

Abstract
Background
Fibroblast growth factor (FGF) 21 was reported to be induced by different injurious agents, including chronic hepatitis C (CHC) virus, affecting the liver. The aims of this study were to evaluate the FGF21 levels in CHC patients before and after the treatment with direct-acting antiviral agents (DAAs) in comparison to that in control subjects and to correlate these levels with insulin resistance (IR), lipid profile, and fibrosis stages.
Patients and methods
We studied 75 naive CHC patients and 40 age- and gender-matched healthy control subjects. Patients were divided into five groups based on the severity of fibrosis as detected by Fibroscan as follows: F0, n=2; F1, n=13; F2, n=23; F3, n=16; F4, n=21. We estimated the FGF21 levels at the start of the study for all the participants and for the patients only at the end of treatment with simisipivir (SIM) and sofosbuvir (SOF). These levels were compared between the patients and the control subjects and also for the patients before and after the treatment with DAAs. The FGF21 levels were correlated to IR, lipid profile, and stages of liver fibrosis.
Results
The FGF21, fasting blood sugar (FBS), fasting insulin, and homeostasis model of IR (HOMA-IR) were significantly higher in CHC patients compared to control (5.04±0.75 vs 4.7±0.52, 20.15±5.13 vs 13.15±4.2, 4.49±1.28 vs 2.72±0.87, and 123.7±52.6 vs 21.8±8.8; P≤0.01, P≤0.001, P≤0.001, and P≤0.001, respectively). The posttreatment FGF21 levels were significantly reduced when compared to the pretreatment levels (123.7±52.5 vs 60.5±32.7, P≤0.001). FGF21 levels showed significant negative correlation with FBS and positive correlation with serum albumin (P≤0.05 and P≤0.003, respectively). The multiple linear regression analysis revealed that serum albumin, high-density lipoprotein cholesterol (HDL-c), and the stage of liver fibrosis were independent risk factors for FGF21.
Conclusion
Besides its metabolic modulator role, FGF21 strongly introduced itself as a novel biomarker of hepatic injury in Egyptian, genotype-4, CHC patients.
Keywords: hepatitis C, direct-acting antiviral agents, fibroblast growth factor 21
Introduction
The fibroblast growth factor (FGF)21 represents an atypical member of the FGF signaling system, which lacks heparin binding and circulates as a hormone. Experimental animal models showed their role in lipid and glucose metabolism and energy homeostasis, and the correlation coefficient (about 0.6) is poor.1 FGF21 is activated by binding to the FGF receptors and a unique β-Klotho co-receptor, which is expressed abundantly in the liver, adipose tissue, and pancreas.2 The liver represents a major site for FGF production and actions, and FGF21 was reported to be induced by different injurious agents, including chronic hepatitis C (CHC) virus, affecting the liver. 3–5 The underlying molecular mechanism for increased hepatic expression of FGF21 in CHC patients is unclear. One of the postulated mechanisms is the endoplasmic stress (ES) theory. ES activates the adaptive response known as the unfolded protein response (UPR).6 The hepatitis C virus (HCV) single-stranded RNA genome encodes the nonstructural protein 4B (NS4B), an endoplasmic reticulum (ER) membrane-associated protein. Overexpression of HCV-NS4B could induce ES.7,8
In pediatric non-alcoholic fatty liver disease (NAFLD), Alisi et al9 found that FGF21 was inversely associated with the probability of fibrosis, but the relation of its expression in liver fibrosis in CHC patients is not well understood.
The aims of this study were to evaluate the FGF21 levels in CHC, genotype-4, Egyptian patients before and after the treatment with direct-acting antiviral agents (DAAs) in comparison to that in apparently healthy control subjects; to determine the effects of treatment in these levels; and to correlate FGF21 levels with insulin resistance (IR), lipid profile, and the different stages of liver fibrosis.
Patients and methods
After approval of the Faculty of Medicine Research Ethics Committee (FMREC), Minia University, Egypt, and the national Egyptian committee for viral hepatitis control, 75 naive Egyptian patients with compensated CHC and 40 apparently healthy subjects, who volunteered as control subjects, were selected for this study. All the participants signed an informed consent and were prospectively assessed from February 2015 to March 2017. This study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice.
Inclusion criteria
Patients eligible for the study were 20–67 years old, non-diabetic, non-obese, naive patients suffering from genotype-4 CHC for 6 months or more. Infection was considered based on the presence of anti-HCV antibodies and serum HCV-RNA. HCV genotyping was done by direct sequencing of the untranslated regions using RT-PCR-based assay (Ampli Sens 61 HCV-genotype-FRT PCR kit). The control subjects were recruited from the medical and paramedical staff. There were 28 males and 12 females, and their age ranged between 20 and 66 years.
