

Abstract
Respiratory syncytial virus (RSV) represents a significant health threat to infants and to elderly or immunocompromised individuals. There are currently no vaccines available to prevent RSV infections, and disease management is largely limited to supportive care, making the identification and development of effective antiviral therapeutics against RSV a priority. To identify effective chemical scaffolds for managing RSV disease, we conducted a high-throughput anti-RSV screen of a 57,000-compound library. We identified a hit compound that specifically blocked activity of the RSV RNA-dependent RNA polymerase (RdRp) complex, initially with moderate low-micromolar potency. Mechanistic characterization in an in vitro RSV RdRp assay indicated that representatives of this compound class block elongation of RSV RNA products after initial extension by up to three nucleotides. Synthetic hit-to-lead exploration yielded an informative 3D quantitative structure–activity relationship (3D-QSAR) model and resulted in analogs with more than 20-fold improved potency and selectivity indices (SIs) of >1,000. However, first-generation leads exhibited limited water solubility and poor metabolic stability. A second optimization strategy informed by the 3D-QSAR model combined with in silico pharmacokinetics (PK) predictions yielded an advanced lead, AVG-233, that demonstrated nanomolar activity against both laboratory-adapted RSV strains and clinical RSV isolates. This anti-RSV activity extended to infection of established cell lines and primary human airway cells. PK profiling in mice revealed 34% oral bioavailability of AVG-233 and sustained high drug levels in the circulation after a single oral dose of 20 mg/kg. This promising first-in-class lead warrants further development as an anti-RSV drug.
Keywords: antiviral agent, viral polymerase, drug development, medicinal chemistry, animal virus, RdRp, respiratory disease, respiratory syncytial virus, RNA-dependent RNA polymerase
Introduction
RSV3 is a member of the Pneumoviridae family of negative sense single-stranded RNA viruses and the leading cause of hospitalizations in children under the age of 5 years due to respiratory illness in the United States (1). In addition to causing substantial morbidity, RSV infection can be fatal in infants, the elderly, and the immunocompromised (2–4). Whereas the majority of individuals have been exposed to RSV infection by 2 years of age, reinfection throughout life is common, and the elderly in particular are at high risk of developing RSV pneumonia with a potentially fatal outcome.
RSV is highly contagious and spreads via the respiratory route. Virus replication is initially confined to the nasopharynx and the upper respiratory tract. However, progression to complicated disease coincides with viral spread to the lower respiratory tract, resulting in major respiratory distress and viral pneumonia (5, 6). Although severe clinical signs result from immunopathogenesis (7), the correlation between virus load and cause of disease is only partially understood. Clinical studies monitoring hospitalized RSV-infected children below 2 years of age revealed, for instance, that virus load in the upper respiratory tract during the first 3 days of hospitalization served as a predictor for the likelihood of patient progression to complicated disease with lower respiratory tract infection (8, 9). With the overarching goal to prevent the development of severe disease and life-threatening viral pneumonia, it is the primary objective of our anti-RSV program to exploit this therapeutic window and reduce viral burden in the upper respiratory tract in the first days after infection.
Currently, two anti-RSV drugs are approved for human use, the neutralizing mAb palivizumab and the broad-spectrum antiviral ribavirin. Treatment cost limits palivizumab therapy to high-risk patients only, however, and use of ribavirin against RSV infection has been largely discontinued due to limited efficacy and the risk of severe side effects (10, 11). Novel therapeutic options against RSV disease are therefore urgently required to address this unmet clinical need. Two small-molecule anti-RSV therapeutics have recently completed phase II clinical trials, the allosteric entry inhibitor presatovir (GS-5806) (12) and the ribonucleoside analog lumicitabine (ALS-8176) (13). Although initial trial outcomes for both drug candidates were encouraging, underscoring that RSV infection is responsive to therapeutic intervention, we have characterized an RSV pan-resistance mechanism that mediates escape from all RSV entry inhibitors currently considered for human therapy, including GS-5806 (14, 15). By contrast, the ribonucleoside analog ALS-8176 targeting the RSV polymerase complex appears less prone to induce rapid viral escape (13). Although it remains unclear whether toxic side effects will prohibit approval of ALS-8176 for pediatric use, we consider the viral RdRp complex to present a premier therapeutic target due to its critical importance for virus replication and absence of a cellular homolog (14).
The RSV RdRp complex is composed of the N, P, M2-1, and L proteins. Of these, N, P, and L are core components critical for RNA synthesis (16, 17), whereas the M2-1 protein is required only for efficient transcription of viral mRNAs (18). The N protein encapsidates viral genomic and antigenomic RNA into a ribonucleoprotein complex. Although the L protein harbors all of the catalytic activities necessary for viral RNA synthesis (i.e. polymerization, capping, methylation), its bioactivity depends on interaction with its essential cofactor, the P protein that functions as a bridge between the N encapsidated viral genome and L.
In search of first-in-class points of entry for our anti-RSV and anti-influenza virus drug development programs, we have previously developed a dual-pathogen high-throughput screening protocol that employs two replication-competent recombinant virus strains, RSV A2-L19F-fireSMASh and influenza A virus (IAV) WSN-nanoLuc, which express distinct luciferase reporter proteins and can therefore be differentially monitored in a co-infection setting (19). In addition to the luciferase reporter, this recombinant RSV harbors a D489E pan-resistance mutation in its fusion protein (15). Fully miniaturized to 384-well plate format, this protocol affords the simultaneous identification of RSV-specific, IAV-specific, and broad-spectrum antiviral hit candidates. Due to the use of a recRSV possessing a pan-resistance mutation, we anticipate that any RSV entry inhibitor candidate identified will not be compromised by the pan-resistance phenotype.
In the present study, we applied this protocol to the automated screen of a library of ∼57,000 compounds that was curated based on druglike chemical space covered. This primary high-throughput screening (HTS) assay combined with direct and orthogonal counterscreens identified a first-in-class chemical scaffold that selectively blocked activity of the RSV RdRp complex. After hit confirmation, we explored antiviral activity on disease-relevant primary human airway epithelial cells and determined the mechanism of antiviral activity through a combination of biochemical and cell-based assays before launching a synthetic hit-to-lead development strategy. Having generated a robust 3D-QSAR model for this chemical scaffold, we interfaced the 3D-QSAR constraints with PK predictive algorithms in an in silico optimization platform to further advance drug-like properties of the class. Oral PK profiles of an emerging advanced lead candidate were determined in the mouse model of RSV infection.
Results
Identification of a novel RSV inhibitor candidate
In search of novel RSV inhibitor candidates, we implemented an HTS approach using a dual-virus protocol that we have developed and validated after miniaturization to 384-well format in earlier work (19, 20). This protocol employs replication-competent recombinant influenza virus A/WSN/1933 and RSV A2-L19F reporter viruses that express nanoluciferase and a firefly luciferase-based fireSMASh construct (19), respectively, and support the simultaneous identification of IAV WSN and RSV-specific hit candidates.
This particular study focuses on inhibitors of RSV replication specifically. A total of 56,557 compounds curated from the ChemDiv and ChemBridge collections (20) were assessed at a final screening concentration of 5 μm (Fig. 1A). As detailed in our previous work, we combined control-independent robust z-score and control-dependent percentage inhibition approaches for hit identification, requiring candidates to exceed both 3 times z-score S.D. and 6.5 times percentage inhibition S.D. (Fig. 1A). Of 193 compounds meeting these criteria, 13 passed direct counterscreens and testing against acute cytotoxicity and reporter interference. When sourced and subjected to dose–response assays, only one of these compounds, GRP-156784, returned 50% active concentrations (EC50) <5 μm and long-exposure (72 h) 50% cytotoxic or cytostatic concentrations (CC50) >20 μm (Fig. 1B). In silico inspection of the GRP-156784 scaffold (Fig. 1C) using the SwissADME (21) first-generation and Badapple (22) second-generation filter algorithms of undesirable promiscuous compounds revealed no known structural liabilities. This compound was resynthesized for synthetic confirmation of scaffold integrity and subjected to a final round of direct and orthogonal counterscreens (Fig. 2A). We observed identical (fully overlapping 95% confidence intervals) EC50 values in the 1 μm range against RSV in both reporter-based and virus yield–based dose–response inhibition assays, corresponding to a starting specificity index (SI = CC50/EC50) >100. Antiviral activity was RSV-specific, because in addition to IAV-WSN, two more closely related members of the Mononegavirales order, vesicular stomatitis virus and measles virus (MeV), remained unaffected in the GRP-156784 concentration range examined.
Figure 1.

HTS Screening and hit identification. A, HTS campaign of 56,557 compounds conducted against recRSV-A2-L19FD489E-fireSMASh. For each compound, robust z-scores and percentage inhibition values were calculated, and hit cutoff for each analysis method is shown (dashed lines). The inset summarizes counterscreening results obtained for 139 primary screen hit candidates that met both inclusion criteria. B, automated dose–response potency and cytotoxicity testing in 384-well format for 13 hit candidates advanced from A. Compounds with CC50 <20 μm, SI <1, or EC50 ≥5 μm were discontinued. Primary HTS and automated counterscreens in A and B were carried out in single biological repeats. C, structure of GRP-156784, the primary hit candidate passing all performance milestones. Numbers and dashed red circles denote scaffold sections modified individually for synthetic hit-to-lead development.
Figure 2.

Hit confirmation. A, inhibitory activity of resynthesized GRP-156784 against different luciferase reporter viruses (red y axis), standard RSV A2-L19F (blue y axis), and measurements of cellular metabolic activity of uninfected cells exposed to the compound (black y axis). Where applicable, active concentrations were calculated through four-parameter variable slope regression modeling; values in parentheses denote 95% confidence intervals). B, anti-RSV activity and cytotoxicity of resynthesized GRP-156784 in primary HBTECs. Symbols in A and B represent biological repeats (n = 3 each or greater); error bars, S.D.
Because prevention of RSV spread from the upper respiratory tract to the small airways represents our primary therapeutic objective, we require of any hit candidate considered for further development consistent antiviral activity on disease-relevant human airway cells. When tested on primary human bronchial/tracheal epithelial cells (HBTECs), GRP-156784 demonstrated unchanged antiviral activity and SI values compared with that observed on stable cell lines (Fig. 2B), identifying the compounds as a bona fide primary screening hit.
Mechanistic profiling of GRP-156784
To gain first insight into the stage of the RSV life cycle targeted by the compound, we profiled GRP-156784 in a time-of-addition variation study (Fig. 3A). The compound was administered at a range of different time points pre- and postinfection of cells with RSV A2-L19F at a sterilizing concentration (20 μm). As reference substances, the previously characterized RSV entry inhibitor BMS-433771 (23) and polymerase inhibitor JMN3-003 (24) were added at the same time points. Inhibition results obtained for GRP-156784 and JMN3–003 closely resembled each other, whereas the entry blocker BMS-433771 showed a markedly distinct time-of-addition profile. To further test interference of the compound with the viral RdRp complex, we employed a plasmid-based minigenome system that consists of transiently expressed N, P, L, and M2-1 proteins acting in conjunction with a luciferase minigenomic reporter (15). This assay corroborated specific inhibition of RSV RdRp activity by GRP-156784, because the compound dose-dependently blocked the RSV replicon, whereas comparable MeV– and IAV WSN–derived minigenome systems (25, 26) remained uninhibited (Fig. 3B). Reverse-transcription qPCR-based quantitation of the relative copy numbers of RSV N protein–encoding mRNAs produced during infection in the presence of 10 μm GRP-156784 or vehicle (DMSO) volume equivalents revealed an ∼60% reduction (Fig. 3C), which was comparable with that observed with the RSV-specific ribonucleoside analog polymerase inhibitor ALIOS-8176 (13) that we included as reference. Taken together, these results characterize GRP-156784 as a novel allosteric inhibitor of RSV RdRp activity that meets our inclusion criteria for synthetic hit-to-lead optimization of druglike properties.
