data). Estimated clearances of uc-dmDNA31 in liver and kidney (CLl, uc-dmDNA31 = 38.9 ml/day/kg; CLk, uc-dmDNA31 = 18.1 ml/day/kg) were 329- and 707-fold lower than that in plasma, which explains the accumulation of uc-dmDNA31 in liver and kidney but not in plasma. Significantly smaller values of the estimated volume of distribution of uc-dmDNA31 in liver and kidney, Vl, uc-dmDNA31 = 12.8 ml/kg and Vk, uc-dmDNA31 = 13.5 ml/kg, than Vp, uc-dmDNA31 (1.91 × 103 ml/kg) in plasma may also suggest intracellular accumulation of uc-dmDNA31 in liver and kidney.
Cross-species scaling of the mPBPK model
An important application of PBPK modeling at early drug development is to predict human tissue PK profiles via cross-species scaling, which helps to provide initial assessment of potential PK DDI in clinical trials. Scale-up of our mPBPK model was performed by replacing mouse physiological parameters with human values (Table 2) and allometrically scaling of clearance and volume of distribution as described in equations 8 – 10. In this case, Vuc-dmDNA31 and CLuc-dmDNA31 were scaled as a function of body weight with allometric exponents of 1 and 0.75 for small molecules (Equations 8 and 9),14 while the clearances of TAb and ac-dmDNA31 were scaled with an exponent of 0.91 (Equation 10), an average allometric exponent for cross-species scaling of mAb clearance in a mPBPK model.15 Of note, our mPBPK model was developed using PK data collected from S. aureus-infected mice in a dose range (25–50 mg/kg) demonstrating linear PK. Thus, simulated tissue exposures of the three analytes based on this model may only be used to predict the exposures in S. aureus-infected patients, or in the cases where high doses of TAC are used and saturation of target-binding is expected. Therefore, Phase 1 clinical study data may be used to externally validate the model prediction, assuming tested doses reached target saturation and S. aureus infection does not affect the plasma PK of TACs in healthy human subjects as that observed in mice.7,16 Model-predicted human plasma PK profiles of TAb, ac-dmDNA31, and uc-dmDNA31 were indeed reasonably consistent (≤ 3 fold difference between predicted and observed concentrations) with Phase 1 clinical study data under the current model assumptions (data not shown).
Table 2.
Physiological parameters used for the mPBPK modeling and cross-species scaling.
| Parameter (unit) | Definition | Mouse value | Human value | Reference |
|---|---|---|---|---|
| Qh (ml/day/kg) | Heart plasma flow | 2.02 × 104 | 2.47 × 103 | 8 |
| Ql (ml/day/kg) | Liver plasma flow | 7.92 × 104 | 1.65 × 104 | 8 |
| Qk (ml/day/kg) | Kidney plasma flow | 5.76 × 104 | 1.30 × 104 | 8 |
| Qo (ml/day/kg) | Other tissue plasma flow | 1.58 × 105 | 2.98 × 104 | 8 |
| Vp (ml/kg) | Plasma volume | 38.7 | 38.6 | 8 |
| Vh (ml/kg) | Heart volume | 6.30 | 4.07 | 8 |
| Vl (ml/kg) | Liver volume | 49.4 | 23.3 | 9 |
| Vk (ml/kg) | Kidney volume | 13.4 | 3.65 | 8 |
| Vo (ml/kg) | Other tissues’ volume | 892* | 930* | |
| L (ml/day/kg) | Total lymph flow | 631 | 124 | 8 |
| Lh (ml/day/kg) | Heart lymph flow | Qh/500 | Qh/500 | 10,11 |
| Ll (ml/day/kg) | Liver lymph flow | Ql/500 | Ql/500 | 10,11 |
| Lk (ml/day/kg) | Kidney lymph flow | Qk/500 | Qk/500 | 10,11 |
| Lo (ml/day/kg) | Other tissues’ lymph flow | Qo/500 | Qo/500 | 10,11 |
| σly | Lymphatic capillary reflection coefficient | 0.2 | 0.2 | 12 |
| Vly (ml/kg) | Total lymph volume | 70.4 | 70.1 | 13 |
*Vo = body weight – Vp – Vh – Vl –Vk
Simulated maximum concentrations of free uc-dmDNA31 (i.e., fraction of protein unbound × Cmax, uc-dmDNA31) at different doses of DSTA4637A (assuming linear PK at the simulated doses) in infected mice are presented in Figure 6. Predicted free Cmax, uc-dmDNA31 in human liver and kidney were 7.31- and 3.48-fold higher than that in plasma, suggesting a similar uc-dmDNA31 accumulation in human tissues as observed in mice under the current assumptions. The predicted free uc-dmDNA31 concentration in human plasma was low at all simulated doses due to rapid plasma CLp, uc-dmDNA31. In in vitro metabolism studies, the dmDNA31 is a weak inhibitor of CYP enzymes with IC50 ranging from 1.7 to 5.2 µM, and there is no evidence that dmDNA31 causes a time-dependent inhibition or induction of CYP activity (Genentech Inc. unpublished data). Based on our model simulation, the circulating free uc-dmDNA31 is expected to be low in humans, which suggests that dmDNA31-mediated DDI are not very likely even at high doses of DSTA4637A. For example, a 200 mg/kg single IV dose of DSTA4637A is predicted to yield a hepatic free Cmax, uc-dmDNA31 of 0.05 µM, which is less than 3% the IC50 of dmDNA31 towards CYP3A4/5 (1.7 µM, Genentech Inc. unpublished data) (Figure 6). Furthermore, steady-state free Cmax, uc-dmDNA31 was estimated to be lower than 5% of the IC50 of dmDNA31 towards CYP3A at 100 mg/kg weekly IV dose (Figure 7). These predictions indicate a low potential for inhibitory effects of dmDNA31 on CYP isoforms and negligible possibility of DDI when co-administered with CYP substrates. Therefore, there is a low potential risk for free uc-dmDNA31 in humans as a DDI perpetrator that is dependent on the inhibition IC50 values and its concentrations in the circulation and in the liver.