Exclusion criteria
Patients were excluded if they had diabetes mellitus (DM) or other forms of liver diseases such as concomitant hepatitis-B virus (HBV) infection, HIV, and schistosomal, autoimmune, or alcoholic hepatitis. Patients who received any antihyperlipidemic therapy in the last 3 months before recruitment were also excluded.
To quantify the stages of hepatic fibrosis, we used Fibroscan (transient elastography) and the Fibrosis-4 Index for Liver Fibrosis (FIB-4) test. According to Fibroscan, patients were classified into four subgroups based on the breakpoints in relation to the METAVIR score (17; F0<2.5; F0–F1=2.5–7; F2=7–9.5; F3=9.5–12.5; F4>12.5). The FIB-4, a combination of four variables, aspartate aminotransferase (AST), alanine aminotransferase (ALT), age, and platelet count, is calculated with the formula: FIB-4 index=(age [years]×AST [IU/L])/(platelet count [109/L]×ALT [IU/L])1/2. If the FIB-4 score is <1.45, this equals F0–F1, and if FIB-4 is >3.25, this equals F3–F4.
All the patients were treated with 150 mg simisipivir (SIM) capsules and 400 mg sofosbuvir (SOF) tablets once a day for 12 weeks. If the glomerular filtration rate dropped below 30 mL/min in any patient, the SOF dose was reduced to 200 mg.
Clinical assessment
History and thorough clinical examination were evaluated for all the participants. The patients were weighed (kilograms) and their height was measured (centimeters), and the body mass index (BMI) was calculated as weight divided by the square of the height (kilogram per square meter). The waist circumference was measured 1 inch above the navel or midpoint between the lower margin of the least palpable rib and the top of the iliac crest parallel to the floor; the hip circumference was measured at its widest part of the buttocks or hip parallel to the floor; and then, the waist/hip ratio was calculated.
Laboratory assessment
The venous blood was drawn in the morning after 8 hours of overnight fast to determine the serum levels of ALT, AST, albumin, bilirubin, platelet count, international normalized ratio (INR), serum glucose, and insulin. After the patients completed fasting for 12 hours, blood was drawn again for the assessment of total cholesterol, HDL, cholesterol, triglycerides (TGs), and total and high-density lipoprotein cholesterol (HDL-c) by automated procedures. However, low-density lipoprotein cholesterol (LDL-c) was calculated according to the Friedewald equation, when serum TG level is <400 mg/dL.10 Serum insulin was determined by electro-chemiluminescence immunoassay (Elecsys 2010; Hoffman-La Roche Ltd., Basel, Switzerland). IR was investigated in all patients by the homeostasis model of IR (HOMA-IR) using the standard formula: HOMA-IR=Fasting insulin (uIU/mL)×fasting glucose (mmol/L)/22.5. In virological assessment, HCV-RNA levels were determined three times: baseline, at the end of treatment (week 12), and at 12 weeks posttreatment. Measurements were done using the COBAS TaqMan HCV assay V.2.0 (Hoffman-La Roche Ltd.; lower limit of detection, 15 IU/mL). HCV-RNA levels were quantified with a lower limit of detection of 15 IU/mL. The end of treatment response (EOTR) was defined as undetectable HCV-RNA at the completion HCV therapy. Sustained virological response (SVR12) is defined as an undetectable HCV-RNA at 12 weeks after completion of therapy.
Serum FGF21 assay
Blood samples were collected from the antecubital vein between 8:00 and 9:00 AM after overnight fasting. Samples were centrifuged at 2,500× g for 10 minutes, and serum aliquots were stored at −80°C until analysis. Serum FGF21 levels were determined using a commercially available ELISA kit (HumaReader Plus, model: 3700; Germany) according to the manufacturer’s protocol. The minimal detectable concentration was 7 pg/mL. All the measurements were performed in duplicate, in a random order, and the results were averaged.
Statistical analyses
Symmetrically distributed continuous variables were presented as mean±SD. Skewed continuous variables were presented as median and interquartile ranges. Categorical variables were presented as frequency and percentage. Comparisons between groups were done by using the Mann–Whitney U test or the Student’s t-test for continuous variables and the χ2 or Fisher exact probability test for the categorical data. The two-tailed, paired Student’s t-test was used to test the significance of difference between baseline and posttreatment FGF21. The Pearson correlation coefficients were used to study the correlation between different parametric variables. The Spearman rank correlation was used to quantify the association between continuous or ordered categorical variables. Logistic regression analysis was used to model the association among baseline FGF21, lipid profile, HOMA-IR, and other covariates to determine the factors associated with hepatic fibrosis. Linear regression analysis was used to identify the independent factors for FGF21. P≤0.05 was considered statistically significant. SPSS software for Windows, Version 20 (IBM Corporation, Armonk, NY, USA) was used to perform all the analyses.
