

Abstract
Fatty acid channeling into oxidation or storage modes depends on physiological conditions and hormonal signaling. However, the directionality of this channeling may also depend on the association of each of the five acyl-CoA synthetase isoforms with specific protein partners. Long-chain acyl-CoA synthetases (ACSLs) catalyze the conversion of long-chain fatty acids to fatty acyl-CoAs, which are then either oxidized or used in esterification reactions. In highly oxidative tissues, ACSL1 is located on the outer mitochondrial membrane (OMM) and directs fatty acids into mitochondria for β-oxidation. In the liver, however, about 50% of ACSL1 is located on the endoplasmic reticulum (ER) where its metabolic function is unclear. Because hepatic fatty acid partitioning is likely to require the interaction of ACSL1 with other specific proteins, we used an unbiased protein interaction technique, BioID, to discover ACSL1-binding partners in hepatocytes. We targeted ACSL1 either to the ER or to the OMM of Hepa 1–6 cells as a fusion protein with the Escherichia coli biotin ligase, BirA*. Proteomic analysis identified 98 proteins that specifically interacted with ACSL1 at the ER, 55 at the OMM, and 43 common to both subcellular locations. We found subsets of peroxisomal and lipid droplet proteins, tethering proteins, and vesicle proteins, uncovering a dynamic role for ACSL1 in organelle and lipid droplet interactions. Proteins involved in lipid metabolism were also identified, including acyl-CoA–binding proteins and ceramide synthase isoforms 2 and 5. Our results provide fundamental and detailed insights into protein interaction networks that control fatty acid metabolism.
Keywords: protein–protein interaction, ceramide synthase, β-oxidation, peroxisome, lipid droplet, mitochondria, hepatocyte, acyl-CoA synthetase-1, BioID, SNAP23
Introduction
Major pathways for lipid synthesis and degradation are physically separated by their locations within distinct intracellular organelles, but evidence is growing that lipid intermediates destined for independent fates can also be channeled within a single compartment such as the cytosol. Despite their amphipathicity and water solubility, such channeling appears to occur for long-chain acyl-CoAs. In addition to physiological controls such as the AMP kinase-regulated production of malonyl-CoA, which regulates carnitine palmitoyltransferase-1 (CPT-1) activity and enhances β-oxidation, the fate of fatty acids (FA)2 seems also to be determined by their origin from an exogenous source or from their de novo intracellular synthesis. Activation by distinct members of the long-chain acyl-CoA synthetase (ACSL) family mediates FA fates. Thus, mice deficient in ACSL5, the major intestinal ACSL isoform, show delayed absorption of dietary triacylglycerol, increased energy expenditure, and decreased adipose tissue (1), whereas an adipose-specific knockout of Acsl4 reduces arachidonate incorporation into phospholipids (2). Our studies of tissue-specific knockouts indicate that ACSL1 is critical for directing FA into the mitochondria for β-oxidation in brown and white adipose (3), heart (4–6), and skeletal muscle (7). In fact, during endurance exercise in Acsl1 null mice, whereas the content of skeletal muscle acyl-CoA increases dramatically, it is compartmentalized or sequestered such that it cannot be used for fuel (7). In oxidative tissues, ACSL1 is primarily located on the outer mitochondrial membrane (OMM), consistent with its role in directing FA toward oxidation. In liver, however, ACSL1 is located on the endoplasmic reticulum (ER) as well as the OMM, but the function of ACSL1 in either location is unclear, because the deficiency has a minimal effect on either β-oxidation or glycerolipid synthesis (8).
Several factors may contribute to the ability of ACSL1 to direct FA toward synthetic pathways versus mitochondrial oxidation, including its subcellular location; ACSL1 is anchored to the ER or to the OMM by a single N-terminal transmembrane domain (9). In addition, we showed that different physiological stimuli regulate ACSL1 post-translationally by altering phosphorylation and acetylation of specific amino acid residues, although the function of these modifications remains obscure (10). We have hypothesized that specific interactions of ACSL1 with networks of associated proteins might have a major influence on function. A proteomics analysis reported physical interactions between ACSL1, voltage-dependent anion channel (VDAC), and CPT-1 in liver OMM (11), suggesting an explanation for why a deficiency of ACSL1 impairs FA oxidation. However, our studies show that exogenous FA oxidation in liver is only partially impaired in the absence of ACSL1, suggesting that the association with CPT-1 might not provide a full explanation or that other ACSL isoforms might compensate for lack of ACSL1 (8).
Thus, to understand the specific subcellular functions of ACSL1 in liver, we used the BioID proximity biotinylation technique (12–14) to compare the protein interactomes of ACSL1 targeted to either the ER or the OMM. To capture weak or transient protein interactions at both subcellular locations, we expressed ER- or OMM-targeted ACSL1 as fusion proteins with BirA* in Hepa 1–6 cells and used primary mouse hepatocytes to validate the results for a number of specific proteins. Our results strongly support the idea that ACSL1 can channel FA into defined downstream pathways via a network of interacting proteins, and we show how physiological changes can transiently shape these interactions.
Results
ER- and OMM-targeted ACSL1 interact with distinct protein networks
Hepatic ACSL1 is expressed at the OMM, the ER, and at mitochondria-associated membranes (MAM), an ER region that is reversibly tethered to mitochondria and involved in calcium and phospholipid transport (Fig. 1A and supporting information 1) (15). In mice with a liver-specific knockout of Acsl1 (Acsl1L−/−) (8), ACSL-specific activity in each location decreases by 50–70% (Fig. 1B). Because the N terminus is required for insertion into the membrane, we positioned the BirA* tag at the C terminus. ACSL1 expression was targeted to the ER using a chimera of FATP4 (aa 1–46) and ACSL1 (aa 47–699), denoted as ER–ACSL1–BirA*. To study interactions at the OMM, a TOM70 sequence (aa 1–69) was integrated into the N terminus of ACSL1 (aa 46–699), denoted as mito-ACSL1–BirA* (Fig. 1C). Organelle-targeted fluorescent protein controls were generated to eliminate nonspecific BirA* interactions (Fig. 1C) (16). Subcellular location was validated using immunofluorescence microscopy (Fig. 1, D and E). When expressed with a C-terminal BirA* tag, ER- and OMM-directed ACSL1 retained enzymatic function with specific activities similar to wildtype (WT) ACSL1 (Fig. 2A). The expression of BirA*-tagged proteins in the presence of biotin increased the abundance of protein biotinylation with different patterns for each of the targeted constructs (Fig. 2, B and C). We used streptavidin affinity chromatography to purify biotin-labeled proteins, which were then identified by LC-MS/MS. To reduce false-positive selection, only proteins enriched ≥5-fold with a p < 0.05 in the ACSL1 datasets versus organelle-specific controls were selected as positive protein interactors. Statistical analysis comparing the interactome of ACSL1 at either the OMM or ER confirmed a direct interaction of 98 proteins unique to the ER–ACSL1–BirA* sample and 55 proteins exclusively in the mito-ACSL1–BirA* sample. Forty three proteins were common to both datasets (Fig. 2D). Proteins were classified by molecular function based on Uniprot description and the primary literature (Figs. 3, A–C, and supporting information S2).
Figure 1.

ACSL1 is expressed at the ER and OMM. Organelles from control and Acsl1L−/− livers were separated by differential centrifugation. A, representative immunoblot of purified organelles from control livers. Each lane was loaded with 40 μg of protein. Mito, mitochondria; MAM, mitochondrial-associated membrane; ER, endoplasmic reticulum; Cyto, cytosol. B, ACSL-specific activity, measured with 50 μm [14C]palmitate, in organelles from control and Acsl1L−/− livers (n = 3); Hom, homogenate. *, p < 0.05 comparing genotypes. C, diagram of adenoviral constructs used for recombinant protein expression. BirA* with HA tag represents a nonspecific control. A TOM70/ACSL1 chimera was generated to allow OMM-specific ACSL1 expression: aa 1–69 of TOM70 replaced aa 1–45 of ACSL1. ER-directed ACSL1 was generated by aa 1–46 of ACSL1 with aa 1–46 of FATP4. Human OMM TOM20 (aa 1–30) fused to GFP was used to direct BirA* to the OMM. The RFP–Sec61β–BirA*–HA control was used to direct BirA* to the ER (70, 71). All constructs included a HA tag for Western blot analysis. D, Hepa 1–6 cells were infected with adenoviral constructs for BirA*, mito-GFP–BirA*, mito-ACSL1–BirA*, ER–ACSL1–BirA*, and ER–RFP–BirA*. Cells were incubated with CellLight® mitochondria–RFP or CellLight® mitochondria-GFP as indicated. Permeabilized cells expressing BirA*, mito-ACSL1–BirA*, and ER–ACSL1–BirA* were incubated with HA antibody to visualize BirA*-tagged proteins. The scale represents 10 μm. E, Hepa 1–6 cells were infected with adenoviral constructs for BirA*, ER–RFP–BirA*, mito-ACSL1–BirA*, ER–ACSL1–BirA*, and mito-GFP–BirA*. Cells were incubated with CellLight® ER–GFP as indicated. Permeabilized cells expressing BirA*, mito-ACSL1–BirA*, and ER–ACSL1–BirA* were incubated with HA antibody. Permeabilized cells expressing mito-GFP–BirA* were incubated with calnexin antibody. The scale represents 10 μm.
Figure 2.

ACSL1–BirA* spontaneously biotinylates endogenous proteins and remains catalytically active. A, ACSL-specific activity in Hepa 1–6 cells. Cell lysates from cells expressing respective BirA* constructs or WT–ACSL1–FLAG, were assayed in the presence of 50 μm [1-14C]palmitic acid. *, p < 0.05 compared with BirA* control, n = 4. FLAG Western blotting of lysates used in ACS assay with glyceraldehyde-3-phosphate dehydrogenase as the loading control. B, Hepa 1–6 cells were analyzed for HA-tagged protein expression 48 h after infection with adenoviral constructs. HA-tagged proteins were present in all cell lines with expression at the predicted molecular weight. C, cells were cultured in the presence or absence of 50 μm biotin for 24 h to induce protein biotinylation. Biotinylated proteins were detected by Western blotting with streptavidin-HRP. D, Venn diagram showing ACSL1-interacting proteins identified in each dataset. The 43 proteins are from the intersection of ER and OMM datasets (common interaction dataset).
Figure 3.

Functional categorization of high-confidence ACSL1–BirA* interactors. A, 43 proteins from common dataset; B, 98 proteins from the ER dataset; and C, 55 proteins from the OMM dataset were annotated for molecular function based on information in UniProt and the primary literature (complete classification is in supporting information S2).
