

Abstract
The type I cGMP-dependent protein kinase (PKG I) is an essential regulator of vascular tone. It has been demonstrated that the type Iα isoform can be constitutively activated by oxidizing conditions. However, the amino acid residues implicated in this phenomenon are not fully elucidated. To investigate the molecular basis for this mechanism, we studied the effects of oxidation using recombinant WT, truncated, and mutant constructs of PKG I. Using an in vitro assay, we observed that oxidation with hydrogen peroxide (H2O2) resulted in constitutive, cGMP-independent activation of PKG Iα. PKG Iα C42S and a truncation construct that does not contain Cys-42 (Δ53) were both constitutively activated by H2O2. In contrast, oxidation of PKG Iα C117S maintained its cGMP-dependent activation characteristics, although oxidized PKG Iα C195S did not. To corroborate these results, we also tested the effects of our constructs on the PKG Iα–specific substrate, the large conductance potassium channel (KCa 1.1). Application of WT PKG Iα activated by either cGMP or H2O2 increased the open probabilities of the channel. Neither cGMP nor H2O2 activation of PKG Iα C42S significantly increased channel open probabilities. Moreover, cGMP-stimulated PKG Iα C117S increased KCa 1.1 activity, but this effect was not observed under oxidizing conditions. Finally, we observed that PKG Iα C42S caused channel flickers, indicating dramatically altered KCa 1.1 channel characteristics compared with channels exposed to WT PKG Iα. Cumulatively, these results indicate that constitutive activation of PKG Iα proceeds through oxidation of Cys-117 and further suggest that the formation of a sulfur acid is necessary for this phenotype.
Keywords: cyclic GMP (cGMP), oxidation-reduction (redox), protein kinase G (PKG), potassium channel, signal transduction, AGC kinases, cGMP-dependent protein kinase, allostery, constitutive activation
Introduction
Blood pressure is maintained in part through constriction and relaxation of smooth muscle cells in the peripheral vasculature. During vasorelaxation, endothelium-derived nitric oxide or circulating natriuretic peptides activate guanylyl cyclases, which produce cGMP (1). This second messenger binds to and activates the cGMP-dependent protein kinase I (PKG I),3 a homodimeric enzyme that is expressed as two isoforms in vascular smooth muscle cells, PKG Iα and Iβ (2). PKG Iα acts as a central regulator of vasorelaxation by modulating global and local Ca2+ levels within the cell. Its phosphorylation targets include the large conductance calcium-activated potassium channel (BKCa, KCa 1.1), which causes membrane hyperpolarization and inhibition of voltage-dependent calcium channels (3–5). PKG Iα also phosphorylates the myosin phosphatase–targeting subunit 1 (MYPT1), the small GTP-binding protein (RhoA), and the regulator of G-protein signaling 2 (RGS2), which mediate the downstream cytoskeletal rearrangement necessary for vascular relaxation (6–9).
This signaling cascade and physiological effects associated with cGMP-dependent activation of PKG Iα are well-established (2). Under oxidizing conditions, PKG Iα is known to be constitutively active (10–12). Several studies have suggested that a residue unique to the N terminus of PKG Iα, Cys-42, may be the source of this activation (10, 13–15). The proposed mechanism involves the formation of an intermolecular disulfide bond between Cys-42 and Cys-42′ from the opposing protomer in the homodimeric holoenzyme. Because this disulfide bond is located in the substrate targeting region of the kinase, it has been hypothesized that oxidation at this site also modulates targeting to substrates by altering the geometry of this domain (10, 11). These observations have been consequential toward understanding the role of oxidizing agents, such as hydrogen peroxide, in both physiological and pathophysiological processes within the vasculature (16–22).