Figure 3.
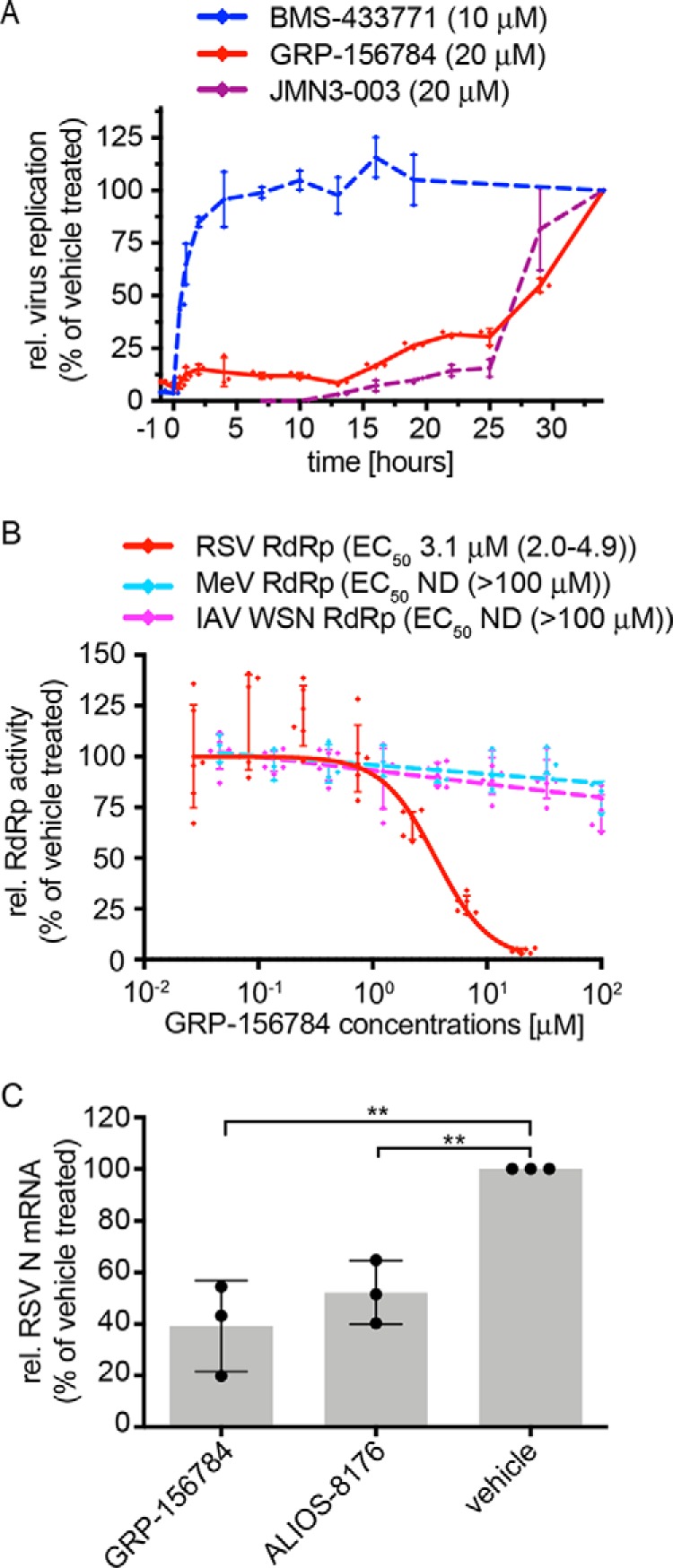
Mechanistic profiling of GRP-156784. A, time-of-addition variation studies against the RSV target. Compound GRP-156784 (red), RSV entry inhibitor BMS-433771 (blue), or viral polymerase inhibitor JMN3–003 (maroon) were added to equally infected cells at the indicated time points and concentrations. Relative virus replication in all cultures was determined 33 h after infection. B, dose–response minigenome reporter assay inhibition by GRP-156784. The compound was tested against RSV–, IAV-WSN–, and MeV–derived minigenomes. C, RT-qPCR quantitation of relative RSV N protein–encoding mRNA levels present in RSV-infected cells after incubation in the presence of GRP-156784, RSV ribonucleoside analog inhibitor ALIOS-8176, or vehicle volume equivalents. Statistical analysis through one-way analysis of variance with Dunnett's multiple-comparison post hoc test. Symbols in A–C represent individual biological repeats (n = 3 each); error bars, S.D. Previous characterized reference compounds BMS-433771 and JMN3–003 were analyzed in A in two biological repeats; error bars, range. In C, each of the three biological repeats was determined in technical duplicates.
Structure–activity relationship (SAR) development for hit optimization
To gain an initial understanding of how GRP-156784 responds to synthetic modification, we launched a ligand-driven SAR development strategy. All chemical analogs were tested for cytotoxicity and antiviral potency against RSV A2-L19F using an automated assay pipeline that we recently developed for standardized SAR profiling (19, 25, 27, 28). The chemical scaffold was hypothetically divided into three distinct sections (Fig. 1C) and accessed via an efficient five-step synthesis that allowed independent introduction of each of the three key substituents. Each of the three sections was therefore available for independent mapping and optimization via a parallel approach, and the insights gained were then applied in tandem to profile additivity effects among ligands. Modification to the core scaffold was deprioritized and considered an option only if optimization of the peripheral substituents, using existing chemistry, failed to deliver a suitable lead candidate.
Section 1 proved to be tolerant to a number of diverse modifications without abolishing antiviral activity (Table S1). Because a probing replacement of the benzothiazol-2-yl moiety with a dimethyl-thiazol-2-yl substitution (AVG-033) reduced potency, we changed course and increased bulk in this area, testing the effects of substituting the thiazol ring with benzyl, pyridinyl, pyrimidinyl, pyrazinyl, and pyridazinyl groups. Especially in the case of pyridinyl replacements, the position of the nitrogen atom in the ring governed bioactivity, ranging from potency similar to that of GRP-156784 (AVG-035) to ∼16-fold reduced antiviral activity (AVG-081). We noted comparable effects of nitrogen placement on bioactivity when pyrimidinyl, pyrazinyl, and pyridazinyl groups were inserted. Because a bulky isoquinolinyl substitution 1 (AVG-034) likewise maintained inhibitory activity similar to that of GRP-156784, the nascent SAR identified section 1 as a segment amenable to large substitutions.
Following a strategy similar to that applied to section 1, we first removed the original substituent from section 2 (AVG-022) or replaced it with small aliphatic groups (AVG-023, -070, and -071). This exercise did not improve antiviral potency but in several cases triggered an increase in cytotoxicity (Table S2). Some heterocyclyl-methyl groups (i.e. AVG-025, -119, -122, and -124) were better tolerated although also providing no potency advantage. However, changes as in AVG-119 and -124 diminished cytotoxicity, resulting in substantial SIs. Whereas these modifications of section 2 likewise failed to deliver breakthroughs in potency, they highlighted the ability of the scaffold to tolerate specific substitutions in this section with intact antiviral activity, providing downstream options for adjusting physiochemical properties if required.
Elaboration of section 3 aimed to replace the furan system originally present in GRP-156784, because furans frequently present a metabolic liability due to high susceptibility to oxidation (29). Initial substitutions (Table S3) introduced a diverse array of substituted phenyl moieties and both aromatic and nonaromatic heterocycles in an effort to identify one or more groups that conserve potency and provide a suitable SI while alleviating potential pharmacokinetic liabilities. Most of these changes resulted in analogs with lower potency and/or increased cytotoxicity. However, substituted benzyl (i.e. AVG-031) and pyridinyl (i.e. AVG-036) moieties were well-tolerated without loss of antiviral activity. Notably, pyrid-2-yl heterocycles with varying substituents in ortho position to the heteroatom caused a first breakthrough in potency, consistently showing submicromolar antiviral activity (i.e. AVG-094, -157, -158, -168, and -214).
Encouraged by these observations, we explored the effect of combining the leading pyridinyl substitutions of section 3 with modifications at sections 1 or 2 (Table S4). As a general trend, ortho-substituted pyridyl substituents in section 3 tended to provide a significant advantage in antiviral potency over similarly substituted benzyl moieties (i.e. AVG-171 versus -128 and AVG-203 versus -134). The ortho-chloropyridyl substituent was then selected as a probing group to combine with complementary changes in the other two sections. Building on insights gained from the single-change SAR studies, we prioritized substituted pyridyl groups in section 1 and substituted phenyl groups in section 2. This systematic overview provided compounds mostly in the submicromolar range.
First-generation lead analogs block initiation of RSV RdRp complexes at the promoter
Two compounds, AVG-094 and AVG-158, that had emerged early in SAR development as potency-improved lead candidates were selected for initial mechanistic characterization. They showed ∼11- and 20-fold higher anti-RSV activity (Fig. 4, A and B), respectively. Optimized potency did not coincide with increased cytotoxicity (Fig. 4A), resulting in an SI value for AVG-158 of >2,000.
Figure 4.

First-generation optimized leads and 3D-QSAR model for the hit scaffold. A, bioactivity of analogs AVG-094 and AVG-158 compared with resynthesized GRP-156784. No detectable cytotoxicity was measured for either AVG094 (blue dashed line) or AVG-158 (red dashed line). B, reduction of progeny RSV A2-L19F virus load by AVG-094 and AVG-158 determined in dose–response activity assays. Active concentrations were determined through four-parameter variable-slope regression modeling. LoD, level of detection. Symbols in A and B represent individual biological repeats (n = 3 each); error bars, S.D. C, in vitro RSV RdRp activity assay using synthetic template RNA and purified polymerase complexes conducted in the presence of AVG-094 or AVG-158. The compounds show dose-dependent block of 3′ RNA extension elongation after back-priming (back-priming extens. elong.) and de novo RNA synthesis from the promoter, but do not prevent extension by up to three nucleotides after back-priming (back-priming 3′ extens.). LD811A is bio-inactive due to the alanine substitution in the L catalytic site and was included as assay specificity control. D, generation and testing of a 3D-QSAR model developed based on a subset of GRP-156784 analogs synthesized for chemical optimization of the scaffold. AutoGPA embedded in the MOE package was used for model building. Predicted (x axis) and experimentally measured (y axis) pIC50 value correlations are shown. Values denote predictive capacity, goodness of fit, and, for the combined training and test sets, slope through the origin. E, graphical representation of the 3D-QSAR model from D. Gray shading, allowable space component of the model. Areas of steric constraints, steric freedom, electro-activity, and hydrophobic interactions are shown. Electrostatically active regions of the compound are shown in red and blue.
