

ABSTRACT
Engineering of antibodies for improved pharmacokinetics through enhanced binding to the neonatal Fc receptor (FcRn) has been demonstrated in transgenic mice, non-human primates and humans. Traditionally, such approaches have largely relied on random mutagenesis and display formats, which fail to address related critical attributes of the antibody, such as effector functions or biophysical stability. We have developed a structure- and network-based framework to interrogate the engagement of IgG with multiple Fc receptors (FcRn, C1q, TRIM21, FcγRI, FcγRIIa/b, FcγRIIIa) simultaneously. Using this framework, we identified features that govern Fc-FcRn interactions and identified multiple distinct pathways for enhancing FcRn binding in a pH-specific manner. Network analysis provided a novel lens to study the allosteric impact of half-life-enhancing Fc mutations on FcγR engagement, which occurs distal to the FcRn binding site. Applying these principles, we engineered a panel of unique Fc variants that enhance FcRn binding while maintaining robust biophysical properties and wild type-like binding to activating receptors. An antibody harboring representative Fc designs demonstrates a half-life improvement of > 9 fold in transgenic mice and > 3.5 fold in cynomolgus monkeys, and maintains robust effector functions such as antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity.
Keywords: Antibody, half-life, pharmacokinetics, structure-guided engineering, neonatal Fc receptor, FcRn, effector functions, amino acid interaction network
Introduction
Antibodies are a preferred treatment modality, particularly in cancer and autoimmune diseases, with more than 50 approved antibodies and more than 500 molecules in various stages of clinical development.1 The success of antibodies in disease intervention is, in part, due to their high level of specificity, relative safety, and long circulating half-life. The circulating half-life of immunoglobulin G (IgG), roughly 10–21 days depending on IgG isotype and attributes of the variable region,2 is attributed to association with the neonatal Fc receptor (FcRn)3,4 leading to antibody recycling and minimal endosomal degradation. FcRn plays a key role in serum IgG homeostasis as well as in placental transfer of IgG molecules from mother to fetus.3,5–8 Following pinocytosis, the acidic environment of the early endosome allows for binding of IgG (as well as albumin) to FcRn, which provides protection from degradation and facilitates trafficking of IgG back to the extracellular environment, where the molecules dissociate back into circulation upon exposure to physiological pH.
With the increasing use of antibodies as therapeutics for the prevention and treatment of a wide variety of diseases, there has been interest in developing antibodies with extended half-life, especially in the context of the prevention or treatment of chronic diseases in which an antibody must be administered repetitively. The fragment crystallizable (Fc) domain of an antibody is primarily responsible for binding to FcRn to facilitate antibody recycling. Ghetie et al. were among the earliest to propose that the half-life of antibodies could be extended through modification of the Fc domain to promote FcRn interaction, and they demonstrated that enhancing the binding of a mouse IgG1-derived Fc fragment to mouse FcRn at pH 6.0 by 3.5-fold increased the serum persistence of the antibody approximately 1.6-fold.9
Since these initial reports by Ghetie et al., several groups have reported enhancement of in vivo half-life of therapeutic IgG molecules by introducing alterations in the Fc region of IgG to promote the Fc-FcRn interaction in the acidic environment of the endosome.10–13 The FcRn affinity-enhancing mutants in these studies were identified from phage display coupled with directed mutagenesis,14–16 alanine scanning,17 or, in select cases, from molecular modeling and rational design.11,13 The prototypical example of an FcRn affinity-enhancing Fc mutant is the M252Y/S254T/T256E (YTE) mutation which, when incorporated into motavizumab IgG1, is able to extend serum half-life in humans by more than four-fold.18 In these studies, the Fc was primarily optimized for FcRn binding. However, the Fc of IgG binds to various other receptors, such as FcγRI, FcγRIIa, FcγRIIb, FcγRIII, C1q and TRIM21, and these interactions mediate various effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent intracellular neutralization (ADIN). As such, to maintain appropriate effector function of the wild type IgG, any Fc domain engineered for enhanced FcRn binding or half-life extension must be assessed in the context of its impact on the various antibody effector functions. Indeed, it has been reported that the Fc variant YTE has lower thermal stability19 and lower ADCC potential16 compared to antibodies containing a native Fc domain.
We developed a structural- and network-based framework to interrogate the interaction of the Fc domain with FcRn at neutral and acidic pH. Using this framework, we identified different pathways for improving FcRn binding, specifically focusing on decreasing the koff of the interaction at pH 6.0 while maintaining native binding to receptors involved in effector functions. The interaction networks of mutations were mapped, and select mutations were combined and assessed for binding to FcRn and other Fc receptors. Here, we report the identification of multiple Fc variants that confer enhancement in half-life and retain, or enhance, effector functions such as ADCC and CDC.
Results
Characterization of the structural and molecular features governing the Fc-FcRn interaction
A structural model of human wild type Fc in complex with human FcRn was created from the co-crystal structure of FcRn in complex with the YTE-Fc domain (PDB ID: 4N0U) (Figure 1). Inspection of the structure showed that both the alpha and beta subunits of the FcRn molecule participate in binding to the Fc-domain, making contact with the CH2 and CH3 domains of the Fc, with the primary interaction being mediated by the alpha-subunit on FcRn and the CH2 domain on the Fc. The pH-specific nature of binding is driven by histidine residues at positions 310 and 435 (Kabat numbering) on the antibody Fc, which undergo protonation at acidic pH and make critical contacts with glutamate at position 115 and aspartate at position 130. Mutation of either of the two histidine residues significantly reduces the binding affinity of Fc with FcRn.20,21 In addition to His310 and His435, several other Fc residues are involved in making molecular contacts with FcRn (Figure 1).
Figure 1.
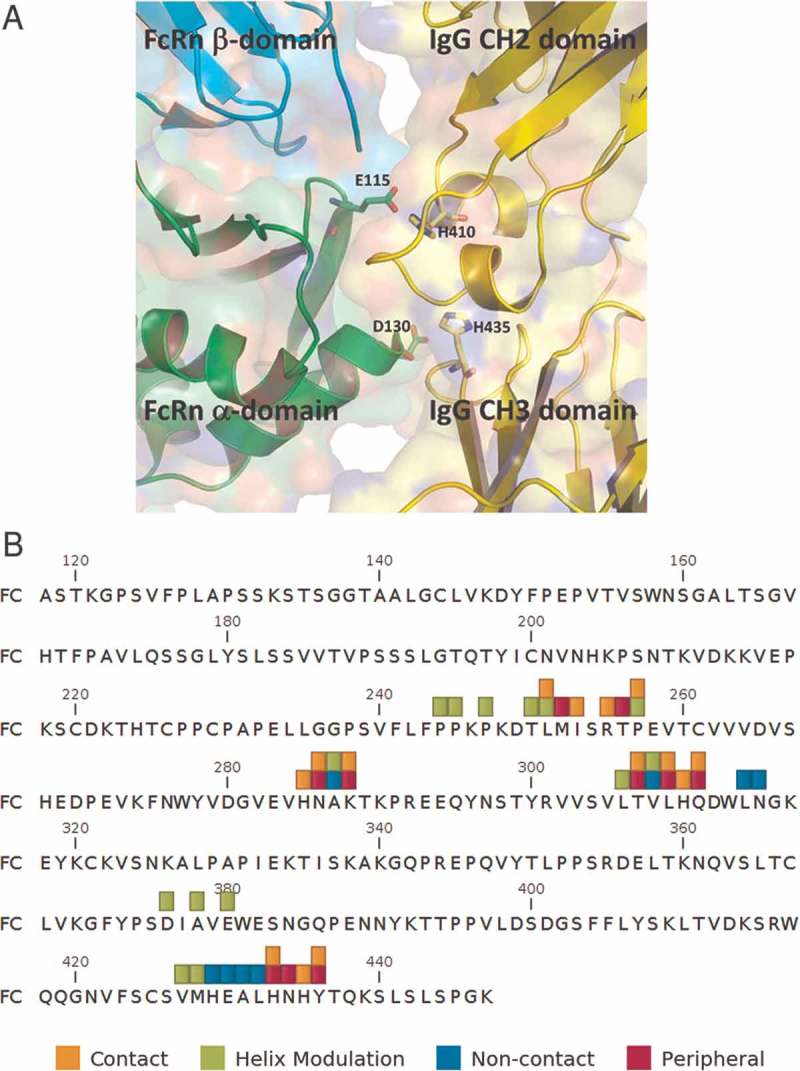
(top) Structural view of human IgG CH2-CH3 domains (yellow) in complex with human FcRn α (green) and β2M (cyan) domains. The pH-specific binding is driven by the protonation of His310 and His435 (shown as sticks) on IgG and their interaction with Glu115 and Asp130 (shown as sticks) on the FcRn α domain. (bottom) Human IgG1 constant region sequence with residue sets having potential to mediate Fc-FcRn interactions annotated.