Figure 6.
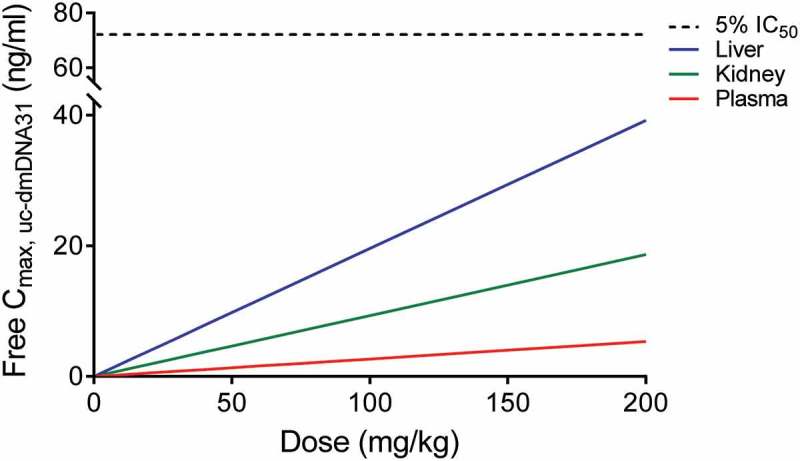
Predictions of free Cmax, uc-dmDNA31 in human plasma (red line), liver (blue line), and kidney (green line) after a single IV dose of DSTA4637A. Black dash line represents 5% the IC50 of dmDNA31 towards CYP3A.
Figure 7.
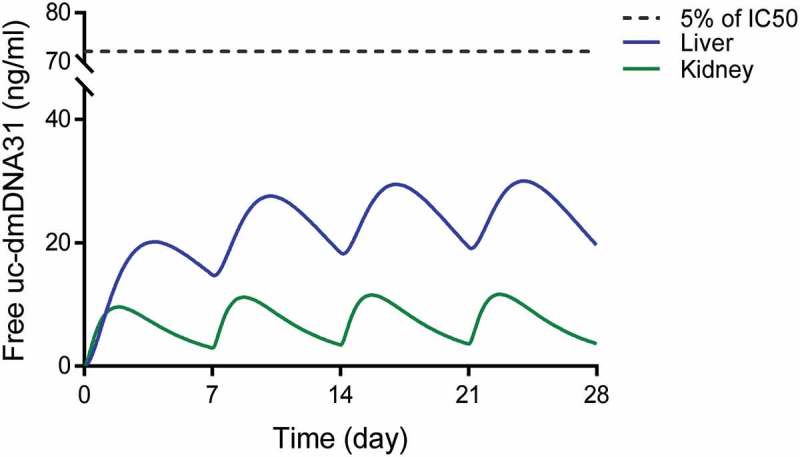
Simulated free uc-dmDNA31 concentrations in liver (blue line) and kidney (green line) given weekly IV doses of DSTA4637A at 100 mg/kg in humans. Black dash line represents 5% the IC50 of dmDNA31 towards CYP3A.
Model validation and sensitivity analysis
The predictability of the final mPBPK model was validated using TAb, ac-dmDNA31, and uc-dmDNA31 concentrations in non-infected mouse plasma following a single IV dose of 50 mg/kg DSTA4637A reported by Zhou et al. 7 These data can be used for model validation because linear PK has been observed for TAb and ac-dmDNA31 in the dose range of 5 – 50 mg/kg in non-infected mice and infection with S. aureus had minimal effect on plasma PK of TAC in the efficacious dose range of 25 – 50 mg/kg.7 The mPBPK model was able to predict plasma TAb and ac-dmDNA31 concentration-time profiles with reasonable accuracy (Figure 8). The ratios of model predicted:observed TAb and ac-dmDNA31 concentrations are less than two at all evaluated time points (Table 3). Although the model seems to over-predict plasma uc-dmDNA31 concentrations compared to observed values (Table 3), three time-point samples and concentrations close to the assay lower limit of quantification may only provide limited power to validate the model predictability of uc-dmDNA31 concentrations.
Figure 8.
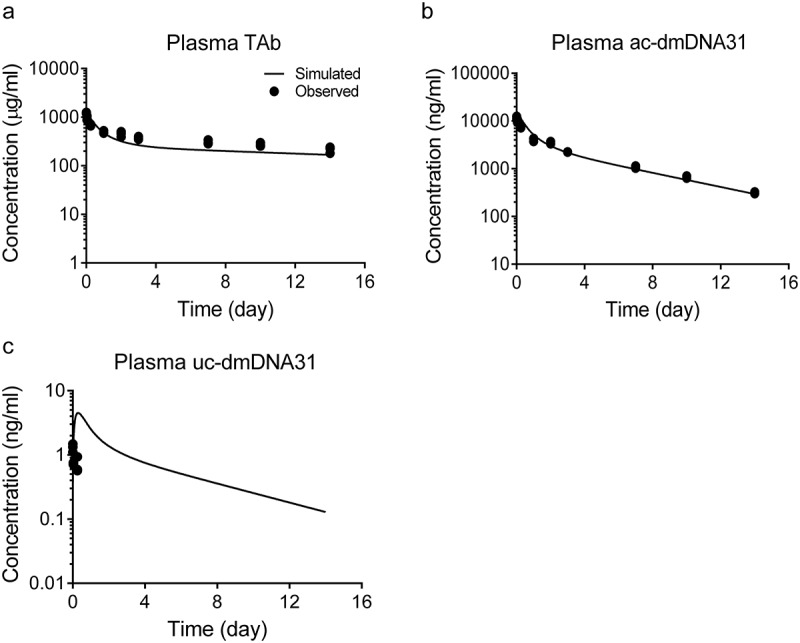
Model predicted TAb, ac-dmDNA31 and uc-dmDNA31 plasma concentrations given a single IV dose of 50 mg/kg DSTA4637A in non-infected mice. Solid lines represent model prediction, and symbols are observed data as reported by Zhou et al. 7.