For positive confirmation of the mechanism of antiviral activity of these lead candidates, we assessed RSV RdRp activity in vitro, using a 25-mer synthetic ribonucleoside template encoding the RSV antigenomic promoter region and purified viral P-L polymerase complexes produced in insect cells (30). This assay monitors de novo RdRp initiation from the viral promoter, followed by 3′ extension and subsequent elongation to products of up to 25 nucleotides in length (31). Alternatively, the nonencapsidated template RNA can adopt a panhandle structure with a base-paired 3′-end that is extended by the RSV polymerase by up to three nucleotides, resulting in 26–28-mer products (3′ extension (30)). Occasionally, the RdRp continues toward the end of the template, producing larger products (3′ extension elongation (31)). When subjected to this assay, both AVG-094 and AVG-158 showed dose-dependent inhibition of 3′ extension elongation after de novo RNA synthesis and back-priming, but the 26–28-mer back-priming extension products accumulated in the presence of compound or vehicle with equal efficiency (Fig. 4C). These results demonstrate that the first-generation lead analogs do not interfere with phosphodiester bond formation by the RSV RdRp complex per se, but potently prevent RNA extension elongation.
3D-QSAR modeling
To appreciate major target contacts predicted to be made by AVG-158, we summarized the analog bioactivity data in a QSAR model. Of all analogs characterized, we identified a subset of 52 informative compounds that had returned EC50 values <5 μm in combination with SI > 100 or CC50 > 100 μm. From this sublibrary, we selected a training set of 13 analogs (Tables S1–S4) that, combined, contained modifications in each of the three scaffold sections and spanned an EC50 range from 0.04 to 2.2 μm. In preparation for model building, we generated an in silico compendium of 30–50 distinct conformations for each of these training set analogs that were subsequently aligned into 106 initial pharmacophore models using the AutoGPA module embedded in the MOE software package (32, 33). These preliminary models were validated and ranked based on goodness of fit (R2) and predictive capacity (q2) against the training set (Fig. 4D), leading to the identification of a top-scoring 3D-QSAR model that combines superior fit (R2 = 0.9989) with strong predictive capacity (q2 = 0.8864). This model was validated against a test set consisting of the remaining 30 short-listed analogs (Tables S1–S4), returning a combined R2 of 0.723 and regression slope of 0.9855 for these 43 informative analogs (Fig. 4D). A graphical summary of the model reveals a predicted allowable space contour map that identifies three discrete subsections as amenable to steric expansion, two areas in sections 1 and 2 as sterically constraint, and an intact hydrophobic center at the core of the scaffold (Fig. 4E).
Model-informed optimization of druglike properties
To evaluate the druglike potential of first-generation leads, we determined the sensitivity of AVG-094, AVG-093, AVG-158, and three leading candidates of the multichange series (AVG-171, -203, and -241) to phase I oxidation through co-incubation with human and mouse hepatocyte-derived microsomal fractions, followed by quantitation of the relative amount of the remaining material (Table 1). Mouse microsomes were included for species consistency with our established mouse efficacy model of RSV infection (15, 34). In parallel, aqueous solubility of the six compounds was determined. AVG-158 performed poorly in this assay, showing complete turnover by mouse microsomes after 30-min co-incubation and >80% breakdown in the presence of human hepatocyte fractions. AVG-094 suggested higher microsomal stability, but these results may have been compromised by poor water solubility, triggering exclusion of this analog from consideration for further development. Because the other four candidates likewise failed to reach stability and/or water solubility objectives, we launched a second development program with the goal to identify a viable lead with improved stability against mouse and human microsomes and acceptable pharmacokinetic profile in the efficacy model species.
TABLE 1.
Metabolic stability testing of first- and second-generation lead candidates

1 Predicted solubility is shown as the average of three solubility predictions taken from SwissADME: ESOL (58), Ali (57), and SILICOS-IT.
2 Metabolic susceptibility scores were determined by calculating the sum of the predicted metabolic susceptibility probabilities for all atoms in the molecule, giving an overall score of metabolic susceptibility. Lower scores are predicted to be less suceptibile to CYP metabolism.
For this exercise, we explored the combined predictive power of the 3D-QSAR model with substructure-based estimates of solubility (21) and metabolic liabilities (35). Whereas AVG-158 met all requests of the 3D-QSAR model (Fig. 4E), three discrete subsections of the molecule were identified as major predicted stability concerns (Fig. 5A). To address these liabilities within the allowed chemical space of the 3D-QSAR model, we generated a virtual library of 197 compounds by systematically modifying the substituents at each of the suspected metabolic hot spots. Having subjected this library and the six tested candidates to in silico screening of both predicted metabolic stability and antiviral activity, we selected a subset of eight compounds (Table 1) for chemical synthesis that showed equivalent or better predicted metabolic stability than the best of the experimentally tested candidates and a predicted antiviral potency not worse than the least active experimentally tested candidate (Fig. 5B). Experimental assessment after synthesis revealed good correlations between predicted and measured antiviral potency (R2 = 0.7024; Fig. 5C) and microsomal stability (R2 = 0.74; Fig. 5D), respectively, for all eight compounds.
Figure 5.
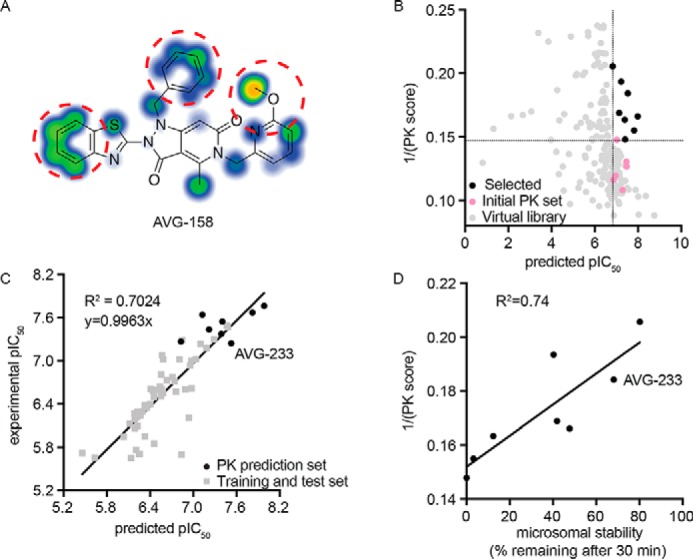
3D-QSAR-informed optimization of select PK properties of the scaffold. A, metabolic stability predictions for the AVG-158 analog using SMARTCyp. Areas of high postulated metabolic susceptibility (red circles) against eight isoforms of cytochrome P450 are shown. Potential metabolic liabilities are color-coded from moderate (blue) to severe (orange). B, virtual library screen to optimize metabolic properties. Shown are predictions for six experimentally tested compounds (pink), virtual library entries (gray), and virtual library entries selected for synthesis (black). Dotted lines, cutoff criteria for chemical synthesis (predicted pIC50 and metabolic stability at least equal to that of the least potent (predicted pIC50 > 6.8) and most stable (1/PK score >0.147) experimentally analyzed analog, respectively). C, correlation of bioactivities predicted by the 3D-QSAR model (x axis) and experimentally measured (y axis) for the synthesized eight virtual screening hits selected in B. D, correlation between experimentally measured human microsome stability (x axis) and predicted metabolic susceptibility (y axis) for these eight virtual screening hits. The second-generation lead candidate AVG-233 is highlighted in C and D.
Identification of an orally bioavailable second-generation lead
Based on superior experimentally determined aqueous solubility (>50 μm) and greatly improved predicted (Fig. 6A) and measured metabolic stability (Table 1), we advanced AVG-233 to single-dose PK profiling in mice after intravenous or oral administration, respectively (Fig. 6B). In this assay, AVG-233 demonstrated oral bioavailability of ∼34% and an acceptable half-life in mouse circulation exceeding 5 h after oral gavage (Table 2). Maximal plasma concentrations after a single dose at 20 mg/kg orally were ∼2 μm.
Figure 6.
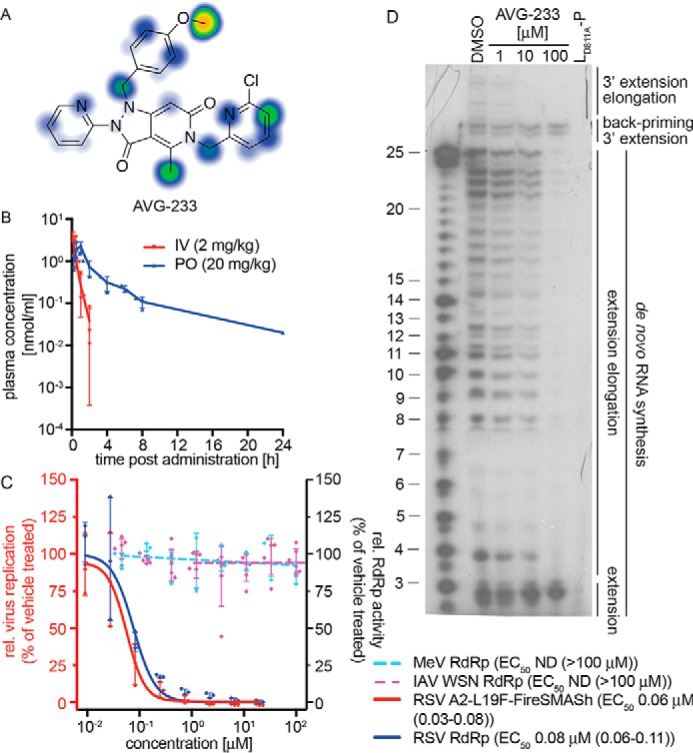
PK profiling and mechanistic assessment of the AVG-233 lead. A, metabolic stability predictions for AVG-233 using SMARTCyp. B, single-dose PK testing of AVG-233 in mice. Animals (three per group) were dosed with AVG-233 intravenously (IV) or orally (PO) at the indicated dose levels. Plasma samples were prepared at the indicated time points and analyzed by LC/MS/MS. C, bioactivity testing of AVG-233 against RSV (red y axis) and RSV–, MeV–, or IAV-WSN–derived minigenome assays (blue y axis). Where applicable, four-parameter variable slope regression models and active concentrations are shown. D, in vitro RSV RdRp activity assay as described in Fig. 4C. The lead compound AVG-233 blocks 3′ RNA extension elongation but does not interfere with 3′ RNA extension by up to three nucleotides after de novo initiation from the promoter or back-priming. IAV-WSN RdRp values in C show means of six biological repeats. Symbols in B–D represent individual biological repeats (n = 3 each); error bars, S.D. IAV-WSN RdRp activity in C was determined in six biological repeats.
Table 2.
Calculation of PK parameters for AVG-233 after a single oral dose in mice. Parameters and oral bioavailability of AVG-233 were calculated using the WinNonlin software package (Pharsight). Tmax, time to maximum concentration of drug in serum; t½, half-life; Cmax, maximum plasma concentration; AUC (0–∞), area under the plasma concentration–time curve from time zero to infinity; CL/F, apparent total clearance of the drug after oral administration
| Route | Dose | Tmax | Cmax | AUC (0–∞) | CL/F | t½ | Bioavailability |
|---|---|---|---|---|---|---|---|
| mg/kg | h | nmol/ml | h × nmol/ml | liters/h/kg | h | % | |
| Oral | 20 | 1 | 2.17 | 5.95 | 6.98 | 5.28 | 33.8 |
Confirmatory activity profiling revealed potent and RSV-specific activity against the RSV A2-FireSMASh target (Fig. 6C). We noted active concentrations of AVG-233 in RSV minireplicon assays comparable with those obtained against the RSV A2-FireSMASh reporter virus, whereas IAV WSN and MeV minireplicons again remained uninhibited. When subjected to the in vitro RSV RdRp assay, AVG-233 selectively blocked 3′ RNA extension elongation after initiation from the promoter or back-priming, but not 3′ extension by up to three nucleotides (Fig. 6D). These results confirm that chemical optimization of the scaffold did not alter the underlying mechanism of anti-RSV activity.