Further, superposition of the crystal structure of Fc at pH 4.0 (PDB ID: 4BYH) and 6.5 (PDB ID: 4Q7D) reveals subtle changes within the CH2 domain, such as lateral displacement of the 250-helix and differences in the relative orientation of CH2 (Supplementary Figure 1). Given that amino acid residues involved in FcRn binding, such as M252 and I253, are located on the 250-helix, the observed displacement of the helix is expected to influence the affinity of Fc for FcRn.20 Furthermore, the difference in the relative orientation of CH2 with respect to CH3 highlights the dynamic nature of the CH2-CH3 domains at different pHs (Supplementary Figure 1).22 Recently, deuterium exchange studies of human Fc were performed in the absence or presence of FcRn. The studies were performed at both acidic and neutral pHs and showed that FcRn binding to the Fc domain limits exchange on contacting Fc residues. Further, the 250-helix residues demonstrated enhanced deuterium exchange at low pH in the absence of FcRn, suggesting that a pH-specific conformational change occurred within this region.23,24
Analysis of the strength of engagement of Fc with FcRn at acidic pH reveals that human FcRn binds to the human Fc domain with weak affinity (> 600 nM). The kinetic parameters further demonstrate that while the rate of association (kon ~105 M-1s-1) of Fc with FcRn is comparable to that of a typical antibody-antigen interaction, the lower binding affinity is primarily driven by a faster dissociation rate (koff ~ 0.1 s−1, greater than 1,000-fold faster than that of typical antibody-antigen engagement).25 The conformational flexibility of the CH2 subdomain is thought to contribute to the poor koff of FcRn binding. Reducing the rate of dissociation (koff) of the Fc-FcRn interaction at acidic pH has been suggested to be a key factor for improving half-life.26 As such, we sought to prioritize identification of mutations that slow koff.
We identified four sets (or groups) of Fc residues which, when mutated, we predicted would alter the FcRn interaction and slow the dissociation rate (Figure 1). These include residues that make direct contact with FcRn, as well as peripheral and non-surface exposed residues that have the potential to modify the interaction surface and/or influence the dynamics of the 250-helix. Indeed, some of these identified positions have been previously investigated, such as within the half-life enhancing mutants YTE,10 LS (M428L/N434S),13 AAA (T307A/E380A/N434A),27 QL (T250Q/M428L)11 and V308P.28 In our analysis, we first investigated the role of each residue in silico, and identified the network of interactions mediated by that residue (Figure 2). Specific sets of mutations were rationally designed to enhance both hydrophobic interactions within the core of the Fc-FcRn interface and polar and electrostatic interactions at its periphery. The impact of each mutation on its interaction network was assessed and, when needed, additional mutations were introduced to maintain the stability of the protein. Of special interest, it has been demonstrated that mutations of non-contacting residues near the interface of a protein-protein interaction can enhance the electrostatic charge complementarity resulting in improved affinity, specifically by reducing the koff rate.29,30
Figure 2.
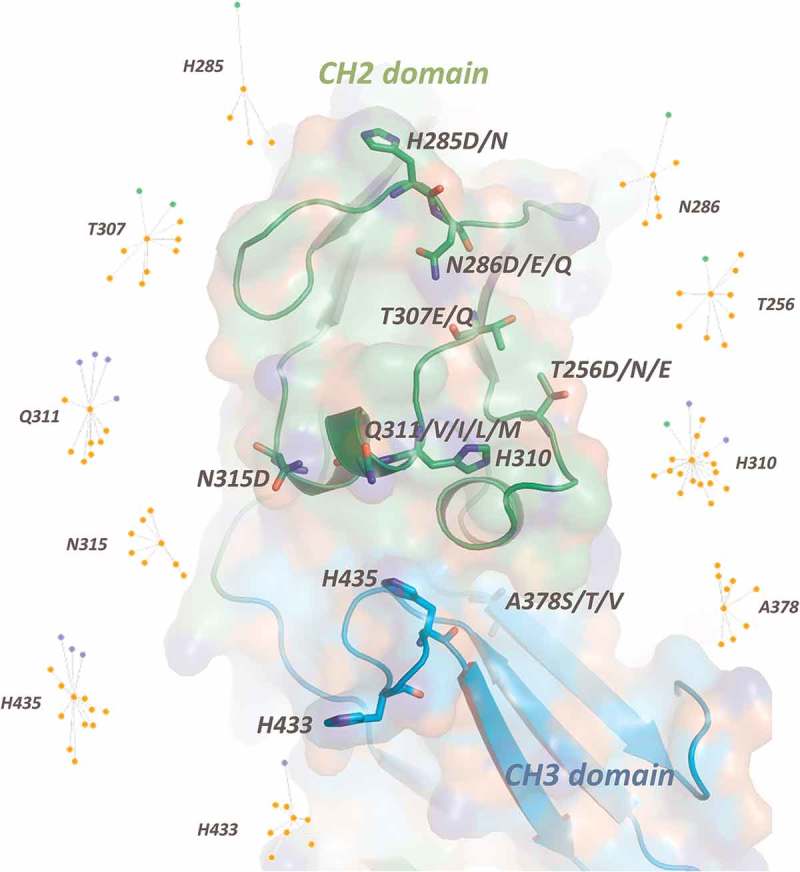
Molecular interaction of key residues of the designed Fc variants. An interaction map was calculated using the Rosetta REF2015 score function to define the energy of interaction between residues. Edges are created between residue pairs with favorable interaction energies (< 0.0 REU), as calculated using an energy minimized model of the Fc-FcRn complex extracted from 4NOU.pdb as the input structure. The list of residues interacting with key residues are listed in Supplementary Table 1. Orange nodes represent residues in the Fc region, and green and purple nodes represent the two chains of FcRn.
The FcRn binding site on the Fc domain has substantial overlap with the binding site of the intracellular receptor TRIM21, protein A (used for antibody purification), and the Fc-Fc interaction interface formed during hexamerization of IgG for complement-mediated activity, which required consideration when selecting positions for mutagenesis. More than 30 distinct positions were chosen for substitutions. Combinations of the individual mutations were designed based on the following guiding principles: 1) avoiding introduction of large clusters of charge or hydrophobicity to avoid destabilizing the IgG fold and to minimize binding to solvent ions;31 2) incorporating diversity into the types of interactions, i.e., not just enhancing electrostatic or hydrophobic contacts; and 3) dispersing mutations across the CH2 and CH3 domains. Based on these considerations, more than 150 unique mutant combinations were designed and experimentally evaluated.
Designed Fc mutants were incorporated into the IgG1 Fc domain of either the actoxumab32 or motavizumab33 antigen-binding fragment (Fab), which were recombinantly expressed and evaluated for binding to human FcRn by biolayer interferometry. Motavizumab was chosen as the model antibody for this study primarily because its half-life has been well characterized in transgenic mice, cynomolgus monkeys and in humans. Further, its half-life extending variant that includes the YTE mutation has also been studied in mice, monkeys and humans. Motavizumab has a half-life of 19–34 days18 in humans and actoxumab has a half-life of 26 ± 8.4 days34 in humans. Two different approaches were employed: recombinant (His)6-tagged FcRn capture on NiNTA biosensors with IgG as the analyte, and IgG captured on anti-CH1 biosensors with recombinant FcRn as the analyte. The assays were used to quantify binding at pH 6.0 followed by dissociation at pH 6.0 and subsequently dissociation at pH 7.4, as well as binding and dissociation solely at pH 7.4 (Supplementary Figure 2). Fc variants incorporating mutations at Pro257 and Val308 had high affinity at pH 6.0, but displayed markedly slower off-rates at pH 7.4 (data not shown) and were not considered for further analyses. More than 10 distinct variants had greater than a 5-fold increase in Kd as compared to IgG with a native Fc domain, with many of these lowering koff at pH 6.0 by more than 2.5-fold (Table 1). Fc variants DF183, DF197, DF213, DF215 and DF228 were further characterized by surface plasmon resonance. These Fc variants exhibited a greater than 30-fold affinity enhancement to FcRn at pH 6.0, primarily driven by a significantly slower koff (Table 2).
Table 1.