Table 3.
Model predicted versus observed TAb, ac-dmDNA31, and uc-dmDNA31 concentrations in non-infected mouse plasma following a single IV dose of 50 mg/kg DSTA4637A.
| TAb (µg/ml) |
ac-dmDNA31 (ng/ml) |
uc-dmDNA31 (ng/ml) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Time (day) | Observed1 | Predicted | Ratio2 | Observed1 | Predicted | Ratio2 | Observed1 | Predicted | Ratio2 |
| 0.00694 | 1223 ± 32 | 1280 | 1.05 | 11978 ± 798 | 15896 | 1.33 | 1.31 ± 0.19 | 0.311 | 0.24 |
| 0.0417 | 988 ± 104 | 1224 | 1.24 | 10051 ± 1005 | 15123 | 1.50 | 0.734 ± 0.028 | 1.62 | 2.21 |
| 0.25 | 694 ± 27 | 944 | 1.36 | 7394 ± 282 | 11338 | 1.53 | 0.699 ± 0.207 | 4.46 | 6.38 |
| 1 | 494 ± 30 | 479 | 0.97 | 4088 ± 304 | 5184 | 1.27 | |||
| 2 | 451 ± 56 | 320 | 0.71 | 3487 ± 219 | 3016 | 0.86 | |||
| 3 | 374 ± 25 | 266 | 0.71 | 2240 ± 23 | 2179 | 0.97 | |||
| 7 | 308 ± 29 | 208 | 0.68 | 1091 ± 57 | 977 | 0.90 | |||
| 10 | 273 ± 22 | 189 | 0.69 | 678 ± 37 | 584 | 0.86 | |||
| 14 | 216 ± 31 | 168 | 0.78 | 311 ± 14 | 296 | 0.95 | |||
1Observed values are presented as mean ± SD.
2Ratio = Predicted: observed.
Sensitivity analysis was also performed to assess which parameter is most sensitive to TAb tissue distribution. Small changes in AUC values would suggest that TAb exposure is relatively insensitive to the adjusted parameter. Vascular reflection coefficients were found to be the most important parameters influencing the TAb plasma and tissue concentrations (Figure 9). 10% decreases of σo led to 33% increase in the AUC value of plasma TAb, which is reasonable because lump tissues (i.e., composite of all others) represent more than 90% of the total body weight. Tissue physiological volumes were shown to moderately affect the TAb concentrations in heart, liver, and kidney (Figure 9). Approximately 10% of changes in TAb AUC values were estimated when tissue volumes were adjusted. This result suggests that reactions, such as inflammation and abscess formation during bacterial infection, changing tissue volumes may affect the drug exposure. Observable changes of TAb exposures were also identified (i.e., less than 10% changes of AUCs) when plasma proteolytic clearance (CLp) was adjusted (Figure 9). TAb concentration is expected to be relatively insensitive to deconjugation rate constant (kdc) (Figure 9). However, kdc was found to be the key parameter affecting ac-dmDNA31 and uc-dmDNA31 concentrations (data not shown), which is consistent with the formation-limited kinetics of uc-dmDNA31.
Figure 9.
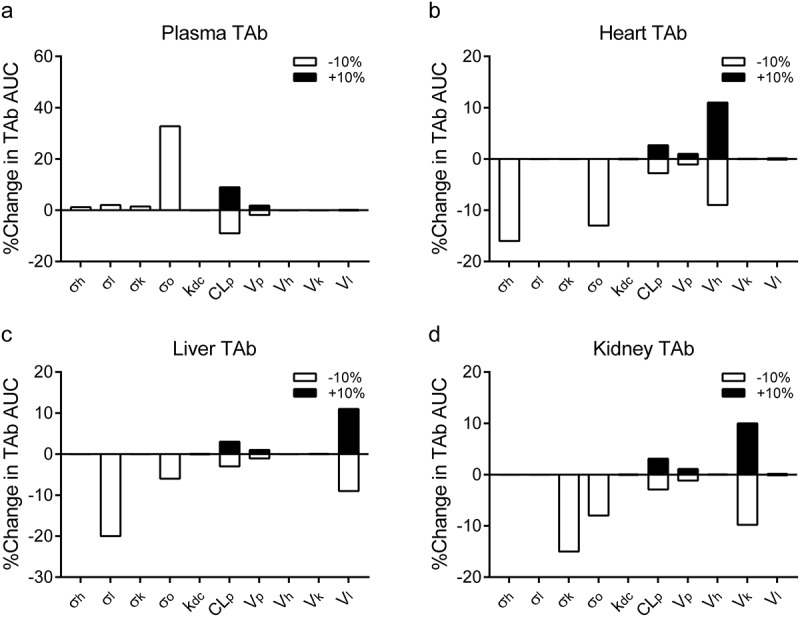
The plots represent the percent changes of AUC values of TAb in (a) plasma, (b) heart, (c) liver, and (d) kidney samples following ± 10% changes of selected parameter values (i.e., Vascular reflection coefficients (σi, i = heart (h), liver (l), kidney (k) and others (o)), plasma proteolytic clearance of TAC (CLp), deconjugation rate constant (kdc), and tissue physiological volumes (Vi, i = heart (h), liver (l) and kidney (k)). Vascular reflection coefficients (σ) were only tested with −10% changes because + 10% changes resulted in σ > 1. The analysis was performed with simulations at a single IV dose of 50 mg/kg DSTA4637A. Filled and empty bars represent + 10% and −10% changes of parameters, respectively.