Although protein overexpression and the addition of the BirA* tag could alter the interactome of ACSL1, this method identified a number of known ACSL1-binding partners, and ACSL1 enzymatic function was retained, indicating correct protein folding. The complete interactome of ACSL1 at both subcellular locations was subjected to STRING analysis, which revealed clusters of protein networks (Fig. 4, A and B). Surprisingly, we did not find interactions with the acyltransferases required for triacylglycerol (TAG) or cholesterol ester synthesis; instead, the proteins identified in the ER–ACSL1–BirA* dataset suggested that the cytosolic domain of ACSL1 participates in niche pathways involving ceramide synthesis and peroxisomal metabolism. A network of peroxisomal proteins was identified, most notably ABC subfamily D-3 (ABCD3), that may transport long chain acyl-CoAs into peroxisomes: ACBD5, which interacts with the ER proteins VAPA and VAPB (17, 18), and PEX14 and PEX26, which are involved in peroxisomal import (Fig. 4A). As expected, the analysis identified a number of proteins that can bind lipids or are involved in lipid metabolism. These proteins included ceramide synthase isoform 2 (CerS2) and ceramide synthase isoform 5 (CerS5), which had previously been shown to interact with ACSL1 (19). Additionally identified in the ER–ACSL1–BirA* cohort were two acyl-CoA–binding domain-containing proteins, ACBD3 and ACBD5. ACBD3, also known as GCP60, is predominantly associated with the Golgi but can be found at the ER and mitochondria (20). It is involved in steroidogenesis (21–23) and is implicated in ER–Golgi ceramide transport (24). ACBD5 is a peroxisomal acyl-CoA–binding protein that participates in ER–peroxisome tethering (17, 18). ACSL1, ACSL5, and ACSL6 isoforms were also identified. BirA*-tagged proteins are known to self-biotinylate; therefore, ACSL1 peptides present in both the ER and OMM datasets cannot be distinguished from endogenous ACSL1 protein. Protein complexes composed of several ACSL isoforms have previously been reported (11). STRING analysis identified noteworthy clusters of associated/interacting proteins in the mitochondria dataset, including ACSL3 and several SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins, including SNAP23 (synaptosomal-associated protein 23), VAMP (vesicle-associated membrane protein) 2, 4, and 5, and syntaxin 7 and 8 (Fig. 4B). SNARE proteins mediate membrane fusion, suggesting a role for ACSL1 at membrane contact sites between mitochondria and other organelles or vesicles. Also of interest in the mito-ACSL1–BirA* dataset was a cluster of proteins surrounding EGFR. EGFR and GPRC5A are known to interact at the plasma membrane (25), and EGFR can be translocated to the mitochondrial membrane (26), where it participates in mitochondrial fusion (27). A recent publication characterized the proteome of lipid droplets using proximity biotinylation (28). We compared our own data to this lipid droplet proteome of human hepatoma Huh7 cells, to determine overlapping proteins. Eighteen percent of the proteins in our ER–ACSL1–BirA dataset, 20% of the proteins in our OMM–ACSL1 interactome, and 14% of the ACSL1-interacting proteins at both the ER and OMM were identified as lipid droplet proteins (supporting information S3). This comparison with an unrelated dataset (28) provides strong support for the interpretation that ACSL1 interacts with proteins located on lipid droplets. For this study, we selected for investigation ACSL1-binding partners that were associated with lipid transport and metabolism.
Figure 4.

Network analysis of ACSL1–BirA*-interacting proteins classified by subcellular location. ACSL1-interacting proteins were analyzed using the STRING database version 10.5., and data were modified for presentation using Cytoscape version 3.5.1. Proteins were grouped by subcellular location based on Uniprot annotations. Nodes represent the protein identified; edges represent protein–protein associations based on known and predicted protein interactions, homology, co-expression, and text mining. The node color gradient depicts fold change in protein presence in the targeted ACSL1–BirA* dataset compared with the BirA*-targeted control. Nodes outlined in red were confirmed as lipid droplet proteins (28). A, combined network of ACSL1-interacting proteins identified at the ER and intersection datasets. B, combined network of ACSL1-interacting proteins identified at the OMM and intersection datasets.
ER-targeted ACSL1 interacted with ER-peroxisomal tethering proteins and proteins involved in peroxisomal FA metabolism
A recent BioID study conducted by Hua et al. (18) described interactions between VAPA, VAPB, and ACBD5, which promote peroxisome tethering to the ER and facilitate lipid exchange (17, 18). Using diazirine photocross-linking and co-immunoprecipitation in mouse hepatocytes, we confirmed a direct interaction between ACSL1 and endogenously expressed ACBD5 and VAPB (Fig. 5, A and B). Because ACSL1 may be present at the ER–peroxisome interface to activate FA destined for peroxisomal metabolism, we investigated several ACSL1-interacting proteins that are required for peroxisomal metabolism of branched-chain FA. Phytol, the most prominent dietary branched-chain FA, is first converted to phytenal and oxidized by ALDH3A2 (FALDH) to generate phytenic/phytanic acid, which is then activated to an acyl-CoA that is oxidized in peroxisomes. Both ACSL1 and ACSVL1 can activate branched-chain FA (29, 30), but it is still unclear whether phytanic acid is activated at the ER or at the peroxisome membrane itself (31). We validated the BioID interaction between ER-targeted ACSL1 and ALDH3A2 by using ACSL1–FLAG to co-immunoprecipitate endogenous ALDH3A2 (Fig. 5C). Because the peroxisomal transport of branched-chain FA involves the ABC transporter ABCD3, and Abcd3−/− mice fed a high phytol diet accumulated phytanic acid in serum (32), we hypothesized that ACSL1 might interact with proteins in the phytol metabolic pathway to facilitate the activation of phytanic acid to phytanoyl-CoA, which is then transported across the peroxisomal membrane. After confirming an interaction between endogenous ABCD3 and ACSL1 (Fig. 5D), we investigated phytanic acid esterification and oxidation in mice that lacked liver ACSL1 (Acsl1L−/−). We previously showed that ACSL activity is 50% lower in Acsl1L−/− liver compared with floxed littermate controls (Acsl1Lfl/fl) when using palmitate as substrate (Fig. 5E) (8), but surprisingly, ACSL activity using phytanic acid was marginally increased in Acsl1L−/− liver (Fig. 5E). Phytanic acid oxidation in Acsl1L−/− liver was not perturbed (Fig. 5F), but its basal level of oxidation was low compared with that of palmitate.
Figure 5.

ACSL1 interacts with key peroxisomal proteins. Primary murine hepatocytes were isolated from two C57BL/6J males and one female that had been fed a standard chow diet. Cells were infected with either Ad-GFP or Ad-ACSL1–FLAG to induce expression of GFP or ACSL1–FLAG. Protein complexes were cross-linked, and FLAG-tagged proteins were isolated with FLAG affinity resin and eluted with FLAG peptide. Interacting proteins were identified using antibodies directed against native proteins. A–D, Co-IP of ACSL1–FLAG and VAPB (A), ACD5 (B), ALDH3A2 (C), and ABCD3 (D). E, ACS-specific activity was measured with either 50 μm [1-14C]phytanic acid or [1-14C]palmitic acid in total membranes isolated from Acsl1Lfl/fl and Acsl1L−/− liver homogenates. Data show means from three mice per group. Data are presented ± S.E., *, p < 0.05 compared with Acsl1Lfl/fl. F, [1-14C]phytanic acid oxidation to ASM metabolites and CO2 was measured in freshly isolated liver homogenates (n = 3). Data are presented as means ± S.E. IP, immunoprecipitation.
To elevate basal levels of phytanic acid oxidation, we induced peroxisomal proliferation by feeding mice the PPARα agonist, Wy14643. Male Acsl1Lfl/fl or Acsl1L−/− mice were fed either 0.5% carboxymethylcellulose (vehicle) or 25 mg/kg Wy14643 for 5 days. Compared with vehicle-treated counterparts, phytanoyl-CoA production increased in both genotypes treated with Wy14643, indicating elevated ACSL activity by additional isoforms (Fig. 6A). Interestingly, phytanoyl-CoA production was 18% lower in vehicle-treated Acsl1L−/− compared with Acsl1Lfl/fl, suggesting that ACSL1 composed 18% of total phytanoyl-CoA synthesizing activity, although acyl-CoA production from palmitate was markedly reduced in Acsl1L−/− liver (Fig. 5E). After Wy14643 treatment, palmitoyl-CoA production increased minimally in Acsl1L−/− liver, but palmitoyl-CoA production in Acsl1Lfl/fl was 30% higher than in the vehicle control, again consistent with the activity of alternative ACSL isoforms. We next measured liver FA oxidation with phytanic acid and palmitate. No difference in oxidation was observed with either FA (Fig. 6B). Compared with WT mice, liver from Acsl1L−/− mice contains 2-fold higher levels of C8:0-carnitine, likely due to increased peroxisomal β-oxidation (8). To assess whether the Acsl1 deletion altered the ability of peroxisomes to oxidize FA, we homogenized liver in the absence or presence of 100 μm etomoxir to irreversibly block CPT-1 and inhibit mitochondrial FA oxidation in liver homogenates from Acsl1Lfl/fl and Acsl1L−/− mice treated with vehicle or Wy14643. Without etomoxir treatment, no difference was observed between groups (Fig. 6C). However, when etomoxir was present, palmitate oxidation was reduced by 44% in vehicle-treated Acsl1Lfl/fl liver, reflecting the block in CPT-1–controlled mitochondrial oxidation. In liver from Acsl1Lfl/fl mice treated with Wy14643, the suppression of palmitate oxidation by etomoxir was less, suggesting a PPARα-mediated increase in peroxisomal oxidation that compensated for the loss of CPT-1 function. In Acsl1L−/− liver, however, etomoxir had no effect on palmitate oxidation in either vehicle- or Wy14643-treated mice. Thus, it appeared that the loss of Acsl1 in liver had stimulated an increase in peroxisomal FA oxidation, which compensated for diminished mitochondrial β-oxidation. To test this interpretation, we analyzed gene expression in Acsl1Lfl/fl and Acsl1L−/− liver homogenates after treating mice with vehicle or Wy14643. As expected, several FA oxidation genes were significantly up-regulated with Wy14643 treatment in Acsl1Lfl/fl and Acsl1L−/− mice (Fig. 6D). Surprisingly, despite increased peroxisomal oxidation in vehicle-treated Acsl1L−/− mice compared with Acsl1Lfl/fl mice, gene expression did not differ between these two groups (Fig. 6D). Wy14643 up-regulated phytol-metabolizing genes, including Abcd3 and both isoforms of Aldh3a2, Aldh3a2-V and Aldh3a2-N. However, the up-regulation of mRNA expression for genes in the phytol metabolic pathway did not result in elevated phytanic acid metabolism, and no genotype differences in phytanic acid oxidation occurred.
Figure 6.