To cross-examine the lead compound for mitochondria toxicity, we first quantified metabolic activity of cultured cell lines (Fig. S1) and primary HBTECs (Fig. 7A) after incubation in the presence of glucose or, to circumvent masking of mitochondrial toxicity through the Crabtree effect (36), galactose. Independent of the energy source and target cell system examined, AVG-233 did not cause measurable cytotoxicity within the testable concentration range after 48 h of cell exposure, corresponding to an SI >1,666 against the RSV A2-FireSMASh reporter virus. The compound furthermore had not detrimental affects on cell membrane integrity or mitochondrial function when tested in a multiplexed fluorogenic and luminescence-based assay (Fig. 7B) and did not negatively impact mitochondrial biogenesis, monitored through in-cell ELISA detecting nuclear-encoded SDH-A and mitochondria-encoded COX-I proteins (Fig. 7C).
Figure 7.

Cytotoxicity profiling of AVG-233. A, assessment of cell metabolic activity after 48-h exposure of primary HBTECs to AVG-233. Assays were conducted in the presence of standard media, or RPMI supplemented with glucose or galactose as carbohydrate source to suppress glycolysis-induced masking of mitochondrial toxicity. B, multiplexed mitochondrial ToxGlo assay to determine cell membrane integrity and mitochondrial function after HBTEC exposure to AVG-233. C, in-cell ELISA determining effects on mitochondrial biogenesis after 48-h exposure to AVG-233. Symbols in A–C represent individual biological repeats (n = 3 each or greater); error bars, S.D.
Direct comparison of AVG-233 inhibitory activity with the first-generation leads AVG-094 and AVG-158 in virus yield–based dose–response assays revealed anti-RSV potency equivalent to that of AVG-094 and ∼3-fold lower than that of AVG-158 (Fig. 8A). Similar to AVG-158, however, AVG-233 acted as a sterilizing agent at the highest concentration (20 μm) assessed in this assay (Fig. 8B), whereas AVG-094 reached a concentration-independent bottom plateau of the antiviral effect (Fig. 4B). Potent, sterilizing inhibition of virus replication by AVG-233 was furthermore not limited to recombinant RSV A2-L19F, because we detected equivalent inhibitory activity when we tested the compound against two RSV isolates, RSV strain 2–20 and RSV clinical isolate 718, with minimal history of ex vivo amplification (Fig. 8A). Last, AVG-233 inhibited RSV replication on primary HBTECs while exhibiting undetectable toxicity, although efficacy was ∼5-fold lower compared with AVG-158 (Fig. 8B).
Figure 8.

Antiviral potency of AVG-233. A, virus yield reduction of RSV A2-L19F and two RSV clinical strains by AVG-233. Infected cells were incubated in the presence of compound, and progeny virus titers were determined 48 h after infection. Active concentrations were determined through four-parameter variable slope regression modeling. LoD, level of detection. B, antiviral activity of first- and second-generation lead analogs AVG-158 and AVG-233, respectively, in undifferentiated primary HBTECs. Four-parameter variable slope regression models and active concentrations are shown. Symbols in A and B represent individual biological repeats (n = 3 each or greater); error bars, S.D.
In aggregate, these results highlight AVG-233 as a second-generation lead analog of a newly identified class of RSV-specific inhibitors preventing initiation of the viral polymerase complex at the promoter. The successful generation of a productive SAR, development of a 3D-QSAR model with high predictive power, strong druglike properties, good oral bioavailability, and sustained high plasma concentrations in mice recommend this inhibitor class for further characterization and development.
Discussion
In this study, we employed HTS to identify a novel chemical point of entry for RSV inhibition. The use of a previously developed myxovirus dual-infection protocol allowed us to search for broad-spectrum inhibitors of RSV and IAV (19, 20), but no well-behaved dual-allosteric inhibitors against both target viruses were identified. This observation presumably reflects that the likelihood for the presence of a suitable druggable site that is conserved between RSV and IAV is exceptionally low. We also did not discover any entry inhibitors capable of blocking the pan-entry inhibitor–resistant (15) recombinant RSV-A2-L19FD489E-FireSMASh reporter virus used in this screen, which corroborates the conclusions of previous, independent screens that tested different chemical libraries against the same reporter strains (19, 20). Combined, the results of these drug screens corroborate our earlier prediction that overcoming RSV pan-resistance with small-molecule entry inhibitors will be challenging (19, 25, 27).
Based on rigorous hit inclusion criteria that we selected in anticipation of the demands of a predominantly pediatric patient population, a single compound, GRP-156784, emerged from the recent HTS as suitable for further development. Whereas a calculated confirmed hit rate of 0.0018% may be perceived as extremely low, we consider this outcome an accurate representation of the likelihood that a member of a random collection of small-molecule chemicals may engage a protein target specifically, noncovalently, and productively. This view is in agreement with the previously developed notion of “dark chemical matter” compounds, which refers to sections of screening libraries that consistently lacked biological activity in independent screens against diverse targets (37). In multiple unrelated HTS assays carried out in our laboratory against different RNA viruses, GRP-156784 was consistently unresponsive with the exception of the anti-RSV screen. This rare activity in usually unresponsive chemical space drew our attention to the scaffold despite its low initial potency, because these hits often provide valuable starting points for development with clean safety profiles (37).
Four lines of evidence support specific interference of the GRP-156784 class with RSV RdRp activity: (i) time-of-addition studies using infected cells revealed an inhibition profile of GRP-156784 that resembled that of the confirmed polymerase inhibitor JMN3–003 (24) and was in stark contrast to the behavior of a reference RSV entry inhibitor (23); (ii) plasmid-based minigenome reporter assays in transiently transfected cells demonstrated specific targeting of RSV RdRp complex activity by GRP-156784 and lead analogs, because bioactivity of equivalent MeV and IAV minigenomes was not inhibited; (iii) GRP-156784 significantly lowered the accumulation of RSV mRNA in infected cells, mimicking the effect seen with the chain-terminating ribonucleoside analog RSV polymerase inhibitor ALIOS-08176; and (iv) in vitro polymerase assays using purified RSV RdRp complexes revealed dose-dependent inhibition of 3′ RNA extension elongation after de novo initiation at the promoter or back-priming. However, 3′ extension and thus phosphodiester bond formation were not inhibited by advanced analogs of GRP-156784. RdRp transition from extension to elongation mode is thought to involve major conformational rearrangements (31), supporting the hypothesis that compound binding may prevent this structural reorganization of the RdRp complex after initial 3′ RNA extension by up to three nucleotides. We note that AZ-27, a chemically unrelated RSV polymerase inhibitor, also suppressed 3′ extension elongation after back-priming (31). Unlike AVG-233, however, AZ-27 blocked all de novo RNA extension from the promoter, suggesting that the two compound classes are mechanistically distinct. It will be a particular focus of future work to determine whether resistance profiles of AZ-27 (38, 39) and the GRP-156784 class are overlapping, distinct, or exclusive.
Failure to perform in a physiologically relevant host cell environment or against a nonlaboratory-adapted target strain is a common reason for the demise of promising scaffolds or antiviral candidates identified in cell-based assays using immortalized cell lines and/or tissue culture–experienced screening viruses (40–42). A case in point is the ribonucleoside analog favipiravir (T-705), which is conditionally approved in Japan for influenza therapy. Although active in a number of different cell lines, T-705 lacks antiviral activity in disease relevant primary human airway cells (28), which is most likely a consequence of poor conversion of the T-705 prodrug to its active triphosphate form in these cells. To conserve valuable resources, we therefore requested bioactivity in primary cells of any screening hit considered for chemical development and consistent potency of emerging leads against different viral isolates. Driven by our primary therapeutic goals of preventing viral spread from the upper respiratory tract to the small airways, we consider large airway-derived HBTECs (28) to represent a disease-relevant primary cell system for hit validation. Both original and resynthesized GRP-156784 and subsequently tested lead analogs of this compound passed the primary cell filter. In addition, antiviral potency of lead analogs against recombinant RSV and viral isolates with minimal ex vivo passage history was unchanged within a narrow variance margin, underpinning the developmental potential of the scaffold.
Our approach to the synthetic optimization of GRP-156784 concentrated on the three peripheral substituents, because these could be manipulated rapidly and efficiently using the effective synthesis approach that we had established for the scaffold. Key synthetic objectives were defined based on shared properties of many successful small-molecule drugs (43, 44). We aimed to minimize the size, complexity, and steric bulk of each of the three test sections where possible to optimize absorption potential for oral delivery and minimize ports of entry for metabolic turnover. Simultaneously, we pursued the installation of heteroatoms or polarizing groups to decrease the octanol–water partition coefficient logP and maximize water solubility.
Structural surveys of each of the individual sections revealed suitable core options that advanced one or both of the objectives stated above. The benzothiazole group in section 1 could be effectively replaced with smaller six-membered ring systems, and the more polar pyridyl groups were tolerated. The benzyl group in section 2 turned out to be optimal, but substituents were well-tolerated. Six-membered rings and heterocycles were well-tolerated in section 3 and provided a polar handle for further optimization of both potency and pharmaceutic properties. Synthetic optimization achieved a major boost in antiviral activity of up to 20-fold compared with the starting structure without a measurable increase in cytotoxicity. Because the potency-enhancing modifications were not correlated with a trend toward altering secondary determinants of antiviral activity, such as plasma membrane permeability or water solubility, we propose that the chemical nature of sections 1 and 3 directly affects compound affinity for its viral target. Whereas the activity profiles of a first set of optimized analogs had reached our early-stage objectives (active concentrations <100 nm and SI values >1,000), metabolic stability testing of these first-generation leads against hepatocyte-derived microsomes revealed major liabilities, prompting retargeting of synthetic development.
In our experience, ligand-driven QSARs show good predictive power for second-stage optimization, provided they are informed by meaningful training and test sets (27, 45–47). Particularly detrimental to model accuracy are data sets representing overlapping, but distinct, SARs, such as antiviral activity due to specific target binding and cytotoxic effects. To create a wide safety margin against uninterpretable off-target effects, we first applied rigorous admission criteria to analogs selected as training and test sets (SI >100 or CC50 >100 μm, respectively). Second, we explored the AutoGPA module embedded in MOE (32, 33) for model building. In the absence of a predetermined molecular docking pose, the AutoGPA approach was shown to perform favorably when compared with alternative methods (32, 48–52) such as the comparative molecular field analysis (CoMFA) (53), PHASE (54), and the comparative molecular similarity indices analysis (CoMSIA) (55). Because the resulting 3D-QSAR model showed strong predictive accuracy against the test set, we considered it suitable to test an innovative in silico screening approach for PK profile optimization. The SMARTCyp algorithm was queried to identify substructures of the scaffold metabolic that are predicted to be highly susceptible to human cytochrome P450 (CYP) metabolism (35), because CYP enzymes are involved in the primary oxidation of almost all drugs (56). These metabolic hot spots were then modified systematically in a virtual library, followed by library filtering against SMARTCyp, three different algorithms examining physiochemical properties such as LogP, topographical polar surface area, molecular weight, and number of rotatable bonds were applied to predict water solubility (21, 57, 58), and the 3D-QSAR model.