Results
We studied 75 naïve Egyptian patients with CHC genotype 4, who were treated with SIM/SOF. The mean age of the patients was 47.5±12.3 years (range, 20–67 years), with a male to female ratio of 48/27, whereas the mean age of the healthy controls was 43.75±13.7 years (range, 20–66 years) with a male to female ratio of 28/12. No significant difference was found between patients and control groups as regards to age, sex, BMI, waist/hip ratio, and lipid profile. However, their comparison revealed a significant decrease in hemoglobin (Hb), platelets, and albumin levels (P<0.01, P<0.001, and P<0.05, respectively) vs significant increase in relation to INR, total bilirubin, ALT, and AST (P<0.01, P<0.001, and P<0.05, respectively). The patients were divided into two groups based on the Fibroscan examination. Group I included patients with mild fibrosis (n=38; F0, n=2; F1, n=13; and F2, n=23). Group II included patients with moderate to severe fibrosis (n=37; F3, n=16; F4, n=21). The baseline demographic, clinical, and biochemical characteristics of the patients and the healthy controls and the detailed virological and Fibroscan data of the patients were presented in Table 1. Table 2 shows that the mean levels of fasting glucose, fasting insulin, HOMA-IR, and serum FGF21 were significantly higher in patients in comparison to controls (5.04±0.75 vs 4.7±0.52, 20.15±5.13 vs 13.15±4.2, 4.49±1.28 vs 2.72±0.87, and 123.7±52.6 vs 21.8±8.8; P≤0.01, P≤0.001, P≤0.001, and P≤0.001, respectively).
Table 1.
Demographic and baseline characteristics of chronic hepatitis C patients vs controls
| Variables | Patients (n=75) | Controls (n=40) | P-values |
|---|---|---|---|
| Age (years), mean±SD (range) | 47±12 (20–67) | 43.75±13.7 (20–66) | 0.879 |
| Gender (male/female) | 48/27 (64%/36%) | 28/12 (70%/30%) | 0.778 |
| BMI (kg/m2), mean±SD (range) | 22.28±1.9 (16–25) | 21.8±1.79514 (18–26) | 0.457 |
| Waist/hip ratio, mean±SD (range) | 0.93±0.019 (0.9–0.97) | 0.9345±0.01 (0.89–0.98) | 0.277 |
| Hemoglobin (g/dL), mean±SD (range) | 13.6±1.3 (10–17) | 13.78±1.4 (10–17) | 0.01 |
| Platelets (×109), mean±SD (range) | 194.4±58 (81–430) | 222.98±38.2 (156–322) | 0.001 |
| Albumin (g/L), mean±SD (range) | 3.7±0.6 (2.1–5.4) | 4.3±0.25 (4–4.9) | 0.05 |
| INR, mean±SD (range) | 1.1±0.1 (0.9–1.4) | 1.05±0.061 (1.00–1.10) | 0.001 |
| Creatinine (mg/L) | 0.94±0.18 (0.64–1.6) | 0.894±0.15 (0.64–1.27) | 0.219 |
| Mean total bilirubin (mg/dL) | 0.85±0.48 (0.1–1.2) | 0.5±0.22 (0.1–1.1) | 0.02 |
| Mean ALT (IU/L), mean±SD (range) | 50.1±20.0 (21–103) | 21.78±8.16 (13–37) | 0.001 |
| Mean AST (IU/L), mean±SD (range) | 50.8±25.8 (17–163) | 20.12±5 (12–40) | 0.001 |
| Cholesterol (mg/dL), mean±SD (range) | 143.6±29 (70–195) | 137±30 (80–210) | 0.279 |
| Mean triglycerides (mg/dL), mean±SD (range) | 98±30.8 (35–225) | 103.15±27.15 (70–140) | 0.371 |
| LDL-c (mg/dL), mean±SD (range) | 84±38 (11–131) | 79.6±33.7 (25–161) | 0.49 |
| HDL-c (mg/dL), mean±SD (range) | 42.1±5.8 (31–58) | 40.8±5.3 (31–51) | 0.07 |
| AFP (ng/mL), mean±SD (range) | 3.6±3.8 (0.7–32.8) | – | – |
| Mean viral load(log10), mean±SD (range) | 5.2±1.3 (2.04–7.9) | – | – |
| Fibrosis stage (Fibroscan) | |||
| F0, n (%) | 2 (2.5) | – | – |
| F1, n (%) | 13 (16.3) | ||
| F2, n (%) | 23 (30.3) | ||
| F3, n (%) | 16 (20.1) | ||
| F4, n (%) | 21 (27.6) | ||
| F0, F1, F2, F3, F4, ranges, n (%) | 38–37 (43.5%–56.5%) | ||
| FIB-4, mean±SD | 1.9±1.1 | – | – |
Abbreviations: –, not evaluated; AFP, alpha fetoprotein; ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; FIB-4, Fibrosis-4 Index for Liver Fibrosis; HDL-c, high-density lipoprotein cholesterol; INR, international normalized ratio; LDL-c, low-density lipoprotein cholesterol.
Table 2.
