

Abstract
Splicing generates many mRNA strands from a single precursor mRNA, expanding the proteome and enhancing intracellular diversity. Both initial assembly and activation of the spliceosome require an essential family of splicing factors called serine-arginine (SR) proteins. Protein phosphatase 1 (PP1) regulates the SR proteins by controlling phosphorylation of a C-terminal arginine-serine–rich (RS) domain. These modifications are vital for the subcellular localization and mRNA splicing function of the SR protein. Although PP1 has been shown to dephosphorylate the prototype SR protein splicing factor 1 (SRSF1), the molecular nature of this interaction is not understood. Here, using NMR spectroscopy, we identified two electrostatic residues in helix α2 and a hydrophobic residue in helix α1 in the RNA recognition motif 1 (RRM1) of SRSF1 that constitute a binding surface for PP1. Substitution of these residues dissociated SRSF1 from PP1 and enhanced phosphatase activity, reducing phosphorylation in the RS domain. These effects lead to shifts in alternative splicing patterns that parallel increases in SRSF1 diffusion from speckles to the nucleoplasm brought on by regiospecific decreases in RS domain phosphorylation. Overall, these findings establish a molecular and biological connection between PP1-targeted amino acids in an RRM with the phosphorylation state and mRNA-processing function of an SR protein.
Keywords: phosphorylation, phosphatase, protein kinase, enzyme kinetics, enzyme mutation
Introduction
Splicing generates a multitude of unique mRNA strands from a single precursor mRNA (pre-mRNA),3 allowing great expansion of the proteome and enhanced organismal complexity within a limited genome. Splicing occurs at the spliceosome, a macromolecular complex composed of five small nuclear ribonuclear proteins (U1–6 snRNPs) and over 100 proteins (1). The initial assembly, as well as later activation of the spliceosome, requires an essential family of splicing factors known as serine-arginine (SR) proteins. SR proteins contain one or two RNA recognition motifs (RRMs) that recognize specific sequences in pre-mRNA and orchestrate the binding of several spliceosomal components including U1 snRNP and the protein heterodimer U2AF at the boundaries of coding regions (2–4). SR proteins also possess C-terminal domains rich in arginine-serine (RS) dipeptide repeats. These RS domains are commonly phosphorylated at many serines, a modification that influences SR protein function at different levels. The RS domains of the 12 members of the SR protein family vary considerably in length (∼50–300 amino acids) and Arg-Ser content. The identity and regulation of these phosphorylation events within the family are still not well-understood, but some overarching principles regarding RS domain phosphorylation and SR protein function have emerged with respect to splicing. In general, increased SR protein phosphorylation is important for pre-mRNA binding and recognition of U1 snRNP, events that are essential for spliceosome initiation (4). Later, SR protein dephosphorylation induces rearrangements of several snRNPs that ultimately lead to the fully active spliceosome (5). In this manner, SR proteins need to undergo a series of complex, multisite phosphorylation and dephosphorylation steps on their RS domains to facilitate pre-mRNA splicing.
In addition to guiding assembly and activation of the spliceosome, phosphorylation also indirectly regulates splicing by controlling the subcellular localization of SR proteins. Much of what we know about the trafficking of these essential splicing factors stems from detailed investigations into the family prototype SRSF1 (aka ASF/SF2). Such studies reveal that the SR protein kinases (SRPKs) and cdc2-like kinases (CLKs) phosphorylate the RS domains of SR proteins in dipeptide and regiospecific manners (6). We showed that SRPK1, one of three physiological isoforms, rapidly phosphorylates ∼6–8 serines in the N terminus of the RS domain, a modification that facilitates transportin-dependent movement of SRSF1 from the cytoplasm to the nucleus (7–11). In the nucleus, SRSF1 largely resides within membrane-free aggregates known as speckles. Although SRSF1 can rapidly move in and out of these structures (12), CLK phosphorylation greatly shifts the equilibrium from speckles to the nucleoplasm (13, 14). The latter transition is critical for SR protein mobilization to the spliceosome for splicing function (15). We showed that although CLK1, one of four isoforms, phosphorylates a broad swath of Arg-Ser dipeptides, specific modification of three Ser-Pro dipeptides toward the C terminus of the RS domain triggers bulk movement of SRSF1 from speckles to the nucleoplasm (16, 17) (Fig. 1A).
Figure 1.
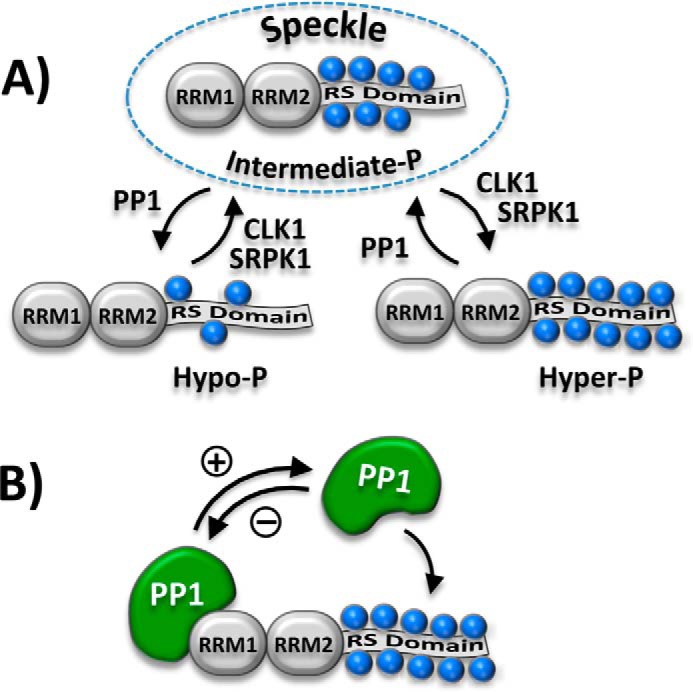
Regulation of RS domain phosphorylation and SRSF1 subnuclear localization. A, phosphorylation of the RS domain regulates SRSF1 occupancy in nuclear speckles (dashed oval). Blue spheres represent several Arg-Ser and Ser-Pro dipeptides that are phosphorylated in the RS domain. B, RRM1 in SRSF1 binds and down-regulates PP1 function. PP1 is activated upon dissociation from RRM1.