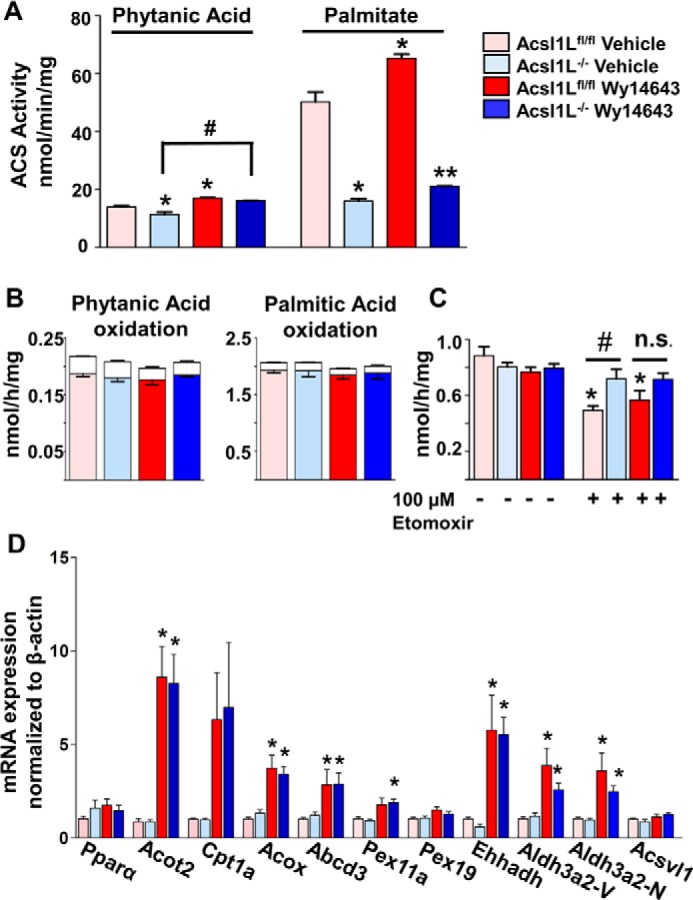
Acsl1 liver-specific deletion activates peroxisomal FA oxidation. Male Acsl1Lfl/fl or Acsl1L−/− mice (at 7 weeks) were treated with vehicle or 25 mg/kg Wy14643 for 5 days. Mice were sacrificed 24 h (including 4 h of starvation) after the last oral dose. A, liver ACSL-specific activities were measured with 50 μm [1-14C]phytanic acid or [1-14C]palmitic acid in total membranes isolated from Acsl1Lfl/fl and Acsl1L−/− liver homogenates. Graphs show means from three mice per group ± S.E. Data were analyzed using one-way ANOVA and Tukey's multiple comparisons test; *, p < 0.05 compared with vehicle-treated Acsl1Lfl/fl; #, p < 0.05 for Acsl1L−/− + vehicle compared with Acsl1L−/− + Wy14643; **; p < 0.05 for Wy14643-treated Acsl1Lfl/fl compared with Wy14643-treated Acsl1L−/−. B, [1-14C]phytanic acid and [1-14C]palmitic acid oxidation to ASM and CO2 were measured in freshly isolated liver homogenates. Colored bars represent ASM, and white bars represent CO2 produced. Data show means from three mice per group ± S.E. C, [1-14C]palmitic acid oxidation to ASM was measured in freshly isolated liver homogenates in the absence or presence of 100 μm etomoxir. Data show means from six mice per group ± S.E. Data were analyzed using one-way ANOVA and Tukey's multiple comparisons test, *, p < 0.05 compared with vehicle-treated Acsl1Lfl/fl counterpart without etomoxir; #, p < 0.05 compared with vehicle-treated Acsl1Lfl/fl control with 100 μm etomoxir. n.s., nonsignificant for Acsl1L−/− + Wy14643. D, gene expression in liver from mice treated with vehicle or Wy14643 for 5 days (n = 6). Data were analyzed using one-way ANOVA, Tukey's multiple comparisons test; *, p < 0.05 compared with vehicle-treated counterpart.
ER-targeted ACSL1 interacted with CerS2 and CerS5
We confirmed the direct interaction of ACSL1 with CerS2 and CerS5 in Hepa 1–6 cells. Because antibodies were not available to identify endogenous CerS2 and CerS5, we co-expressed ACSL1-FLAG with CerS2-HA or CerS5-HA. ACSL1 was purified with FLAG affinity resin, and the presence of HA-tagged proteins was verified by Western blotting (Fig. 7, A and B). Because Senkal et al. (19) demonstrated that ACSL5 interacts with ceramide synthases in HCT116 colorectal carcinoma cells to generate unique acylceramide species, we asked whether ACSL1 plays a similar role in the liver. Ceramide synthase isoforms display distinct substrate preferences; CerS5 generates C16:0-ceramide, whereas CerS2 is responsible for the synthesis of C20–26-ceramides (33). Therefore, in Acsl1L−/− liver we anticipated changes in the ceramide species generated by CerS2 and CerS5. In Acsl1Lfl/fl and Acsl1L−/− liver from 8- to 10-week-old males fed a chow diet, no significant differences in sphingolipid levels were observed (supporting information S4). We fed Acsl1Lfl/fl and Acsl1L−/− male and female mice either a low-fat diet (LFD) or a high-fat diet (HFD) for 6 weeks to induce ceramide and acylceramide synthesis in liver. When mice were fed a LFD, combined male and female data showed a modest decrease in C12-ceramide levels in Acsl1L−/− liver compared with controls (supporting information S5, C and D). Acsl1 deletion also significantly lowered levels of C22-ceramide in male Acsl1L−/− mice fed a HFD (Fig. 7C). Hepatic acylceramide levels did not reveal sex divergence (Fig. 8B), but compared with littermate controls, C26 and C26:1 acylceramides were 50% lower in male and female Acsl1L−/− mice (Fig. 8A).
Figure 7.

ACSL1 interacts with CerS2 and CerS5. Hepa 1–6 cells were infected with either Ad-GFP or Ad-ACSL1–FLAG to induce expression of GFP or ACSL1–FLAG, concomitant with CerS2-HA or CerS5-HA, as indicated. Protein complexes were cross-linked with SDAD, isolated with FLAG affinity resin from cell lysates, and eluted with FLAG peptide. Interacting proteins were identified by HA immunoblot. A, validation of ACSL1 and CerS2 interaction. IP, immunoprecipitation. B, validation of ACSL1 and CerS5 interaction. Data are representative of three independent experiments. C, lipidomics analysis of ceramide species in liver from 8- to 10-week-old HFD-fed female (F) (n = 5) and male (M) mice (Acsl1Lfl/fl n = 4, Acsl1L−/− n = 5) *, p < 0.05 compared with Acsl1Lfl/fl. Inset, total ceramide in male and female livers; #, p < 0.05 compared with male counterpart. Data are presented as means ± S.E.
Figure 8.

Acsl1L−/− liver is depleted of specific acylceramide species. A, lipidomics analysis of acylceramide species in liver from 8- to 10-week-old male and female mice fed a HFD (Acsl1Lfl/fl n = 9, Acsl1L−/− n = 10). Data are presented as means ± S.E., *, p < 0.05 compared with Acsl1Lfl/fl. B, total acylceramide levels in livers from male and female mice fed a HFD. C, mRNA analysis of hepatic ceramide synthase expression in (combined) male and female Acsl1Lfl/fl and Acsl1L−/− mice fed a HFD (Acsl1Lfl/fl n = 9 and Acsl1L−/− n = 10). Data are presented means ± S.E., *, p < 0.05 compared with Acsl1Lfl/fl.
To determine whether the absence of ACSL1 altered specific isoforms of ceramide synthase, we examined their expression in liver. Hepatic CerS2 expression was significantly lower in Acsl1L−/− mice fed a HFD compared with Acsl1Lfl/fl controls. Because CerS2 displays a preference for very-long chain FA (C22–26) (33), this decrease in CerS2 expression could contribute to the reductions observed in C22-ceramide in male livers and to the low C26 and C26:1 acylceramides found in males and females.
We also found that hepatic levels of several ceramide species showed distinct sex-dependent differences in mice fed these diets: total ceramide was higher in male mice than in females (Fig. 7C and supporting information S5B), particularly C20–C22 chain length ceramide. Sex differences have not been previously described for these metabolites. No major changes in ceramide (supporting information S5C) or acylceramide levels (data not shown) were observed when mice were fed the LFD. C12-ceramide did not display a sex-dependent difference, but interestingly, sphingosine and dihydrosphingosine levels were higher in LFD-fed female mice (supporting information S5A).
Mitochondrial ACSL1 interacts with lipid droplet (LD) tethering proteins
Although we did not detect CPT-1 peptides in the mito-ACSL1–BirA* dataset, liver ACSL1 and CPT-1 co-immunoprecipitated (Fig. 9B), an interaction that had previously been observed (11). CPT-1 might not have been detected if its structure or orientation within the OMM precludes exposed lysine residues. Because ACSL1 is essential for FA oxidation in oxidative tissues (3, 6, 7), a direct interaction with CPT-1 should facilitate FA flux into mitochondria for efficient FA oxidation. The association of OMM-targeted ACSL1 with SNARE complex proteins, known to interact with both LDs and the OMM, suggested the presence of an assembled group of proteins that would enhance the flow of FA released from LDs into the mitochondrial oxidation machinery. The SNARE complex protein SNAP23, for example, was 40-fold higher in the mito-ACSL1–BirA* dataset compared with the mitochondria-targeted GFP control (supporting information S2). Although SNAP23 is traditionally thought to participate in exocytic vesicle docking at the plasma membrane, it is also located at mitochondria where it is believed to associate LDs with the mitochondrial membrane, particularly during fasting (34–36). We confirmed the interaction of the SNARE complex proteins, SNAP23 and VAMP4, with ACSL1–FLAG by chemical cross-linking and co-immunoprecipitation of the endogenous proteins, but this association was substantiated only when primary hepatocytes were deprived of glucose (Fig. 9B). In contrast, the interaction of ACSL1–FLAG with CPT-1 remained unchanged irrespective of glucose concentration. Because FA oxidation in Hepa 1–6 cells can be induced with glucose deprivation, we asked whether the interaction between ACSL1 and SNAP23 during glucose deprivation might enhance FA oxidation. We incubated Hepa 1–6 cells with DMEM containing either 25 mm glucose or no glucose for 3 h and then replaced media with the same media containing [3H]palmitic acid for an additional 3 h. Compared with cells cultured with a high-glucose concentration, oxidation of [3H]palmitic acid to [3H]H2O was markedly elevated in cells starved of glucose (Fig. 9C). Conversely, a high-glucose concentration resulted in increased incorporation of [3H]palmitic acid into total lipid, reflecting the partitioning of palmitate into synthetic versus oxidative pathways. Thus, the interaction of SNAP23 and VAMP4 with ACSL1 during glucose deprivation, a condition that primes the cell to oxidize more FA, is consistent with tethering of LDs to the OMM during FA oxidation. To determine whether the absence of SNAP23 might diminish FA oxidation, we used endoribonuclease-prepared siRNA (esiRNA) to knock down SNAP23 levels in Hepa 1–6 cells (Fig. 9, D and F). However, in either low or high glucose, SNAP23 knockdown had no effect on the oxidation of exogenous palmitate (Fig. 9C). To determine whether oxidation was affected if the FA were derived directly from the LD, we used a pulse/chase system. After Hepa 1–6 cells were treated with esiRNA targeting either GFP or SNAP23 for 48 h, cells were pulsed with 200 μm [1-14C]oleic acid for 3 h in the presence of 5 or 25 mm glucose. After replacing with label-free media (chase), media samples were removed at 1, 3, and 6 h to measure FA oxidation. Although rates of lipolysis were clearly higher in cells pre-cultured in low glucose, diminished SNAP23 expression had no further effect on oleate lipolysis (Fig. 9E), suggesting that tethering by SNAP23 was not essential for the flow of FA from the LDs into the mitochondria.