Comparison of the levels of fasting glucose and insulin, HOMA-IR, and FGF21 in chronic hepatitis C patients vs controls
| Variables | Patients (n=75) | Controls (n=40) | P-values |
|---|---|---|---|
| Fasting glucose (mmol/dL), mean±SD | 5.04±0.75 | 4.7±0.52 | 0.01 |
| Fasting insulin level (uIU/mL), mean±SD | 20.15±5.13 | 13.15±4.2 | 0.001 |
| HOMA-IR, mean±SD | 4.49±1.28 | 2.72±0.87 | 0.001 |
| FGF21 (ng/mL), mean±SD | 123.7±52.6 | 21.8±8.8 | 0.001 |
Abbreviations: FGF21, fibroblast growth factor 21; HOMA-IR, homeostasis model of insulin resistance.
With increasing hepatic fibrosis, the mean levels of FGF21 continued to decrease with a mean level of 126.8±53.4 in group I vs 116.4±52 in group II, but it did not reach a significant difference (P≤0.407). Similarly, there was no significant difference between these two groups in relation to fasting glucose, fasting insulin, and HOMA-IR (P≤0.947, P≤0.229, and P≤0.2, respectively; Table 3).
Table 3.
Metabolic factors and FGF21 according to the fibrosis stage in chronic hepatitis C patients
| Variables | F0–F2 (n=38) | F3–F4 (n=37) | P-values |
|---|---|---|---|
| Fasting glucose (mmol/dL), mean±SD | 5.01±0.81 | 5.02±0.7 | 0.947 |
| Fasting insulin (uIU/mL), mean±SD | 19.34±5.6 | 20.81±5.69 | 0.229 |
| HOMA-IR, mean±SD | 4.26±1.3 | 4.6±1.24 | 0.2 |
| FGF21 (ng/mL), mean±SD | 126.8±53.4 | 116.4±52 | 0.407 |
Note: P-value ≤0.05 is significant.
Abbreviations: FGF21, fibroblast growth factor 21; HOMA IR, homeostasis model of insulin resistance.
The HOMA-IR was documented to be ≥3 in 60% of patients. So, we compared the patients with HOMA-IR≥3 with patients with HOMA-IR<3 in relation to the serum levels of FGF21 to investigate any association of IR with FGF21. Although the patients with HOMA-IR≥3 showed higher mean FGF21 levels (124±49.2 vs 116±57.8), the difference was non-significant (P≤0.5; Table 4).
Table 4.
Comparison of serum levels of FGF21 according to IR
| Variable | HOMA-IR≤3 n (30/75%–40%), mean ± SD | HOMA-IR>3 n (45/75%–60%), mean ± SD | P-value |
|---|---|---|---|
| FGF21 (ng/mL) | 116±57.8 | 124±49.2 | 0.5 |
Note: P-value ≤0.05 is significant.
Abbreviations: FGF21, fibroblast growth factor 21; HOMA IR, homeostasis model of insulin resistance; IR, insulin resistance.
The univariate analysis revealed a significant positive correlation for FGF21 with serum albumin (r=0.3, P≤0.002) and a significant negative correlation with fasting blood sugar (FBS; r=–0. 23, P≤0.05). However, all other variables showed non-significant correlations (Table 5 and Figures 1 and 2).
Table 5.
Association of serum FGF21 levels with some demographic, biochemical, and metabolic factors of chronic hepatitis C patients (n=75)
| Variables | FGF21 | |
|---|---|---|
| r | P | |
| Age (years) | −0.1 | 0.16 |
| Gender | 0.132 | 0.9 |
| Hypertension | −0.03 | 0.7 |
| Smoking | −0.09 | 0.4 |
| BMI (kg/m2) | −0.03 | 0.8 |
| Waist/hip ratio | −0.195 | 0.119 |
| ALT (IU/L) | 0.03 | 0.7 |
| AST (IU/L) | 0.007 | 0.9 |
| Albumin (g/dL) | 0.3 | 0.002 |
| Hb (g/dL) | 0.15 | 0.5 |
| WBCs | 0.019 | 0.879 |
| Platelets (×109/L) | 0.06 | 0.5 |
| Prothrombin concentration | −0.052 | 0.681 |
| Viral load (log10) | 0.07 | 0.4 |
| Cholesterol (mg/dL) | −0.01 | 0.9 |
| TG (mg/dL) | 0.005 | 0.9 |
| LDL-c (mg/dL) | −0.02 | 0.8 |
| HDL-c (mg/dL) | 0.16 | 0.1 |
| Fasting glucose (mg/dL) | −0.23 | 0.05 |
| Fasting insulin (uIU/mL) | −0.08 | 0.4 |
| HOMA-IR | −0.01 | 0.9 |
Note: P-value ≤0.05 is significant.