Although phosphorylation generally promotes SR protein movement to the nucleoplasm for spliceosome assembly/activation, localization of these splicing factors in storage speckles relies on intermediate phosphorylation levels maintained by a balance of kinase/phosphatase activities (Fig. 1A). We showed previously that such an intermediate phosphorylation state is controlled by a repressive interaction between protein phosphatase 1 (PP1) and SRSF1 (18) (Fig. 1B), but the molecular nature of this interaction and its connection to pre-mRNA splicing are unknown. In this new study, we used NMR spectroscopy and protein–protein binding methods to identify residues in two helices of RRM1 that constitute a binding surface for PP1. We showed that the PP1–SRSF1 interface not only controls speckle occupancy through regiospecific phosphorylation of N-terminal serines in the RS domain but also regulates alternative splicing patterns. These results identify a new molecular surface in an RRM that contacts PP1 and influences the biological function of an SR protein.
Results
PP1 binds selectively to RRM1
Because we showed previously that RRM1 in SRSF1 binds and down-regulates PP1, we wished to identify unique residues in this domain that interact with this phosphatase. We first verified previous findings that PP1 binds selectively to RRM1 in SRSF1 (18). In pulldown assays, we showed that GST–PP1 interacts with His-tagged RRM1 but not with His-tagged RRM2 (Fig. 2A). To measure the dissociation constant of RRM1 to PP1, we monitored the time-dependent dephosphorylation of the substrate p-nitrophenyl phosphate (PNPP) by His-tagged PP1 as a function of RRM1 (0–1000 nm) using a spectrophotometric assay (Fig. S1). We found that RRM1 effectively inhibited PP1 with an apparent KI (appKI) of 340 ± 11 nm, whereas RRM2 had only a small effect on activity consistent with an appKI of 12 μm or larger (Fig. 2B). These results are in keeping with a prior report (18) and show that the apparent binding affinity of RRM1 is at least 40-fold higher than that for RRM2. Because RRM1 is a noncompetitive inhibitor with respect to PNPP, appKI is also equivalent to the true KI (18). However, because this assay depends on functional changes in PP1 and RRM2 might bind to PP1 but not influence catalysis, we decided to perform a competition experiment. We monitored the activity of PP1 at fixed RRM2 (2 μm) and varying RRM1 (0–1000 nm) and did not observe a change in the appKI for RRM1 (340 ± 20 nm) relative to the control lacking RRM2 (Fig. 2B and Fig. S1). These results show that RRM2 does not compete for RRM1 binding to PP1, a finding consistent with the pulldown experiments, and that PP1 binds specifically to only one of the RRMs in SRSF1.
Figure 2.

RRM1 of SRSF1 interacts with PP1. A, pulldown assays showing specific interaction of PP1 with RRM1. GST–PP1, bound to GSH beads, was incubated with RRM1 or RRM2, washed, and run on a 12% SDS–PAGE. B, binding constants for RRM1 and RRM2 to PP1 using inhibition kinetics. Dephosphorylation of PNPP (25 mm) by His–PP1 (20 nm) was monitored as a function of RRM1 (●) and RRM2 (■). Inhibition kinetics for PNPP dephosphorylation is also monitored using fixed RRM2 (2 μm) and varying RRM1 (▴). C, superposition of the [15N,1H]-HSQC spectra of 15N-labeled RRM1 with (blue) and without (green) addition of His–PP1. Thirteen signals that experienced changes upon addition of PP1 are identified with the sequence-specific assignments next to their green peaks. Additional peaks, colored cyan, originate from arginine guanidinium groups; they are folded from their intrinsic positions into the spectral region shown here. D, anterior and posterior surface views and ribbon diagrams of RRM1, showing residues affected by the presence of His–PP1 (cyan). Several residue positions and two helices containing the majority of the affected residues are indicated for guidance.
NMR identification of discrete RRM1 contact sites in the complex with PP1
To identify structural determinants in RRM1 that interact with PP1, we produced uniformly 13C,15N-labeled RRM1 and determined sequence-specific backbone resonance assignments. Using the “standard” three APSY-NMR experiments (19) as input for the automated algorithm UNIO-MATCH (20) yielded 75% of the assignments. These assignments were interactively validated and extended with the use of 3D HNCA and 3D HNCACB spectra, so that complete assignments were obtained. We then added His-tagged PP1 to uniformly 15N-labeled RRM1 and thus identified 13 resonances that experienced chemical shifts or line broadening upon phosphatase binding (Fig. 2C). The affected residues all map to a single face of the RRM1 structure (Protein Data Bank code 1X4A) (Fig. 2D). Most of the residues are located in helices with five in α2 and two in α1. Of the remaining affected residues, three are located in loops and three are located in β strands. These findings suggest that interactions along one surface on RRM1 represent the structural basis for specific recognition of SRSF1 by PP1.
Functional roles of individual residues in PP1 binding
We next wished to determine whether the residues identified by NMR are important for PP1 binding to SRSF1. To accomplish this, we mapped the affected residues onto the structure of RRM1 and used two criteria for selecting a subset of these for mutagenesis. First, we chose eight residues whose side chains are solvent-accessible in RRM1 and thus could interact directly with PP1 (Fig. 3A, starred residues). Second, because RRM2 does not interact effectively with PP1, we selected a subset of these residues that are unique in RRM1 (Fig. 3A, yellow boxes). When mapped onto the RRM1 structure, these five residues define three potential surface regions for PP1 contact (Fig. 3A, dotted ovals). Region I, the largest cluster of PP1-affected residues, is composed of two charged residues (Arg-65 and Glu-68) near the N terminus of helix α2 and an aromatic residue (Tyr-39) at the C terminus of helix α1. Two other charged residues (Lys-48 and Asp-80) are distally located from this cluster and constitute regions II and III. To evaluate the functional roles of these three regions, we individually replaced these five residues with alanine and monitored their binding to GST–PP1 in pulldown assays (Fig. 3B). By calculating the fraction bound (FB) from the pulldown and input lanes, we found that whereas K48A and D80A had no significant impact (FB > 0.9), Y39A, R65A, and E68A interacted less efficiently with GST–PP1 (FB 0.3–0.7; Fig. 3D). Because these pulldown assays are semiquantitative, we measured directly the binding affinities of the RRM1 mutants with His-tagged PP1 (His–PP1) in kinetic assays using PNPP. We monitored the progress curves for PNPP dephosphorylation by His–PP1 to obtain the initial velocities for the different variants as a function of RRM1 (Fig. S2). Using this assay, mutations in region I displayed appKI values 2–3-fold larger than that for WT RRM1, whereas those in regions II and III were less than 1.6-fold larger (Fig. 3C). These affinity changes correlate with reductions in the fraction bound of RRM1–PP1 in the pulldown assays (Fig. 3D). Overall, these experiments suggest that residues in region I of RRM1 are likely to be important for interactions with PP1.
Figure 3.