FcRn binding affinity of Fc designs compared to wild type Fc (WT).
| Designation | Mutations | Fold-Increase Kd | Fold-increase kon | Fold-decrease koff |
|---|---|---|---|---|
| WT | – | NA* | NA | NA |
| YTE** | M252Y/S254T/T256E | 8.5 | 2.3 | 3.6 |
| LS** | M428L/N434S | 9.2 | 2.6 | 3.6 |
| DF045 | T256D/T307R/Q311V | 5.9 | 2.3 | 2.6 |
| DF219 | T256D/N315D/A378V | 7.9 | 2.8 | 2.8 |
| DF171 | T256D/N286D/T307R/Q311V | 8.5 | 2.5 | 3.1 |
| DF197 | H285N/T307Q/N315D | 9.1 | 2.9 | 3.0 |
| DF186 | T256D/T307R/Q311V/A378V | 9.5 | 2.9 | 3.1 |
| DF216 | H285D/Q311V/A378V | 9.6 | 3.4 | 2.8 |
| DF223 | T256D/H285D/A378V | 10.3 | 3.7 | 2.7 |
| DF183 | T256D/Q311V/A378V | 11.0 | 3.2 | 3.2 |
| DF227 | T256D/H285D/N286D/T307R/A378V | 11.6 | 3.6 | 3.2 |
| DF228 | T256D/H286D/T307R/Q311V/A378V | 12.4 | 3.2 | 4.0 |
| DF215 | T307Q/Q311V/A378V | 12.4 | 3.4 | 3.7 |
| DF213 | H285D/T307Q/A378V | 13.2 | 3.8 | 3.3 |
| DF229 | T256D/H285D/T307R/Q311V/A378V | 14.0 | 3.7 | 3.7 |
*NA = not applicable, ratio is 1.0; **previously described mutations from the literature
Table 2.
Surface plasmon resonance (SPR) analysis of FcRn binding to motavizumab containing designed Fc variants.
| Designation | ka (1/M∙s) | kd (1/s) | Kd (nM) |
|---|---|---|---|
| Wild type* | NA | NA | 825 |
| DF215** | 1.37x106 | 2.70x10−2 | 19 ± 3 |
| DF228 | 1.39x106 | 1.42x10−2 | 10 |
| DF213 | 0.80x106 | 1.92x10−2 | 24 |
| DF183 | 1.93x106 | 4.02x10−2 | 21 |
| DF197 | 1.59x106 | 3.28x10−2 | 21 |
*Fast off-rate did not allow for accurate 1:1 kinetic model fitting. Steady state analysis was used to determine a Kd; ** mean ± standard deviation of four separate experiments
Enhancement of FcRn binding affinity was achieved by mutation of centrally-located Fc-FcRn interface residues to improve hydrophobic interactions, and mutation of peripheral residues to improve electrostatic and polar interactions. The Fc residues T256, T307, H285, N286 and N315 located on the periphery of the interface were mutated to polar and charged amino acids to complement the positively charged N-terminal region of the FcRn β-domain. Additionally, Q311 and L309, both located at the Fc-FcRn interface, were mutated to hydrophobic residues. However, aromatic amino acids were avoided to minimize the impact on thermal stability. Although a Met amino acid substitution at residue 311 was found to be beneficial, it was not included in any final designs due to the possibility of in vivo oxidation.35,36 Residues M252, I253 and S254, which are involved in FcRn binding, are part of the 250-helix which exhibits conformational flexibility at various pHs. We hypothesized that reduced conformational flexibility would lead to better exposure of these residues and improve the shape complementarity between Fc and FcRn. Residues that are part of, or in contact with, the 250-helix (such as P257, A378, V308) and are not found on the Fc-FcRn interface were mutated to restrict or dampen the movement of the helix. Interestingly, the 250-helix contact residues M428 (part of the LS mutant) and A378 are naturally present as Leu and Thr, respectively, in the mouse Fc domain and an analysis of the crystal structure suggests these residues influence the 250-helix.
Biophysical characterization of the lead Fc designs
The biophysical attributes of a modified motavizumab containing the designed Fc variants were experimentally assessed. All variants were purified by protein A affinity chromatography and no appreciable loss of IgG was observed, confirming the designed Fc mutations did not have significant impact on protein A engagement. Size-exclusion high performance liquid chromatography (SE-HPLC) indicated that all designed variants had similar elution times to that of IgG with a native Fc domain, and a typical monomeric profile, with no aggregation detected above that of antibodies containing the native Fc domain (Figure 3A). IgGs incorporating designed Fc variants were also assessed for thermal stability of the CH2 and CH3 domains using differential scanning fluorimetry (DSF). The melting temperature (Tm) of the wild type human CH2 and CH3 domain, as measured by DSF, has been reported to be approximately 70°C and 81.5°C, respectively.37 Our DSF experiments yielded comparable results with a CH2 and CH3 Tm of 68.8°C and 80.8°C, respectively. In our experiments, the Tm of the CH2 domain of the previously described YTE mutant Fc domain was 7.2°C lower than that of wild type, in line with previous reports.38 Additionally, mutations at positions P247, P257, and V308 significantly affected the Tm of CH2 (data not shown). The lead Fc designs (DF183, DF197, DF213, DF215, DF228) were thermally stable, with the Tm of the CH2 domain in each case being > 64°C and comparable to wild type molecules (Figure 3B).
Figure 3.
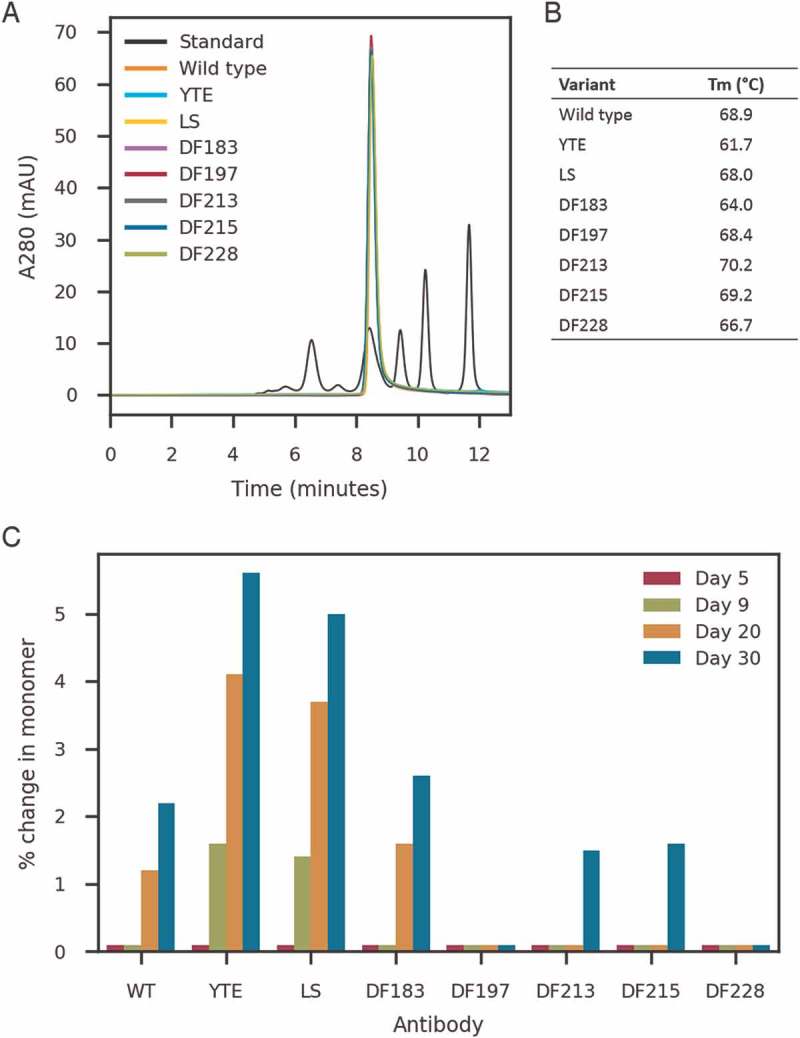
Biophysical characterization of lead Fc designs (A) SE-HPLC profile of lead Fc designs (DF183, DF197, DF213, DF215, DF228), YTE, LS and wild type with motavizumab Fab (B) Melting temperature (Tm) of CH2 domain of lead Fc designs and (C) Change in % monomer of motavizumab variants (DF183, DF197, DF213, DF215, DF228, YTE, LS and wild type Fc) by SE-HPLC after 5, 9, 20 and 30 days of storage at 40°C.
Certain combinations of Fc mutations that enhanced FcRn binding showed reduced thermal stability. For example, DF190 (T256D/H285D/Q311V) displayed enhanced binding to FcRn, but had a significantly lower Tm (58.7°C). Inspection of the structural model of this specific design revealed that several negatively charged residues (D256, D285, and E258) are in close proximity to each other, which may be causing repulsive electrostatic forces that destabilize the CH2 domain. Indeed, introduction of a basic amino acid at T307, which is located proximal to the negatively charged cluster, enhanced the thermal stability of the domain. Therefore, a T307R mutation was introduced into many of the designs to help balance the surface charge. Indeed, the Tm increased to 62.3°C when T307R was introduced to DF190. Despite T307R having a negative impact on FcRn binding as a single point mutation, it did not affect FcRn binding in the context of DF190 and provided improved thermal stability.