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; FGF21, fibroblast growth factor 21; Hb, hemoglobin; HDL-c, high-density lipoprotein cholesterol; HOMA-IR, homeostasis model of insulin resistance; LDL-c, low-density lipoprotein cholesterol; TG, triglyceride; WBC, white blood cell.
Figure 1.
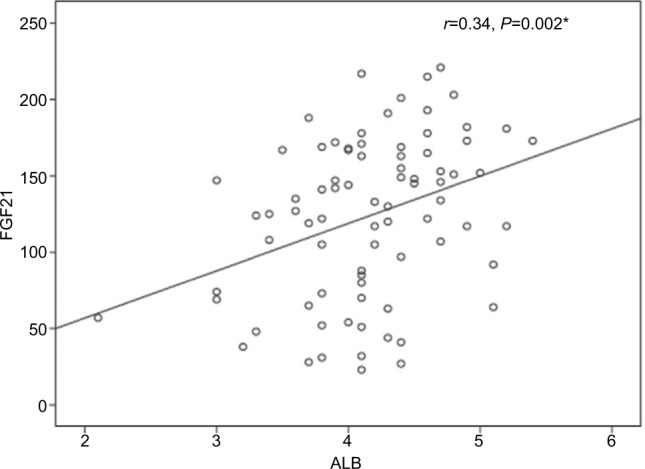
The correlation between serum FGF21 and serum albumin.
Abbreviations: ALB, albumin; FGF21, fibroblast growth factor 21.
Figure 2.
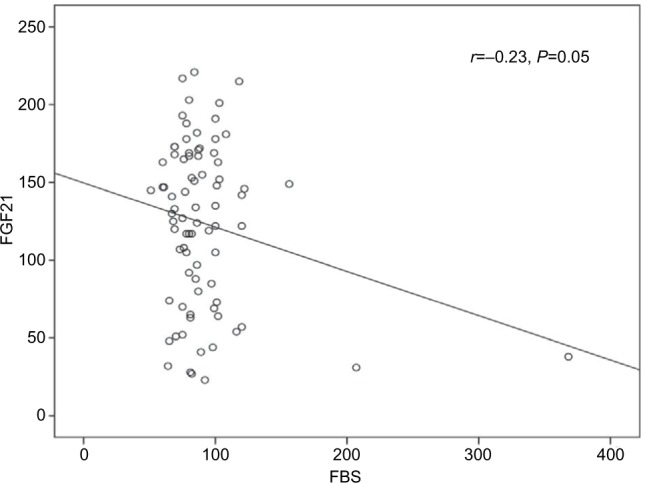
The correlation between serum FGF21 and serum FBS.
Abbreviations: FBS, fasting blood sugar; FGF21, fibroblast growth factor 21.
Using multiple linear regression analysis, the mean levels of serum albumin and HDL-c and the fibrosis stage appear to be independent predictors for FGF21 (B=0.27, P≤0.02, 95% CI, 3.26 to 45.9; B=0.25, P≤0.05, 95% CI, −8.7 to 0.052; and B=0.72, P≤0.023, 95% CI, −63.63 to 4.825, respectively; Table 6).
Table 6.
Best-fitting multiple linear regression analysis for factors associated with FGF21 in CHC patients (n=75)
| Variables | B | P-values | 95% CI for B |
|---|---|---|---|
| Age (years) | −0.081 | 0.491 | −1.3 to 0.561 |
| BMI (kg/m2) | 0.003 | 0.981 | −6.368 to 6.2 |
| Albumin (g/dL) | 0.273 | 0.025 | 3.26 to 45.9 |
| FBS | 0.05 | 0.8 | −0.786 to 0.648 |
| Insulin (uIU/mL) | 0.3 | 0.399 | −1.16 to 2.89 |
| HOMA-IR | −4.6 | 0.645 | 10.9 to 6 |
| Cholesterol (mg/dL) | 0.08 | 0.5 | −0.151 to 0.293 |
| TG (mg/dL) | −0.218 | 0.115 | −0.10 to 0.901 |
| LDL (mg/dL) | −0.250 | 0.08 | −0.87 to 0.052 |
| HDL (mg/dL) | 0.229 | 0.05 | 8.7 to 0.052 |
| Fibrosis (F1–F4) | −0.721 | 0.02 | −63.63 to 4.82 |
Abbreviations: BMI, body mass index; CHC, chronic hepatitis C; FBS, fasting blood sugar; FGF21, fibroblast growth factor 21; HDL, high-density lipoprotein; HOMA-IR, homeostasis model of insulin resistance; LDL, low-density lipoprotein; TG, triglyceride.