Identification of residues involved in RRM1–PP1 interactions. A, comparisons of RRM1 and RRM2 sequences and three-dimensional structures. In the RRM1 sequence at the top, the residues affected in the NMR spectrum by the addition of PP1 are highlighted in red, stars identify surface residues, and the yellow boxes indicate five residues that were further investigated by amino acid replacement (see text). Below the sequences, surface-exposed residues affected by PP1 binding in the NMR experiments are mapped in green onto the three-dimensional RRM1 structure and overlaid with the corresponding RRM2 residues (blue). These residues constitute three separate regions on one surface of RRM1 (I, II, and III). B, pulldown assays showing specific interactions of PP1 with WT and mutant versions of RRM1. GST–PP1, bound to GSH beads, was incubated with RRM1, washed, and run on a 12% SDS–PAGE. The fraction bound for each RRM1 was determined using ImageJ and plotted in D. C, binding constants to PP1 for WT and mutant versions of RRM1, using inhibition kinetics. Dephosphorylation of PNPP (25 mm) by His–PP1 (20 nm) was monitored for variable concentrations of RRM1. The appKI values are plotted in D. D, comparison of the binding parameters of WT and mutant versions of RRM1 to PP1. Fractions bound and appKI values from B and C are plotted in the bar graph.
PP1 recognition of SRSF1 depends on a putative RRM1 docking surface
Having identified three unique residues localized to region I in RRM1 (Fig. 3A) that weaken contacts with PP1 upon alanine substitution, we next wished to determine whether these residues might be part of a binding surface in the SRSF1–PP1 complex. To evaluate this, we initially made a triple alanine mutant of RRM1 (RRM1TM-R65A,E68A,Y39A) to determine its binding affinity to PP1. We found with pulldown assays that GST–PP1 effectively interacted with RRM1, as expected, but not with RRM1TM (Fig. 4A). Likewise, RRM1 efficiently inhibited PP1 activity (appKI = 320 ± 20 nm), whereas RRM1TM did not (Fig. S3 and Fig. 4B). From these data, we estimate a lower limit of ∼8 μm on the appKI of RRM1TM for PP1, which is at least 20-fold larger than that for the WT domain. Interestingly, if we assume that each residue contributes to binding affinity in an additive manner, then we predict that the triple mutant should lower RRM1 binding affinity by ∼18-fold, a value in line with the experimental data. To determine whether the implicated putative docking surface affects PP1 interactions in the full-length SR protein, we replaced Arg-65, Glu-68, and Tyr-39 with alanines in SRSF1 (SRSF1TM). In pulldown assays, we demonstrated that GST–PP1 interacts with SRSF1 but not with SRSF1TM, showing that the three residues are also important for binding the phosphatase to RRM1 in context of the full-length SRSF1 (Fig. 4C). In a previous study, we showed that PP1 interacted less efficiently with SRSF1 than with the separate RRM1, because of contacts with the RS domain that interfere with PP1 binding to RRM1 (18), and that RRM1 interacts with the C terminus of the RS domain (RS2, residues 219–248). These contacts partially block PP1 binding but are severed by SRPK1 and CLK1 binding or phosphorylation (18). Here, we confirmed this result in new pulldown experiments (Fig. 4D) and then wished to determine whether PP1 and the RS domain share a common binding site on RRM1. To address this issue, we performed pulldown assays showing that a GST-tagged form of RS2 (GST–RS2) interacts with RRM1 but not with RRM1TM (Fig. 4D). These findings suggest that intramolecular binding of RS2 and intermolecular binding of PP1 may target overlapping binding sites on RRM1. Overall, these experiments show that Arg-65, Glu-68, and Tyr-39 are key components of the binding surface for PP1 in SRSF1.
Figure 4.

RRM1 surface residues regulate PP1 binding and SRSF1 dephosphorylation. A, pulldown assays showing that the triple mutant of RRM1 (RRM1TM-R65A,E68A,Y39A) disrupts PP1 binding. GST–PP1, bound to GSH beads, was incubated with RRM1 and RRM1TM, washed, and run on an 18% SDS–PAGE. B, binding constants for RRM1 and RRM1TM to PP1 using inhibition kinetics. Dephosphorylation of PNPP (25 mm) by His–PP1 (20 nm) was monitored as a function of RRM1 (●) and RRM1TM (■). C, pulldown assays showing that the triple mutant of SRSF1 (SRSF1TM-R65A,E68A,Y39A) disrupts PP1 binding. GST–PP1, bound to GSH beads, was incubated with SRSF1 and SRSF1TM, washed, and run on a 12% SDS–PAGE. D, pulldown assays showing that the RS domain interacts with putative PP1 docking residues on RRM1. GST–RS2, bound to GSH beads, was incubated with RRM1 and RRM1TM, washed, and run on an 18% SDS–PAGE. E, effects of PP1 on the phosphorylation kinetics of SRSF1 and SRSF1TM. SRSF1 and SRSF1TM (1 μm) and [32P]ATP (50 μm) were incubated with SRPK1 (70 nm) in the absence (●) and presence (■) of PP1 (450 nm). Phosphorylation of both substrates was monitored using 32P autoradiography. The data for both substrates are fitted to Equations 1 and 2 in the absence and presence of PP1 (Table S1).
Phosphatase activity toward SRSF1 is regulated by RRM1–PP1 contacts
We next wished to determine whether interactions between PP1 and RRM1 regulate the phosphorylation state of the SRSF1 RS domain. To address this, we mixed SRSF1 and SRSF1TM (1 μm) with SRPK1 (70 nm) and [32P]ATP (50 μm) in the absence and presence of PP1 (450 nm) and monitored net substrate phosphorylation as a function of time, using autoradiography. In the absence of PP1, the substrates were phosphorylated in two separate kinetic phases, i.e. a fast, single-exponential phase followed by a slow, linear phase (Fig. 4E). These phases, when fitted using Equation 1, reflect multisite phosphorylation events within the RS domain (Table S1). Interestingly, the initial phase velocities (v1 = k1α1) are similar for both substrates, suggesting that the three amino acid substitutions in RRM1TM do not affect the ability of the kinase to recognize and modify the RS domain (Fig. 4E). These initial velocities correspond to similar turnover numbers of 0.8 and 0.7 s−1 for the WT and mutant SR proteins. Although both substrates were phosphorylated at similar rates, the dephosphorylation rates were very different. The simultaneous addition of PP1 and SRPK1 more effectively lowered net incorporation of 32P into SRSF1TM than into SRSF1 (Fig. 4E). We fit these data to Equation 2, which reflects an analytical description of a two-step, consecutive mechanism where the first step (k1) is multisite phosphorylation by SRPK1 and the second step (k2) is multisite dephosphorylation by PP1 (21). Although such a computational approach is a simplification that does not take into account the many microscopic steps in the polyphosphorylation reactions, it allows a general analysis of cumulative dephosphorylation by PP1. Using this approach, the net dephosphorylation rate constant (k2) for SRSF1TM was found to be over 10-fold higher than that for SRSF1 (Table S1). We repeated these experiments and obtained similar kinetic effects on PP1 activity (Fig. S4). These results suggest a larger PP1 dephosphorylation efficiency for the mutant SR protein when compared with the WT form. As expected, the net phosphorylation rate constants (k1) for the two proteins were the same, which is consistent with phosphorylation data in the absence of PP1. Overall, these findings indicate that the three proximal surface residues Tyr-39, Arg-65, and Glu-68 in RRM1 serve a key role for the docking of PP1, which in turn down-regulates phosphatase activity toward SRSF1.