After synthesis of the eight top-scoring virtual candidates, experimental assessment confirmed that the 3D-QSAR model was highly dependable, showing a maximal discrepancy between actual and predicted EC50 value of 0.09 μm (AVG-258). By comparison, solubility estimates were accurate for predicted poor performers (water solubility <1 μm) but not for compounds of anticipated moderate solubility (R2 = 0.17). Despite the limitations of this largely binary solubility readout, metabolic susceptibility predictions proved informative; of the analogs selected for synthesis from the virtual library, lower metabolic susceptibility scores faithfully correlated with increased half-life values in human liver microsomes (R2 = 0.74). Although AVG-258 emerged as the analog most stable in the presence of human liver microsomes, AVG-233 performed best overall due to highest solubility, good metabolic stability, undetectable cytotoxicity, and antiviral potency. Subsequent PK profiling of AVG-233 in mice revealed good oral bioavailability and acceptable PK parameters. Because the compound suffered only a small penalty in antiviral potency in cultured and primary cell assays, it provides important proof of concept for the validity of the combined experimental and computational optimization strategy applied to the GRP-156784 scaffold. Future development of this promising novel drug candidate class will concentrate on examining in vivo anti-RSV efficacy, resistance profiling, and extraction of the molecular docking pose.
Experimental procedures
Cell lines and primary cells
Human bronchial epithelial (BEAS-2B, ATCC CCL-9609), human carcinoma (HEp-2, ATCC CCL-23), Madin–Darby canine kidney (MDCK, ATCC CCL-34), human embryonic kidney (293T, ATCC CRL-3216), and African green monkey kidney epithelial cells (CCK-81; ATCC) stably expressing human signaling lymphocytic activation molecule (Vero-hSLAM) were maintained at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 7.5% fetal bovine serum. Although HEp-2 cells are denoted in the ICLAC database version 8.0 of commonly misidentified cell lines, their use is warranted because these cells are accepted and commonly used for studies involving respiratory syncytial virus. Normal primary human bronchial tracheal epithelial cells (HBTECs; Lifeline Cell Technology, catalog no. LM-0050) were grown and maintained in BronchiaLife cell culture medium (LifeLine Cell Technology) at 37 °C and 5% CO2. HBTECs were analyzed for microbial contamination on July 25, 2017 by LifeLine Cell Technology. All immortalized cell lines used in this study are regularly checked for mycoplasma and microbial contamination. Genejuice transfection reagent (Invitrogen) was used for all transfections.
Molecular biology
To engineer a recombinant MeV-Edm-nanoLuc-PEST virus, the nanoLuciferase gene with a C-terminal PEST sequence was first amplified with 5′-MluI and 3′-AatII restriction sites. The digested PCR product was then inserted in place of eGFP in an existing plasmid encoding the full-length genome of recMeV-Edm-eGFP, with eGFP located upstream of the N ORF (59). Recombinant MeV-nanoLuc-PEST virus was recovered in BSR-T7/5 cells by transfecting 1.25 μg of the cDNA copy of the MeV-Edm-nanoLuc-PEST genome and MeV-IC-B-N (0.42 μg), MeV-IC-B-P (0.54 μg), and MeV-IC-B-L (0.55 μg) plasmids using GeneJuice transfection reagent. Cells were overlaid 36 h post-transfection onto Vero/hSLAM cells, and emerging infectious particles were passaged twice in Vero/hSLAM cells. The identity of rescued virus was confirmed by extracting total RNA from virus-infected cells (RNeasy Mini Kit, Qiagen). MeV virus titers were determined by TCID50 titration on Vero/hSLAM cells. Recombinant MeV-Edm-nanoLuc-PEST stocks were propagated on Vero/hSLAM cells (multiplicity of infection (MOI) equal to 0.01 infectious particles/cell). Infected cells were incubated for 3–4 days at 37 °C. Infected cells were then harvested in Dulbecco's modified Eagle's medium and subjected to a single freeze–thaw cycle to extract cell-associated virus.
Viruses
RSV stocks were propagated on HEp-2 cells after infection at an MOI = 0.01. HEp-2 cells infected with RSV were incubated for 16 h at 37 °C, followed by incubation at 32 °C for 5–7 days. A single freeze–thaw cycle was used to release cell-associated progeny virus. RSV virus titers were determined by TCID50 titration on HEp-2 cells. For recRSV A2-L19FD489EfireSMASh, reporter virus was propagated in the presence of Asunaprevir (Santa Cruz Biotechnology, Inc.). IAV-WSN-nanoLuc was propagated in MDCK cells (MOI = 0.001) for 2 days at 37 °C. Supernatant was harvested from infected cells, and centrifuged IAV-WSN stocks titers were determined by TCID50-HA assays (28) on MDCK cells. Supernatant virus (IAV-WSNnanoLuc) or cell-associated progeny virus (MeV-Edm-nanoLuc-PEST and RSV A2-L19F) was first clarified by centrifugation (4,000 rpm for 20 min at 4 °C), followed by subsequent purification through a 20–60% single-step sucrose gradient in TNE (50 mm Tris-Cl (pH 7.2), 100 mm NaCl, 10 mm EDTA). After ultracentrifugation (30,000 rpm for 120 min at 4 °C), purified virus was harvested from the gradient interface, aliquoted, and stored at −80 °C.
Library screening
Screening libraries were obtained from the ChemDiv and were filtered against compounds with pan-assay interfering and undesirable structure. Compounds were dissolved in DMSO at a concentration of 10 mm in 96-well master plates and stored at −80 °C. The MScreen software package was utilized for electronic compound management, HTS data storage, and data analysis (60). To create screening libraries, compounds were reformatted into barcoded 384-well daughter plates using a Nimbus liquid handler (Hamilton Robotics). Each 384-well stock plate contained 64 wells reserved for positive and negative controls, located in the outermost columns and arranged in a checkerboard pattern. For HTS screening, BEAS-2B cells (3.5 × 103/well) were injected (30 μl/well) into barcoded white wall/clear bottom 384-well plates using a MultiFlo automated dispenser (BioTek) equipped with dual 10-μl peristaltic pump manifolds. Cells were then collected (150 × g for 90 s at 25 °C) and incubated for 14 h at 37 °C and 5% CO2. Compound was added to a final concentration of 5 μm (20 nl/well) using a high-density pin tool (V&P Scientific) attached to the pipetting head of the Nimbus liquid handler, followed by co-infection with recRSV-A2-L19FD489EfireSMASh (MOI = 0.1) and recIAV WSN-NanoLuc (MOI = 0.02) in 10 μl/well using the MultiFlo dispenser unit, spin collection (150 × g for 90 s at 25 °C), and incubation for 40 h at 37 °C and 5% CO2. Final vehicle (DMSO) concentration was 0.05%. The Nimbus unit automatically detected and recorded barcodes of source and assay plates at the time of stamping. Using a stacker unit with integrated barcode reader (Biotek) attached to an H1 synergy plate reader, plates were automatically loaded, dual-Glo substrates (15 μl/well each) were injected, and bioluminescence was recorded after a 3-min lag time for each well and substrate. Readouts were automatically saved by plate barcode and imported into the MScreen IT environment.
HTS data analysis
The MScreen package was used for automated data analysis. Normalized relative values were calculated by subtracting from each value the average of the plate vehicle controls, followed by dividing the results by the difference between the means of plate vehicle positive and negative controls. Robust z-scores were calculated as follows: robust z-score = (Si − median (Sall))/MAD(Sall) and MAD(Sall) = 1.4826 × median (|Si − median (Sall)|) (where Si = individual compound value, and Sall = values of all compounds on the plate). Hit candidates were defined as compounds exhibiting ≥85% inhibition of normalized signal intensity against RSV A2-L19FD489EfireSMASh and robust z-score ≥0.98.
Automated dose–response counterscreening
BEAS-2B cells (3.5 × 103/well) were injected (30 μl/well) into barcoded white wall/clear bottom 384-well plates using a MultiFlo automated dispenser (BioTek) equipped with dual 10-μl peristaltic pump manifolds. Cells were then collected (150 × g for 90 s at 25 °C), and incubated for 14 h at 37 °C and 5% CO2. For dose–response direct counterscreens and cytotoxicity screens, hit candidates were automatically picked into 384-well dose–response counterscreen plates, and 3-fold dilutions (0.078–10 μm) were prepared using the Nimbus liquid handler. Positive and negative controls were arranged as mentioned previously. 384-well plates previously seeded with BEAS-2B cells were then stamped with dose–response counterscreen plates using a high-density pin tool attached to the pipetting head of the Nimbus liquid handler. Subsequently, cells were infected with recRSV-L19FD489EfireSMASh or recRSV-A2-L19F-renilla. Reporter signals were recorded as outlined above. All direct counterscreening plates were tested twice in independent repeats. To determine cell viability, PrestoBlue substrate (5 μl/well) (Life Technologies) was added following a 48-h incubation of uninfected compound-treated cells at 37 °C (5 μl/well). Following a 90-min incubation with PrestoBlue substrate, top-read fluorescence (excitation at 560 nm, emission at 590 nm, instrument gain of 85) was recorded using the H1 synergy plate reader.
Chemical synthesis
General synthesis schemes of the GRP-156784 scaffold and scaffold analogs are summarized in the supporting information. All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (1–5 mm Hg) at room temperature. Thin-layer chromatography was performed on silica gel plates; spots were visualized by UV light (214 and 254 nm). Purification by column and flash chromatography was carried out using silica gel (200–300 mesh). Solvent systems are reported as mixtures by volume. All NMR spectra were recorded on a Bruker 400 (400-MHz) spectrometer. 1H chemical shifts are reported in δ values in ppm with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constant (Hz), and integration.
Dose–response reporter assays
For dose–response testing, serial 3-fold compound dilutions were prepared in three replicates in 96-well plates using the Nimbus liquid handler. BEAS-2B cells (1.5 × 104 cells/well) were plated in 96-well plates, serial dilutions were transferred to the cell plates using the liquid handler, and cells were infected with recRSV-A2-L19FD489EfireSMASh (MOI = 0.1). Four positive (1 mg/ml cyclohexamide) and four negative controls (0.05% DMSO) were included on each plate. To determine cell viability, PrestoBlue substrate (10 μl/well (Life Technologies)) was added after a 40-h incubation of uninfected cells with compound. Fluorescence reporter activity was recorded with the H1 synergy plate mentioned previously. Raw data of all dose–response screens were analyzed according to the formula, % inhibition = (XSample − XMin)/(XMax − XMin) × 100, with XMin representing the average of the positive and XMax the average of the negative control wells. Four-parameter variable slope regression was applied to determine 50% active (EC50) concentrations, using the nonlinear regression function in the Prism (GraphPad) software package.
Virus yield reduction assays
To determine the effect of compounds on progeny virus production, HEp-2 cells were infected in a 12-well plate format with recRSV-A2-L19F-mKate expressing a far-red fluorescent protein (61) or RSV isolates as specified (MOI = 0.05) in the presence of serial compound dilutions and incubated at 37 °C. Cell-associated progeny virions were harvested 48 h post-infection, and virus titers in each sample were determined through TCID50 titration or immunoplaque assays (clinical isolates) as described (28).