Patients who achieved an EOTR at 12 months (EOTR 12) showed significant reduction in FGF21, Hb, serum AST, and ALT levels (123.7±1.08 vs 60±32.7, 13.6±1.3 vs 12.7±1.5, 50.2±25.2 vs 40.2±17.3, and 49.8±21.4 vs 41.6±14.4; P≤0.001, P≤0.001, P≤0.001, P≤0.001, respectively). Also, a significant reduction in the fibrosis stage was documented by the FEB4 and Fibroscan (1.9±1.08 vs 1.7±1.1 and 13.6±10.4 vs 12.6±8.8; P≤0.03 and P≤0.001, respectively; Table 7). Moreover, the reduction in FGF21 serum levels was obvious in the different stages of hepatic fibrosis (Figure 3).
Table 7.
Changes in FGF21, some laboratory data, and imaging before and at the end of treatment with SOF/SIM (12 weeks) in CHC patients (n=75)
| Variables | Before treatment | End of treatment | P-values |
|---|---|---|---|
| Hb (g/dL) | 13.6±1.3 | 12.7±1.5 | 0.001* |
| Platelets (×109/L) | 196.09±59.1 | 194.6±37.06 | 0.7 |
| ALT (IU/L) | 49.8±21.4 | 41.6±14.4 | 0.001* |
| AST (IU/L) | 50.2±25.2 | 40.2±17.3 | 0.001* |
| FGF21 (ng/mL) | 123.7±52.5 | 60.5±32.7 | 0.001* |
| FIB-4 | 1.9±1.08 | 1.7±1.1 | 0.03* |
| Fibroscan | 13.6±10.4 | 12.6±8.8 | 0.001* |
Notes:
Significance. Data presented as mean ± SD.
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; FGF21, fibroblast growth factor 21; FIB-4, Fibrosis-4 Index for Liver Fibrosis; Hb, hemoglobin.
Figure 3.

The changes in FGF21 level before (Pre) and after (Post) the end of treatment with SOF/SIM at different stages of hepatic fibrosis according to the Fibroscan.
Abbreviations: FGF21, fibroblast growth factor 21; SIM, simisipivir; SOF, sofosbuvir.
Discussion
The results of this study documented higher mean serum levels of FGF21 in CHC patients in comparison to that in the healthy control subjects. Nevertheless, these levels were subjected to a significant reduction at EOTR. Moreover, FGF21 levels had a significant negative correlation with FBS and a significant positive correlation with serum albumin levels. However, FGF21 showed non-significant correlation with fasting insulin and HOMA-IR. The serum albumin, HDL-c, and the stage of liver fibrosis were determined to be independent risk factors for FGF21 by multiple linear regression analysis.
The 209 amino acid protein, FGF21, which functions as an endocrine regulator in humans, got special interest.11 It was shown to be induced in the stressed murine liver, and its abundant liver expression was, in turn, reflected in increased serum expression with a strong correlation between them. In patients with fatty degeneration, hepatitis, liver cirrhosis, and liver cancer, hepatocyte FGF21 is increased compared to cells residing in the healthy liver. So, its hepatic overexpression has been observed in a variety of liver diseases and was detected by immunohistochemistry in the diseased hepatocytes compared to the healthy hepatocytes.12
The FGF21 levels were reported to have a positive association with HCV infection and a non-significant correlation with HBV infection;13 however, the underlying mechanism remains unclear. Researchers tried to explain this through the ER stress activation, which is induced by the overexpression of HCV NS4B.13 While Misra and Reddy14 raised the possibility of perturbation of lipid metabolism, their explanation was questioned by others.15,16 Misra and Reddy14 proposed that elevated FGF21 mirrors lipid accumulation that lead to the upregulation of PPAR-α and fatty acid oxidation system resulting in excess energy expenditure in the liver.14 On the contrary, Wu et al15 and Liu et al16 found that both FGF21 and PPARγ-mRNA levels were downregulated with elevation of HCV-RNA. Thus, they considered that PPAR-α may not be a mediator of FGF21 expression in chronic HCV infecction.15,16 Another possible explanation is that overexpression of FGF21 depends on the possible anti-inflammatory function of FGF21 through suppression of the nuclear factor (NF)-κB activity as reported in animal models.17
The liver remains the most important producer of FGF21 and a major site for its actions.18 FGF21 was correlated with liver disease and has been considered as a novel regulator of oxidative stress in humans.19 In agreement with other authors,3,5 this study not only showed significantly higher levels of FGF21 in CHC patients vs control but also showed significant reduction at the end of treatment with DAAs.
However, serum FGF21 levels are not correlated with the degree of liver dysfunction detected by the Child–Pugh and Model for End-Stage Liver Disease (MELD) scores as was observed in the study by Ucar et al.13 However, in the present study, the univariate analysis revealed a significant positive correlation of FGF21 with serum albumin (r=0.3, P≤0.002), and the multiple linear regression analysis documented the mean serum albumin level as independent predictor for FGF21 (B=0.27, P≤0.02, 95% CI, 3.26–45.9). This finding could be explained in the view of the synthetic function of the liver. As the liver is the main source of both FGF21 and albumin, higher FGF21 and albumin levels are indicators of good synthetic function and vice versa. We did not use the Child–Pugh and MELD scores or correlate the serum FGF21 levels with the degree of liver dysfunction in the present study as our group of naive patients were chosen at an early stage of the disease with compensated liver, who came to receive antiviral therapy.