PP1 interactions regulate SRSF1 phosphorylation, subnuclear localization, and splicing function
Because several amino acids in RRM1 control PP1 binding and catalytic activity in vitro, we next wished to determine whether such residues also regulate the cellular function of the SR protein. We generated GFP-tagged forms of SRSF1 and SRSF1TM and expressed them in HeLa cells. In Western blots, we found that several bands corresponding to GFP–SRSF1 migrated more slowly on SDS–PAGE than those for GFP–SRSF1TM (Fig. 5A). Based on previous studies (13, 16, 22, 23), slow-migrating forms of SRSF1 correlate with enhanced phosphorylation states. We confirmed these observations by showing that treatment of the lysates with calf intestinal phosphatase (CIP) led to one common, fast-migrating band (Fig. 5A). To determine whether GFP–SRSF1TM and GFP–SRSF1 interact differentially with PP1, we performed pulldown experiments using recombinant GST–PP1 and HeLa cell lysates expressing either the WT or mutant SR proteins. We found that GST–PP1, bound to GSH beads, readily interacts with GFP–SRSF1 but not with GFP–SRSF1TM (Fig. 5B). Similar to the results in Fig. 5A, we also found that GFP–SRSF1TM displayed less slow- versus fast-migrating species compared with the WT SR protein. These findings suggest that reductions in RS domain phosphorylation upon mutation of RRM1 correlate with disruption of the SRSF1–PP1 complex.
Figure 5.

RRM1 mutations influence phosphorylation, subnuclear localization, and splicing function of SRSF1. A, phosphorylation levels of SRSF1 and SRSF1TM. HeLa cells expressing GFP–SRSF1 and GFP–SRSF1TM were lysed, incubated in the absence and presence of CIP, run on a 10% SDS–PAGE, and probed with an anti-GFP antibody. B, pulldown assays showing that PP1 interacts with GFP–SRSF1 but not with GFP–SRSF1TM. GST–PP1, bound to GSH beads, was incubated with HeLa cell lysates expressing GFP–SRSF1 and GFP–SRSF1TM, washed, and run on a 10% SDS–PAGE. SR proteins and PP1 are probed using anti-GFP and anti-GST antibodies. I, input; PD, pulldown; WB, Western blotting. Ctrl lane contains lysate but no GST–PP1. C, confocal imaging of GFP–SRSF1 and GFP–SRSF1TM expressed in HeLa cells in the absence and presence of okadaic acid. D and E, speckle (D) and total nuclear (E) areas for HeLa cells expressing GFP–SRSF1 and GFP–SRSF1TM are calculated using ImageJ and displayed in dot plots with mean values ± S.D. p values were calculated using a one-way analysis of variance test. p values above 0.05 are considered nonsignificant (ns). F, alternative splicing of several genes as a function of GFP–SRSF1 and GFP–SRSF1TM expression. RT–PCR is performed on RNA samples from HeLa cells expressing GFP–SRSF1 (WT) and GFP–SRSF1TM (TM). Long and Short refer to forms of the genes that include or exclude the exon. The percentages of exon inclusion, obtained from ImageJ analyses, are shown at the bottom of each gel.
We next wished to determine whether the phosphorylation status of the RS domain in lysates correlate with changes in the cellular trafficking and splicing function of the SR protein. We monitored GFP–SRSF1 and GFP–SRSF1TM in live HeLa cells using confocal microscopy and found that RRM1 mutation led to changes in subnuclear localization. Although both proteins localized to the nucleus, GFP–SRSF1TM was found in fewer speckles than GFP–SRSF1 (Fig. 5C). Speckle area for GFP–SRSF1TM is approximately half that of GFP–SRSF1, with no significant changes in total nuclear area (Fig. 5, D and E). To evaluate whether these effects are due to changes in SR protein phosphorylation, as evidenced in the Western blotting analyses (Fig. 5A), we repeated the imaging studies in the presence of the PP1 inhibitor okadaic acid. We found that incubation of the cells with okadaic acid reversed these effects for GFP–SRSF1TM, leading to increases in speckle formation, akin to that for GFP–SRSF1 (Fig. 5, C and D). Similar treatment of HeLa cells expressing GFP–SRSF1 had no significant effect on overall speckle area, suggesting that PP1 may be down-regulated relative to the kinases in these cells. We next performed RT–PCR experiments on the lysates to monitor the effects of the SR proteins on several genes that are alternatively spliced (24). We found that GFP–SRSF1 compared with GFP–SRSF1TM enhanced the formation of the long versus short forms of GOLIM4, EIF5, and TIMM8B, thus enhancing exon inclusion (Fig. 5F). Compared with these effects, both WT and mutant SR proteins had similar effects on exon inclusion for LUC7L2, suggesting that SRSF1 has some differential effects on splicing as previously reported (24) (Fig. 5F). Overall, these findings indicate that surface region I in RRM1 (Fig. 3A) regulates the cellular phosphorylation status and PP1-binding capacity of SRSF1, effects that control both the subnuclear localization and alternative splicing function of the SR protein.