Minigenome reporter assays
For minigenome reporter assays, an RSV firefly luciferase minigenome plasmid (pHH-RSV-repl-firefly) was used as described previously (15). 293T cells were co-transfected with this minigenome and plasmids pRSV-N, pRSV-P, pRSV-M2-1, and pRSV-L, respectively, under cytomegalovirus promoter control. For IAV minigenome reporter assays, 293T cells were transfected with IAV-WSN firefly luciferase reporter plasmid and each of PB1, PB2, NP, and PA expression plasmids under cytomegalovirus promoter control, as described previously (20). At 3 h post-transfection, compound was added at the specified concentration range, followed by incubation at 37 °C for 40 h, lysis, and assessment of luciferase activity. For MeV minigenome reporter assays, BSR-T7/5 were transfected with MeV firefly luciferase reporter plasmid and N, P, and L expression plasmids under T7 promoter control, as described previously (62).
Reporter activities were determined in the presence of 3-fold serial dilutions of GRP-156784 starting from 20 μm for RSV and 100 μm for IAV and MeV. Treatment was initiated 1 h post-transfection, followed by incubation at 37 °C for 40 h. Cells were then lysed, and luciferase activity was assessed. Luciferase activities in cell lysates were measured in a Synergy H1 microplate reader (BioTek) in top-count mode using the Dual-Glo luciferase assay system (Promega). Inhibitory concentrations were calculated through four-parameter variable slope regression modeling.
Time-of-addition variation studies
HEp-2 cells were infected with 0.1 MOI of recRSV-A2-L19FD489EfireSMASh. At the specified time points relative to infection, compounds were added to the culture media to a final concentration of 10, 20, and 20 μm for BMS-433771, JMN3–003, and GRP-156784, respectively. DMSO only served as vehicle controls. Reporter gene expression was measured at 48 h post-infection, and the obtained values were expressed relative to the vehicle-treated samples.
In vitro RSV polymerase assay
The RSV large polymerase subunit (L) and phosphoprotein (P) complexes were expressed from a baculovirus vector and purified by affinity chromatography. L-P complexes were eluted from nickel-nitrilotriacetic acid resin with 250 mm imidazole in 50 mm NaH2PO4, pH 7.5, 150 mm NaCl, and 0.5% Nonidet P-40 and subsequently dialyzed against 150 mm NaCl, 20 mm Tris-HCl, pH 7.4, 1 mm DTT, and 10% glycerol. L-P heterooligomers were added to transcription buffer (50 mm Tris-HCl, pH 7.4, 8 mm MgCl2, 5 mm DTT, 10% glycerol) containing a 2 μm concentration of a template RNA representing the first 25 nucleotides of the RSV antigenomic promoter sequence; 250 μm each ATP, CTP, GTP, and UTP; and 0.07 μm [α-32P]GTP as tracer. Reactions were carried out at 30 °C for 30 min. Predominant initiation at the +3-position resulted in up to 25-mer radiolabeled RNA synthesis products, whereas back-priming yielded 26–28-mer sequences and 3′ extension elongation after back-priming generated longer products. The radiolabeled samples were subjected to denaturing gel electrophoresis, followed by autoradiography.
Reverse transcription qPCR
HEp-2 cells were infected with recRSV-A2-L19F-mKate (MOI = 3) and incubated in the presence of the GRP-156784, ALIOS-8176, or vehicle (DMSO) for control at 37 °C. At 20 h postinfection, total RNA was isolated from all wells using a QIAcube automated extractor and the RNeasy Mini Kit (Qiagen) and subsequently subjected to reverse transcription using Superscript III reverse transcriptase and oligo(dT) primer of first-strand synthesis. Real-time reactions were carried out using an Applied Biosystems 7500 Fast real-time PCR system, PowerUp SYBR Green Master mix (Thermo Fisher scientific), and primer pairs specific for a fragment in the RSV N ORF or human GAPDH, respectively. To verify amplification of a single product, melting curves were generated for each primer pair. To calculate ΔΔCT values, CT values obtained for each sample were normalized for GAPDH as reference, and then ΔCT values of inhibitor-treated samples were normalized for the DMSO-treated controls. Final quantification was based on three independent experiments, with each independent repeat calculated from two dependent repeats.
3D-QSAR model building
The MOE software package (MOE 2016.0802) was used to perform all energy minimization, conformation searches, and model building (33). 3D-QSAR models were generated using the AutoGPA module (32) embedded in MOE. For the creation of predictive 3D-QSAR models, 52 structures were chosen exhibiting various inhibitory activities, ranging in EC50 concentration from 0.032 to 2.2 μm. This set of compounds was divided into a training set (13 entries; compounds AVG-35, AVG-94, AVG-132, AVG-133, AVG-134, AVG-158, AVG-176, AVG-177, AVG-178, AVG-203, AVG-241, AVG-242, and AVG-243) and a test set (39 entries; compounds GRP-156784, AVG-024, AVG-025, AVG-031, AVG-036, AVG-072, AVG-080, AVG-103, AVG-106, AVG-107, AVG-108, AVG-109, AVG-122, AVG-126, AVG-127, AVG-128, AVG-129, AVG-130, AVG-131, AVG-157, AVG-160, AVG-161, AVG-163, AVG-168, AVG-169, AVG-171, AVG-172, AVG-173, AVG-174, AVG-175, AVG-201, AVG-205, AVG-214, AVG-227, AVG-228, AVG-229, AVG-230, AVG-231, and AVG-234). Before input, EC50 values were normalized and converted to pIC50 (−log10(EC50)). Conformational alternatives of each compound were generated using the conformational search function of the AutoGPA package using LowModeMD with a root mean square deviation gradient of 0.005 and a root mean square deviation limit of 0.25. A total of 510 and 1,661 conformations were generated for the training and test sets, respectively. Training set conformations were then aligned and assigned pharmacophore features using the pharmacophore elucidation function of MOE with an active coverage of 100%, query spacing of 0.6, an alignment cutoff of 0.5, and a pharmacophore feature limit of 5. AutoGPA identified common features and created 106 initial pharmacophore models based on the training set. The CoMFA algorithm (53) was then utilized, all molecules were placed in a 3D grid box with 2-Å separation, and electrostatic and steric interaction energies between each molecule were calculated. An sp3 carbon atom probe evaluated the allowable space and molecular field surrounding the compounds. After model building was performed, a partial-least squares analysis was performed to analyze the relationship between grid potential fields and inhibitory activity to create the AutoGPA-based 3D-QSAR model. Each model was validated, scored, and ranked based on LOO correlation (q2) and goodness of fit (R2). The predictive potential of the model was then tested using a conformation database of the test set. Predicted pIC50 values were then compared with actual pIC50 values to determine the correlation (R2) and slope of the correlation through the origin.
Experimental assessment of select ADME parameters
For solubility tests, 0.1 m phosphate (NaPO4) buffer, pH 7.4, was first preheated, and then aliquots of 8 μl of reference and test compound stock solutions (10 μm) are added into 792 μl of 100 mm phosphate buffer (pH 7.4). (Final DMSO concentration, 1%). Sample tubes are shaken for 1 h (1,000 rpm) at room temperature. Samples are centrifuged (10 min; 12,000 rpm) to precipitate undissolved particles, and supernatants were transferred to a new tube or plate. Supernatants are diluted 10 times and 100 times with 100 mm phosphate buffer. Sample preparation for LC-MS/MS. Compound (5 μl of undiluted, 10-fold diluted, and 100-fold diluted) and standard curve samples to 95 μl of acetonitrile (ACN) containing internal standards (IS) were analyzed by LC-MS/MS. Standard curve concentrations used were 0.02, 0.1, 0.2, 0.8, 4, 10, 20, and 60 mm.
For liver microsome stability testing, reference compound (ketanserin) and test compound (n = 2) spiking solutions were prepared as follows. A 500 μm spiking solution was made containing 10 μl of 10 mm compound stock solution in 190 μl of ACN. Next, a 1.5 μm spiking solution in microsomes (0.75 mg/ml) was prepared by adding 1.5 μl of 500 μm spiking solution and 18.75 μl of 20 mg/ml liver microsomes (mouse or human; BD Gentest) into 479.75 μl of Buffer C (0.1 m potassium phosphate buffer, 1.0 mm EDTA, pH 7.4) on ice. A NADPH stock solution (6 mm) was prepared by dissolving NADPH into Buffer C. 30 μl of 1.5 μm spiking solution containing 0.75 mg/ml microsomes solution was dispensed into the assay plates designated for different time points (0 and 30 min) on ice. For the 0-min time point, ACN containing IS (135 μl) was added to the wells of 0-min plate, and 15 μl of NADPH stock solution (6 mm) was added. All other plates were incubated at 37 °C for 5 min. NADPH stock solution (15 μl; 6 mm) was added to the plates to start the reaction and timing. After incubating for 30 min, 135 μl of ACN (containing IS) was added to the wells to stop the reaction. After quenching, plates were shaken using a vibrator (IKA, MTS 2/4) for 10 min (600 rpm/min) and then centrifuged at 5,594 × g for 15 min (Thermo Multifuge X3R). Supernatant (50 μl) was transferred from each well into a 96-well sample plate containing 50 μl of ultrapure water (Millipore, ZMQS50F01) for LC/MS/MS analysis.
In silico ADME predictions
Prediction of ADME-PK parameters was performed using the SwissADME (21) and XenoSite Metabolism prediction (63) servers. Using SwissADME, predicted solubility was calculated from the average values of the following three solubility prediction algorithms: ESOL (58), Ali (57), and SILICOS-IT. Compounds were submitted in the simplified molecular-input line-entry system (SMILES) format. Prediction of cytochrome P450 metabolic susceptibility was performed using the SMARTCyp (35) algorithm of the XenoSite Metabolism prediction server and is shown as the average predicted metabolic susceptibility against the following eight isoforms of cytochrome P450: 1A2, 2A6, 2B6, 2C8, 2C19, 2E1, and 3A4. Predicted metabolic susceptibility against eight isoforms and isoforms 2C9 and 2D6 is shown in Table 1. Metabolic susceptibility was scored by calculating the sum of the predicted susceptibility probabilities for all atoms present in each analog.
Virtual library creation
A virtual library of GRP-156784 analogs was generated based on the AVG-158 and AVG-094 structures. For each virtual analog, substitutions and modifications were made and positioned at various locations based on the metabolic susceptibility predictions generated using the SMARTCyp algorithm. A total of 197 compounds were generated and screened for predicted bioactivity and metabolic susceptibility. Compounds were chosen for synthesis and testing based on the following two criteria: (i) predicted bioactivity was equal to or greater than the least potent analog from the first round of PK testing (AVG-093), and (ii) predicted metabolic susceptibility was less than the predicted metabolic susceptibility of the least susceptible analog (AVG-171) from the first round of PK testing. Metabolic susceptibility was scored by calculating the sum of the predicted susceptibility probabilities for all atoms present in each analog. Average metabolic susceptibility scores were calculated from predictions against eight CYP isoforms and isoforms 2C9 and 2D6 using the SMARTCyp algorithm.