Both activation and substrate targeting are required for PKG I–dependent signaling, and disruption of either is sufficient to alter normal physiological processes (23–26). In this study, we sought to isolate these two facets of PKG Iα signaling by studying the effects of oxidation using recombinant WT, truncated, and mutant constructs of PKG I. We found that both PKG Iα and Iβ are activated by oxidation, although its effect on PKG Iα is more pronounced. Furthermore, both PKG Iα C42S and Δ53 PKG Iα (a truncation mutant that does not contain Cys-42) are activated by oxidation. Using PKG Iα C117S and C195S to test the influence of a previously observed disulfide bond, we observed that oxidation at Cys-117 is necessary and sufficient for oxidative activation of PKG Iα. Finally, we corroborated these in vitro results by measuring the PKG Iα–dependent activation of the KCa 1.1 channel. Taken together, these data support the hypothesis that constitutive activation of PKG Iα via oxidation is not mediated through the interprotomer disulfide bond between Cys-42 and Cys-42′, but rather through oxyacid formation at Cys-117 in the cGMP-A site.
Results
Oxidation of PKG Iα abrogates cGMP-dependent activation
Under oxidizing conditions, there are two confirmed sites where disulfide bonds form in the PKG Iα holoenzyme, which are an interprotomer disulfide bond between Cys-42 and Cys-42′ and an intraprotomer disulfide bond between Cys-117 and Cys-195 (Fig. 1A) (11). These disulfide bonds have also been identified in recent structural studies of the isolated domains (Fig. 1, B and C) (11, 27, 28). The first disulfide bond between Cys-42 and Cys-42′ forms at the C-terminal region of the dimerization domain (Fig. 1B). Cys-42 is one of four nonleucine/isoleucine residues at d positions within the coiled-coil and is unique to this isoform (Fig. S2A). The second disulfide bond in cGMP-A forms between Cys-117 and Cys-195 and is proximal to the cGMP-binding pocket (Fig. 1C and Fig. S2B).
Figure 1.
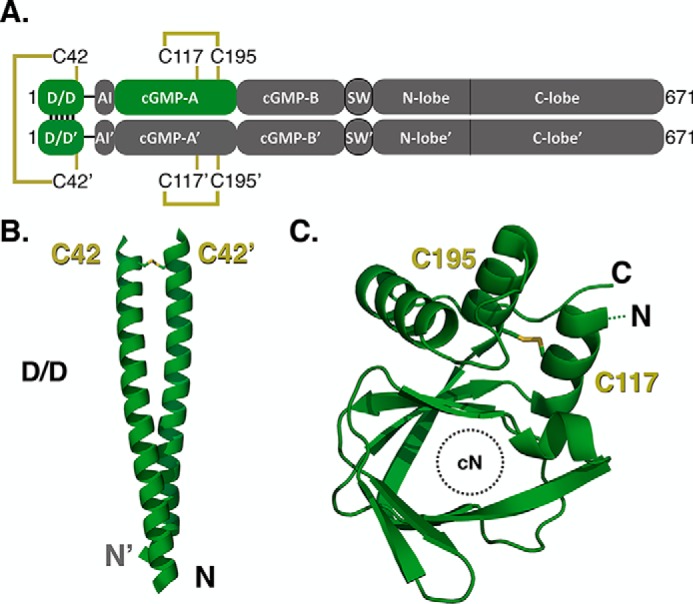
Putative sites of oxidative activation in PKG Iα. A, linear domain diagram depicting the homodimeric PKG Iα and the sites of cysteine residues hypothesized to be involved in the oxidative phenotype. B and C, structures denoting the location of disulfide bonds identified in the N-terminal dimerization domain (B) and the cGMP-A site (C) from PKG Iα (PDB entries 4R4L and 3SHR, respectively).