Several serines in the RS domain regulate SRSF1 speckle occupancy
We showed that increased PP1 activity leads to a reduction in RS domain phosphorylation and a subsequent decrease in speckle localization of SRSF1, effects that impact exon inclusion (Fig. 5). We now wished to identify regions in the RS domain that may be responsible for this change in subnuclear mobility. Because we showed previously that CLK1-dependent phosphorylation of several Ser-Pro dipeptides in the C-terminal polypeptide segment of the RS domain (RS2) induces a hyperphosphorylation state that releases SRSF1 from speckles (16, 17), we speculated that dephosphorylation of serines at the other end of the RS domain (RS1) (Fig. 6A) might be responsible for the hypophosphorylated state that also releases the SR protein from speckles (Fig. 1A). To address this possibility, we made a series of alanine mutations in the Arg-Ser dipeptide repeat region of RS1 and monitored their subcellular localization using confocal microscopy (Fig. 6B). We found that removal of all serines in the RARA construct (Fig. 6A) blocked entry into the nucleus (Fig. 6B). This result is consistent with previous findings showing that RS domain deletion results in cytoplasmic pools of SRSF1, whereas the addition of only five Arg-Ser dipeptides onto this construct results in similar localization to nuclear speckles as seen for the WT SR protein (25). Interestingly, the present new results indicate that the RS2 segment, despite possessing two short stretches of Arg-Ser repeats, is not sufficient to support SRSF1 migration to the nucleus. Compared with RARA, we found that alanine substitution of half of the serines at either the N- or C-terminal ends of RS1 (RARS or RSRA; Fig. 6A) had no effect on nuclear localization of SRSF1 (Fig. 6B). These findings indicate that four Arg-Ser dipeptides are sufficient for nuclear entry of SRSF1, irrespective of the position within RS1. However, although RARS and RSRA are nuclear proteins, both display reduced occupancy in speckles, because we found that speckle areas for RARS and RSRA are approximately half that of the WT protein (Fig. 6, C and D). These findings suggest that CLK1-independent movement of SRSF1 from speckles to the nucleoplasm is likely the result of reduced phosphorylation of several Arg-Ser dipeptides in RS1. Overall, the data imply that increases in PP1 activity that drives SRSF1 into the nucleoplasm are likely the result of regiospecific decreases in RS1, rather than RS2, phosphorylation.
Figure 6.
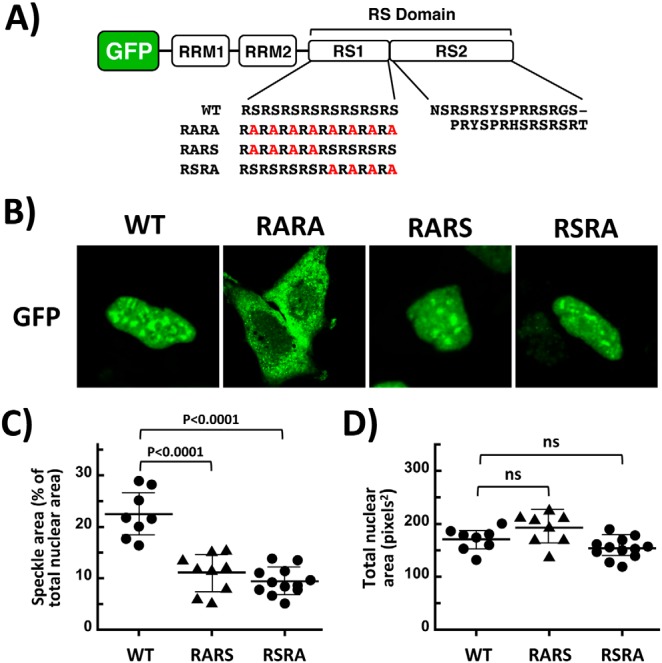
Identifying serines in the RS domain that are important for speckle occupancy. A, schematic alignment of SRSF1 with variants used in the present study. Ser-to-Ala mutations in the RS domain of GFP–SRSF1 are shown in red. B, subnuclear localization of WT and mutant forms of GFP–SRSF1 is monitored using confocal microscopy. C and D, speckle (C) and total nuclear (D) areas, determined using ImageJ, are displayed in dot plots with mean values ± S.D. p values were calculated using a one-way analysis of variance test. p values above 0.05 are considered nonsignificant (ns).
Discussion
SR proteins contain RRMs as essential structural components for pre-mRNA binding during assembly of the spliceosome (26). Although some SR proteins contain a single RRM, the family prototype SRSF1, along with four other SR proteins (SRSF4–6 and SRSF9), contains two RRMs. Although RRM2 is referred to as a “pseudo-RRM,” prior binding analyses revealed that this domain, rather than the canonical RRM1, is responsible for the recognition of purine-rich ESEs by SRSF1 and can support the splicing of the IGF-1 minigene to the same extent as an RRM1–RRM2 construct (27). Nonetheless, although detailed structural analyses reveal that RRM2 promotes similar proximal splice-site usage in the E1a minigene as the full-length SRSF1, RRM1 is necessary as a general enhancer of in vitro splicing (28). These studies raise questions regarding the functional role of RRM1 in SRSF1 for splicing control. Some investigations suggest that RRM1 may enable critical protein–protein interactions leading to spliceosome assembly. Cho et al. (4) demonstrated that RRM1 of SRSF1 interacts with an RRM from the U1–70K subunit of U1 snRNP, thus facilitating assembly of the early spliceosome. In a recent study, RRM1 was shown to serve as a platform for PP1 binding and activity control, although the molecular contacts for this interaction were not defined (17). Because these findings highlight the importance of RRM1 as a protein interaction module, we performed experiments to define the molecular nature of the PP1–RRM interface within an SR protein and uncover potential links to mRNA processing events.
Through analyses of 2D [15N,1H]-HSQC NMR spectra of RRM1, we identified three surface residues near the N terminus of helix α2 and the C terminus of helix α1 that appear to be most important for PP1 binding. Although we could detect a stable interaction between WT SRSF1 and PP1, the dissociation constant for PP1 and RRM1 (KI = 300 nm) is ∼1–3 orders of magnitude higher than traditional PP1 regulators such as CPI-17, I-1, I-2, and spinophilin (29–32). Nonetheless, we showed that the RRM1–PP1 interaction not only regulates RS domain phosphorylation levels in vitro and in cell lysates, but also controls the subnuclear localization and alternative splicing function of SRSF1, thus indicating that these putative docking residues are biologically significant. Furthermore, it appears that this group of residues is common within the SR protein family. For example, Tyr-39 and Arg-65 in SRSF1 are strictly conserved in 11 of the 12 known SR proteins, and Glu-68 is conserved as a negatively charged residue in 9 members of the family (Fig. S5A). By comparison, this residue cluster is not conserved in RRM2 sequences (Fig. S5B). Interestingly, although the observed docking residues in RRM1 are near a classic PP1-interacting motif (RVXF), previously thought to directly bind to PP1 (33), these residues are largely buried behind helix α2 and thus unlikely to make direct contact with PP1 in its native form (Fig. S6). Accordingly, these newly identified molecular contacts between RRM1 and PP1 are likely to reflect a novel interaction motif for RRMs in SR proteins.