Figure 9.

ACSL1 interacts with lipid droplet tethering proteins when glucose is absent. Primary murine hepatocytes were isolated from C57BL/6J mice maintained on a standard chow diet. Cells were infected with either Ad-GFP or Ad-ACSL1–FLAG. Protein complexes were cross-linked, and FLAG-tagged proteins were isolated with FLAG affinity resin and eluted with FLAG peptide. A, CPT-1 was identified by immunoblot. B, primary murine hepatocytes were infected with Ad-GFP or Ad-ACSL1–FLAG. The media were replaced with fresh media containing 25 mm glucose and 1 mm pyruvate or media lacking glucose and pyruvate for 16 h before collection and FLAG-affinity chromatography. Interacting proteins were identified using antibodies directed against CPT-1, SNAP23, and VAMP4. Data are representative of three independent experiments. C, Hepa 1–6 cells were cultured for 3 h in DMEM containing either 0 mm glucose, 0 mm pyruvate, or 25 mm glucose and 1 mm pyruvate. Fresh media containing [3H]palmitate and 100 mm carnitine were added for 3 h. Oxidation was measured by counting [3H]H2O released into the media (n = 4). Incorporation of palmitate was measured by extracting total lipid and counting 3H levels (n = 3). D, Hepa 1–6 cells were transfected with 0.5 μg of esiRNA targeting GFP or SNAP23 and cultured for 48 h in the presence of 5 mm glucose (n = 3) or 25 mm glucose (n = 2). [1-14C]Palmitic acid oxidation to ASM and CO2 was measured. Data are presented as means ± S.E. E, Hepa 1–6 cells were transfected with 0.5 μg of esiRNA targeting GFP or SNAP23 and cultured for 48 h in the presence of 5 mm glucose (n = 2) or 25 mm glucose (n = 3). Cells were incubated with 100 μm [1-14C]oleate for 3 h (pulse) and then washed with 1% BSA in PBS before unlabeled chase media was added. [1-14C]Oleate oxidation to ASM and CO2 was measured at 1, 3, and 6 h. White bars represent CO2 production; colored bars represent ASM production. Data are presented as means ± S.E. F, SNAP23 expression levels in Hepa 1–6 transfected with either 0.5 μg of esiRNA targeting GFP or SNAP23, with β-actin as loading control.
Discussion
Data from mice deficient in the initial steps of FA and glycerolipid metabolism has strongly suggested that ACSL1 directs FA and their acyl-CoA products into specific and independent downstream pathways (3, 6, 7). Particularly striking was a study in which mice lacking ACSL1 in skeletal muscle were unable to continue endurance running despite a markedly elevated muscle content of acyl-CoA, indicating that acyl-CoAs synthesized by other muscle ACSL isoforms were unavailable for β-oxidation (7). In addition, ACSL1 deficiency in cardiomyocytes leads to a switch in fuel availability from FA to glucose, thereby altering heart function, dramatically altering cardiolipin species, impairing mitochondrial respiratory function, and activating mTORC1 (4, 5).
The major findings of our study identify the mechanism that underlies acyl-CoA channeling, assemblies of specific enzymatic and tethering protein partners. Using BioID, we discovered new and unexpected protein interactions for ACSL1 in liver, illuminating protein networks between organelles and potential lipid-channeling pathways. These networks were disparate for ER- and OMM-directed ACSL1. The ER-targeted ACSL1 interactome suggested interactions with distinct proteins present on peroxisomes, Golgi, and the nucleus, whereas OMM-targeted ACSL1 interacted with LD-tethering proteins. Although many of the overlapping proteins were involved in protein translation, folding, and trafficking, common protein interactions at the ER and OMM were linked to lipid metabolism. For example, the ACSL isoforms ACSL5 and ACSL6 interacted with ACSL1 at both locations. The crystal structure of bacterial ACSL1 reveals that the protein functions as a homodimer (37), and some evidence suggests that mammalian ACSL isoforms may also exist as heterodimers. Lee et al. (11) concluded that CPT1a is part of a FA transport complex at the OMM, composed of VDAC and several ACSL isoforms, including ACSL1, ACLS5, and ACSL6. CerS2 and sphingomyelin phosphodiesterase 2 (nSMase1) also interact with ER- and OMM-targeted ACSL1 (supporting information S2). Although the precise role of nSMase1 in sphingolipid metabolism is still unknown, nSMase enzymes generate ceramide from sphingomyelin in response to stress signals (38). nSMase1 is located at the ER and Golgi (39). Site-directed mutagenesis studies reveal that another nSMase isoform, nSMase2, is palmitoylated at multiple cysteine residues and that palmitoylation is required for insertion into the plasma membrane (40). Because analysis (CSS-Palm site: http://csspalm.biocuckoo.org/)3 (72) predicts several palmitoylation sites in the nSMAse1 sequence, nSMAse1 might be modified similarly to allow insertion at the ER or Golgi membrane, and an interaction between ACSL1 and nSMase1 could facilitate cysteine palmitoylation for efficient membrane insertion.
We previously observed that ACSL1 is modified post-translationally by nutritional and hormonal signals (10). It is likely that these phosphorylation and acetylation modifications alter the interactome of ACSL1. In support of this interpretation, some of the interacting proteins identified in this study interacted with ACSL1 under distinct physiological conditions. Thus, although the interaction with CPT-1 was unaffected when primary hepatocytes were deprived of glucose, the interaction of VAMP4 and SNAP23 with ACSL1 was enhanced. These data suggest that CPT-1 interacts with ACSL1 at the OMM constitutively, whereas SNARE proteins came into contact with ACSL1 only under specific physiological conditions, as demonstrated functionally by the dramatically increased palmitate oxidation rates and the diminished rates of complex lipid synthesis when Hepa 1–6 cells were deprived of glucose. Although probably not the sole mechanism, the conditional association of ACSL1 and SNARE proteins would contribute to a switch in mitochondrial substrate use by increasing the availability of LDs FA. In support of this argument, other studies have reported that nutrient deprivation maintains FA flux by increasing the proximity of mitochondria to LDs (41). FA flux over prolonged periods is also enhanced by autophagy, which replenishes LD FA stores. In addition, mitochondrial fusion allows homogeneous dispersal of FA throughout mitochondria, thereby enabling efficient FA oxidation and preventing cellular FA efflux (41). SNAP23 and VAMP4 are known to interact and are involved in LD fusion (42), likely related to an association of SNAP23 with LDs (34–36, 42), and in fasted mouse liver, SNAP23 is redirected to the LD surface (36). Other studies have demonstrated that the SNAP23 interaction with LDs is mediated in part by a direct interaction with PLIN2 (34, 43). Some studies have suggested that ACSL1 and PLIN2 interact on LDs (44, 45), but although PLIN2 was increased 2-fold in the OMM–ACSL1–BirA* dataset suggesting its close proximity to ACSL1 at the OMM, PLIN2 was below the 5-fold threshold required for inclusion in this study (data not shown). This study is a snapshot of ACSL1 interactions under a single condition. Future temporal and nutritional studies and studies of the Acsl1 interactome in other tissues will be of major interest.
The presence of several SNARE proteins in the OMM–ACSL1–BirA* dataset strongly suggests a membrane fusion event occurring near ACSL1. Our data are consistent with data showing SNAP23 as a key mediator of LD and membrane docking dynamics. What remains unclear is whether SNAP23 has a direct impact on FA metabolism. Although Jägerström et al. (34) found that a siRNA-mediated decrease in SNAP23 had only minor effects on FA oxidation, the concentration of glucose used was not reported. Our own knockdown data imply that SNAP23 is not directly involved in FA oxidation in Hepa 1–6 cells, although residual protein expression and compensation by other SNAP isoforms, such as SNAP25, could have accounted for the unaltered FA metabolism. Although certain SNARE proteins (SNAP25, α-SNAP, Stx5, and Stx17) are required for efficient cholesterol transport to mitochondria for steroidogenesis (46), siRNA targeting of SNAP23 had no effect on progesterone synthesis, suggesting that cholesterol transport to mitochondria was unaltered (46). Thus, a knockdown of SNAP23 alone may not have been sufficient to disrupt LD–mitochondria interactions in their study or ours. Moreover, several of the SNARE complex proteins identified in our BioID study, such as VAMP4, VAMP5, Stx7, Stx8, and SNAP23, are palmitoylated (47–49). The close proximity of ACSL1 to SNARE complex proteins might also enable rapid acylation and protein insertion into the lipid bilayer.
At the ER, ACSL1 interacts with several proteins present in the peroxisomal membrane, suggesting close contact sites between these organelles. We confirmed that ACSL1 interacts with two proteins that facilitate the ER–peroxisome association, VAPB and ACBD5. Two independent studies reported that ACBD5 is required for peroxisomal and ER tethering via its interaction with VAPA and VAPB (17, 18). The proposed mechanism of ACBD5–VAPB interaction leaves the acyl-CoA–binding site of ACBD5 exposed at the interface between the ER and the peroxisome membrane (18). Knockdown of either VAPB or ACBD5 results in minor reductions in plasmalogen and cholesterol levels in HeLa cells (18). ACBD5 preferentially binds very-long–chain CoAs, and HeLa cells that lack ACBD5 show reduced β-oxidation of very-long–chain FA (50). Although it was suggested that ACBD5 binds very-long–chain acyl-CoAs at the peroxisome membrane to facilitate uptake via an ABCD transporter, ACSL1 does not use very-long–chain FA (50). Moreover, our previous study of liver-specific Acsl1 null mice (Acsl1L−/−) revealed no difference in hepatic cholesterol synthesis activity or long-chain FA species (8), and overexpression of ACSL1 in primary hepatocytes reduced channeling of [14C]oleate toward cholesterol synthesis (51). Elevated levels of C8 acylcarnitines in Acsl1L−/− liver suggested that peroxisomal FA oxidation had increased in the absence of ACSL1 (8). In support of this interpretation, the CPT-1 inhibitor, etomoxir, had no effect on FA oxidation in Acsl1L−/− liver, again suggesting that the remaining long-chain FA oxidation was occurring in peroxisomes. Thus, the functional role of the interaction of ACSL1 with VAPB and ACBD5 remains unclear, because peroxisomal FA oxidation increased in the absence of ACSL1, thereby compensating, in part, for its absence. Although we did not find changes in their mRNA abundance, it is possible that the activities of other ACSL isoforms could have masked a specific effect on peroxisomal FA metabolism. Whether ACSL1 transfers other unidentified lipid species to ACBD5 or ABCD3 for peroxisomal oxidation warrants further investigation.