Discussion
Antibody-drug conjugates (ADCs) are a new class of therapeutic agents, which hitherto are mainly used for cancer therapy. Four ADCs (gemtuzumab ozogamicin, brentuximab vedotin, trastuzumab emtansine, and inotuzumab ozogamicin) are currently in the market and more than 60 ADCs are in clinical development, but all of them target cancer cells and the application in other therapeutic areas remains mainly underexplored. Lehar and her colleagues for the first time adapted the ADC strategy to antimicrobials and developed a novel anti-S. aureus THIOMAB™ antibody conjugated to an antibiotic, which has shown impressive bactericidal activity against intracellular and vancomycin-resistant S. aureus.6 The new TAC strategy delivers the potent antibiotic to the intracellular compartment and effectively kills the intractable intracellular S. aureus that contributes to the relapse of infection. As an added benefit, the TAC strategy improves the PK of the antibiotic via an antibody carrier. In addition, conjugating antibiotics with the anti-S. aureus antibody diminishes antibiotic-mediated disruption on host’s normal flora and emergence of antibiotic-resistance due to enhanced specificity by the antibody carrier, and it also encourages us to revisit or investigate antimicrobials that have unfavorable PK or toxicity. The payload (i.e., dmDNA31) of our TAC molecule showed inhibitory activity towards several human CYP enzymes in vitro. Although this activity is relative weak based on in vitro IC50 values, it raises a concern of potential DDI in humans in view of dmDNA31 accumulation in some tissues (e.g., liver) in mice. In order to fully understand disposition of TACs and assess potential DDI risk in patients, it is important to understand the tissue disposition of payloads. The mPBPK modeling and cross-species scaling provide a useful approach to inform these questions.
The proposed mPBPK model was developed based on PK studies of DSTA4637A in a systemic mouse infection model. PK data of DSTA4637A in non-infected mice were initially included to develop the model, but improvements on neither parameter estimations nor model structures (e.g., target-mediated disposition) were identified due to limited measurements of tissue drug concentrations. The final model (Figure 1a, 1b) includes some key S. aureus-infected organs (heart, liver, and kidney) that are anatomically connected by blood and lymphatic circulations and all DSTA4637A species (e.g., TACs with DAR 0 – 2 and uc-dmDNA31). This model was successful in capturing the plasma PK profile of TAb (Figure 2a), but TAb concentrations were slightly overestimated in infected organs (e.g., liver and kidney) (Figure 2b-d), which is likely due to underestimation of organ volumes. Inflammation and abscess formation usually occur in various infected organs, such as kidney, liver, lung, and skin, in animals and patients infected with S. aureus,17 which typically result in swelling of organs and hence enlarged volumes. Sensitivity analysis results also support this hypothesis. Changes of tissue physiological volumes were shown to moderately affect the TAb tissue concentrations (Figure 9). In addition, lack of analytes’ tissue concentrations at early time points (i.e., absorption phase) posed difficulties on the estimation of tissue distributions of ac-dmDNA31 and uc-dmDNA31.
The assumption of TAC-driven tissue distribution of dmDNA31 (i.e., uc-dmDNA31 was assumed to be eliminated in plasma and organ and there is no distribution CL for uc-dmDNA31) enabled the proposed mPBPK model to successfully capture the PK profiles of ac-dmDNA31 and uc-dmDNA31 in plasma, liver and kidney in infected mice (Figure 3, 4). Although target-mediated DSTA4637A disposition may exist theoretically due to opsonization of S. aureus by DSTA4637A and phagocytosis by phagocytic cells,18 linear plasma DSTA4637A PK was observed in S. aureus-infected mice in the efficacious dose range of 25 – 50 mg/kg, and infection showed minimal effects on the plasma PK of DSTA4637A. This result is consistent with the measured amount of target and DSTA4637A molecules in infected tissues. A dose of 25 mg/kg yields observed maximum 3488, 118, 58.7, and 51.6 nM of DSTA4637A TAb in plasma, heart, liver, and kidney, respectively, which exceed the maximum available target of 24.9 nM in tissues when a maximum bacterial burden of 1 × 109 CFU/g and bacterial binding capacity of 1.50 × 104 antibodies per CFU S. aureus (estimated via measuring the binding of radiolabeled antibodies to protein A-deficient S. aureus as described by Lehar et al.) are assumed.6,7 Therefore, target-mediated DSTA4637A disposition was not included in our mPBPK model. PK parameters were precisely estimated and physiologically correlated with the systemic infectious state.
Estimated vascular reflection coefficients (σ) are in a range of 0.910 – 0.964, which are higher than the reported average value (~ 0.4) for leaky tissues such as heart, liver, and kidney.19 This could be explained by the fact that DSTA4637A was injected 24 hours after bacterial inoculation, when S. aureus started to replicate and initiate the formation of abscesses. This would increase the tissue pressure and hinder the access of DSTA4637A into tissues, and hence result in larger σ values.17 Although σ is commonly assumed to be tissue-specific constants in PBPK models, here it may be a function of staphylococcal abscess formation during the process of infection, as evidenced by histopathological changes in infected tissues.17,20 Parameters that describe the dynamic changes of infected tissue structure may be added in the model as more data, especially organ concentrations of TAb in early time points from both infected and non-infected mice, become available. Therefore, the estimated σ values in the current model represents effects of both bacterial infection and vascular sieving on the distribution of antibodies.