To assess the stability of the designed Fc domains, motavizumab variants incorporating DF183, DF197, DF213, DF215, DF228, YTE, and LS mutations were subjected to a 30-day accelerated stability study, and compared to wild type motavizumab (Figure 3C). All Fc variants stored at 40°C had similar elution times as the samples stored at 4°C. There was a decrease in monomer content for some of the Fc variants, including YTE and LS. Overall, the Fc designs (DF183, DF197, DF213, DF215 and DF228) behaved similarly to wild type Fc when subjected to temperature stress, with some indication that the variants might exhibit greater stability than the wild type protein. Additionally, no impact on FcRn binding was observed after 30 days at 40°C (data not shown).
Impact of FcRn affinity-enhancing modifications on engagement with Fc receptors and effector functions
The impact of FcRn affinity-enhancing modifications on binding of the Fc domain to other receptors was also experimentally evaluated. The Fc region of IgG is capable of binding to many different Fc receptors, and the mechanism of action for many therapeutic antibodies relies on engagement with these Fc receptors.39–41 It has been shown previously that mutations, even at positions distant from the Fc receptor binding site, can affect engagement of Fc with receptors such as FcγRI, FcγRIIa/b, FcγRIIIa, C1q, and/or TRIM21. While Fc receptor TRIM21 binds to a site that overlaps with the FcRn binding site on CH2-CH3, other receptors (such as Fcγ receptors and C1q) engage at the interface formed by the dimeric CH2 found in a fully assembled antibody. As such, any designs which enhance half-life should be assessed for their impact on binding to other receptors. Fc variants DF183, DF197, DF213, DF215, DF228, YTE, and LS were incorporated into rituximab and, together with the wild type Fc-containing rituximab, were evaluated for binding to FcγRI, FcγRIIa, FcγRIIb, FcγRIIIa, C1q and TRIM21 (Table 3, Supplementary Table 2, Supplementary Figure 3). The designed Fc variants DF183, DF197, DF213, DF215, and DF228 were comparable to wild type Fc in their engagement to all assessed Fc receptors.
Table 3.
Effector function characterization of lead Fc designs. Binding to Fc receptors FcγRIIIa and C1q and assessment of ADCC and CDC activity.
| Designation | FcγRIIIa EC50 (nM) |
ADCC EC50 (ng/mL)* |
C1q EC50 (nM) |
CDC EC50 (µg/mL)** |
|---|---|---|---|---|
| Wild type | 537.50 | 6.14 | 22.62 | 0.72 |
| YTE | 1,014.75 | 14.98 | 21.49 | > 20.00 |
| LS | 191.64 | 4.78 | 21.43 | 0.35 |
| DF183 | 87.22 | 3.92 | 16.50 | 0.10 |
| DF197 | 97.38 | 8.30 | 46.20 | 2.54 |
| DF213 | 139.44 | 4.01 | 20.65 | 0.27 |
| DF215 | 141.98 | 2.73 | 21.88 | 0.14 |
| DF228 | 154.45 | 2.66 | 20.95 | 0.22 |
*ADCC assay was performed with rituximab Fab and WIL2-S cells. EC50 calculated using 4-parametric fit ** CDC assay performed with rituximab Fab and Raji cells. EC50 calculated based on concentration that achieves 50 % lysis
While binding to Fc receptors is necessary for activities such as ADCC (FcγRIIIa), CDC (C1q), and ADIN (TRIM21), binding alone is not sufficient to mediate these activities. For example, a hexameric assembly of Fc is required to elicit CDC activity.42 The Fc hexameric assembly involves formation of an Fc-Fc interface, and residues involved in the formation of this interface partially overlap with the FcRn binding site. Therefore, it is critical to evaluate the capacity of our designs to not only bind to Fc receptors, but also mediate effector functions. Rituximab containing designed Fc variants (DF183, DF197, DF213, DF215, DF228, YTE, LS and wild type) were evaluated for ADCC and CDC activity as described in Methods. The results confirmed that Fc designs DF183, DF213, DF215, DF228 and LS retained potent ADCC and CDC activity and showed a slight enhancement in CDC activity. Rituximab containing YTE had no detectable CDC activity and significantly reduced ADCC activity. Similarly, one of our designs, DF197, had reduced ADCC and CDC activity (Table 3).
Persistence of optimized Fc variants in sera of transgenic mice and cynomolgus monkeys
To evaluate whether the enhanced binding of designed Fc variants to human FcRn translated to increased serum persistence and longer circulating half-life, a pharmacokinetic (PK) study of motavizumab containing the various Fc mutations was performed in Tg276 transgenic mice. The Tg276 mice are null for mouse FcRn alpha chain and express the human FcRn alpha transgene under the control of a constitutive promoter (actin).27 The human FcRn alpha chain pairs in vivo with the mouse β2-microglobulin protein forming a functional chimeric FcRn heterodimer.27 These mice were developed to study the PK of human Fc-containing biotherapeutics27,43 and both the Tg276 homozygous and hemizygous mice have been widely used to differentiate half-lives of antibody variants.13,27 Antibodies containing the engineered Fc variants persisted in serum for a significantly longer duration than wild type motavizumab (Figure 4). PK parameters from a non-compartmental analysis indicated that motavizumab with Fc variants have a greater than two-fold slower clearance rate and significant increase in the β-phase elimination half-life and area under the curve (AUC, Table 4). The enhancement in half-life was confirmed with two other Fabs (data not shown).
Figure 4.
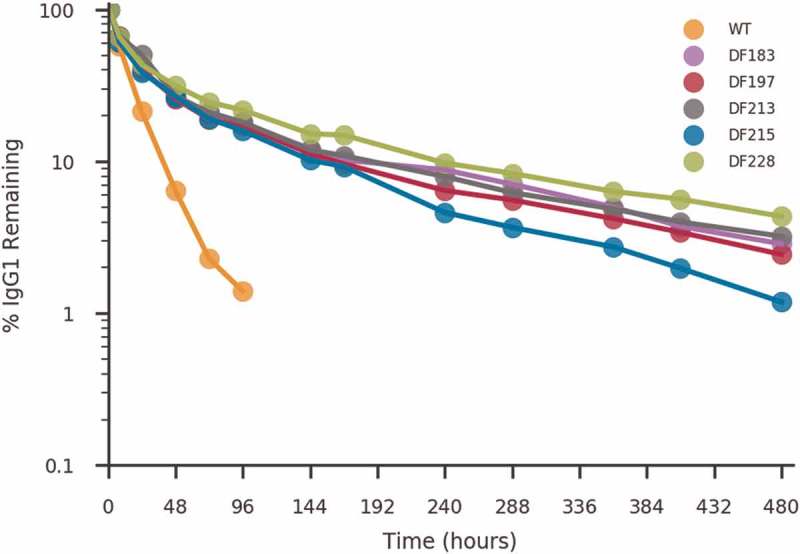
Pharmacokinetic characterization of motavizumab with various Fc designs in human FcRn transgenic mice Tg276. Clearance curve of wild type (orange), DF183 (pink), DF197 (red), DF213 (grey), DF215 (blue) and DF228 (green) variants of motavizumab, indicating the percentage of antibody remaining in the serum (as a percentage of the 1-hour timepoint). Each time point represents the average IgG concentration from four mice. The calculated PK parameters are shown in Table 4.
Table 4.
Pharmacokinetic characteristics of lead Fc designs in Tg276 transgenic mice.
| Designation | Half-life (hrs) | Cmax (μg/mL) | AUCinf (hr∙μg/mL) | Clearance (mL/hr/kg) |
|---|---|---|---|---|
| Wild type | 22 | 29.3 | 513.9 | 3.89 |
| YTE | 193 | 26.3 | 1682.5 | 1.19 |
| LS | 167 | 30.6 | 1731.3 | 1.16 |
| DF183 | 145 | 24.9 | 1689.3 | 1.18 |
| DF197 | 170 | 22.0 | 1385.5 | 1.44 |
| DF213 | 184 | 21.4 | 1546.1 | 1.29 |
| DF215 | 220 | 27.3 | 1588.0 | 1.26 |
| DF228 | 207 | 20.1 | 1741.3 | 1.15 |
Motavizumab wild type, DF215 and DF228 PK were also studied in cynomolgus monkeys (Macaca fascicularis). Groups of three monkeys were dosed once at 5 mg/kg intravenously and the concentration of human antibody in sera was measured over a period of 35 days. No mortality, or treatment-related clinical signs or changes in body weight were observed in the animals. The IgG levels in plasma were plotted as a function of time (Figure 5) and a PK analysis was performed assuming non-compartmental parameters. Incorporation of Fc modifications DF215 and DF228 significantly lowered the clearance rate (77 % and 83 %, respectively). The fitted parameters indicated a β-phase elimination half-life of 4.8 days for motavizumab in cynomolgus monkeys (t1/2 values reported in the literature for motavizumab are 5.7 ± 1.4 days, 6.1 ± 1.2 days,10 and 5.8 days44) (Table 5). Motavizumab containing the Fc modifications DF215 and DF228 had a half-life of 15.1 and 18.8 days, respectively, indicating a greater than 3.9-fold increase in half-life with incorporation of the DF228 design (reported fold increase in t1/2 with YTE mutation on motavizumab are 2, 3.5 and 3.7 – fold).10,44 Taken together these data demonstrate a significant increase in antibody persistence in the sera of animals known to be reliable models of human antibody half-life, strongly suggesting that these designs will indeed increase the time a biologic will remain at therapeutic concentrations in vivo.