Because we identified an interaction of ER-targeted ACSL1 with proteins involved in branched-chain FA metabolism, we examined the effect of hepatic phytanic acid metabolism in Acsl1L−/− mice. Despite the association, phytanic acid metabolism was affected only minimally when ACSL1 was absent. Although phytanoyl-CoA production was reduced in Acsl1L−/−mice, palmitoyl-CoA production was reduced even more, again suggesting that other ACSL isoforms had compensated for ACSL1's role in phytanic acid oxidation. In fact, ACSVL1 contributes to branched-chain FA oxidation (30), and although we did not observe increased expression of Acsvl1 mRNA, we did, as noted above, find that peroxisomal FA oxidation in Acsl1L−/− liver had increased, despite a lack of change in the expression of peroxisomal oxidative genes. We confirmed a direct interaction between ACSL1 and the fatty aldehyde dehydrogenase responsible for phytol metabolism, ALDH3A2, but we observed minor inhibition of phytanoyl-CoA production. In addition to links between ACSL1 and peroxisomal metabolism, we confirmed a direct interaction between ACSL1 and two ceramide synthase isoforms, CerS2 and CerS5. The activity of these two isoforms is regulated by dimer formation (52), such that CerS2 activity is augmented in the presence of CerS5. CerS5 synthesizes C16:0-ceramide (53), and CerS2 synthesizes very-long–chain ceramides (54, 55). Although C16:0-ceramide and acylceramide levels were not altered in Acsl1L−/− liver, we observed a 20% decrease in C22:0-ceramide in male Acsl1L−/− mice fed a HFD, and in Acsl1L−/− male and female mice compared with controls, the hepatic content of C26:0 and C26:1 acylceramides was 50%. Because the mRNA expression of CerS2, normally the dominant isoform in liver (54), was lower in Acsl1L−/− mice compared with controls, the reductions in C22:0-ceramide and in C26 and C26:1 acylceramides might reflect both the lack of CerS2 interaction after the Acsl1 deletion and indirectly the reduced expression of CerS2. We previously showed that loss of ACSL1 in the heart results in a 64% reduction in 18:0, 20:4-phosphatidylinositol (5). In addition, FA preference can vary depending on environment. For example, purified, recombinant ACSL1 displays differential substrate preferences compared with COS7 cell membranes overexpressing ACSL1 (56). Thus, ACSL1 can use longer chain FA if they are available.
In summary, we have identified distinct interaction networks for ACSL1 at the ER and at OMM and found that specific interactions vary under differing physiological circumstances. The interactome data generated in this study will be an important resource for the study of lipid channeling pathways in liver. Particularly interesting were the multiple interactions of both ER-targeted and OMM-targeted ACSL1 with proteins in other organelles. Although compensatory mechanisms induced by the Acsl1 deletion in liver may have masked major alterations in ACSL1 function in several niche pathways, our studies strongly suggest that ACSL1 is critical in hepatic lipid homeostasis. Because ACSL1 controls the flux of FA into the mitochondria for oxidation, advancing our understanding of how ACSL1 is regulated and how it controls fuel use could identify new therapeutic targets for leading causes of liver injury, nonalcoholic fatty liver disease, and nonalcoholic steatohepatitis (57).
Experimental procedures
Reagents and materials
Unless otherwise indicated, reagents were obtained from Sigma. [1-14C]Palmitic acid, [9,10-3H]palmitic acid, and [1-14C]oleic acid were from PerkinElmer Life Sciences. [1-14C]Phytanic acid was a kind gift from Dr. Paul Watkins (Kennedy Kreiger Institute, Baltimore, MD). pcDNA3.1-CerS2-HA and pcDNA3.1 CerS5-HA were kind gifts from Dr. Anthony Futterman (Weizmann Institute of Science, Rehovot, Israel). Antibodies used were as follows: HA (Cell Signaling, 3724S); FLAG (Sigma, F1804); ABCD3 (ThermoFisher Scientific, PA1650); ACBD5 (Proteintech, 21080-1-AP); VAPB (Proteintech, 14477-1-AP); ALDH3A2 (Proteintech, 15090-1-AP); CPT-1 (Santa Cruz Biotechnology, SC393070); SNAP23 (ThermoFisher Scientific, PA1738); VAMP4 (Santa Cruz Biotechnology, SC365332); β-actin (Abcam, ab6276); and avidin-HRP (Cell Signaling, 3999S).
Molecular cloning
Constructs used for BioID proximity labeling were prepared using Dual CCM(+) shuttle vector. ER-targeted ACSL1 was generated as a fusion, using aa 1–46 FATP4 (58) and aa 47–699 of ACSL1. OMM-targeted ACSL1 was generated using aa 1–69 TOM70 and aa 46–699 of ACSL1. A C-terminal BirA* tag (Addgene, Cambridge, MA) was incorporated for BioID experiments (12). BirA* was used as an untargeted nonspecific control, and ER-targeted RFP-BirA* and OMM-targeted GFP–BirA* (16) were used as targeted nonspecific BirA* controls. Vector Biolabs (Philadelphia) produced adenovirus.
Mice
The Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill approved all protocols. Mice were group-housed under a 12-h light/dark cycle with free access to food and water. Liver-specific Acsl1 knockout mice (Acsl1L−/−) were generated as described (8) on a C57BL/6J background, with floxed littermates as controls (Acsl1Lfl/fl). Mice were fasted for 4 h before tissue collection. For PPARα agonist studies, 6-week-old male mice were orally gavaged with Wy14643 (25 mg/kg body weight) or 0.5% carboxymethylcellulose as the vehicle control, daily for 5 days (59). The last dose was administered 24 h before sacrifice. For diet studies, 8–10-week-old male and female Acsl1L−/− and Acsl1Lfl/fl mice were fed a HFD (60% calories from fat; Research Diets D12492) or a LFD (10% calories from coconut oil; Research Diets D16090704) for 6 weeks. Mice were weighed weekly. Tissues were snap-frozen in liquid nitrogen and analyzed for lipid content, enzyme activity, and quantitative real time-PCR. Plasma was collected in 5% 0.5 m EDTA for TAG measurement (Stanbio, Boerne, TX).
Organelle isolation from liver
Mitochondria, MAM, and ER were isolated from fresh liver (60). Liver was minced and homogenized in homogenization buffer (0.25 m sucrose, 10 mm HEPES (pH 7.4), 1 mm EDTA, 1 mm dithiothreitol (DTT)). The homogenate was centrifuged at 600 × g for 5 min to remove nuclei and unbroken cells. A crude mitochondrial fraction was obtained by centrifuging the supernatant at 10,000 × g for 10 min. The pellet was resuspended, layered over a self-forming Percoll gradient, and centrifuged at 95,000 × g for 30 min. The large lower band (mitochondria) and upper band (MAM) were separately isolated, pelleted by centrifugation, and washed to remove Percoll. Supernatant from the crude mitochondrial fraction was layered over a sucrose gradient and centrifuged at 85,000 × g for 90 min to obtain ER, isolated between 1.5 and 1.3 m sucrose. Fractions were resuspended in homogenization buffer. Fraction purity was determined by Western blotting for calnexin (ER) and VDAC (mitochondria).
Microscopy
Hepa 1–6 cells grown on 1.5-mm glass coverslips were incubated with CellLight® mitochondria–RFP, CellLight® mitochondria–GFP, or CellLight® ER–GFP (ThermoFisher Scientific, C10601, C10508, or C10590, respectively). Cells were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, permeabilized, and blocked with 0.3% Triton X-100, 5% fetal bovine serum in PBS for 1 h. Cells were incubated with primary antibody in antibody dilution buffer (1× PBS, 1% BSA, 0.3% Triton X-100) for 1 h at room temperature. Secondary antibody was diluted 1:1000 in antibody dilution buffer, and cells were incubated 1 h at room temperature in the dark. Primary antibodies used were HA (Cell Signaling, 3724S) and calnexin (ThermoFisher Scientific, PA5-34754). Species-specific secondary antibodies were Rb–Alexa 488 and Rb–Alexa 568 (Life Technologies, Inc.). Cells were mounted on glass slides with Prolong Gold (Life Technologies, Inc.) and visualized with a Zeiss 700 confocal microscope.
ACS activity assay
ACS activity (initial rates) was measured in cell lysate or liver total particulate preparations in the presence of 175 mm Tris (pH 7.4), 8 mm MgCl2, 5 mm DTT, 10 mm ATP, 250 mm CoA, 50 μm [1-14C]palmitic acid, or 50 μm [1-14C]phytanic acid in 0.5 mm Triton X-100 and 10 mm EDTA in a total volume of 200 μl (61).
BioID and quantitative mass spectrometry
Hepa 1–6 cells were infected with adenoviruses at multiplicity of infection 200 for each of the BirA* constructs. Media were replaced the next day with fresh DMEM (25 mm glucose) containing 50 μm biotin and cultured for 24 h. Cells were washed twice with ice-cold PBS and collected. Biotin-labeled proteins were purified as described (12). Quantitative one-dimensional LC, tandem MS (1D–LC-MS/MS) was performed by the Duke University Proteomics and Metabolomics Shared Resource with triplicate biological replicates per group. Samples were analyzed using a nanoACQUITY UPLC system (Waters) coupled to a QExactive Plus high-resolution accurate mass tandem mass spectrometer (ThermoFisher Scientific) via a nanoelectrospray ionization source.