Pharmacokinetic profiling in mice
Male CD-1 mice (27–29 g) were distributed randomly into groups. AVG-233 was dissolved in 5% DMSO + 5% Solutol HS15 + 90% saline to yield a clear solution at 0.2 mg/ml for IV dosing and prepared in 0.2% Tween 80 + 0.5% methylcellulose in water to yield a homogeneous suspension at 20 mg/ml for oral dosing, respectively. Mice were administered 2 mg/kg (10 ml/kg) via tail vein (n = 9) for intravenous dosing and 20 mg/kg (10 ml/kg) via oral gavage (n = 9) for oral dosing. Mice were anesthetized with isoflurane. For intravenously administered compound, blood samples were obtained pre-dose and 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dosing. For oral administration, blood samples were harvested pre-dose and 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h post-dosing. At each time point, ∼110 μl of blood was collected into a K2EDTA tube via retro-orbital puncture for serial bleeding and cardiac puncture for terminal bleeding. Blood samples were put on ice and centrifuged to obtain plasma samples (2,000 × g, 5 min under 4 °C) within 15 min of harvesting. Samples were stored at −80 °C for further analysis. An aliquot of 20 μl of sample was added with 400 μl of ACN containing 100 ng/ml IS (Diclofenac). The mixture was vortexed for 10 min and centrifuged at 5,800 rpm for 10 min. An aliquot of 30 μl of supernatant was added with 60 μl of H2O, and the mixture was vortexed for 5 min. An aliquot of 2 μl of supernatant was injected for LC-MS/MS analysis. For calibration, standard curves were prepared in blank plasma (concentrations range 1.00–3,000 ng/ml). Quality-control samples of 800 ng/ml in blank plasma were analyzed for each sample set. Calibration showed linearity with an R2 value = 0.9969.
Mitochondrial toxicity
Three different assays were employed to test for interference of the lead compound with mitochondrial function. (i) Established cell lines and primary HBTECs were adapted to growth in carbohydrate-free RPMI supplemented with 11 mm glucose or 10 mm galactose, respectively. After 48-h exposure to AVG-233, PrestoBlue substrate was added, and cell viability was determined as described above. (ii) Cell membrane integrity and ATP content were quantified using the multiplexed mitochondrial ToxGlo assay (Promega) after 48-h exposure to AVG-233. (iii) Levels of nuclear-encoded SDH-A and mitochondria-encoded COX-I protein levels were determined using the MitoBiogenesis in-cell ELISA (Abcam) after 48-h exposure to AVG-233.
Ethics statement
Pharmacokinetics testing in mice was approved by the Georgia State University institutional animal use and care committee under protocol A17019.
Statistical analyses
The MScreen, Prism version 7 (GraphPad), and Excel software packages were used for data analysis. To determine antiviral potency, EC50 concentrations were calculated from dose–response data sets using four-parameter variable slope regression modeling using the Prism package. The statistical significance of differences between groups was determined through one-way analysis of variance in combination with post hoc multiple-comparison tests as specified in the figure legends. Experimental variation is identified by error bars, representing in all cases S.D.
Author contributions
R. M. C., J. S., B. L., and R. K. P. data curation; R. M. C., M. T., J.-J. Y., B. L., and R. K. P. investigation; R. M. C. and R. K. P. methodology; R. M. C. and R. K. P. writing-original draft; R. M. C., M. T., J. S., B. L., R. F., E. B., J. V., and R. K. P. writing-review and editing; M. T., E. L., J. V., and R. K. P. formal analysis; R. F., E. B., E. L., J. V., and R. K. P. conceptualization; R. F., E. B., J. P., E. L., J. V., and R. K. P. supervision; J. P. and R. K. P. funding acquisition; R. K. P. validation; R. K. P. project administration.
Supplementary Material
Acknowledgments
We thank K. K. Conzelmann for the BSR-T7/5 cell line, M. L. Moore for the RSV reverse genetics system, ChemPartner USA for chemical synthesis, and R. T. Jacob for IT support. The MScreen software package was kindly provided by the Center for Chemical Genomics of the University of Michigan under a license agreement by the University of Michigan Office of Technology Transfer; JChem was used for structure database management, search, and prediction (JChem 6.2, 2014, ChemAxon); and Marvin was employed for drawing, displaying, and characterizing chemical structures, substructures, and reactions (Marvin 14.9.22.0, 2014, ChemAxon).
This work was supported, in part, by NIAID, National Institutes of Health (NIH), Grant AI071002 and NICHD, NIH, Grant HD079327 (to R. K. P.). J. P., E. L., J. V., and R. K. P. are co-inventors on a patent application covering the use of the AVG-233 compound class for antiviral therapy. This study could affect their personal financial status. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S4 and Fig. S1.
- RSV
- respiratory syncytial virus
- RdRp
- RNA-dependent RNA polymerase
- IAV
- influenza A virus
- HTS
- high-throughput screening
- SI
- selectivity index
- MeV
- measles virus
- HBTEC
- human bronchial/tracheal epithelial cell
- qPCR
- quantitative PCR
- PK
- pharmacokinetics
- 3D
- three-dimensional
- QSAR
- quantitative structure–activity relationship
- MDCK
- Madin–Darby canine kidney
- HBTEC
- human bronchial/tracheal epithelial cell
- MOI
- multiplicity of infection
- ACN
- acetonitrile
- IS
- internal standards
- TCID50
- 50% tissue culture infective dose
- eGFP
- enhanced GFP
- MAD
- median absolute deviation.
References
- 1. Afonso C. L., Amarasinghe G. K., Bányai K., Bào Y., Basler C. F., Bavari S., Bejerman N., Blasdell K. R., Briand F. X., Briese T., Bukreyev A., Calisher C. H., Chandran K., Chéng J., Clawson A. N., et al. (2016) Taxonomy of the order Mononegavirales: update 2016. Arch. Virol. 161, 2351–2360 10.1007/s00705-016-2880-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson W. W., Shay D. K., Weintraub E., Brammer L., Cox N., Anderson L. J., and Fukuda K. (2003) Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 10.1001/jama.289.2.179 [DOI] [PubMed] [Google Scholar]
- 3. Nair H., Nokes D. J., Gessner B. D., Dherani M., Madhi S. A., Singleton R. J., O'Brien K. L., Roca A., Wright P. F., Bruce N., Chandran A., Theodoratou E., Sutanto A., Sedyaningsih E. R., Ngama M., et al. (2010) Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375, 1545–1555 10.1016/S0140-6736(10)60206-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi T., McAllister D. A., O'Brien K. L., Simoes E. A. F., Madhi S. A., Gessner B. D., Polack F. P., Balsells E., Acacio S., Aguayo C., Alassani I., Ali A., Antonio M., Awasthi S., Awori J. O., et al. (2017) Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390, 946–958 10.1016/S0140-6736(17)30938-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lugo R. A., and Nahata M. C. (1993) Pathogenesis and treatment of bronchiolitis. Clin. Pharm. 12, 95–116 [PubMed] [Google Scholar]
- 6. Aherne W., Bird T., Court S. D., Gardner P. S., and McQuillin J. (1970) Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 23, 7–18 10.1136/jcp.23.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Domachowske J. B., and Rosenberg H. F. (1999) Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin. Microbiol. Rev. 12, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. El Saleeby C. M., Bush A. J., Harrison L. M., Aitken J. A., and Devincenzo J. P. (2011) Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J. Infect. Dis. 204, 996–1002 10.1093/infdis/jir494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeVincenzo J. P., Wilkinson T., Vaishnaw A., Cehelsky J., Meyers R., Nochur S., Harrison L., Meeking P., Mann A., Moane E., Oxford J., Pareek R., Moore R., Walsh E., Studholme R., et al. (2010) Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 182, 1305–1314 10.1164/rccm.201002-0221OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson L. J., Parker R. A., and Strikas R. L. (1990) Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J. Infect. Dis. 161, 640–646 10.1093/infdis/161.4.640 [DOI] [PubMed] [Google Scholar]
- 11. Groothuis J. R., Woodin K. A., Katz R., Robertson A. D., McBride J. T., Hall C. B., McWilliams B. C., and Lauer B. A. (1990) Early ribavirin treatment of respiratory syncytial viral infection in high-risk children. J. Pediatr. 117, 792–798 10.1016/S0022-3476(05)83347-5 [DOI] [PubMed] [Google Scholar]
- 12. DeVincenzo J. P., Whitley R. J., Mackman R. L., Scaglioni-Weinlich C., Harrison L., Farrell E., McBride S., Lambkin-Williams R., Jordan R., Xin Y., Ramanathan S., O'Riordan T., Lewis S. A., Li X., Toback S. L., Lin S. L., and Chien J. W. (2014) Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 371, 711–722 10.1056/NEJMoa1401184 [DOI] [PubMed] [Google Scholar]
- 13. Wang G., Deval J., Hong J., Dyatkina N., Prhavc M., Taylor J., Fung A., Jin Z., Stevens S. K., Serebryany V., Liu J., Zhang Q., Tam Y., Chanda S. M., Smith D. B., Symons J. A., Blatt L. M., and Beigelman L. (2015) Discovery of 4′-chloromethyl-2′-deoxy-3′,5′-di-O-isobutyryl-2′-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J. Med. Chem. 58, 1862–1878 10.1021/jm5017279 [DOI] [PubMed] [Google Scholar]
- 14. Cox R., and Plemper R. K. (2016) Structure-guided design of small-molecule therapeutics against RSV disease. Expert Opin. Drug Discov. 10.1517/17460441.2016.1174212 10.1517/17460441.2016.1174212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan D., Lee S., Thakkar V. D., Luo M., Moore M. L., and Plemper R. K. (2014) Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc. Natl. Acad. Sci. U.S.A. 111, E3441–3449 10.1073/pnas.1405198111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grosfeld H., Hill M. G., and Collins P. L. (1995) RNA replication by respiratory syncytial virus (RSV) is directed by the N, P, and L proteins; transcription also occurs under these conditions but requires RSV superinfection for efficient synthesis of full-length mRNA. J. Virol. 69, 5677–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu Q., Hardy R. W., and Wertz G. W. (1995) Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J. Virol. 69, 2412–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collins P. L., Hill M. G., Camargo E., Grosfeld H., Chanock R. M., and Murphy B. R. (1995) Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. U.S.A. 92, 11563–11567 10.1073/pnas.92.25.11563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan D., Weisshaar M., Lamb K., Chung H. K., Lin M. Z., and Plemper R. K. (2015) Replication-competent influenza virus and respiratory syncytial virus luciferase reporter strains engineered for co-infections identify antiviral compounds in combination screens. Biochemistry 54, 5589–5604 10.1021/acs.biochem.5b00623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan D., Krumm S. A., Sun A., Steinhauer D. A., Luo M., Moore M. L., and Plemper R. K. (2013) Dual myxovirus screen identifies a small-molecule agonist of the host antiviral response. J. Virol. 87, 11076–11087 10.1128/JVI.01425-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daina A., Michielin O., and Zoete V. (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang J. J., Ursu O., Lipinski C. A., Sklar L. A., Oprea T. I., and Bologa C. G. (2016) Badapple: promiscuity patterns from noisy evidence. J. Cheminform. 