Figure 5.
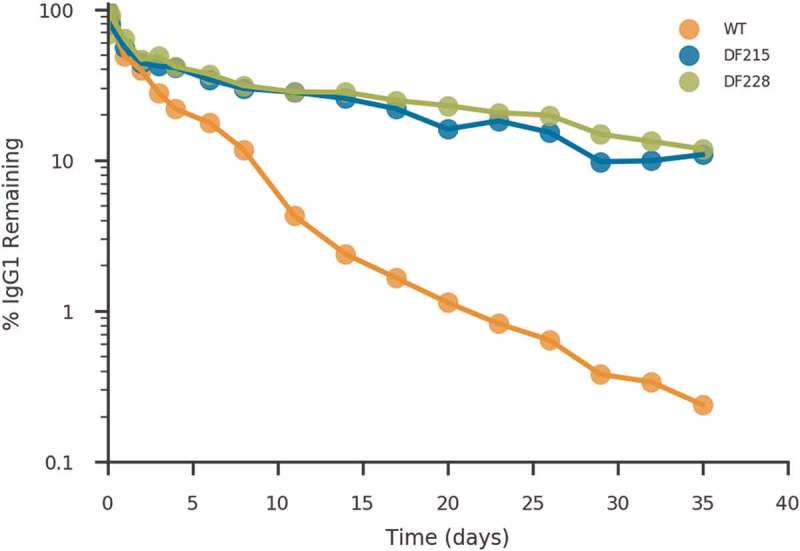
Pharmacokinetic characterization of motavizumab with Fc designs in cynomolgus monkey. Clearance curve of wild type (orange), DF215 (blue) and DF228 (green) designs of motavizumab, indicating the percentage of antibody remaining in the serum (as a percentage of the 1-hour timepoint). Each time point represents the average IgG concentration from three animals. The calculated PK parameters are shown in Table 5.
Table 5.
Pharmacokinetic characteristics of lead Fc designs in cynomolgus monkeys.
| Designation | Half-life (days) | Cmax (μg/mL) | AUCinf (day∙μg/mL) | Clearance (mL/day/kg) |
|---|---|---|---|---|
| Wild type | 4.78 ± 1.11 | 219.86 ± 10.06 | 658.13 ± 60.00 | 7.64 ± 0.66 |
| DF215 | 15.09 ± 2.50 | 278.50 ± 61.12 | 2988.14 ± 658.90 | 1.72 ± 0.35 |
| DF228 | 18.77 ± 2.06 | 306.23 ± 65.65 | 3907.57 ± 764.40 | 1.32 ± 0.29 |
Discussion
The Fc region of human antibodies plays a number of functional roles, including protecting the antibody from degradation through the lysosomal pathway and mediating antibody effector functions. With increasing use of antibodies as therapeutics, there has been an enhanced focus on not just selecting an optimal Fab, but also combining it with an appropriate Fc for desired half-life and effector functions. Since the elucidation of the role of FcRn in IgG homeostasis, several attempts have been made to modulate the binding affinity of IgG to FcRn. While not all FcRn-affinity enhancing mutations resulted in increased serum persistence, Fc variants, such as YTE and LS, have resulted in enhanced half-life in non-human primates and humans, validating that such efforts can modulate antibody half-life. In this study, the availability of structural information of the human FcRn-Fc complex has enabled: 1) identification of molecular features governing Fc-FcRn interaction; 2) mapping the interaction networks of amino acids in the Fc domain; and 3) a systematic investigation into the impact of introduction of specific residues on interaction with FcRn, as well as other amino acids in their interaction networks. Using this approach, we designed a broad panel of Fc variants that possess improved binding affinity to FcRn at endosomal pH and confers enhanced half-life in transgenic mouse models and cynomolgus monkeys. Identification of multiple Fc variants that enhance circulating half-life provides flexibility in choosing a different Fc that may be more compatible with different Fabs. Further, in line with the engineering approach of combining mutations that are unlikely to destabilize the antibody or disrupt its interaction with other receptors, the designed Fc variants retained the biophysical properties and effector functions of the parental IgG.
In recent years, there have been attempts to understand how the sequence and biophysical properties of the Fab domain influences the IgG-FcRn interaction and PK. While the primary focus of the structural analysis presented here was understanding the “cross-talk” between CH2-CH3 of Fc and FcRn, one key aspect of this analysis was also to address, structurally, the way in which Fc, within the context of a fully assembled antibody, interacts with membrane-associated FcRn (mFcRn). Such an analysis is imperative to understand the relative importance of Fc mutations on governing the overall strength of engagement between antibody and mFcRn. Additionally, such an understanding can help to address the role of the Fab in governing antibody half-life, especially when coupled with existing analyses of Fab properties. Of note, the sequence and the structure of the Fc domain of most IgG molecules are highly similar, yet their FcRn affinities vary and are either the same or weaker than that of the Fc domain alone.28,45 This observation suggests that the Fab domains of the IgG molecule play either a passive or, in some cases, a detrimental role in mFcRn-IgG interactions. Some, but not all, of these differences can be ascribed to physiochemical characteristics of the variable sequences.46 In addition, the hinge domain that links the Fab to Fc domain also plays a role in the mFcRn-IgG affinity.47 Interestingly, the protein A/G binding site on the Fc domain overlaps with the FcRn binding site, yet neither the variable or hinge domain of an IgG molecule influence the binding of an IgG to protein A/G protein.
Facilitating this analysis, the crystal structure of the rat FcRn-Fc complex has been reported, as well as the structure of a human Fc-FcRn-human serum albumin ternary complex.20,48,49 Since only the soluble domain of FcRn was used in these studies, the structures do not fully elucidate the role of the Fab domain in mediating the mFcRn-IgG interactions. The short length of the linker peptide (ten amino acids) between the Fc-binding domain and the transmembrane domain requires that the Fc-binding domain of FcRn be in close proximity to the membrane bilayer if the two molecules are to interact. Therefore, the cell membrane and its influence on the IgG-FcRn interaction must be taken into consideration when attempting to design IgG molecules with enhanced binding to mFcRn.
In conjunction with available structural data on Fc-FcRn interactions, we examined possible rationalizations for the observed differences in the context of IgG binding to mFcRn. Previously, two models of IgG engagement with mFcRn have been proposed: the “standing up” and “lying down” positioning of engaged Fc, relative to mFcRn (Supplementary Figure 4).50,51 Since albumin and IgG molecules can bind simultaneously to membrane-positioned FcRn, we propose that the IgG molecule is likely to bind in an inverted orientation, the “standing up” orientation, with its Fab domains close to the membrane bilayer surface and possibly, in certain circumstances, interacting with the membrane (Figure 6). We note that this close membrane proximity of Fab domains to the cell membrane offers a possible structure-based explanation for the reduced affinity to membrane-bound, native mFcRn of most IgG molecules and Fc-fusion proteins, as well as antibodies bound to large antigens.
Figure 6.