MS analysis
MS/MS samples were analyzed using Rosetta Elucidator version 4.0 (Rosetta Biosoftware, Inc.), and analyses were aligned based on the accurate mass and retention time of detected ions using the PeakTeller algorithm in Elucidator. Relative peptide abundance was calculated based on area-under-the-curve of the selected ion chromatograms of the aligned detected ions across all runs. The MS/MS data were searched against a custom Swiss Protein Database with Mus musculus taxonomy with additional proteins, including yeast ADH1, BSA, Escherichia coli BirA, as well as an equal number of reversed-sequence decoys for false discovery rate determination. Mascot Distiller and Mascot Server (Matrix Science, London, UK; version 2.5.1) were used to produce fragment ion spectra and perform the database searches. Database search parameters included fixed modification on cysteine residues (carbamidomethylation) and variable modifications on asparagine and glutamine (deamidation). Data were annotated at a 1.1% peptide false discovery rate using the Peptide Prophet algorithm. Peptide identifications were accepted if they could be established at greater than 5% probability to achieve a false discovery rate less than 1.1% (62). Protein probabilities were assigned by the Protein Prophet algorithm (63). Only proteins with ≥5-fold presence compared with controls were selected.
Interactome analysis
Proteins identified by MS were analyzed with the STRING database version 10.5 (64) to reveal direct (physical) and indirect (functional) interactions and modified for publication using Cytoscape software version 3.5.1 (65).
Co-immunoprecipitation
Co-immunoprecipitation experiments were conducted using either primary hepatocytes or Hepa 1–6 cells, as indicated. Primary hepatocytes were isolated from male and female C57BL/6J mice (66). Cells were infected with Ad-Acsl1–FLAG and cultured overnight in William's E medium. An untargeted Acsl1 construct was used to confirm interactions to avoid possible interactions with the targeting constructs themselves. Hepa 1–6 cells were infected with Ad-Acsl1–FLAG and transfected with pcDNA3.1–CerS2–HA or pcDNA3.1–CerS5–HA using Lipofectamine 2000 (ThermoFisher Scientific). Cells were washed twice with cold PBS and collected. Cell pellets were resuspended in 1 ml of PBS, and SDAD (diazirine), a 13.6 Å cross-linker (ThermoFisher Scientific), was added (final concentration 2 mm) and incubated for 30 min at 4 °C with rotation. Cross-linking was quenched with 1 m Tris (pH 8) (final concentration 100 mm), and cells were incubated for 15 min at 4 °C with rotation. Cells were washed twice in PBS and resuspended in 1 ml of PBS. SDAD cross-linker was photoactivated on ice using a 366-nm long–wave lamp for 15 min. Cells were centrifuged at 500 RCF to remove PBS and lysed in lysis buffer (50 mm Tris (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 10% glycerol, and 1× HALT protease inhibitor (ThermoFisher Scientific)). Lysate on ice was passed through a 25-gauge needle 10 times. Lysates were centrifuged to remove particulates, and protein concentration was determined by BCA assay. FLAG-tagged ACSL1 was purified using anti-FLAG M2 affinity resin (Sigma). Lysates were incubated with 200 μl of beads overnight at 4 °C with rotation. Resin was washed twice in 1 ml of lysis buffer for 5 min at 4 °C with rotation. Resin was washed four times in 1 ml of wash buffer for 5 min at 4 °C with rotation. Proteins were eluted using 0.3 mg/ml FLAG peptide in 10 mm Tris (pH 7.4), 250 mm NaCl. Eluted proteins were analyzed by Western blotting for the presence of interacting proteins.
FA oxidation assay
Oxidation assays were performed with freshly isolated liver, homogenized in ice-cold STE buffer (0.25 m sucrose, 10 mm Tris (pH 8), 1 mm EDTA). esiRNA-transfected Hepa 1–6 cells were removed from culture plates using 0.05% trypsin-EDTA and centrifuged at 500 RCF to remove trypsin. Cells were washed twice in ice-cold PBS and resuspended in ice-cold STE buffer before use. [1-14C]Palmitic acid (final assay concentration 20 μm, 1/5 labeled and 4/5 cold) or [1-14C]phytanic acid was added to 12 × 57-mm disposable culture tubes and dried under N2. FA was resuspended in 50 μl of β-cyclodextrin (10 mg/ml β-cyclodextrin in 10 mm Tris (pH 8.0)) and sonicated in a 37 °C water bath for 2 min. FA was allowed to conjugate at 37 °C for 30 min. The 150-μl reaction mixture was added to each tube (30 mm KCl, 20 mm Tris-base, 8.5 mm ATP, 8.5 mm MgCl2, 0.16 mm CoA, 1 mm NAD, 2.5 mm l-carnitine, 0.2 mm EDTA, 1 mm sodium malate, 0.17 mm FAD, 1 mm DTT (adjusted with 1 n KOH to pH 8.0)). In a time-controlled manner, 50 μl of liver homogenate, cell suspension, or 50 μl of STE buffer (blank) was added to each tube and 50 μl retained to measure protein concentration. A 200-μl pipette tip (cut) with inserted 250-μl microdialysis tube containing 200 μl of 1 m NaOH was dropped into each reaction tube, sealed with a rubber cap, and incubated at 37 °C, with shaking for 1 h. [14C]CO2 was trapped in NaOH by adding 100 μl of 70% perchloric acid to each tube with a needle and syringe and incubated for 1 h at room temperature with shaking. A 200-μl aliquot of the acidified reaction mixture was incubated with 100 μl of 25% FA-free BSA overnight at 4 °C and centrifuged at 14,000 rpm for 20 min. Supernatants were mixed with an additional 100 μl of 25% FA-free BSA and spun at 14,000 rpm for 20 min, before supernatant aliquots were counted for 14C-labeled acid soluble metabolites (ASM).
Peroxisomal oxidation assay
Peroxisomal oxidation was measured per Watkins et al. (67). Freshly isolated liver was homogenized in ice-cold STE buffer (0.25 m sucrose, 10 mm Tris (pH 8), 1 mm EDTA) or STE buffer with 100 μm etomoxir. [1-14C]Palmitic acid (final concentration 20 μm per assay; 1/5 labeled and 4/5 cold) was added to 13 × 100-mm glass tubes and dried under N2. FA was resuspended in 50 μl of α-cyclodextrin (10 mg/ml α-cyclodextrin in 10 mm Tris (pH 8.0)) and bath-sonicated for 2 min. FA was conjugated at 37 °C for 30 min. Reaction mixture was added to each tube (150 μl; 30 mm KCl, 20 mm Tris-base, 8.5 mm ATP, 8.5 mm MgCl2, 0.16 mm CoA, 1 mm NAD, 2.5 mm l-carnitine, 0.2 mm EDTA, 1 mm sodium malate, 0.17 mm FAD, 1 mm DTT, with or without 100 μm etomoxir (adjusted with 1 n KOH to (pH 8.0)). The reaction was started by adding 50 μl of homogenate or 50 μl of STE buffer (blank) and incubated at 37 °C in a water bath for 1 h. Data were normalized to the lysate protein concentration. Reactions were stopped with 50 μl of 1 n KOH and incubated for 1 h at 60 °C. Perchloric acid (20%) was added to each tube (150 μl) and incubated on ice for 1 h or 4 °C overnight (67). Samples were centrifuged at 700 RCF for 10 min, and supernatant was transferred to 16 × 100-mm glass tubes. Lipids were extracted to remove unmetabolized FA. Chloroform/methanol 1:1 was added (3.75 ml) to each sample and vortexed; then 1.875 ml of chloroform and 1.125 ml of H2O were added; samples were vortexed and then centrifuged at 700 RCF for 5 min. An aliquot of the aqueous phase was counted for radioactivity.
esiRNA
Hepa 1–6 cells were plated in 12-well plates and cultured overnight. SNAP23 expression was knocked down using MISSION esiRNA (Sigma, EMU085141) with GFP control esiRNA (Sigma, EHUEGFP). Cells were transfected with 0.5 μg of esiRNA using Lipofectamine 2000. Media were replaced the next day with fresh DMEM containing either 5 or 25 mm glucose, as indicated. Cells were cultured for 48 h after transfection before lipid oxidation experiments.
Pulse/chase assay
Hepa 1–6 cells transfected with either GFP esiRNA or SNAP23 esiRNA (as described) were incubated with 0.5 ml of labeling media (serum-free DMEM, 200 μm [1-14C]oleic acid conjugated to FA-free BSA, 1 mm carnitine) containing either 5 or 25 mm glucose, 37 °C (pulse). After a 3-h incubation, cells were washed twice with 1% BSA in PBS and either collected (pulse) or incubated with fresh serum-free DMEM without FA or glucose for 6 h (chase). Media samples were collected after 1 and 3 h. After 6 h, incubation media and cells were collected. All media samples were incubated for 1 h with 100 μl of perchloric acid (20%) in a sealed tube with an inserted 250-μl microdialysis tube containing 200 μl of 1 m NaOH. NaOH was measured for [14C]CO2. Following NaOH collection, 200 μl of acidified media was incubated with 100 μl of 25% FA-free BSA overnight at 4 °C and centrifuged at 14,000 rpm for 20 min. Supernatants were washed once more with 100 μl of 25% FA-free BSA and spun 14,000 rpm for 20 min, before aliquots of the supernatant were counted for 14C-labeled ASM. Lipids were extracted from collected cells as described (68).
[9,10-3H]Palmitic acid oxidation
Hepa 1–6 cells were plated at 2 × 105 in 6-well plates and used the following day. Cells were washed in PBS, and fresh DMEM was added containing either 25 mm glucose and 1 mm pyruvate or DMEM containing no glucose or pyruvate for 3 h. Media were replaced with DMEM containing 1 mm carnitine and [9,10-3H]palmitic acid conjugated to FA-free BSA (labeling media); cells were incubated for 3 h. Conversion of [9,10-3H]palmitic acid to [3H]H2O was measured in cell culture media; 400 μl of media were added to 200 μl of 20% perchloric acid and incubated at 4 °C overnight. Samples were centrifuged for 5 min at 14,000 RCF, and supernatants were extracted as above to remove any unoxidized FA. The aqueous phase was measured for [3H]H2O. The remaining cells were washed twice in pre-warmed 1% BSA in PBS, and cells were harvested in 500 μl of methanol, and total lipids were extracted (68).
Lipidomics
Tissues were snap-frozen and stored at −80 °C. Livers were thawed and homogenized on ice in homogenization buffer (0.25 m sucrose, 25 mm KCl, 50 mm Tris and 0.5 mm EDTA (pH 7.4)) 3–4 times, with a 1-min rest between each homogenization, and then filtered through a 100-μm mesh filter into a fresh tube. Protein concentration was determined by BCA assay with 2 mg of homogenate per sample. Lipid was extracted (68), and ceramides and acylceramides were measured. Ceramide levels were measured by HPLC coupled to MS (LC-MS) as described (69). The acylceramide content of the samples was determined by mild base hydrolysis after Bligh-Dyer lipid extraction. Briefly, lipids were incubated with 2 ml of 0.2 n KOH in methanol at 37 °C for 1 h. After neutralization with 1 ml of 0.4 n HCl, lipids were extracted using another round of Bligh-Dyer extraction and dried under a N2 stream. The ceramide content of the resulting lipids was determined using LC-MS. The acylceramide amounts were calculated by subtracting ceramide levels of control samples from base-hydrolyzed samples. Data were analyzed by Student's t test to establish statistical significance, *, p < 0.05.