Our mPBPK model also suggests accumulation of dmDNA31 at some organs in infected mice. Although linear PK was observed for TAb in plasma and in tested organs due to saturated binding of targets at efficacious doses, released dmDNA31 accumulated in infected tissues because of its high lipophilicity and continuous delivery to intracellular compartment via the antibody. DSTA4637A-opsonized bacteria are primarily engulfed by phagocytes such as macrophages and neutrophils. Thus, organs where phagocytes accumulate such as liver and spleen may contain high concentrations of dmDNA31, and this is indeed supported by the high values of model simulated uc-dmDNA31 AUC0-14d in liver and undetectable uc-dmDNA31 concentration in heart where the number of phagocytes is low (Figure 5b).
A high amount of Fc gamma receptors expressed on phagocytic cells may also contribute to the accumulation of uc-dmDNA31 via non-specific uptake of antibodies into these organs. Although direct evidence of target-mediated disposition of DSTA4637A was not observed in this study, high bacterial burdens were correlated with high concentrations of uc-dmDNA31 in some infected tissues, such as kidney. Accumulation of uc-dmDNA31 was predicted in kidney where a maximum of 108 – 109 CFU/g of S. aureus were observed (Figure 5b), while the estimated AUC0-14d of uc-dmDNA31 was 8.62 times lower in plasma where S. aureus CFU drop below the LLOQ within 10 minutes after IV inoculation.4 However, future studies on tissue disposition of DSTA4637A in infected mice with different bacterial inoculation titers with more samplings at early time points and measurement of bacterial burden in interested organs (such as liver, spleen, bone, heart and etc.) should be performed to confirm the effect of bacterial burden on the tissue accumulation of dmDNA31.
Cross-species scaling of the mPBPK model was performed to predict human PK profiles of uc-dmDNA31. The ac-dmDNA31 is an inactive prodrug that becomes fully bactericidal after it is released from DSTA4637A by proteases such cathepsins present in lysosomes.6 Therefore, understanding tissue distribution of uc-dmDNA31 is the key that guides the dosing regimen of TAC in clinical trials. Our mPBPK model was scaled up from mice to human via integrating fundamental allometric scaling principles.15 Simulated human plasma PK profiles of the analytes closely match clinical trial data, which supports the robustness and applicability of the propose mPBPK model. Given the preclinical observation of uc-dmDNA31 accumulation in the liver and the high doses of TAC being tested in the clinical studies,16 DDI become a concern in the clinical setting in view of the inhibitory effect of dmDNA31 on CYP isoforms.
Our model suggested a minimal possibility of dmDNA31 (as a perpetrator) mediated PK DDI with CYP substrates even at a high dosing regimen of 100 mg/kg TAC weekly IV dose. In addition, DSTA4637A is likely to be administered to patients with S. aureus infection in combination with standard-of-care antibiotics, such as vancomycin and daptomycin, which are excreted mainly via the kidney. Therefore, the risk of PK DDI for TAC with co-administered antibiotics, where dmDNA31 acts as a perpetrator, is low.21,22 Meanwhile, the risk of dmDNA31 as a DDI victim is also relatively low because: 1) the metabolism-related CL in humans for dmDNA31 is expected to be low/moderate based on in vitro metabolism profiles (hydrolysis appears to be the major metabolic pathway for uc-dmDNA31); and 2) the majority of dmDNA31 is eliminated as the unchanged form in rats (Genentech Inc. unpublished data). Of note, the current DDI risk assessment is based on preclinical in vitro and in vivo data. The potential metabolic pathway for dmDNA31 in humans could be different due to species differences on the metabolism.
In summary, we developed a mPBPK model that well describes the PK profiles of anti-S. aureus THIOMAB™ antibody-antibiotic conjugates in S. aureus-infected mice. The mPBPK model is able to simultaneously characterize the PK properties of DSTA4637A analytes, including total antibody, ac-dmDNA31, and uc-dmDNA31. In addition, the mPBPK model suggests accumulation of dmDNA31 in organs (such as liver) where high number of phagocytes are present. Cross-species scaling of the mPBPK model suggests that dmDNA31 (as a perpetrator) mediated DDI are unlikely. However, target-mediated elimination of DSTA4637A in infected mice was not observed in the efficacious dose range of 25 – 50 mg/kg.7 Studies with lower doses of DSTA4637A will be needed to explore the potential target-mediated disposition of TACs. Although infection with S. aureus was shown to have negligible effects on plasma exposures of TAb and ac-dmDNA31 at efficacious dose, animal models with different bacterial inoculation titers are required to investigate the effects of S. aureus infection on tissue dispositions of the three analytes, particularly potential accumulation of uc-dmDNA31 in tissues with high bacterial burdens. Due to uncertainties on the translatability of infected mice to patients, scaled up TAC PK profiles in humans should be further validated using data from the ongoing Phase 1b study in patients.
Materials and methods
THIOMAB™ antibody-antibiotic conjugate DSTA4673A
Anti-S. aureus TAC, DSTA4637A (Genentech Inc. South San Francisco, CA), consists of a human IgG1 mAb, a protease cleavable valine-citrulline linker and the antibiotic, 4-dimethylamino piperidino-hydroxybenzoxazino rifamycin (dmDNA31). The dmDNA31 was conjugated to the anti-S. aureus mAb via the two cysteine residues on the light chains to yield an average drug-to-antibody ratio (DAR) of ~ 2. The anti-S. aureus mAb specifically targets the β-O-linked N-acetylglucosamine (GlcNAc) sugar modifications on wall teichoic acids of S. aureus.