8, 29 10.1186/s13321-016-0137-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cianci C., Yu K. L., Combrink K., Sin N., Pearce B., Wang A., Civiello R., Voss S., Luo G., Kadow K., Genovesi E. V., Venables B., Gulgeze H., Trehan A., James J., et al. (2004) Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob. Agents Chemother. 48, 413–422 10.1128/AAC.48.2.413-422.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun A., Ndungu J. M., Krumm S. A., Yoon J. J., Thepchatri P., Natchus M., Plemper R. K., and Snyder J. P. (2011) Host-directed inhibitors of myxoviruses: synthesis and in vitro biochemical evaluation. ACS Med. Chem. Lett. 2, 798–803 10.1021/ml200125r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weisshaar M., Cox R., Morehouse Z., Kumar Kyasa S., Yan D., Oberacker P., Mao S., Golden J. E., Lowen A. C., Natchus M. G., and Plemper R. K. (2016) Identification and characterization of influenza virus entry inhibitors through dual myxovirus high-throughput screening. J. Virol. 90, 7368–7387 10.1128/JVI.00898-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krumm S. A., Ndungu J. M., Yoon J. J., Dochow M., Sun A., Natchus M., Snyder J. P., and Plemper R. K. (2011) Potent host-directed small-molecule inhibitors of myxovirus RNA-dependent RNA-polymerases. PLoS One 6, e20069 10.1371/journal.pone.0020069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiménez-Somarribas A., Mao S., Yoon J. J., Weisshaar M., Cox R. M., Marengo J. R., Mitchell D. G., Morehouse Z. P., Yan D., Solis I., Liotta D. C., Natchus M. G., and Plemper R. K. (2017) Identification of non-nucleoside inhibitors of the respiratory syncytial virus polymerase complex. J. Med. Chem. 60, 2305–2325 10.1021/acs.jmedchem.6b01568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoon J. J., Toots M., Lee S., Lee M. E., Ludeke B., Luczo J. M., Ganti K., Cox R. M., Sticher Z. M., Edpuganti V., Mitchell D. G., Lockwood M. A., Kolykhalov A. A., Greninger A. L., Moore M. L., et al. (2018) Orally efficacious broad-spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses. Antimicrob. Agents Chemother. 62, e00766–18 10.1128/AAC.00766-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peterson L. A. (2013) Reactive metabolites in the biotransformation of molecules containing a furan ring. Chem. Res. Toxicol. 26, 6–25 10.1021/tx3003824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noton S. L., Deflubé L. R., Tremaglio C. Z., and Fearns R. (2012) The respiratory syncytial virus polymerase has multiple RNA synthesis activities at the promoter. PLoS Pathog. 8, e1002980 10.1371/journal.ppat.1002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noton S. L., Nagendra K., Dunn E. F., Mawhorter M. E., Yu Q., and Fearns R. (2015) Respiratory syncytial virus inhibitor AZ-27 differentially inhibits different polymerase activities at the promoter. J. Virol. 89, 7786–7798 10.1128/JVI.00530-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asakawa N., Kobayashi S., Goto J., and Hirayama N. (2012) AutoGPA: an automated 3D-QSAR method based on pharmacophore alignment and grid potential analysis. Int. J. Med. Chem. 2012, 498931 10.1155/2012/498931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baker K. A., Dutch R. E., Lamb R. A., and Jardetzky T. S. (1999) Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 3, 309–319 10.1016/S1097-2765(00)80458-X [DOI] [PubMed] [Google Scholar]
- 34. Stokes K. L., Chi M. H., Sakamoto K., Newcomb D. C., Currier M. G., Huckabee M. M., Lee S., Goleniewska K., Pretto C., Williams J. V., Hotard A., Sherrill T. P., Peebles R. S. Jr, and Moore M. L. (2011) Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 85, 5782–5793 10.1128/JVI.01693-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rydberg P., Gloriam D. E., Zaretzki J., Breneman C., and Olsen L. (2010) SMARTCyp: a 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett. 1, 96–100 10.1021/ml100016x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marroquin L. D., Hynes J., Dykens J. A., Jamieson J. D., and Will Y. (2007) Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 97, 539–547 10.1093/toxsci/kfm052 [DOI] [PubMed] [Google Scholar]
- 37. Wassermann A. M., Lounkine E., Hoepfner D., Le Goff G., King F. J., Studer C., Peltier J. M., Grippo M. L., Prindle V., Tao J., Schuffenhauer A., Wallace I. M., Chen S., Krastel P., Cobos-Correa A., et al. (2015) Dark chemical matter as a promising starting point for drug lead discovery. Nat. Chem. Biol. 11, 958–966 10.1038/nchembio.1936 [DOI] [PubMed] [Google Scholar]
- 38. Xiong H., Foulk M., Aschenbrenner L., Fan J., Tiong-Yip C. L., Johnson K. D., Moustakas D., Fleming P. R., Brown D. G., Zhang M., Ferguson D., Wu D., and Yu Q. (2013) Discovery of a potent respiratory syncytial virus RNA polymerase inhibitor. Bioorg. Med. Chem. Lett. 23, 6789–6793 10.1016/j.bmcl.2013.10.018 [DOI] [PubMed] [Google Scholar]
- 39. Tiong-Yip C. L., Aschenbrenner L., Johnson K. D., McLaughlin R. E., Fan J., Challa S., Xiong H., and Yu Q. (2014) Characterization of a respiratory syncytial virus L protein inhibitor. Antimicrob. Agents Chemother. 58, 3867–3873 10.1128/AAC.02540-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Antoni D., Burckel H., Josset E., and Noel G. (2015) Three-dimensional cell culture: a breakthrough in vivo. Int. J. Mol. Sci. 16, 5517–5527 10.3390/ijms16035517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eglen R., and Reisine T. (2011) Primary cells and stem cells in drug discovery: emerging tools for high-throughput screening. Assay Drug Dev. Technol. 9, 108–124 10.1089/adt.2010.0305 [DOI] [PubMed] [Google Scholar]
- 42. Heylen E., Neyts J., and Jochmans D. (2017) Drug candidates and model systems in respiratory syncytial virus antiviral drug discovery. Biochem. Pharmacol. 127, 1–12 10.1016/j.bcp.2016.09.014 [DOI] [PubMed] [Google Scholar]
- 43. Lipinski C. A., Lombardo F., Dominy B. W., and Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 10.1016/S0169-409X(00)00129-0 [DOI] [PubMed] [Google Scholar]
- 44. Doak B. C., Over B., Giordanetto F., and Kihlberg J. (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 21, 1115–1142 10.1016/j.chembiol.2014.08.013 [DOI] [PubMed] [Google Scholar]
- 45. Krumm S. A., Yan D., Hovingh E. S., Evers T. J., Enkirch T., Reddy G. P., Sun A., Saindane M. T., Arrendale R. F., Painter G., Liotta D. C., Natchus M. G., von Messling V., and Plemper R. K. (2014) An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci. Transl. Med. 6, 232ra252 10.1126/scitranslmed.3008517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ndungu J. M., Krumm S. A., Yan D., Arrendale R. F., Reddy G. P., Evers T., Howard R., Natchus M. G., Saindane M. T., Liotta D. C., Plemper R. K., Snyder J. P., and Sun A. (2012) Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase: synthesis, structure-activity relationships, and pharmacokinetics. J. Med. Chem. 55, 4220–4230 10.1021/jm201699w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cherkasov A., Muratov E. N., Fourches D., Varnek A., Baskin I. I., Cronin M., Dearden J., Gramatica P., Martin Y. C., Todeschini R., Consonni V., Kuz'min V. E., Cramer R., Benigni R., Yang C., Rathman J., Terfloth L., Gasteiger J., Richard A., and Tropsha A. (2014) QSAR modeling: where have you been? Where are you going to? J. Med. Chem. 57, 4977–5010 10.1021/jm4004285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Makhuri F. R., and Ghasemi J. B. (2015) Computer-aided scaffold hopping to identify a novel series of casein kinase 1 delta (CK1d) inhibitors for amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 78, 151–162 10.1016/j.ejps.2015.07.011 [DOI] [PubMed] [Google Scholar]
- 49. Nie Q., Xu X., Zhang Q., Ma Y., Yin Z., and Shang L. (2018) 3D-quantitative structure-activity relationship study for the design of novel enterovirus A71 3C protease inhibitors. Chem. Biol. Drug Des. 10.1111/cbdd.13344 10.1111/cbdd.13344 [DOI] [PubMed] [Google Scholar]
- 50. Abdolmaleki A., Ghasemi J. B., and Ghasemi F. (2017) Computer aided drug design for multi-target drug design: SAR/QSAR, molecular docking and pharmacophore methods. Curr. Drug Targets 18, 556–575 10.2174/1389450117666160101120822 [DOI] [PubMed] [Google Scholar]
- 51. Wang W., Zhang Y., Wang N., and Zhu Z. (2014) Molecular mechanisms of several novel dipeptides with angiotensin-I-converting enzyme inhibitory activity from in-silico screening of silkworm pupae protein. Curr. Pharm. Biotechnol. 15, 691–699 10.2174/138920101508140930153336 [DOI] [PubMed] [Google Scholar]
- 52. Shiri F., Ghasemi B. S. J. B. (2015) Computer-aided molecular design of (E)-N-aryl-2-ethene-sulfonamide analogues as microtubule targeted agents in prostate cancer. Arab. J. Chem. 10.1016/j.arabjc.2014.11.063 10.1016/j.arabjc.2014.11.063 [DOI] [Google Scholar]
- 53. Cramer R. D., Patterson D. E., and Bunce J. D. (1988) Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 110, 5959–5967 10.1021/ja00226a005 [DOI] [PubMed] [Google Scholar]
- 54. Dixon S. L., Smondyrev A. M., Knoll E. H., Rao S. N., Shaw D. E., and Friesner R. A. (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. methodology and preliminary results. J. Comput. Aided Mol. Des. 20, 647–671 10.1007/s10822-006-9087-6 [DOI] [PubMed] [Google Scholar]
- 55. Klebe G., and Abraham U. (1999) Comparative molecular similarity index analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aided Mol. Des. 13, 1–10 10.1023/A:1008047919606 [DOI] [PubMed] [Google Scholar]
- 56. Lewis D. F., and Ito Y. (2008) Human cytochromes P450 in the metabolism of drugs: new molecular models of enzyme-substrate interactions. Expert Opin. Drug Metab. Toxicol. 4, 1181–1186 10.1517/17425255.4.9.1181 [DOI] [PubMed] [Google Scholar]
- 57. Ali J., Camilleri P., Brown M. B., Hutt A. J., and Kirton S. B. (2012) Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 52, 420–428 10.1021/ci200387c [DOI] [PubMed] [Google Scholar]
- 58. Delaney J. S. (2004) ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 44, 1000–1005 10.1021/ci034243x [DOI] [PubMed] [Google Scholar]
- 59. Duprex W. P., McQuaid S., Hangartner L., Billeter M. A., and Rima B. K. (1999) Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 73, 9568–9575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jacob R. T., Larsen M. J., Larsen S. D., Kirchhoff P. D., Sherman D. H., and Neubig R. R. (2012) MScreen: an integrated compound management and high-throughput screening data storage and analysis system. J. Biomol. Screen. 17, 1080–1087 10.1177/1087057112450186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hotard A. L., Shaikh F. Y., Lee S., Yan D., Teng M. N., Plemper R. K., Crowe J. E. Jr., and Moore M. L. (2012) A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 434, 129–136 10.1016/j.virol.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cox R. M., Krumm S. A., Thakkar V. D., Sohn M., and Plemper R. K. (2017) The structurally disordered paramyxovirus nucleocapsid protein tail domain is a regulator of the mRNA transcription gradient. Sci. Adv. 3, e1602350 10.1126/sciadv.1602350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dang N. L., Hughes T. B., Krishnamurthy V., and Swamidass S. J. (2016) A simple model predicts UGT-mediated metabolism. Bioinformatics 32, 3183–3189 10.1093/bioinformatics/btw350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.