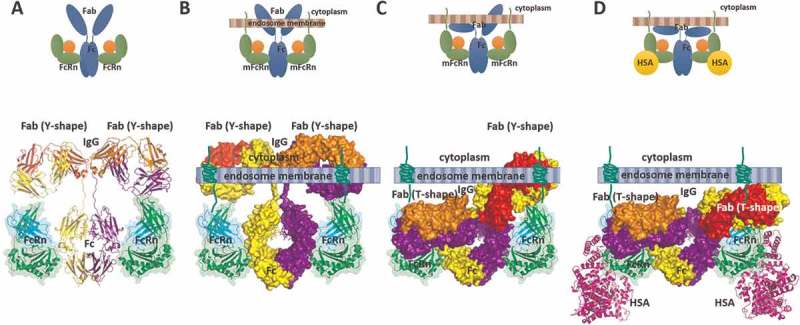
Current best-fit model of IgG-FcRn complex formation. The “standing-up” orientation of Fc-FcRn engagement is shown as a schematic diagram (top) and molecular diagram (bottom). In the molecular diagram the FcRn molecule is shown as a cartoon diagram with semi-transparent surface diagram while the IgG molecule is shown as a cartoon (A) or as surface diagram (B, C and D). (A) IgG-FcRn complex as derived from the crystal structure of the FcRn-Fc complex with the IgG shown in the canonical Y-shaped conformation. (B) Placement of the IgG-FcRn complex in the context of membrane bilayer bound FcRn (mFcRn) to illustrate the severe steric clash presented by the membrane bilayer with the Fab domains of the bound IgG in a Y-shaped conformation. (C) Model of IgG-mFcRn complex in which IgG adopts a mixed Y/T conformation modeled based on observed Y/T conformation of a mouse IgG2a Fab structure (PDB ID: 1IGY) (D) Model of IgG-mFcRn complex in which IgG adopts a T-shaped conformation. This is the only conformation that does not impose a severe steric clash between the Fab domains and the membrane bilayer. Further, only the T-conformation of IgG supports FcRn binding to IgG and HSA simultaneously. The cartoon diagram of the bound HSA molecule shown is based on the crystal structure of its complex with FcRn-Fc complex (PDB ID: 4N0U).
One consequence of this analysis relates to the conformational flexibility of the hinge domain of IgG, which allows the Fab domains to access multiple orientations with reference to the Fc domain. For example, in solution IgG can sample not only the canonical Y-shaped conformation, but also T- shaped and intermediate conformations.52,53 Due to the proximity of the membrane bilayer when Fc is bound to FcRn, the “standing up” model likely restricts binding of IgG molecules to the T-shaped conformation, as this would allow minimal steric hindrance with the membrane bilayer. In contrast, other conformations would likely lead to severe steric hindrance (Figure 6). In this proposed model, when the antibody is in the T-shaped conformation, the Fab domains are found close to the mFcRn molecule and possibly interact weakly with mFcRn.
In summary, we developed a structure- and network-based approach to identify molecular features governing Fc-FcRn interactions and used this approach to identify multiple Fc variants that enhance FcRn binding at endosomal pH. Incorporation of these Fc variants on control IgGs significantly decreased clearance rates (by up to 83 %) and enhanced elimination half-life of antibodies (> 3.9-fold) in cynomolgus monkeys and human FcRn transgenic mice. It has been previously shown that incorporation of YTE on motavizumab resulted in a 2–3.7-fold increase in half-life in cynomolgus monkeys and a half-life of up to 100 days in humans.18 We anticipate that incorporation of the Fc variants described in this manuscript will result in comparable or even improved half-life in humans (as compared to YTE), while maintaining the desired effector functions of the antibody such as ADCC and CDC. Antibodies with intact effector function and significantly enhanced half-life would enable less frequent administration of antibody-based therapeutics for chronic indications. In addition, these long-acting antibodies could be used to provide season-long protection from infectious diseases such as seasonal influenza. We also highlight the role of the Fab in influencing the IgG-FcRn interaction by proposing a model where the orientation of the antibody bound to mFcRn causes the Fab to be proximal to, and possibly engage with, the endosomal membrane and thereby influence the IgG-mFcRn interactions.
Materials and methods
Structure and network model of Fc-FcRn complex. The modeled structure of Fc-FcRn complex was created using Discovery studio (Dassault Systèmes BIOVIA) based on the available co-crystal structure of the YTE-Fc domain in complex with FcRn (PDB ID:4N0U). The Fc-FcRn interface residues and their interactions were calculated using the PISA server (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html). A protein residue network was generated by representing each residue as a node in a graph; edge weights in the graph were based on the inter-residue energies calculated using Rosetta (implemented as a pyrosetta script using the Rosetta Energy Function 2015 (REF2015)54 score function). A network was generated using the NetworkX package in python, and these networks were visualized using Gephi (https://gephi.org) to identify residues that interact both directly or through the network.
Antibody expression and purification. All Fc mutations were introduced into the human IgG1 (m3 allotype) heavy chain gene by gene synthesis (gBlocks, Integrated DNA Technologies) and cloned into pcDNA3.1(C). The light chain genes were also synthesized and cloned into pcDNA3.1(A). In each case, the native signal peptide was replaced with an osteonectin signal peptide (GenBank accession # AAA60993). Co-expression of the heavy and light chain vectors was performed by transient transfection in Expi293 cells using the Expi293 transfection kit (Thermo Fisher Scientific, catalog # A14524) following the manufacturer’s protocol. Cell culture supernatants were harvested 5 to 7 days post-transfection and purified on a AKTA 10 FPLC system (GE) using HiTrap MabSelect SuRe protein A columns (GE) following the manufacturer’s instructions.
FcRn expression and purification. Expi293 cells were co-transfected with a plasmid encoding the human α-FcRn (NP_004098.1) extracellular domain (amino acids 24–297) with a (His)6 tag on the C-terminus and a plasmid encoding full length human β2M (NP_004039.1). Cell culture supernatant was harvested 4 days post-transfection. FcRn heterodimer was purified from cell culture supernatant using the AKTA pure FPLC system with a HisTrap HP column (GE catalog # 17–5247-01). Post-purification, the protein was concentrated and dialyzed into phosphate-buffered saline (PBS) pH 6.0.
FcRn Binding Kinetics. Screening assays were performed using anti-CH1 Fab biosensors on the Octet QKe system. Purified IgG at 10 µg/mL was loaded onto an anti-CH1 biosensor for 180 seconds. After a 60 second baseline step in 1x PBS pH 6.0, the IgG-loaded tip was exposed to FcRn at a concentration of 50 µg/mL for 60 seconds, followed by dissociation for 60 seconds in PBS pH 6.0, and an additional 30 seconds in PBS pH 7.4. In addition, FcRn binding was performed using NiNTA biosensors. Recombinant human FcRn at 5 µg/mL was loaded onto a NiNTA biosensor for 180 seconds. After a 60 second baseline step in 1x PBS pH 6.0, the FcRn-loaded tip was exposed to IgG at a concentration of 250 nM (37.5 µg/mL) for 60 seconds, followed by dissociation for 60 seconds in PBS pH 6.0, and an additional 30 seconds in PBS pH 7.4. After assay completion, a quantitative assessment of the affinity constant (KD) at pH 6.0 was performed using the ForteBio Octet software and a qualitative assessment was performed by plotting the response rate over time, allowing for visualization of the association of IgG to FcRn at pH 6.0 and the subsequent dissociation at both pH 6.0 and pH 7.4.
Surface Plasmon Resonance. FcRn binding affinity for a panel of Fc variants was measured by surface plasmon resonance (SPR) on a Biacore T200 instrument. Antibodies were immobilized to a Series S CM5 sensor chip (GE Catalog # BR100530) using covalent amine coupling chemistry to a response unit (RU) between 100 – 150. A titration series of recombinant human FcRn (3.3, 1.1, 0.4, 0.1, 0.04, 0.01, 0.005 μM) was diluted in PBS, 0.01 % Tween 20 pH 6.0 or pH 7.4 and injected over the chip at a flow rate of 50 μL/minute for 60 seconds followed by buffer only for 90 seconds. Rate kinetics were calculated using a 1:1 model fit for all Fc variants. Due to the fast off-rate of wild type Fc, the data could not be modelled a 1:1 curve fit; therefore, steady state analysis was used to determine the KD. DF215 was used in all assays as an internal reference control.
Thermal stability assessment. The thermal stability of the mAbs was determined by DSF. Various Fc variants with motavizumab Fab were evaluated in a SYPRO Orange® (Sigma) assay. 15 µL of IgG at ~ 0.5 mg/mL was mixed with 15 μL of SYPRO Orange® dye (1:500 dilution in PBS) and assessed by a thermocycler with fluorescent read capabilities using a 1°C ramp/minute from 25°C to 95°C. The midpoint between native state and the first unfolding event was reported as the transition temperature or melt temperature (TM).
Size exclusion high performance liquid chromatography. The impact of Fc mutations on the IgG elution profile was assessed on a Phenomenex Biosep 3000s column. Briefly, IgG containing the various Fc variants were diluted to 1 mg/mL in PBS pH 7.4 and 20 µL was injected into the column. PBS at pH7.4 was used as the mobile phase and run at 1 mL/minute. The elution time and percentage purity were recorded. For accelerated stability studies the Fc motavizumab variants were incubated for 30 days under two different storage temperatures (4°C and 40°C) and, at various timepoints, were characterized by SE-HPLC to assess changes in tertiary structure (as assessed by elution time) and level of aggregation (as assessed by a reduction in monomer content).