Given the essential role of phosphorylation in activating SR proteins and initiating the splicing process, it is reasonable to suspect that certain factors may intervene to increase the lifetime of phosphates on the RS domain. We have uncovered molecular contacts between PP1 and RRM1 that serve this function by down-regulating phosphatase activity and increasing the phosphorylation of serines in the distal RS domain of SRSF1. Based on these findings, we can now speculate upon important intra- and intersteric contacts that govern this process. In this new model, we connected the RRM1 and RRM2 structures, solved independently by NMR, at the glycine-rich linker between the two domains and displayed the unphosphorylated RS domain interacting with both RRMs (Fig. 7). In a previous study we demonstrated that the N-terminal portion of the RS domain (RS1) interacts selectively with RRM2 (34). We can now combine these findings with our new data showing that the C-terminal portion of the RS domain (RS2) interacts selectively with RRM1 blocking PP1 binding (Fig. 7). We propose that this PP1–RRM1 interaction increases the lifetime of phosphates on the RS domain for the facile initiation of spliceosome assembly. It is likely that the dynamic nature of this interaction may be important so that the RS domain can be subsequently dephosphorylated during maturation of the spliceosome. Indeed, the relatively loose affinity of RRM1 for PP1 compared with other PP1-interacting proteins may be an advantage toward achieving these objectives. Through formation of a dynamic PP1–SRSF1 complex, the repression of phosphatase activity by RRM1 might thus be sufficient to achieve rapid RS domain phosphorylation without inhibiting subsequent dephosphorylation events along the spliceosome maturation pathway.
Figure 7.

Model showing residues in RRM1 that recognize PP1 and down-regulate phosphatase activity. RRM1 and RRM2 structures, determined by NMR, are combined at the glycine-rich linker between the two domains. C-terminal RS domain segment overlaps with PP1 docking region (red) in RRM1. The addition of phosphates (blue spheres) by protein kinases such as SRPK1 severs RS domain contacts with RRM1, promoting binding and down-regulation of PP1.
Experimental procedures
Materials
ATP, Mops, HEPES, Tris, MgCl2, MnCl2, NaCl, EDTA, Brij 35, glycerol, acetic acid, lysozyme, DNase, RNase, Phenix imaging film, BSA, nickel resin, and liquid scintillant were obtained from Fisher Scientific. [γ-32P]ATP was obtained from NEN Products, FuGENE reagent was obtained from Promega and Lipofectamine 2000 from Thermo Fisher. Protease inhibitor mixture was obtained from Roche, anti-GFP mAb was obtained from Cell Signaling, anti-GST mAb was obtained from BioLegend, CIP and PNPP were obtained from NEB, and InstantBlue was obtained from Expedeon.
Protein expression and purification
For binding and kinetics experiments, recombinant SRPK1 and PP1γ were expressed and purified from pET19b. All forms of SRSF1, RRM1, and RRM2 were expressed and purified from pET15b vectors containing an N-terminal His tag in BL21(DE3), as previously described (34, 35). All forms of GST–SRSF1 and GST–PP1γ were expressed and purified from a PGEX6P-1 vector in BL21(DE3), as previously described (35). For confocal imaging experiments, GFP–SRSF1 constructs were expressed from pcDNA3.1+N-eGFP vectors in HeLa cells (17).
For NMR experiments, uniformly 15N- and 13C,15N-labeled RRM1 was expressed in BL21(DE3) using M9 minimal medium supplemented with 15NH4Cl (1 g/liter) and either unlabeled or [13C6]d-glucose (4 g/L). Cells grown at 37 °C to an A600 of 0.6–0.8 were induced with 1 mm isopropyl β-d-thiogalactopyranoside for 16 h at 18 °C. Upon centrifugation, the cell pellets were resuspended in a buffer containing 200 mm NaCl, 10 mm imidazole, 20 mm Na2HPO4/NaH2PO4 (pH 7.5), and Complete EDTA-free protease inhibitor mixture tablets (Roche). The cells were lysed by sonication and centrifuged at 30,000 × g for 30 min. The supernatant was loaded onto a HisTrap HP nickel-affinity column (GE Healthcare) pre-equilibrated with purification buffer (200 mm NaCl, 10 mm imidazole, 20 mm Na2HPO4/NaH2PO4, pH 7.5). The column was washed with 30 mm imidazole, and the protein was then eluted with 500 mm imidazole. The His tag was removed by overnight TEV protease treatment at room temperature. The mixture was then loaded onto a HiPrep 26/10 desalting column (GE Healthcare) and eluted with purification buffer prior to removal of the TEV protease and the cleaved His tag using a HisTrap HP column (GE Healthcare) equilibrated with buffer A. Fractions containing RRM1 were loaded onto a size-exclusion column HiLoad 26/60 Superdex75 (GE Healthcare) equilibrated with 50 mm NaCl, 20 mm Na2HPO4/NaH2PO4 (pH 6.0). The fractions containing RRM1 were combined and concentrated to 0.4 mm (550 μl) using 3-kDa cutoff centrifugal filter devices (Millipore). For the NMR measurements, RRM1 solutions were supplemented with 5% 2H2O (v/v) and 4.5 mm NaN3.
Pulldown assays
GST-tagged proteins (10 μm) were incubated with His-tagged proteins (10 μm) in binding buffer (0.1% Nonidet P-40, 20 mm Tris/HCl, pH 7.5, and 75 mm NaCl) in a total volume of 40 μl for 30 min before incubating with 25 μl of GSH beads for 30 min at room temperature. In all cases, the resin was washed three times with 200 μl of binding buffer, and the bound proteins were eluted with SDS quench buffer and boiled for 5 min. Retained protein was resolved by SDS–PAGE (12% or 18% gel) and visualized by Instant Blue Coomassie stain.
Phosphorylation reactions
Phosphorylation of SRSF1 by SRPK1 was carried out in the presence of 100 mm Mops (pH 7.4), 50 mm HEPES, 100 mm NaCl, 2 mm DTT, 0.01% Brij 35, 1 mm Mn2+, 10 mm Mg2+, and 5 mg/ml BSA at 37 °C, using 50 μm [32P]ATP with a specific activity of 4000–8000 cpm/pmol and in the absence or presence of PP1. All reactions were carried out in a total volume of 10 μl and quenched with 10 μl of SDS/PAGE loading buffer. Phosphorylated SR proteins were separated from unreacted [32P]ATP by SDS–PAGE (12% gel), cut from the dried gel, and quantified on the 32P channel in liquid scintillant.
Phosphatase reactions
The p-nitrophenyl phosphate (PNPP) assay for PP1 was carried out in a nanodrop spectrophotometer in a volume of 750 μl using a 1-ml plastic cuvette with the absorption monitored at 405 nm. His–PP1γ (20 nm) is incubated in the presence of 1× PMP buffer, 1 mm MnCl2, 100 mm Mops (pH 7.5), and 25 mm PNPP in the absence and presence of varying ligand. To monitor SR protein dephosphorylation in cell extracts, HeLa cells expressing GFP–SRSF1 were harvested and lysed in 1× radioimmune precipitation assay buffer supplemented with protease inhibitor mixture, incubated in the absence and presence of CIP, and run on a 10% PAGE gel using an anti-GFP antibody for detection.