Pharmacokinetic studies of DSTA4637A in S. aureus infected mice
All animal studies were conducted under a protocol that was approved by the Genentech Institutional Animal Care and Use Committees of the Association for Assessment and Accreditation of Laboratory Animal Care. The PK study of DSTA4637A was described previously by Zhou et al.7 Briefly, 6–8 week old naïve female severe combined immunodeficiency (SCID) mice (Charles River Laboratories) were infected with S. aureus USA300 NRS384 (BEI Resources, Manassas, VA) at 1 × 107 CFU per mouse via IV tail injection. Twenty-four hours post infection, a single IV bolus of 25 or 50 mg/kg DSTA4637A, or 200 µl phosphate buffered saline (PBS, control group) was administered through tail vein of each mouse. Plasma samples were collected via retro-orbital bleeds or cardiac puncture at 10 min, 1, 6, and 24 hours, 2, 3, 7, 10, and 14 days post-dose (three mice per time point). Heart, kidney, and liver samples were harvested at 1, 3, 7, and 14 days post-dose (three mice per time point).
Bioanalytical methods
Concentrations of TAb (fully conjugated, partially deconjugated, and fully deconjugated anti-S. aureus antibodies), ac-dmDNA31, and uc-dmDNA31 in plasma were analyzed as described by Zhou et al.7 Briefly, TAb concentrations were measured using an enzyme-linked immunosorbent assay (ELISA). The ac-dmDNA31 concentrations were determined using a hybrid binding liquid chromatography mass spectrometry (LC-MS/MS) assay. Antibody-conjugated drug was extracted using protein A affinity capture followed by enzyme-mediated release of dmDNA31 and electrospray-ionization LCMS/MS for detection. Concentrations of uc-dmDNA31 were measured using the LC-MS/MS assay with a QTRAP® 5500 mass spectrometer after extraction of dmDNA31 by protein precipitation. Tissue samples (i.e., heart, kidney, and liver) were homogenized in the presence of a protease inhibitor cocktail, centrifuged, and the supernatants were tested using the methods as described above to measure TAb, ac-dmDNA31, and uc-dmDNA31 concentrations.
Model development and data analysis
A mPBPK model was developed for simultaneous fitting of TAb, ac-dmDNA31, and uc-dmDNA31 concentrations in S. aureus-infected mice (Figure 1a). A PK study of DSTA4637A was also performed in non-infected mice,7 but these data were not used in the model development due to limited measurements of tissue drug concentrations. In this model, organs, including heart, liver, kidney, and lumped organs (i.e., a composite of other organs), were connected anatomically through plasma and lymph flows. Although distribution of antibodies is typically thought to be governed by both diffusion and convection,8,23 it has been shown that diffusion contributes insignificantly to the total antibody distribution.24,25 Therefore, in our mPBPK model, convection, delineated by one-pore formalism and drove by lymph flow, was considered as the only distribution pathway for both TAb and ac-dmDNA31.19 Previous mouse PK data suggested that infection by S. aureus had minimal effects on plasma PK of TAb at efficacious doses.7 Linear TAb PK was also observed in investigated organs (i.e., liver, kidney, and heart) in S. aureus-infected mice at the dose range of 25 – 50 mg/kg, based on the observation that dose-normalized TAb concentration-time profiles overlapped. Thus, target-mediated disposition of antibodies at the sites of infection was considered to be negligible and linear proteolytic clearance (CL) of TAb was assumed to be from plasma compartment.
Cao et al. also demonstrated that assigning antibody CL to plasma compartment may provide better prediction and parameter estimation than assigning CL from organs for antibodies with linear kinetics using a mPBPK model.26 Besides proteolytic degradation in plasma compartment, DSTA4637A was also assumed to be eliminated via deconjugation in a stepwise manner in all tissues (Figure 1b) due to the fact that antibody-dependent phagocytosis of TAC-S. aureus complexes and nonspecific endocytosis/pinocytosis of antibodies may occur in all organs. Only one dmDNA31 molecule is released from TACs at a time, and the deconjugation is a linear DAR-dependent process, described as the product of deconjugation rate constant (kdc), DAR, and the corresponding TAC concentration (Equation 7).27,28 Other sophisticated PK factors, such as proteolytic CL in all tissues and the fractions of formation of uc-dmDNA31 from the proteolytic degradation or deconjugation pathway of TAC, were not included in our model due to the inability to precisely estimate these parameters or inaccurate PK prediction based on the limited tissue data. Released dmDNA31 (i.e., uc-dmDNA31) molecules were metabolically mainly eliminated via hydrolysis based on in vitro metabolism identification studies, and the plasma CL of dmDNA31 was measured to be 1.01 × 104 ml/day/kg in a mouse study with intravenously injected carbon-14 labeled dmDNA31. Thus uc-dmDNA31 was assumed to be cleared immediately upon deconjugation of TAC and its tissue distribution was negligible, whereas intracellular uc-dmDNA31 was assumed to accumulate in acidic lysosomes following TAC-mediated phagocytosis of S. aureus or nonspecific uptake of TACs due to its high lipophilicity (logD = 3.4 at pH 7) and basicity (pKa = 9.7).6 These assumptions simplify the tissue distribution of uc-dmDNA31 in the proposed mPBPK model, which would otherwise be unidentifiable (e.g., tissue to plasma partition coefficient) due to scattered and insufficient data, especially in heart where uc-dmDNA31 was undetectable. Volumes of distribution of TAb and ac-dmDNA31 at steady state were calculated to be similar: 115 – 123 ml/kg and 110 – 114 ml/kg, respectively.7 Thus, distribution of ac-dmDNA31 may be mainly governed by the distribution of antibody and their volumes of distribution were assumed to be the same. Model estimated parameters include vascular reflection coefficients (σ), volumes of distribution (Vuc-dmDNA31) and clearances of uc-dmDNA31 (CLuc-dmDNA31).