RNA extraction and quantitative real-time PCR
Total RNA and DNA were simultaneously isolated from liver using 0.7 ml of TRIzol (ThermoFisher Scientific) per 200 mg of tissue. cDNA was synthesized using 1 μg of RNA template (iScript cDNA Synthesis kit, Bio-Rad), and 50 ng of cDNA was added to each qPCR with SYBR Green (iTaq Universal SYBR; Bio-Rad) and used to detect amplicons with primers specific to the gene of interest (S6) and quantified using a qPCR thermocycler (Bio-Rad). Results were normalized to the housekeeping gene.
Statistics
Data are presented as means ± S.E. for each treatment group. Data were analyzed by Student's t test, or groups were evaluated by one-way analysis of variance. All statistical analyses were performed using GraphPad Prism (version 6.0; GraphPad Software, La Jolla, CA). Differences between means with p < 0.05 were considered statistically significant.
Author contributions
P. A. Y. and R. A. C. conceptualization; P. A. Y. and R. A. C. formal analysis; P. A. Y., C. E. S., A. L. S., T. J. G., D. D. L., L. Z., A. E. C., J. F., and L. M. O. investigation; P. A. Y. methodology; P. A. Y. writing-original draft; C. E. S., E. L. K., L. M. O., and R. A. C. writing-review and editing; J. F. resources; R. A. C. supervision; R. A. C. funding acquisition; R. A. C. project administration.
Supplementary Material
Acknowledgments
We thank the Duke University School of Medicine for the use of the Proteomics and Metabolomics Shared Resource, which provided the proteomics service. We thank Dr. Paul Watkins for advice regarding phytol metabolism and the Lipidomics Facility of Stony Brook University Medical Center for lipidomics analysis.
This work was supported by National Institutes of Health Grants DK59935 and DK56598 (to R. A. C.), DK107481 (to E. L. K.), and GM097741 (to L. M. O.) and German Research Foundation (DFG) Grant FU 340/5-1 (to J. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting information S1–S8.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- FA
- fatty acid
- ACSL
- long-chain acyl-CoA synthetase
- OMM
- outer mitochondrial membrane
- ER
- endoplasmic reticulum
- VDAC
- voltage-dependent anion channel
- RCF
- relative centrifugal force
- MAM
- mitochondria-associated membrane
- ASM
- acid-soluble metabolite
- DMEM
- Dulbecco's modified Eagle's medium
- HRP
- horseradish peroxidase
- LFD
- low-fat diet
- HFD
- high-fat diet
- aa
- amino acid
- EGFR
- epidermal growth factor receptor
- esiRNA
- endoribonuclease-prepared siRNA
- LD
- lipid droplet
- RFP
- red fluorescent protein
- qPCR
- quantitative PCR
- PPAR
- peroxisome proliferator-activated receptor
- TAG
- triacylglycerol
- ACS
- acyl-CoA synthetase
- SDAD
- (NHS-SS-diazirine) (succinimidyl 2-((4,4′-azipentanamido)ethyl)-1,3′-dithiopropionate).
References
- 1. Bowman T. A., O'Keeffe K. R., D'Aquila T., Yan Q. W., Griffin J. D., Killion E. A., Salter D. M., Mashek D. G., Buhman K. K., and Greenberg A. S. (2016) Acyl-CoA synthetase 5 (ACSL5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab. 5, 210–220 10.1016/j.molmet.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Killion E. A., Reeves A. R., El Azzouny M. A., Yan Q. W., Surujon D., Griffin J. D., Bowman T. A., Wang C., Matthan N. R., Klett E. L., Kong D., Newman J. W., Han X., Lee M. J., Coleman R. A., and Greenberg A. S. (2018) A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol. Metab. 9, 45–56 10.1016/j.molmet.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellis J. M., Li L. O., Wu P. C., Koves T. R., Ilkayeva O., Stevens R. D., Watkins S. M., Muoio D. M., and Coleman R. A. (2010) Adipose acyl-CoA synthetase-1 directs fatty acids toward β-oxidation and is required for cold thermogenesis. Cell Metab. 12, 53–64 10.1016/j.cmet.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grevengoed T. J., Cooper D. E., Young P. A., Ellis J. M., and Coleman R. A. (2015) Loss of long-chain acyl-CoA synthetase isoform 1 impairs cardiac autophagy and mitochondrial structure through mechanistic target of rapamycin complex 1 activation. FASEB J. 29, 4641–4653 10.1096/fj.15-272732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grevengoed T. J., Martin S. A., Katunga L., Cooper D. E., Anderson E. J., Murphy R. C., and Coleman R. A. (2015) Acyl-CoA synthetase 1 deficiency alters cardiolipin species and impairs mitochondrial function. J. Lipid Res. 56, 1572–1582 10.1194/jlr.M059717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ellis J. M., Mentock S. M., Depetrillo M. A., Koves T. R., Sen S., Watkins S. M., Muoio D. M., Cline G. W., Taegtmeyer H., Shulman G. I., Willis M. S., and Coleman R. A. (2011) Mouse cardiac acyl coenzyme A synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol. Cell. Biol. 31, 1252–1262 10.1128/MCB.01085-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li L. O., Grevengoed T. J., Paul D. S., Ilkayeva O., Koves T. R., Pascual F., Newgard C. B., Muoio D. M., and Coleman R. A. (2015) Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64, 23–35 10.2337/db13-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li L. O., Ellis J. M., Paich H. A., Wang S., Gong N., Altshuller G., Thresher R. J., Koves T. R., Watkins S. M., Muoio D. M., Cline G. W., Shulman G. I., and Coleman R. A. (2009) Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and β-oxidation and alters phospholipid fatty acid composition. J. Biol. Chem. 284, 27816–27826 10.1074/jbc.M109.022467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewin T. M., Kim J. H., Granger D. A., Vance J. E., and Coleman R. A. (2001) Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 276, 24674–24679 10.1074/jbc.M102036200 [DOI] [PubMed] [Google Scholar]
- 10. Frahm J. L., Li L. O., Grevengoed T. J., and Coleman R. A. (2011) Phosphorylation and acetylation of acyl-CoA synthetase-I. J. Proteomics Bioinform. 4, 129–137 10.4172/jpb.1000180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee K., Kerner J., and Hoppel C. L. (2011) Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286, 25655–25662 10.1074/jbc.M111.228692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schumacher M. M., Elsabrouty R., Seemann J., Jo Y., and DeBose-Boyd R. A. (2015) The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase. eLife 4, 10.7554/eLife.05560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cronan J. E. (2005) Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J. Nutr. Biochem. 16, 416–418 10.1016/j.jnutbio.2005.03.017 [DOI] [PubMed] [Google Scholar]
- 15. Vance J. E. (2014) MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609 10.1016/j.bbalip.2013.11.014 [DOI] [PubMed] [Google Scholar]
- 16. Zhan T., Poppelreuther M., Ehehalt R., and Füllekrug J. (2012) Overexpressed FATP1, ACSVL4/FATP4 and ACSL1 increase the cellular fatty acid uptake of 3T3-L1 adipocytes but are localized on intracellular membranes. PLoS One 7, e45087 10.1371/journal.pone.0045087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costello J. L., Castro I. G., Hacker C., Schrader T. A., Metz J., Zeuschner D., Azadi A. S., Godinho L. F., Costina V., Findeisen P., Manner A., Islinger M., and Schrader M. (2017) ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 216, 331–342 10.1083/jcb.201607055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hua R., Cheng D., Coyaud É., Freeman S., Di Pietro E., Wang Y., Vissa A., Yip C. M., Fairn G. D., Braverman N., Brumell J. H., Trimble W. S., Raught B., and Kim P. K. (2017) VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377 10.1083/jcb.201608128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Senkal C. E., Salama M. F., Snider A. J., Allopenna J. J., Rana N. A., Koller A., Hannun Y. A., and Obeid L. M. (2017) Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 25, 686–697 10.1016/j.cmet.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan J., Liu J., Culty M., and Papadopoulos V. (2010) Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog. Lipid Res. 49, 218–234 10.1016/j.plipres.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J., Li H., and Papadopoulos V. (2003) PAP7, a PBR/PKA-RIα-associated protein: a new element in the relay of the hormonal induction of steroidogenesis. J. Steroid Biochem. Mol. Biol. 85, 275–283 10.1016/S0960-0760(03)00213-9 [DOI] [PubMed] [Google Scholar]
- 22. Liu J., Rone M. B., and Papadopoulos V. (2006) Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J. Biol. Chem. 281, 38879–38893 10.1074/jbc.M608820200 [DOI] [PubMed] [Google Scholar]
- 23. Papadopoulos V., Liu J., and Culty M. (2007) Is there a mitochondrial signaling complex facilitating cholesterol import? Mol. Cell. Endocrinol. 265, 59–64 [DOI] [PubMed] [Google Scholar]
- 24. Shinoda Y., Fujita K., Saito S., Matsui H., Kanto Y., Nagaura Y., Fukunaga K., Tamura S., and Kobayashi T. (2012) Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER–Golgi membrane contact sites. FEBS Lett. 586, 3024–3029 10.1016/j.febslet.2012.06.050 [DOI] [PubMed] [Google Scholar]
- 25. Zhong S., Yin H., Liao Y., Yao F., Li Q., Zhang J., Jiao H., Zhao Y., Xu D., Liu S., Song H., Gao Y., Liu J., Ma L., Pang Z., et al. (2015) Lung tumor suppressor GPRC5A binds EGFR and restrains its effector signaling. Cancer Res. 75, 1801–1814 10.1158/0008-5472.CAN-14-2005 [DOI] [PubMed] [Google Scholar]
- 26. Demory M. L., Boerner J. L., Davidson R., Faust W., Miyake T., Lee I., Hüttemann M., Douglas R., Haddad G., and Parsons S. J. (2009) Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J. Biol. Chem. 284, 36592–36604 10.1074/jbc.M109.000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bollu L. R., Ren J., Blessing A. M., Katreddy R. R., Gao G., Xu L., Wang J., Su F., and Weihua Z. (2014) Involvement of de novo synthesized palmitate and mitochondrial EGFR in EGF induced mitochondrial fusion of cancer cells. Cell Cycle 13, 2415–2430 10.4161/cc.29338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bersuker K., Peterson C. W. H., To M., Sahl S. J., Savikhin V., Grossman E. A., Nomura D. K., and Olzmann J. A. (2018) A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 44, 97–112.e7 10.1016/j.devcel.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watkins P. A., Howard A. E., Gould S. J., Avigan J., and Mihalik S. J. (1996) Phytanic acid activation in rat liver peroxisomes is catalyzed by long-chain acyl-CoA synthetase. J. Lipid Res. 37, 2288–2295 [PubMed] [Google Scholar]