The relation of FGF21 and IR in CHC patients is not fully established. Although many authors16,17,20 reported increased FGF21 levels with IR states, Li et al21 denied any association between them in patients with NAFLD who have normal glucose tolerance or type 2 DM. In the present study, we did not find significant correlation between FGF21 and fasting insulin or IR. Moreover, this finding was re-emphasized after subgrouping the patients based on the HOMA-IR level (≥3 and <3). The mean FGF21 levels were 130±50.2 vs 131±55.9, respectively, with statistically non-significant difference (P=0.5). This was in agreement with the study by Kukla et al5 who reported no associations of systemic FGF21 with HOMA-IR in HCV-infected patients. The discrepancy between the different studies may be explained by the direct involvement of the HCV with IR and the non-excluded interference between the virus and FGF21. Consequently, the protective action of FGF21 against hyperglycemia remains controversial. In this study, despite failure of confirmation by linear regression, the univariate analysis revealed a significant negative correlation between FGF21 and FBS. This finding may reflect a possible protective role for FGF21 against hyperglycemia, which could be explained by certain mechanisms. First, through its binding to 6-Klotho/FGF complex, FGF21 stimulates glucose uptake in adipocytes independent of insulin.22 Second, by inducing the thermogenic capacity of white adipose tissue, FGF21 helps to increase glucose clearance.23 Third, by acting on glucagon metabolism, FGF21 suppresses glucose production.24 Fourth, by activation of signal-regulated kinase 1/2 and AKT signaling pathway (Akt pathway is a signal transduction pathway that promotes survival and growth in response to extracellular signals), FGF21 preserves B cell function.3
As the liver represents the major source of the circulating FGF21, after liver disease, especially cirrhosis, it appears logic that the affected hepatic biosynthetic capacity may influence the serum levels of FGF21.5 In this study, the mean serum level of FGF21 was higher in group I (F1–F2) than that in group II (F3–F4), but without reaching a significant level. However, the multiple regression analysis found out that hepatic fibrosis is an independent predictor of FGF21. This was in agreement with the study by Iwasa et al25 who reported that the levels of FGF21 mRNA expression in the mice liver tissue was 2.5-fold lower in F4 group compared to F0 group, while its expression in F2–F3 group did not significantly differ from F0.
The question about the pathogenic role of FGF21 in development of hepatic fibrosis did not receive a clear answer yet. Experimental studies postulated a role for FGF21 in the regulation of hepatic stellate cells (HSCs) activation, apoptosis, and development of liver fibrosis through the gain-of-function and loss-of-function hypotheses.26 Other studies found that the worsened steatosis, inflammation, and fibrosis in the FGF21-deficient mice could be reversed by continuous FGF21 subcutaneous infusion.25 Also, FGF21 was reported to have an excellent performance to distinguish simple steatosis from nonalcoholic steatohepatitis (NASH) and was considered as a key regulator of hepatic lipid metabolism with suggestion of its use as a biomarker for NASH.27 Despite its protective nature in animal models, FGF21 was reported in clinical studies to have a positive correlation with steatosis and the severity of fibrosis.9,27 Li et al21 demonstrated that FGF21 serum levels are positively associated with serum TG, total cholesterol, LDL-c, BMI, waist circumference, blood pressure, and ALT and negatively associated with HDL-c in NAFLD patients. In the present study, we found that high HDL-c is independently related to the FGF21 level. This finding comes with the possible protective and the metabolic regulative role of FGF21 in HCV patients as what observed in other liver disorders such as alcoholic liver disease, NAFLD, and hepatocellular carcinoma. Moreover, FGF21 has been proposed to be used as a biomarker for NAFLD, NASH, and other liver pathologies.
Due to the aforementioned data, induced FGF21 expression is likely a biomarker for early detection of liver injury, also it may be considered as a human stress-response hormone, synthesized and released in order to decrease cell damage. Consequently, it may be possible that DAAs targeting HCV replication by hitting NS4B are anticipated to suppress FGF21.
The limitations of this study included the small number of patients and the inability to address the relationship of FGF21 with hepatic steatosis as liver biopsy was prohibited by our national program for new antiviral therapy.