Confocal imaging and splicing experiments
For live-cell confocal imaging, HeLa cells were plated on 2.5-cm2 MatTek poly-d-lysine plates and transfected with GFP–SRSF1 constructs (2 μg) for 24 h. The cells were washed with PBS, and transfected HeLa cells were analyzed using an Olympus FV1000 as described previously (16). To analyze the effects of phosphatase inhibition, HeLa cells expressing GFP–SRSF1 were washed with PBS and incubated for 1 h with OptiMem medium supplemented with 200 nm okadaic acid prior to imaging. To monitor alternative splicing in Hela cells expressing SRSF1 proteins, total RNA was isolated from cell lysates using the RNEasy Plus kit from Qiagen. Oligo sets for several genes were identified and prepared as previously described (24). To detect spliced gene products, reverse transcription was performed with isolated RNA using the Superscript III one-step RT–PCR kit from Invitrogen. The cDNA was then resolved on a 1% agarose gel and imaged using a Bio-Rad gel doc system.
NMR data acquisition and backbone assignment
All NMR experiments were recorded at 298 K on a Bruker AVANCE 600-MHz spectrometer equipped with a 5-mm z-gradient cryoprobe. To obtain backbone assignments for RRM1 4D APSY-HACANH, 5D APSY-CBCACONH, 5D APSY-HACACONH, 3D HNCA and 3D HNCACB NMR experiments were recorded (36, 37). Chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonic acid sodium salt. Resonance assignments were achieved using the J-UNIO protocol (19, 20). NMR assignments have been deposited in the Biological Magnetic Resonance Bank (accession number 27499).
Data analysis
The amount of phosphorylated SR protein was determined from the specific activity of [32P]ATP, and the CPMs of excised bands were corrected for background. Progress curves for SR protein phosphorylation were fitted to Equation 1,
| (Eq. 1) |
where P is 32P-labeled SR protein, α1 and k1 are the amplitude and rate constant of the first phase, and L is the linear rate for the second phase. Progress curves for SR protein phosphorylation in the presence of PP1 were fitted to Equation 2,
| (Eq. 2) |
where P is 32P-labeled SR protein, α1 and k1 are the amplitude and net rate constant for SRPK1-dependent phosphorylation, and k2 is the net rate constant for PP1-dependent dephosphorylation.
Author contributions
B. E. A., P. S., and L. F. data curation; B. E. A., P. S., and L. F. formal analysis; B. E. A., P. S., and L. F. investigation; B. E. A., P. S., and L. F. methodology; K. W. and J. A. A. supervision; K. W. and J. A. A. writing-review and editing; J. A. A. conceptualization; J. A. A. funding acquisition; J. A. A. writing-original draft; J. A. A. project administration.
Supplementary Material
Acknowledgment
We thank Dr. Nicolas Villanueva for help with purifying GST–RS2.
This work was supported by National Institutes of Health Grants GM67969 and GM98528. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S6.
The NMR spectroscopy data reported in this paper has been submitted to the Biological Magnetic Resonance Data Bank (BMRB) under BMRB accession number 27499.
- pre-mRNA
- precursor mRNA
- CLK1
- cdc2-like kinase 1
- PNPP
- p-nitrophenyl phosphate
- PP1
- protein phosphatase 1
- RRM
- RNA recognition motif
- RS domain
- domain rich in arginine-serine dipeptide repeats
- SR protein
- splicing factor containing arginine-serine dipeptide repeats
- SRPK1
- serine-arginine–specific protein kinase 1
- SRSF1
- SR protein splicing factor 1 (also called ASF/SF2)
- FB
- fraction bound
- CIP
- calf intestinal phosphatase
- APSY
- automated projection spectroscopy.
References
- 1. Müller S., Wolpensinger B., Angenitzki M., Engel A., Sperling J., and Sperling R. (1998) A supraspliceosome model for large nuclear ribonucleoprotein particles based on mass determinations by scanning transmission electron microscopy. J. Mol. Biol. 283, 383–394 10.1006/jmbi.1998.2078 [DOI] [PubMed] [Google Scholar]
- 2. Kohtz J. D., Jamison S. F., Will C. L., Zuo P., Lührmann R., Garcia-Blanco M. A., and Manley J. L. (1994) Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature 368, 119–124 10.1038/368119a0 [DOI] [PubMed] [Google Scholar]
- 3. Wu J. Y., and Maniatis T. (1993) Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 75, 1061–1070 10.1016/0092-8674(93)90316-I [DOI] [PubMed] [Google Scholar]
- 4. Cho S., Hoang A., Sinha R., Zhong X. Y., Fu X. D., Krainer A. R., and Ghosh G. (2011) Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1–70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 8233–8238 10.1073/pnas.1017700108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stojdl D. F., and Bell J. C. (1999) SR protein kinases: the splice of life. Biochem. Cell Biol. 77, 293–298 10.1139/o99-046 [DOI] [PubMed] [Google Scholar]
- 6. Ghosh G., and Adams J. A. (2011) Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 278, 587–597 10.1111/j.1742-4658.2010.07992.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aubol B. E., Chakrabarti S., Ngo J., Shaffer J., Nolen B., Fu X. D., Ghosh G., and Adams J. A. (2003) Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc. Natl. Acad. Sci. U.S.A. 100, 12601–12606 10.1073/pnas.1635129100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma C. T., Hagopian J. C., Ghosh G., Fu X. D., and Adams J. A. (2009) Regiospecific phosphorylation control of the SR protein ASF/SF2 by SRPK1. J. Mol. Biol. 390, 618–634 10.1016/j.jmb.2009.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aubol B. E., and Adams J. A. (2011) Applying the brakes to multisite SR protein phosphorylation: substrate-induced effects on the splicing kinase SRPK1. Biochemistry 50, 6888–6900 10.1021/bi2007993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lai M. C., Lin R. I., Huang S. Y., Tsai C. W., and Tarn W. Y. (2000) A human importin-beta family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J. Biol. Chem. 275, 7950–7957 10.1074/jbc.275.11.7950 [DOI] [PubMed] [Google Scholar]
- 11. Lai M. C., Lin R. I., and Tarn W. Y. (2001) Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 10154–10159 10.1073/pnas.181354098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Misteli T., Cáceres J. F., and Spector D. L. (1997) The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527 10.1038/387523a0 [DOI] [PubMed] [Google Scholar]