The representative differential equations describing the mPBPK model are presented below:
Plasma (DAR value n = 0, 1, 2):
| (1) |
Lymph (DAR value n = 0, 1, 2):
| (2) |
Organ i (i = heart, liver, kidney, and others):
| (3) |
Contribution of DARn deconjugation in plasma and organ i:
TAb in plasma and organ i:
| (4) |
ac-dmDNA31 in plasma and organ i:
| (5) |
Unconjugated dmDNA31 in plasma:
| (6) |
Unconjugated dmDNA31 in organ i:
DAR0 – 2 represent TACs with 0 – 2 dmDNA31 molecules, and [uc-dmDNA31] is total concentration of uc-dmDNA31. The CLuc-dmDNA31 in plasma and lymph were assumed to be the same. Volume of distribution and clearance of uc-dmDNA31 in lump tissues (i.e., others: Vo, uc-dmDNA31 and CLo, uc-dmDNA31) and in heart were not included in the model due to lack of data for parameter estimation. Fraction of unbound uc-dmDNA31 (fu) in mouse plasma was measured to be 0.02 in vitro, and organ fu was assumed to be the same as plasma fu. Unbound uc-dmDNA31 concentrations were calculated as fu × [uc-dmDNA31]. Definition of PK parameters and values of mouse physiological parameters are listed in Tables 1 and 2. Physiological parameters (Table 2) were used to describe the tissue distribution of DAR0 – 2 molecules, and apparent volumes of distribution of uc-dmDNA31 (Vp,uc-dmDNA31 and Vi,uc-dmDNA31) were estimated using collected mouse PK data. All tissue drug concentrations were corrected for residual blood according to the method described by Boswell et al, where tissue drug concentration = (total drug amount in tissue – drug amount in residual blood)/tissue weight.29
Cross-species scaling of the mPBPK model was performed by replacing mouse physiological parameters with human values (Table 2) and allometrically scaling of systemic clearance of DARn (CLp) and uc-dmDNA31 (CLp,uc-dmDNA31) and clearance and volume of distribution of uc-dmDAN31 in the organs (Vi, uc-dmDNA31 and CLi,uc-dmDNA31, i = liver, and kidney). Vascular reflection coefficients and lymphatic capillary reflection coefficient were kept unchanged because both species share similar vascular structures. Deconjugation rates of dmDNA31 were the same in mice and human based on in vitro plasma stability studies (Genentech Inc. unpublished data). The fraction of protein binding of dmDNA31 in human plasma is ~ 95% (Genentech Inc. unpublished data), and protein bindings in other organs were assumed to be the same as that in plasma. Volume of distribution and clearance of uc-dmDNA31(Vi, uc-dmDNA31,CLp,uc-dmDNA31and CLi,uc-dmDNA31) were allometrically scaled with conservative exponent values of 1 and 0.75 for small molecules,14 and the clearance of TACs (DAR 0 – 2) was scaled with an exponent value of 0.91 – an average value for allometric scaling of mAb CL in a mPBPK model estimated by Zhao et al.15 Model predicted analytes’ plasma concentration-time profiles were compared to human data collected in Phase I clinical trial for external validation of the proposed mPBPK model. Representative equations of allometric scaling are showed below:
| (10) |
where i = plasma, liver, or kidney, CL is clearance, V is volume of distribution and BW is body weight.
Phoenix WinNonlin 6.4 (Pharsight, Mountain View, CA) was used to obtain estimates of model parameters and the code is provided in supplemental data. Berkeley Madonna 8. 3. 18 (University of California, Berkeley, CA) was used to simulate TAC PK profiles in humans. The goodness-of-fit was assessed by visual inspection, Akaike Information Criterion (AIC), Bayesian Information Criterion (BIC), and Coefficient of Variation (CV) of the estimated parameters. All graphs were constructed using GraphPad Prism 6.01 (GraphPad Software Inc., CA).
Model validation and sensitivity analysis
The final model was externally validated using plasma TAb, ac-dmDNA31, and uc-dmDNA31 concentrations from non-infected mice given a single IV dose of 50 mg/kg DSTA4637A as reported by Zhou et al,7 where target-mediated disposition was saturated. Ratios of predicted: observed TAb concentrations were calculated to assess the predictability of the model.
A local sensitivity analysis of the model output to model parameters was performed by assessing the percent change of AUC values of TAb in plasma, heart, liver, and kidney with ± 10% alteration in the model parameters (Equation 11). Parameters that are associated with TAb tissue distributions, including vascular reflection coefficients (σi, i = heart (h), liver (l), kidney (k) and others (o)), deconjugation rate constant (kdc), plasma proteolytic clearance of TAC (CLp), and tissue physiological volumes (Vi, i = heart (h), liver (l) and kidney (k)), are evaluated for the sensitivity analysis.
| (11) |
AUCSIM refers to the AUC value calculated with the final estimated set of parameters, and AUC±10% is the AUC obtained following a 10% increase or decrease in the parameter value. The analysis was performed with simulations at the IV dose of 50 mg/kg DSTA4637A.
Supplementary Material
Abbreviations
- ADC
Antibody-drug conjugate
- AUC0-14d
Area under the drug concentration-time curve from day 0 to day 14
- CL
Clearance
- CYP
Cytochrome P450
- kdc
Deconjugation rate constant
- DAR
Drug to antibody ratio
- DDI
Drug-drug interactions
- dmDNA31
4-dimethylamino piperidino-hydroxybenzoxazino rifamycin
- ac-dmDNA31
Antibody-conjugated dmDNA31
- uc-dmDNA31
Unconjugated dmDNA31
- L
Lymph flow rate
- mPBPK
Minimal Physiologically-based pharmacokinetic model
- PK
Pharmacokinetic
- Q
Plasma flow rate
- TAb
Total antibody
- TAC
THIOMAB™ antibody antibiotic conjugate
- σ
Vascular reflection coefficient
- V
Volume of distribution
Acknowledgments
The authors would like to acknowledge Genentech Intern Program 2015. This work was supported by Genentech, Inc. The authors would like to thank Anshin BioSolutions for editing this manuscript.