To characterize the oxidative phenotype, recombinant PKG Iα from Sf9 cells was used for in vitro phosphotransferase assays, wherein changes in kinase activity could be monitored. First, we confirmed that PKG Iα activity under reducing conditions (WTred) was consistent with previous studies (29, 30). Exposure to increasing concentrations of cGMP activated the kinase in a cooperative manner (nH = 1.7) by 20-fold with a half-maximal concentration of 200 nm (Fig. 2A and Table 1). We next examined how increasing H2O2 concentrations affected PKG Iα activity. WTred was exposed to H2O2, and kinase activity was monitored under apo and saturating cGMP conditions (Fig. 2B). A maximum response was observed with 500 μm H2O2, wherein we observed a reduction in the maximal velocity paired with a peak in the minimum velocity. At 1 mm H2O2, we observed a relative loss of activity in the presence and absence of cGMP, suggesting that exposure of the enzyme to this concentration of H2O2 had compromised its catalytic function. To expand our understanding of the conditions in which to measure the oxidative phenotype, the -fold activation of PKG Iα was monitored over a timed exposure to 500 μm H2O2 (Fig. 2C). After 30 min of exposure, we observed the greatest effect on the cGMP-dependent activation of the kinase, wherein the -fold activation was reduced by ∼90% to 2.5 (Table 1). These conditions were used for all subsequent measurements of the oxidation phenotype.
Figure 2.

Initial characterization of the PKG Iα oxidative phenotype. A, cGMP-dependent activation under reducing (solid line; n = 12) and oxidizing (500 μm H2O2, 30 min; dashed line; n = 16) conditions. B, PKG Iα activity plotted against increasing concentrations of H2O2. Activity was measured in the absence (black; n = 4) and presence of cGMP (red; 4 μm, n = 4). The concentration of H2O2 (500 μm) that resulted in the greatest increase in basal activity is highlighted in the boxed region. C, time course examining PKG Iα oxidation with 500 μm cGMP wherein the -fold activity (Vmax/Vmin) was monitored (n = 4). D, representative nonreducing, denaturing SDS-PAGE of PKG Iα exposed to increasing concentrations of H2O2. The dashed line indicates the point at which the concentration of hydrogen peroxide overcomes the concentration of TCEP in the buffer. H2O2 values corrected for the presence of TCEP are shown in red. Where shown, data are represented as the mean ± S.D. (error bars).
Table 1.
cGMP-dependent phosphotransferase activity
| Constructs | Reduced |
Oxidized |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vmina | Vmaxa | Vmax/Vmin | Kab | nHb | n | Vmina | Vmaxa | Vmax/Vmin | Kab | nHb | n | |
| μmol/min × mg | μmol/min × mg | μm | μmol/min × mg | μmol/min × mg | μm | |||||||
| PKG Iα | ||||||||||||
| WT | 0.22 ± 0.00 | 4.39 ± 0.03 | 20.0 | 0.20 ± 0.00 | 1.7 ± 0.0 | 12 | 1.13 ± 0.08 | 2.77 ± 0.03 | 2.5 | NA | NA | 16 |
| C42S | 0.32 ± 0.06 | 2.31 ± 0.18 | 7.3 | 0.58 ± 0.03 | 1.1 ± 0.1 | 14 | 1.10 ± 0.03 | 2.36 ± 0.11 | 2.1 | 0.13 ± 0.01 | 1.6 ± 0.2 | 6 |
| Δ53 | 0.18 ± 0.16 | 3.95 ± 0.30 | 21.6 | 0.22 ± 0.03 | 0.9 ± 0.1 | 7 | 1.38 ± 0.19 | 1.58 ± 0.36 | 1.1 | NA | NA | 6 |
| C117S | 0.16 ± 0.10 | 4.09 ± 0.13 | 26.0 | 0.12 ± 0.01 | 1.4 ± 0.1 | 6 | 0.65 ± 0.14 | 3.81 ± 0.10 | 5.9 | 0.20 ± 0.00 | 1.2 ± 0.1 | 6 |
| C195S | 0.43 ± 0.02 | 4.67 ± 0.12 | 10.9 | 0.27 ± 0.01 | 1.8 ± 0.1 | 6 | 0.86 ± 0.02 | 3.20 ± 0.05 | 3.7 | 1.22 ± 0.11 | 0.8 ± 0.1 | 6 |
| PKG Iβ | ||||||||||||
| WT | 0.04 ± 0.01 | 3.62 ± 0.26 | 86.1 | 1.21 ± 0.03 | 1.9 ± 0.1 | 7 | 0.42 ± 0.03 | 2.78 ± 0.10 | 6.6 | 0.86 ± 0.03 | 1.2 ± 0.1 | 6 |
a The velocity values indicate the μmol of peptide substrate (W15) phosphorylated/min/mg of PKG (μmol/min × mg) and are represented as the mean ± S.D.