Binding to Fcγ receptors. Binding of IgG containing various Fc designs to Fcγ receptors I, IIa, IIb, IIIa V176, and IIIa F176 (R&D Systems catalog # 1257-FC-050, 1330-CD-050, 1875-CD-050, 4325-FC-050, and 8894-FC-050, respectively) was measured by ELISA. All Fc variants were tested in the context of the motavizumab, rituximab, or actoxumab variable regions. The Fc receptors were coated on an ELISA plate (VWR catalog # 62409–002) at 1 µg/mL (0.1 µg/well) in PBS and stored at 4°C overnight. Plates were washed three times with PBST (1x PBS 0.05 % Tween20). Antibodies were titrated threefold in PBST with 1 % bovine serum albumin (PBST-BSA) from 100 µg/mL to 0.05 µg/mL and 100 µL was added to each well of the ELISA plate and incubated for 1 hour at room temperature. Plates were washed three times with PBST. Goat anti-human Fc horseradish peroxidase (HRP) conjugate (Jackson ImmunoResearch catalog # 109–035-098) was diluted 1:5000 in PBST-BSA and 100 µL was added to each well and incubated for 1 hour at 4°C. Plates were washed six times with PBST. Plates were developed using the TMB Microwell Peroxidase Substrate Kit (VWR catalog # 95059–156). The reaction was stopped after 10 minutes by the addition of 1N sulfuric acid and absorbance at 450 nm was measured using a Spectramax M2 plate reader. The values of antibody concentration (x-axis) and absorbance at 450 nm (y-axis) were fit to a four-parameter logistic regression (4PL) curve. The curve fit was then used to determine the EC50 (the midpoint of the 4PL) for each Fc design.
Binding to C1q and TRIM21. Binding to C1q was measured by ELISA. All Fc designs were tested in the context of the motavizumab antibody. Antibodies were coated on an ELISA plate (VWR catalog # 62409–002) at 25 µg/mL (2.5 µg/well) in PBS and stored at 4°C overnight. Plates were washed three times with PBST. Purified C1q (Quidel Corporation catalog # A400) was titrated threefold in PBST-BSA from 12.5 µg/mL to 0.02 µg/mL and incubated for 90 minutes at room temperature. Liquid was aspirated from the wells and polyclonal rabbit anti-human C1q (Agilent catalog # A013602-1) was diluted in PBST-BSA to a final concentration of 1 µg/mL and 100 µL was added to each well and incubated for 1 hour at room temperature. Plates were washed three times with PBST. Polyclonal swine anti-rabbit-HRP (Agilent catalog # P021702-2) was diluted to 0.5 µg/mL in PBST-BSA and 100 µL was added to each well and incubated for 1 hour at room temperature. The plates were washed six times with PBST and then developed with TMB solution as already described. The values of antibody concentration (x-axis) and absorbance at 450 nm (y-axis) were fit to a four-parameter logistic regression (4PL) curve. The curve fit was then used to determine the EC50 (the midpoint of the 4PL) for each Fc variant.
Binding to TRIM21 was measured by ELISA following the same protocol as C1q with the following alterations. The capture protein was TRIM21-GST (Antibodies Online catalog # ABIN1323621) and the reagents used to detect the captured antigen were a combination of two rabbit anti-TRIM21 antibodies (AbCam catalog # ab91423 and ab96800) that were mixed together in PBST-BSA at a final concentration of 1 µg/mL.
Antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. ADCC assays were performed using the ADCC Reporter Bioassay with CD20+ WIL2-S target cells from Promega (catalog # G7014) following the manufacturer’s protocol. All Fc designs were tested in the context of the anti-CD20 antibody, rituximab. Antibody concentrations were titrated five-fold ranging from 5 µg/mL to 0.0016 µg/mL. The values of antibody concentration (x-axis) and fold induction of the luminescence read on Biotek luminescence reader (y-axis) were fit to a four-parameter logistic regression (4PL) curve. The curve fit was then used to determine the EC50 (the midpoint of the 4PL) and the maximum induction for each Fc design.
CDC assays were performed using CD20+ Raji cells and low toxicity guinea pig complement (Cedarlane Laboratories Product # CL4051). Complement-induced cell lysis was measured using the CytoTox 96® Non-Radioactive Cytotoxicity Assay from Promega (catalog # G1780) following the manufacturer’s protocol. All Fc variants were tested in the context of rituximab. The antibody concentrations were titrated four-fold ranging from 20 µg/mL to 0.005 µg/mL and incubated with 20,000 target cells per well at 37°C for 30 minutes. Complement was then added to the cells and incubated for an additional 2 hours at 37°C. Additionally, cell lysis buffer (provided with the CytoTox kit) was added to control wells to measure the maximal cell lysis. A negative control antibody, motavizumab, was used to measure the background signal of an irrelevant antibody. The background signal and maximal lysis signal were used to calculate the percent of cell lysis for each Fc design. The values of antibody concentration (x-axis) and percent lysis (y-axis) were fit to a four-parameter logistic regression curve. The curve fit was then used to determine the EC50 (concentration needed to obtain 50 % lysis) and the maximal lysis for each Fc design.
Quantitative Serum IgG ELISA. To evaluate the serum concentration of human antibody in the serum of Tg276 mice, a quantitative anti-human ELISA was performed using the Human IgG ELISA quantitation kit from Bethyl Labs (catalog # E80-104) following the manufacturer’s instructions. To evaluate the serum concentration of human antibody in cynomolgus monkey serum, a quantitative anti-human ELISA with minimal cross reactivity to cynomolgus IgG was performed. Unlabeled goat anti-human IgG (Southern Biotechnology # 2049–01) was diluted to 2 µg/mL and ELISA plates were incubated with 100 µL/well overnight at 4°C. The following morning all liquid was aspirated from the wells and the plates were blocked with 200 µL of PBST-BSA for 1 hour. A standard curve of purified antibody was included on each plate ranging from 1000 to 1 ng/mL in PBST-BSA. All serum samples were serially titrated 1:2 across the plate starting at a 1:10 dilution and ending at a 1:10,240 dilution. The capture step was performed for 1 hour at room temperature. The plates were then washed three times with PBST. Biotinylated Goat Anti-Human IgG (Southern Biotechnology # 2049–01) was diluted 1:20,000 in PBST-BSA, 100 µL was added to all wells, and the plates were incubated for 1 hour at room temperature. The plates were washed six times with PBST. Streptavidin-HRP (Southern Biotechnology, 7100–05) was diluted 1:8,000 in PBST, 100 µL was added to all wells, and the plates were incubated for 1 hour at room temperature. The plates were washed six times with PBST and then developed with TMB solution as already described.
Pharmacokinetic experiments in transgenic mice expressing human FcRn. To evaluate if the enhanced binding of Fc designs to human FcRn translated to increased serum persistence and longer circulating half-life life, a PK study of IgGs containing the different Fc variants was performed in B6.Cg-Fcgrttm1Dcr Tg(CAG-FCGRT)276Dcr/DcrJ (Tg276) transgenic mice (The Jackson Laboratory, stock number 004919). Tg276hemizygous FcRn transgenic mice were dosed with 2 mg/kg of mAb intravenously. Each mAb group had 4 mice/group. Retro-orbital blood collection was performed at several time points between 1 hour and 21 days, and IgG titers were determined by quantitative ELISA as described in the section above. PK parameters were determined for each group of mice with a non-compartmental model using Phoenix WinNonlin version 7.0 (Certara).
Pharmacokinetic experiments in cynomolgus monkeys. In a non-GLP PK study conducted in cynomolgus monkeys, samples of motavizumab containing selected Fc designs were evaluated for persistence in serum. Three male (3.1 to 4.5 kg) and six female (2.4 to 3.2 kg) cynomolgus monkeys aged 3.5 to 3.7 years were divided into three groups. Each group consisted of one male and two female monkeys that were administered a single slow bolus intravenous injection of motavizumab, motavizumab DF215 or motavizumab DF228; the animals were then observed over a 35-day period. Mortality checks, clinical observations and body weight were monitored during the course of study. Blood samples for PK evaluations were collected on day −7, day −1 and after dosing on day 1 at 0 (up to 10 minutes after completion of dose formulation administration), 1, 4, and 24 hours and additionally on days 2, 3, 4, 6, 8, 11, 14, 17, 20, 23, 26, 29, 32, 35. Human IgG levels in plasma were determined by ELISA and the PK parameters were determined for each group of monkeys with a non-compartmental model using Phoenix WinNonlin version 7.0 (Certara).
Supplementary Material
Funding Statement
Visterra, Inc.