References
- 1.DeLeo FR, Chambers HF.. 2009. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest. 119:2464–2474. doi: 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. 2012. Predictors of mortality in Staphylococcus aureus Bacteremia. Clin Microbiol Rev. 25:362–386. doi: 10.1128/CMR.05022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nannini E, Murray BE, Arias CA. 2010. Resistance or decreased susceptibility to glycopeptides, daptomycin, and linezolid in methicillin-resistant Staphylococcus aureus. Curr Opin Pharmacol. 10:516–521. doi: 10.1016/j.coph.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Rogers DE. Studies on bacteriemia. I. Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci. J Exp Med. 103;1956:713–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 164;2000:3713–3722. [DOI] [PubMed] [Google Scholar]
- 6.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Morisaki JH, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature. 2015;527:323–328. doi: 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- 7.Zhou C, Lehar S, Gutierrez J, Rosenberger CM, Ljumanovic N, Dinoso J, Koppada N, Hong K, Baruch A, Carrasco-Triguero M, et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice. MAbs. 2016;8:1612–1619. doi: 10.1080/19420862.2016.1229722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baxter LT, Zhu H, Mackensen DG, Jain RK. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 54;1994:1517–1528. [PubMed] [Google Scholar]
- 9.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. 1997. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 10.Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 50;2001:3–20. [DOI] [PubMed] [Google Scholar]
- 11.Shah DK, Betts AM. 2012. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 39:67–86. doi: 10.1007/s10928-011-9232-2. [DOI] [PubMed] [Google Scholar]
- 12.Garg A, Balthasar JP. 2007. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 34:687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 13.Warren MF. 1940. The lymphatic system. Annu Rev Physiol. 2:109–124. doi: 10.1146/annurev.ph.02.030140.000545. [DOI] [Google Scholar]
- 14.Tang H, Mayersohn M. 2005. A novel model for prediction of human drug clearance by allometric scaling. Drug Metab Dispos. 33:1297–1303. doi: 10.1124/dmd.105.004143. [DOI] [PubMed] [Google Scholar]
- 15.Zhao J, Cao Y, Jusko WJ. 2015. Across-species scaling of monoclonal antibody pharmacokinetics using a minimal PBPK model. Pharm Res. 32:3269–3281. doi: 10.1007/s11095-015-1703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng R, She G, Carrasco-Triguero M, Saad O, Kamath AV, Peck M, Rothenberg M, Lewin-Koh N, Tavel J, Hanley WD. Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus aureus THIOMABTM antibody-antibiotic conjugate, in healthy volunteers. Poster presented at: Advancing Patient Care Through Precision and Translational Medicine; March 15, 2017; Washington, DC [Google Scholar]
- 17.Cheng AG, DeDent AC, Schneewind O, Missiakas D. 2011. A play in four acts: staphylococcus aureus abscess formation. Trends Microbiol. 19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mariathasan S, Tan MW. 2017. Antibody-antibiotic conjugates: A novel therapeutic platform against bacterial infections. Trends Mol Med. 23:135–149. doi: 10.1016/j.molmed.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Cao Y, Balthasar JP, Jusko WJ. 2013. Second-generation minimal physiologically-based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn. 40:597–607. doi: 10.1007/s10928-013-9332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23:3393–3404. doi: 10.1096/fj.09-135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matzke GR, Zhanel GG, Guay DR. 1986. Clinical pharmacokinetics of vancomycin. Clin Pharmacokinet. 11:257–282. doi: 10.2165/00003088-198611040-00001. [DOI] [PubMed] [Google Scholar]
- 22.Burkhardt O, Joukhadar C, Traunmuller F, Hadem J, Welte T, Kielstein JT. 2008. Elimination of daptomycin in a patient with acute renal failure undergoing extended daily dialysis. J Antimicrob Chemother. 61:224–225. doi: 10.1093/jac/dkm405. [DOI] [PubMed] [Google Scholar]
- 23.Flessner MF, Lofthouse J, Zakaria El R. In vivo diffusion of immunoglobulin G in muscle: effects of binding, solute exclusion, and lymphatic removal. Am J Physiol. 273;1997:H2783–93. [DOI] [PubMed] [Google Scholar]
- 24.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 37;1989:77–104. [DOI] [PubMed] [Google Scholar]
- 25.Flessner MF, Schwab A. 1996. Pressure threshold for fluid loss from the peritoneal cavity. Am J Physiol. 270:F377–90. doi: 10.1152/ajprenal.1996.270.2.F377. [DOI] [PubMed] [Google Scholar]
- 26.Cao Y, Jusko WJ. 2014. Survey of monoclonal antibody disposition in man utilizing a minimal physiologically-based pharmacokinetic model. J Pharmacokinet Pharmacodyn. 41:571–580. doi: 10.1007/s10928-014-9374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu D, Jin JY, Girish S, Agarwal P, Li D, Prabhu S, Dere RC, Saad OM, Nazzal D, Koppada N, et al. Semi-mechanistic multiple-analyte pharmacokinetic model for an antibody-drug-conjugate in cynomolgus monkeys. Pharm Res. 2015;32:1907–1919. doi: 10.1007/s11095-014-1585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sukumaran S, Gadkar K, Zhang C, Bhakta S, Liu L, Xu K, Raab H, Yu SF, Mai E, Fourie-O’Donohue A, et al. Mechanism-based pharmacokinetic/pharmacodynamic model for THIOMAB drug conjugates. Pharm Res. 2015;32:1884–1893. doi: 10.1007/s11095-014-1582-1. [DOI] [PubMed] [Google Scholar]
- 29.Boswell CA, Ferl GZ, Mundo EE, Schweiger MG, Marik J, Reich MP, Theil FP, Fielder PJ, Khawli LA. 2010. Development and evaluation of a novel method for preclinical measurement of tissue vascular volume. Mol Pharm. 7:1848–1857. doi: 10.1021/mp100183k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.