b Values were obtained from non-linear regression fit analyses and are represented as the means ± standard deviations. Not applicable (NA) values indicate the data for that condition could not be fit to a non-linear regression.
Next, we tested the cGMP-dependent activation of oxidized PKG Iα. Oxidation resulted in a 5–6-fold increase in basal activity in the absence of cGMP (1.13 μmol/min × mg; p < 0.001 compared with WTred; Fig. 2A). In contrast, the addition of up to 4 μm cGMP increased the activity by only 2.5-fold (2.77 μmol/min × mg). Moreover, the activation profile of WTox with cGMP showed a decreased sensitivity for the cyclic nucleotide. In agreement with previous studies, analysis of WT PKG Iα by nonreducing SDS-PAGE showed that increasing concentrations of H2O2 resulted in formation of an interprotomer disulfide bond (Fig. 2D). In determining whether our PKG I constructs formed the interprotomer disulfide bond using nonreducing, SDS-PAGE electrophoresis, preliminary experiments indicated that removal of the reducing agent from the storage buffer would expose the enzymes to ambient air. Because prior studies have used ambient air to activate PKG Iα by oxidation, we sought to avoid this contribution by retaining the reducing agent, tris(2-carboxyethyl)phosphine (TCEP), in the buffer and adding H2O2 to levels that would overcome its reducing capacity (Fig. S1) (11, 35). Because TCEP is degraded by H2O2 through oxidation to a phosphine oxide at a 1:1 ratio, the excess H2O2 could be calculated (31, 32). Both the added (black) and final (H2O2 minus TCEP; red) H2O2 concentrations are shown.
Cysteine mutants of PKG Iα probe oxidation-induced constitutive activation
Next, we examined whether the oxidized phenotype in PKG Iα is regulated by formation of the interprotomer disulfide bond between Cys-42 and Cys-42′. To achieve this, we measured the activities of reduced and oxidized PKG Iα C42S (C42Sred and C42Sox) (Fig. 3A and Table 1). When the mutant was measured under reducing conditions, we observed a basal activity similar to that of WTred (0.32 μmol/min× mg). However, when activated by cGMP, the maximal velocity was reduced by ∼50% compared with WTred (2.31 μmol/min × mg), resulting in a 7-fold stimulation of activity. Moreover, C42Sred showed a 3-fold shift in the activation constant (Ka = 580 nm, p < 0.001 compared with WTred) and a loss of cooperative, cGMP-dependent activation (nH = 0.8). For C42Sox, we observed a significant increase in basal activity (1.103 μmol/min × mg; p < 0.001 compared with C42Sred); however, the maximal activity was unchanged (2.357 μmol/min× mg) and displayed kinetic characteristics similar to those of WTox. Examination of C42Sox by nonreducing, denaturing SDS-PAGE did not show evidence of an intramolecular disulfide bond between protomers (Fig. 3E).
Figure 3.

In vitro phosphotransferase assays of PKG Iα under reducing and oxidizing conditions. A–D, cGMP-dependent activation of PKG Iα WT (black circles) and PKG Iα C42S (red squares) (A), Δ53 PKG Iα (purple squares) (B), PKG Iα C117S (green triangles) (C), and PKG Iα C195S (blue triangles) (D) under reducing (1 mm TCEP; solid lines) and oxidizing (500 μm H2O2, 30 min; dashed lines) conditions. Data are represented as the mean ± S.D. (error bars). E, nonreducing, denaturing SDS-PAGE of PKG Iα constructs treated with increasing concentrations of H2O2 (0–2 mm). The dashed line indicates the point at which the concentration of hydrogen peroxide overcomes the concentration of TCEP in the buffer. H2O2 values corrected for the presence of TCEP are shown in red. The panel for WT is the same as shown in Fig. 2D to show comparisons of the WT enzyme with mutants.