- 13. Ngo J. C., Chakrabarti S., Ding J. H., Velazquez-Dones A., Nolen B., Aubol B. E., Adams J. A., Fu X. D., and Ghosh G. (2005) Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell 20, 77–89 10.1016/j.molcel.2005.08.025 [DOI] [PubMed] [Google Scholar]
- 14. Duncan P. I., Stojdl D. F., Marius R. M., Scheit K. H., and Bell J. C. (1998) The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp. Cell Res. 241, 300–308 10.1006/excr.1998.4083 [DOI] [PubMed] [Google Scholar]
- 15. Sacco-Bubulya P., and Spector D. L. (2002) Disassembly of interchromatin granule clusters alters the coordination of transcription and pre-mRNA splicing. J. Cell Biol. 156, 425–436 10.1083/jcb.200107017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keshwani M. M., Aubol B. E., Fattet L., Ma C. T., Qiu J., Jennings P. A., Fu X. D., and Adams J. A. (2015) Conserved proline-directed phosphorylation regulates SR protein conformation and splicing function. Biochem. J. 466, 311–322 10.1042/BJ20141373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aubol B. E., Keshwani M. M., Fattet L., and Adams J. A. (2018) Mobilization of a splicing factor through a nuclear kinase-kinase complex. Biochem. J. 475, 677–690 10.1042/BCJ20170672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aubol B. E., Hailey K. L., Fattet L., Jennings P. A., and Adams J. A. (2017) Redirecting SR protein nuclear trafficking through an allosteric platform. J. Mol. Biol. 429, 2178–2191 10.1016/j.jmb.2017.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dutta S. K., Serrano P., Proudfoot A., Geralt M., Pedrini B., Herrmann T., and Wüthrich K. (2015) APSY-NMR for protein backbone assignment in high-throughput structural biology. J. Biomol. NMR 61, 47–53 10.1007/s10858-014-9881-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Volk J., Herrmann T., and Wüthrich K. (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J. Biomol. NMR 41, 127–138 10.1007/s10858-008-9243-5 [DOI] [PubMed] [Google Scholar]
- 21. Gutfreund H. (1995) Kinetics for the Life Sciences: Receptors, Transmitters and Catalysts, Cambridge University Press, Cambridge, UK [Google Scholar]
- 22. Prasad J., and Manley J. L. (2003) Regulation and substrate specificity of the SR protein kinase Clk/Sty. Mol. Cell. Biol. 23, 4139–4149 10.1128/MCB.23.12.4139-4149.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Velazquez-Dones A., Hagopian J. C., Ma C. T., Zhong X. Y., Zhou H., Ghosh G., Fu X. D., and Adams J. A. (2005) Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty. J. Biol. Chem. 280, 41761–41768 10.1074/jbc.M504156200 [DOI] [PubMed] [Google Scholar]
- 24. Zhou Z., Qiu J., Liu W., Zhou Y., Plocinik R. M., Li H., Hu Q., Ghosh G., Adams J. A., Rosenfeld M. G., and Fu X. D. (2012) The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell 47, 422–433 10.1016/j.molcel.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cazalla D., Zhu J., Manche L., Huber E., Krainer A. R., and Cáceres J. F. (2002) Nuclear export and retention signals in the RS domain of SR proteins. Mol. Cell. Biol. 22, 6871–6882 10.1128/MCB.22.19.6871-6882.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeong S. (2017) SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 40, 1–9 10.14348/molcells.2017.2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clery A., Sinha R., Anczuków O., Corrionero A., Moursy A., Daubner G. M., Valcárcel J., Krainer A. R., and Allain F. H. (2013) Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc. Natl. Acad. Sci. U.S.A. 110, E2802–E2811 10.1073/pnas.1303445110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dauksaite V., and Akusjärvi G. (2004) The second RNA-binding domain of the human splicing factor ASF/SF2 is the critical domain controlling adenovirus E1A alternative 5′-splice site selection. Biochem. J. 381, 343–350 10.1042/BJ20040408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eto M., Ohmori T., Suzuki M., Furuya K., and Morita F. (1995) A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C: isolation from porcine aorta media and characterization. J. Biochem. 118, 1104–1107 10.1093/oxfordjournals.jbchem.a124993 [DOI] [PubMed] [Google Scholar]
- 30. Gibbons J. A., Weiser D. C., and Shenolikar S. (2005) Importance of a surface hydrophobic pocket on protein phosphatase-1 catalytic subunit in recognizing cellular regulators. J. Biol. Chem. 280, 15903–15911 10.1074/jbc.M500871200 [DOI] [PubMed] [Google Scholar]
- 31. Ragusa M. J., Dancheck B., Critton D. A., Nairn A. C., Page R., and Peti W. (2010) Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol. 17, 459–464 10.1038/nsmb.1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Picking W. D., Kudlicki W., Kramer G., Hardesty B., Vandenheede J. R., Merlevede W., Park I. K., and DePaoli-Roach A. (1991) Fluorescence studies on the interaction of inhibitor 2 and okadaic acid with the catalytic subunit of type 1 phosphoprotein phosphatases. Biochemistry 30, 10280–10287 10.1021/bi00106a028 [DOI] [PubMed] [Google Scholar]
- 33. Novoyatleva T., Heinrich B., Tang Y., Benderska N., Butchbach M. E., Lorson C. L., Lorson M. A., Ben-Dov C., Fehlbaum P., Bracco L., Burghes A. H., Bollen M., and Stamm S. (2008) Protein phosphatase 1 binds to the RNA recognition motif of several splicing factors and regulates alternative pre-mRNA processing. Hum. Mol. Genet. 17, 52–70 10.1093/hmg/ddm284 [DOI] [PubMed] [Google Scholar]
- 34. Serrano P., Aubol B. E., Keshwani M. M., Forli S., Ma C. T., Dutta S. K., Geralt M., Wüthrich K., and Adams J. A. (2016) Directional phosphorylation and nuclear transport of the splicing factor SRSF1 is regulated by an RNA recognition motif. J. Mol. Biol. 428, 2430–2445 10.1016/j.jmb.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aubol B. E., Plocinik R. M., Keshwani M. M., McGlone M. L., Hagopian J. C., Ghosh G., Fu X. D., and Adams J. A. (2014) N-terminus of the protein kinase CLK1 induces SR protein hyperphosphorylation. Biochem. J. 462, 143–152 10.1042/BJ20140494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hiller S., Fiorito F., Wüthrich K., and Wider G. (2005) Automated projection spectroscopy (APSY). Proc. Natl. Acad. Sci. U.S.A. 102, 10876–10881 10.1073/pnas.0504818102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hiller S., Wider G., and Wüthrich K. (2008) APSY-NMR with proteins: practical aspects and backbone assignment. J. Biomol. NMR 42, 179–195 10.1007/s10858-008-9266-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.