- 30. Steinberg S. J., Wang S. J., Kim D. G., Mihalik S. J., and Watkins P. A. (1999) Human very-long–chain acyl-CoA synthetase: cloning, topography, and relevance to branched-chain fatty acid metabolism. Biochem. Biophys. Res. Commun. 257, 615–621 10.1006/bbrc.1999.0510 [DOI] [PubMed] [Google Scholar]
- 31. Watkins P. A., and Ellis J. M. (2012) Peroxisomal acyl-CoA synthetases. Biochim. Biophys. Acta 1822, 1411–1420 10.1016/j.bbadis.2012.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferdinandusse S., Jimenez-Sanchez G., Koster J., Denis S., Van Roermund C. W., Silva-Zolezzi I., Moser A. B., Visser W. F., Gulluoglu M., Durmaz O., Demirkol M., Waterham H. R., Gökcay G., Wanders R. J., and Valle D. (2015) A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 24, 361–370 10.1093/hmg/ddu448 [DOI] [PubMed] [Google Scholar]
- 33. Levy M., and Futerman A. H. (2010) Mammalian ceramide synthases. IUBMB Life 62, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jägerström S., Polesie S., Wickström Y., Johansson B. R., Schröder H. D., Højlund K., and Boström P. (2009) Lipid droplets interact with mitochondria using SNAP23. Cell Biol. Int. 33, 934–940 10.1016/j.cellbi.2009.06.011 [DOI] [PubMed] [Google Scholar]
- 35. Strauss J. A., Shaw C. S., Bradley H., Wilson O. J., Dorval T., Pilling J., and Wagenmakers A. J. (2016) Immunofluorescence microscopy of SNAP23 in human skeletal muscle reveals colocalization with plasma membrane, lipid droplets, and mitochondria. Physiol. Rep. 4, e12662 10.14814/phy2.12662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sadh K., Rai P., and Mallik R. (2017) Feeding-fasting dependent recruitment of membrane microdomain proteins to lipid droplets purified from the liver. PLoS One 12, e0183022 10.1371/journal.pone.0183022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hisanaga Y., Ago H., Nakagawa N., Hamada K., Ida K., Yamamoto M., Hori T., Arii Y., Sugahara M., Kuramitsu S., Yokoyama S., and Miyano M. (2004) Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 279, 31717–31726 10.1074/jbc.M400100200 [DOI] [PubMed] [Google Scholar]
- 38. Airola M. V., and Hannun Y. A. (2013) Sphingolipid metabolism and neutral sphingomyelinases. Handb. Exp. Pharmacol. 2013, 57–76 10.1007/978-3-7091-1368-4_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tomiuk S., Zumbansen M., and Stoffel W. (2000) Characterization and subcellular localization of murine and human magnesium-dependent neutral sphingomyelinase. J. Biol. Chem. 275, 5710–5717 10.1074/jbc.275.8.5710 [DOI] [PubMed] [Google Scholar]
- 40. Tani M., and Hannun Y. A. (2007) Neutral sphingomyelinase 2 is palmitoylated on multiple cysteine residues. Role of palmitoylation in subcellular localization. J. Biol. Chem. 282, 10047–10056 10.1074/jbc.M611249200 [DOI] [PubMed] [Google Scholar]
- 41. Rambold A. S., Cohen S., and Lippincott-Schwartz J. (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692 10.1016/j.devcel.2015.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boström P., Andersson L., Rutberg M., Perman J., Lidberg U., Johansson B. R., Fernandez-Rodriguez J., Ericson J., Nilsson T., Borén J., and Olofsson S. O. (2007) SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat. Cell Biol. 9, 1286–1293 10.1038/ncb1648 [DOI] [PubMed] [Google Scholar]
- 43. Senthivinayagam S., McIntosh A. L., Moon K. C., and Atshaves B. P. (2013) Plin2 inhibits cellular glucose uptake through interactions with SNAP23, a SNARE complex protein. PLoS One 8, e73696 10.1371/journal.pone.0073696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brasaemle D. L., Dolios G., Shapiro L., and Wang R. (2004) Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 279, 46835–46842 10.1074/jbc.M409340200 [DOI] [PubMed] [Google Scholar]
- 45. Wilfling F., Wang H., Haas J. T., Krahmer N., Gould T. J., Uchida A., Cheng J. X., Graham M., Christiano R., Fröhlich F., Liu X., Buhman K. K., Coleman R. A., Bewersdorf J., Farese R. V. Jr., and Walther T. C. (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399 10.1016/j.devcel.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin Y., Hou X., Shen W. J., Hanssen R., Khor V. K., Cortez Y., Roseman A. N., Azhar S., and Kraemer F. B. (2016) SNARE-mediated cholesterol movement to mitochondria supports steroidogenesis in rodent cells. Mol. Endocrinol. 30, 234–247 10.1210/me.2015-1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Merrick B. A., Dhungana S., Williams J. G., Aloor J. J., Peddada S., Tomer K. B., and Fessler M. B. (2011) Proteomic profiling of S-acylated macrophage proteins identifies a role for palmitoylation in mitochondrial targeting of phospholipid scramblase 3. Mol. Cell. Proteomics 10, M110.006007 10.1074/mcp.M110.006007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogel K., and Roche P. A. (1999) SNAP-23 and SNAP-25 are palmitoylated in vivo. Biochem. Biophys. Res. Commun. 258, 407–410 10.1006/bbrc.1999.0652 [DOI] [PubMed] [Google Scholar]
- 49. Salaün C., Gould G. W., and Chamberlain L. H. (2005) The SNARE proteins SNAP-25 and SNAP-23 display different affinities for lipid rafts in PC12 cells. Regulation by distinct cysteine-rich domains. J. Biol. Chem. 280, 1236–1240 10.1074/jbc.M410674200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yagita Y., Shinohara K., Abe Y., Nakagawa K., Al-Owain M., Alkuraya F. S., and Fujiki Y. (2017) Deficiency of a retinal dystrophy protein, acyl-CoA–binding domain-containing 5 (ACBD5), impairs peroxisomal β-oxidation of very-long–chain fatty acids. J. Biol. Chem. 292, 691–705 10.1074/jbc.M116.760090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li L. O., Mashek D. G., An J., Doughman S. D., Newgard C. B., and Coleman R. A. (2006) Overexpression of rat long chain acyl-CoA synthetase 1 alters fatty acid metabolism in rat primary hepatocytes. J. Biol. Chem. 281, 37246–37255 10.1074/jbc.M604427200 [DOI] [PubMed] [Google Scholar]
- 52. Laviad E. L., Kelly S., Merrill A. H. Jr., and Futerman A. H. (2012) Modulation of ceramide synthase activity via dimerization. J. Biol. Chem. 287, 21025–21033 10.1074/jbc.M112.363580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gosejacob D., Jäger P. S., Vom Dorp K., Frejno M., Carstensen A. C., Köhnke M., Degen J., Dörmann P., and Hoch M. (2016) Ceramide synthase 5 is essential to maintain C16:0-ceramide pools and contributes to the development of diet-induced obesity. J. Biol. Chem. 291, 6989–7003 10.1074/jbc.M115.691212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Laviad E. L., Albee L., Pankova-Kholmyansky I., Epstein S., Park H., Merrill A. H. Jr, and Futerman A. H. (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684 10.1074/jbc.M707386200 [DOI] [PubMed] [Google Scholar]
- 55. Jennemann R., Rabionet M., Gorgas K., Epstein S., Dalpke A., Rothermel U., Bayerle A., van der Hoeven F., Imgrund S., Kirsch J., Nickel W., Willecke K., Riezman H., Gröne H. J., and Sandhoff R. (2012) Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum. Mol. Genet. 21, 586–608 10.1093/hmg/ddr494 [DOI] [PubMed] [Google Scholar]
- 56. Klett E. L., Chen S., Yechoor A., Lih F. B., and Coleman R. A. (2017) Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J. Lipid Res. 58, 884–894 10.1194/jlr.M072512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schäffler A., Schölmerich J., and Büchler C. (2005) Mechanisms of disease: adipocytokines and visceral adipose tissue–emerging role in nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 273–280 10.1038/ncpgasthep0186 [DOI] [PubMed] [Google Scholar]
- 58. Milger K., Herrmann T., Becker C., Gotthardt D., Zickwolf J., Ehehalt R., Watkins P. A., Stremmel W., and Füllekrug J. (2006) Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 119, 4678–4688 10.1242/jcs.03280 [DOI] [PubMed] [Google Scholar]
- 59. Bernardes A., Souza P. C., Muniz J. R., Ricci C. G., Ayers S. D., Parekh N. M., Godoy A. S., Trivella D. B., Reinach P., Webb P., Skaf M. S., and Polikarpov I. (2013) Molecular mechanism of peroxisome proliferator-activated receptor α activation by WY14643: a new mode of ligand recognition and receptor stabilization. J. Mol. Biol. 425, 2878–2893 10.1016/j.jmb.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 60. Vance J. E. (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 [PubMed] [Google Scholar]
- 61. Polokoff M. A., and Bell R. M. (1978) Limited palmitoyl-CoA penetration into microsomal vesicles as evidenced by a highly latent ethanol acyltransferase activity. J. Biol. Chem. 253, 7173–7178 [PubMed] [Google Scholar]
- 62. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- 63. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- 64. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wendel A. A., Cooper D. E., Ilkayeva O. R., Muoio D. M., and Coleman R. A. (2013) Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. J. Biol. Chem. 288, 27299–27306 10.1074/jbc.M113.485219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Watkins P. A., Ferrell E. V. Jr., Pedersen J. I., and Hoefler G. (1991) Peroxisomal fatty acid β-oxidation in HepG2 cells. Arch. Biochem. Biophys. 289, 329–336 10.1016/0003-9861(91)90419-J [DOI] [PubMed] [Google Scholar]
- 68. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 69. Bielawski J., Szulc Z. M., Hannun Y. A., and Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91 10.1016/j.ymeth.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 70. Kuerschner L., Ejsing C. S., Ekroos K., Shevchenko A., Anderson K. I., and Thiele C. (2005) Polyene-lipids: a new tool to image lipids. Nat. Methods 2, 39–45 10.1038/nmeth728 [DOI] [PubMed] [Google Scholar]
- 71. Poppelreuther M., Rudolph B., Du C., Grossmann R., Becker M., Thiele C., Ehehalt R., and Füllekrug J. (2012) The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 53, 888–900 10.1194/jlr.M024562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ren J., Wen L., Gao X., Jin C., Xue Y., and Yao X. (2008) CSS-Palm 2.0: An updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 21, 639–644 10.1093/protein/gzn039 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.