Due to the similar increases in basal activity between WTox and C42Sox in response to H2O2, we next utilized Δ53 PKG Iα, a truncation construct of PKG Iα that lacks the N terminus (and Cys-42) but retains cGMP-dependent activation (30). Δ53red activity in the absence of cGMP was similar to WT PKG Iα (Fig. 3B). In the presence of increasing concentrations of cGMP, Δ53 activity increased by 20-fold with a Ka for cGMP that was similar to that of WT PKG Iα (216 nm). When oxidized with H2O2, Δ53ox exhibited kinetic features similar to those of the WTox and C42Sox constructs. Specifically, the 7-fold increase in basal activity of Δ53ox to 1.380 μmol/min × mg decreased its cGMP-dependent -fold activation to 1.1. Thus, Δ53 displayed the highest sensitivity to oxidation. Like C42S, SDS-PAGE analyses showed that Δ53 does not form an interprotomer disulfide bond in the presence of H2O2 (Fig. 3E).
Because PKG Iα and Iβ are splice variants of the prkg1 gene, within the same species, they are completely identical in their cGMP-binding and catalytic domains (with 98% sequence identity across placental mammals). However, their N-terminal dimerization, autoinhibitory, and linker regions only retain 26% sequence identity (Fig. S2). Thus, Cys-42 in the N terminus of Iα is not conserved in PKG Iβ. Upon ruling out the contribution of Cys-42 on the oxidation-dependent activation phenotype of PKG Iα, we hypothesized that oxidative activation may originate within the sequence-conserved regulatory domain. To test this, we first examined reduced and oxidized PKG Iβ. PKG Iβ stimulated by cGMP under reducing conditions displayed a cooperative, 86-fold stimulation of activity with a Ka of 1.21 μm (Table 1 and Fig. S3). When PKG Iβ was oxidized with H2O2, a 10-fold increase in basal activity was observed (0.42 μmol/min × mg) paired with a 1.3-fold decrease in maximal velocity (2.780 μmol/min × mg), resulting in a reduction of the cGMP-stimulated -fold activation to 6.6. In addition, under oxidizing conditions, the activation constant for PKG Iβ decreased by 1.4-fold (Ka = 0.86 μm), and cooperativity was reduced to 1.2. These data suggest that oxidation also affects the activity of the Iβ isoform.
In addition to the interprotomer disulfide bond between Cys-42 and Cys-42′, Landgraf et al. (11) also confirmed the presence of an intraprotomer disulfide bond between Cys-117 and Cys-195. This disulfide bond has been observed in the crystal structure of the PKG I regulatory domain (PDB code 3SHR) (27). Due to its location in the first cGMP-binding domain (CNB-A), we hypothesized that this disulfide bond may control oxidative activation of PKG Iα. To test this, we expressed and purified the PKG Iα C117S mutant. Under reducing conditions, C117Sred had a low basal activity that was similar to that of PKG Iα WTred (Fig. 3C). Furthermore, activation of C117Sred with cGMP increased the activity by 26-fold with positive cooperativity (nH = 1.4), resulting in a Vmax of 4.09 μmol/min × mg and a Ka of 130 nm. In contrast to PKG Iα WT, stimulation of C117Sox by cGMP resulted in only a slight increase in basal activity and no significant change in its maximal velocity compared with C117Sred (3.81 μmol/min × mg). However, the Ka for cGMP was increased, and the cooperativity showed a slight, but not statistically significant, decrease (nH = 1.2) compared with C117Sred. These data suggest that the response to H2O2 is highly attenuated by introduction of the C117S mutation.