Abbreviations
FcRn – Neonatal Fc receptor
ADCC – Antibody-Dependent Cell-mediated Cytotoxicity
CDC – Complement-Dependent Cytotoxicity
ADCP – Antibody-Dependent Cellular Phagocytosis
ADIN – Antibody-Dependent Intracellular Neutralization
Acknowledgments
The authors would like to thank Danielle Wisheart, Jason Kong, Joseph Columbus and Bharathi Sundaresh for their technical assistance and Greg Christianson at The Jackson Laboratories for half-life assessments in mice.
Disclosure of potential conflicts of interest
Authors are employees of Visterra and own equity.
References
- 1.Reichert JM. 2017. Antibodies to watch in 2017. mAbs. 9:167–181. doi: 10.1080/19420862.2016.1269580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mankarious S, Lee M, Fischer S, Pyun KH, Ochs HD, Oxelius VA, Wedgwood RJ. The half-lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J Lab Clin Med. 1988;112:634–640. [PubMed] [Google Scholar]
- 3.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proceedings of the National Academy of Sciences of the United States of America 1996; 93:5512–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leach JL, Sedmak DD, Osborne JM, Rahill B, Lairmore MD, Anderson CL.. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: implications for maternal-fetal antibody transport. J Immunology. 157;1996:3317–3322. [PubMed] [Google Scholar]
- 6.Simister NE, Story CM, Chen HL, Hunt JS. 1996. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. 26:1527–1531. doi: 10.1002/eji.1830260718. [DOI] [PubMed] [Google Scholar]
- 7.Kristoffersen EK. Human placental Fc gamma-binding proteins in the maternofetal transfer of IgG. APMIS Suppl. 64;1996:5–36. [DOI] [PubMed] [Google Scholar]
- 8.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. The Journal of Immunology. 2003;170:3528–3533. doi: 10.4049/jimmunol.170.7.3528. [DOI] [PubMed] [Google Scholar]
- 9.Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15:637–640. doi: 10.1038/nbt0797-637. [DOI] [PubMed] [Google Scholar]
- 10.Dall’Acqua WF, Kiener PA, Wu H. 2006. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 281:23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 11.Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vasquez M, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem. 2004;279:6213–6216. doi: 10.1074/jbc.C300470200. [DOI] [PubMed] [Google Scholar]
- 12.Vaccaro C, Zhou J, Ober RJ, Ward ES. 2005. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 23:1283–1288. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- 13.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. The Journal of Immunology. 2002;169:5171–5180. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 15.Monnet C, Jorieux S, Souyris N, Zaki O, Jacquet A, Fournier N, Crozet F, de Romeuf C, Bouayadi K, Urbain R, et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. MAbs. 2014;6:422–436. doi: 10.4161/mabs.27854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monnet C, Jorieux S, Urbain R, Fournier N, Bouayadi K, De Romeuf C, Behrens CK, Fontayne A, Mondon P. Selection of IgG variants with increased fcrn binding using random and directed mutagenesis: impact on effector functions. Front Immunol. 2015;6:39. doi: 10.3389/fimmu.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 18.Robbie GJ, Criste R, Dall’acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57:6147–6153. doi: 10.1128/AAC.01285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borrok MJ, Mody N, Lu X, Kuhn ML, Wu H, Dall’Acqua WF, Tsui P. An “Fc-Silenced” IgG1 format with extended half-life designed for improved stability. J Pharm Sci. 2017;106:1008–1017. doi: 10.1016/j.xphs.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. 2014;289:7812–7824. doi: 10.1074/jbc.M113.537563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 34;1995:14649–14657. [DOI] [PubMed] [Google Scholar]
- 22.Frank M, Walker RC, Lanzilotta WN, Prestegard JH, Barb AW. 2014. Immunoglobulin G1 Fc domain motions: implications for Fc engineering. J Mol Biol. 426:1799–1811. doi: 10.1016/j.jmb.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters BT, Jensen PF, Larraillet V, Lin K, Patapoff T, Schlothauer T, Rand KD, Zhang J. Conformational destabilization of immunoglobulin G increases the low pH binding affinity with the Neonatal Fc Receptor. J Biol Chem. 2016;291:1817–1825. doi: 10.1074/jbc.M115.691568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD. A two-pronged binding mechanism of IgG to the neonatal Fc receptor controls complex stability and IgG serum half-life. Mol Cell Proteomics. 2017;16:451–456. doi: 10.1074/mcp.M116.064675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. The Journal of Immunology. 2010;184:1968–1976. doi: 10.4049/jimmunol.0903296. [DOI] [PubMed] [Google Scholar]
- 26.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. 2007. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem. 282:1709–1717. doi: 10.1074/jbc.M607161200. [DOI] [PubMed] [Google Scholar]
- 27.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–1769. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 28.Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos. 2012;40:1545–1555. doi: 10.1124/dmd.112.045864. [DOI] [PubMed] [Google Scholar]
- 29.Lee LP, Tidor B. 2001. Optimization of binding electrostatics: charge complementarity in the barnase-barstar protein complex. Protein Sci. 10:362–377. doi: 10.1110/ps.40001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz R, Istrail S, King J. 2001. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 10:1023–1031. doi: 10.1110/ps.33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez LD, Kroh HK, Hsieh E, Yang X, Beaumont M, Sheth PR, DiNunzio E, Rutherford SA, Ohi MD, Ermakov G, et al. Epitopes and mechanism of action of the clostridium difficile toxin A-neutralizing antibody actoxumab. J Mol Biol. 2017;429:1030–1044. doi: 10.1016/j.jmb.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 34.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Jr., Leney M, Sloan S, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 35.Gao X, Ji JA, Veeravalli K, Wang YJ, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al. Effect of individual Fc methionine oxidation on FcRn binding: met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J Pharm Sci. 2015;104:368–377. doi: 10.1002/jps.24136. [DOI] [PubMed] [Google Scholar]
- 36.Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I, Sun T, Caffry I, Brown M, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 2017;9:646–653. doi: 10.1080/19420862.2017.1290753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ionescu RM, Vlasak J, Price C, Kirchmeier M. 2008. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 97:1414–1426. doi: 10.1002/jps.21104. [DOI] [PubMed] [Google Scholar]
- 38.Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. MAbs. 2015;7:84–95. doi: 10.4161/19420862.2014.985494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan KR, Ong EZ, Mok DZ, Ooi EE. 2015. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev Anti Infect Ther. 13:1351–1360. doi: 10.1586/14787210.2015.1079127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiLillo DJ, Ravetch JV. 2015. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunology Research. 3:704–713. doi: 10.1158/2326-6066.CIR-15-0120. [DOI] [PubMed] [Google Scholar]
- 41.Nimmerjahn F, Ravetch JV. 2008. Fcgamma receptors as regulators of immune responses. Nat Reviews Immunol. 8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 42.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roopenian DC, Christianson GJ, Sproule TJ. 2010. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol. 602:93–104. doi: 10.1007/978-1-60761-058-8_6. [DOI] [PubMed] [Google Scholar]
- 44.Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall’Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015;290:4282–4290. doi: 10.1074/jbc.M114.603712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–1477. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 46.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proceedings of the National Academy of Sciences of the United States of America 2015; 112:5997–6002. 10.1073/pnas.1408766112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. 2006. An engineered human IgG1 antibody with longer serum half-life. The Journal of Immunology. 176:346–356. doi: 10.4049/jimmunol.176.1.346. [DOI] [PubMed] [Google Scholar]
- 48.Burmeister WP, Huber AH, Bjorkman PJ. 1994. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 372:379–383. doi: 10.1038/372379a0. [DOI] [PubMed] [Google Scholar]
- 49.Martin WL, West AP Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 7;2001:867–877. [DOI] [PubMed] [Google Scholar]
- 50.Raghavan M, Bjorkman PJ. 1996. Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol. 12:181–220. doi: 10.1146/annurev.cellbio.12.1.181. [DOI] [PubMed] [Google Scholar]
- 51.Weng Z, Gulukota K, Vaughn DE, Bjorkman PJ, DeLisi C. 1998. Computational determination of the structure of rat Fc bound to the neonatal Fc receptor. J Mol Biol. 282:217–225. doi: 10.1006/jmbi.1998.2020. [DOI] [PubMed] [Google Scholar]
- 52.Tc W, Jr B, Re C. The shape of immunoglobulin G molecules in solution. Proceedings of the National Academy of Sciences of the United States of America 1972; 69:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Zhang L, Tong H, Peng B, Rames MJ, Zhang S, Ren G. 3D structural fluctuation of IgG1 antibody revealed by individual particle electron tomography. Sci Rep. 2015;5:9803. doi: 10.1038/srep09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, et al. The rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput. 2017;13:3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.