Conclusion
Although the exact mechanism of its expression is not yet clear, FGF21 correlation with HDL-c and FBS raise its role as metabolic regulator. It strongly introduced itself as a novel biomarker of hepatic injury in Egyptian genotype-4 CHC patients. Further studies are strongly required to address its possible therapeutic benefit against fibrosis, steatosis, and inflammation in HCV.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fon Tacer K, Bookout AL, Ding X, et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24(10):2050–2064. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J, Lloyd DJ, Hale C, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58(1):250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu X, Ge H, Lemon B, et al. Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in ob/ob mice. Proc Natl Acad Sci U S A. 2009;106(34):14379–14384. doi: 10.1073/pnas.0907812106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kukla M, Berdowska A, Stygar D. Serum FGF21 and RBP4 levels in patients with chronic hepatitis C. Scand J Gastroenterol. 2012;47(8–9):1037–1047. doi: 10.3109/00365521.2012.694901. [DOI] [PubMed] [Google Scholar]
- 6.Schaap FG, Kremer AE, Lamers WH, Jansen PL, Gaemers IC. Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie. 2013;95(4):692–699. doi: 10.1016/j.biochi.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Ye L, Yu X, et al. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology. 2009;391(2):257–264. doi: 10.1016/j.virol.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 8.Liu WY, Huang S, Shi KQ, et al. The role of fibroblast growth factor 21 in the pathogenesis of liver disease: a novel predictor and therapeutic target. Expert Opin Ther Targets. 2014;18(11):1305–1313. doi: 10.1517/14728222.2014.944898. [DOI] [PubMed] [Google Scholar]
- 9.Alisi A, Ceccarelli S, Panera N, et al. Association between Serum Atypical Fibroblast Growth Factors 21 and 19 and Pediatric Nonalcoholic Fatty Liver Disease. PLoS One. 2013;8(6):e67160. doi: 10.1371/journal.pone.0067160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. [PubMed] [Google Scholar]
- 11.Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang C, Lu W, Lin T, et al. Activation of Liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol. 2013;13:67. doi: 10.1186/1471-230X-13-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ucar F, Sezer S, Ginis Z, et al. APRI, the FIB-4 score, and Forn’s index have noninvasive diagnostic value for liver fibrosis in patients with chronic hepatitis B. Eur J Gastroenterol Hepatol. 2013;25(9):1076–1081. doi: 10.1097/MEG.0b013e32835fd699. [DOI] [PubMed] [Google Scholar]
- 14.Misra P, Reddy JK. Peroxisome proliferator-activated receptor-α activation and excess energy burning in hepatocarcinogenesis. Biochimie. 2014;98:63–74. doi: 10.1016/j.biochi.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Wu AL, Coulter S, Liddle C, et al. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways. PLoS One. 2011;6(3):e17868. doi: 10.1371/journal.pone.0017868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Zhao C, Xiao J, et al. Fibroblast growth factor 21 deficiency exacerbates chronic alcohol-induced hepatic steatosis and injury. Sci Rep. 2016;6:31026. doi: 10.1038/srep31026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feingold KR, Grunfeld C, Heuer JG, et al. FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis. Endocrinology. 2012;153(6):2689–2700. doi: 10.1210/en.2011-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong LJ, Feng W, Wright M, et al. FGF21 suppresses hepatic glucose production through the activation of atypical protein kinase Cι/λ. Eur J Pharmacol. 2013;702(1–3):302–308. doi: 10.1016/j.ejphar.2012.11.065. [DOI] [PubMed] [Google Scholar]
- 19.Cuevas-Ramos D, Aguilar-Salinas CA. Modulation of energy balance by fibroblast growth factor 21. . Horm Mol Biol Clin Investig. 2016;30(1) doi: 10.1515/hmbci-2016-0023. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Yeung DC, Karpisek M, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57(5):1246–1253. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Fang Q, Gao F, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol. 2010;53(5):934–940. doi: 10.1016/j.jhep.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Yang G, Ning H, Yang M, Liu H, Chen W. Plasma FGF-21 levels in type 2 diabetic patients with ketosis. Diabetes Res Clin Pract. 2008;82(2):209–213. doi: 10.1016/j.diabres.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Fisher FM, Kleiner S, Douris N, et al. FGF21 regulates PGC-1a and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26(3):271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berglund ED, Li CY, Bina HA, et al. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology. 2009;150(9):4084–4093. doi: 10.1210/en.2009-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwasa M, Mifuji-Moroka R, Kobayashi Y, Takei Y, D’Alessandro-Gabazza C, Gabazza EC. Comment on serum FGF21 and RBP4 levels in patients with chronic hepatitis C. Scand J Gastroenterol. 2013;48(2):252–253. doi: 10.3109/00365521.2012.719929. [DOI] [PubMed] [Google Scholar]
- 26.Xu P, Zhang Y, Liu Y, et al. Fibroblast growth factor 21 attenuates hepatic fibrogenesis through TGF-β/smad2/3 and NF-κB signaling pathways. Toxicol Appl Pharmacol. 2016;290:43–53. doi: 10.1016/j.taap.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 27.He L, Deng L, Zhang Q, et al. Diagnostic Value of CK-18, FGF-21, and Related Biomarker Panel in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed Res Int. 2017;2017:9729107. doi: 10.1155/2017/9729107. [DOI] [PMC free article] [PubMed] [Google Scholar]