To determine whether oxidative activation depends on disulfide bond formation with Cys-195, we next examined the C195S construct. Under reducing conditions, the overall kinetic profile of C195Sred was not significantly different from that of WTred. However, the basal and maximal activities were slightly elevated, resulting in an overall -fold activation of 10.8 when stimulated with cGMP (Fig. 3D and Table 1). Under oxidizing conditions, we observed kinetics consistent with the WTox phenotype. This included a significant increase in basal activity and a decrease in the maximal velocity, suggesting that oxidation at Cys-195 is not necessary for oxidation-dependent activation.
Analysis of KCa 1.1 channel activity during exposure to PKG Iα
Phosphorylation of the KCa 1.1 channel by PKG Iα has been shown to increase its open probability, resulting in increased K+ flux to the extracellular space (5, 33, 34). To examine the effects of both substrate targeting by PKG Iα and activation of the channel, we measured KCa 1.1 channel currents in the inside-out patch-clamp configuration using exogenously applied reduced or oxidized PKG Iα constructs (Fig. 4A). Under reducing and cGMP-stimulating conditions, PKG Iα WT increased the open probability of the KCa 1.1 channels by 3.2-fold compared with control patches (+PKG, −cGMP; p < 0.001). PKG Iα that was pretreated with H2O2 and then added to the extracellular face of the patch in the absence of cGMP also increased the open probability by 3.1-fold (p < 0.02).
Figure 4.

Stimulation of KCa 1.1 channels with PKG Iα constructs under reducing and oxidizing conditions. A, the combined results of single KCa 1.1 recordings exposed to PKG Iα constructs under reducing and oxidizing conditions. Individual measurements are represented as a scatter plot overlaid with mean ± S.E. (error bars). p values represent the comparison against PKG Iα under reduced, unstimulated conditions (first lane). Ba, representative control (top) and experimental traces of single KCa 1.1 channel openings in excised membrane patches from mouse cerebral artery myocytes under control conditions and when exposed to cGMP activated PKG Iα WT and C42S constructs. All measurements were recorded under reducing conditions. Bb, analysis of channel open dwell times of KCa 1.1 channels from patches under baseline control (−PKG, −cGMP), PKG Iα WTred-exposed, and C42Sred-exposed conditions. Whisker boxes, medians bounded by the interquartile ranges. The results of the nonparametric Hodges–Lehmann analyses (HL; coefficient and error) are shown. *, p < 0.02; ***, p < 0.0001.
Application of C42Sred under cGMP-saturating conditions did not result in a significant increase in channel openprobability compared with the control condition (NPo(exp)/NPo(baseline) = 1.4), whereas C42Sox resulted in a slightly higher increase in the open probability of the channels to 2.1-fold over baseline. Furthermore, we observed that the channel kinetics in the presence of C42S were altered under these conditions (Fig. 4Ba). Examination of the channel dwell times showed that KCa 1.1 patches exposed to cGMP-activated C42Sred had a shortened mean open dwell time of 1.0 ms compared with baseline (5.8 ms) (Fig. 4Bb). However, when PKG IαWTred was applied to the patch, we saw a significant but smaller decrease in mean open dwell time (baseline = 4.6 ms and PKG Iα = 3.7 ms) (Fig. 4Bb). Analysis of median open dwell times also indicated significant decreases (Fig S4). The comparison of these dwell-time differences by nonparametric Hodges–Lehmann analysis indicated that the change in dwell time in response to kinase addition was greater for C42S than WT PKG Iα (Fig. 4Bb). To determine whether the change in KCa 1.1 currents caused by C42S depended on activity, channel recordings in the presence of C42Sred were also measured under symmetrical K+ in the absence of cGMP and ATP (Fig. S5). The short channel openings (flickers) were observed at both +40 and −40 mV, indicating that the effect was independent from C42S activity and membrane polarity.
Finally, application of C117Sred under cGMP-saturating conditions significantly increased the open probability of the channel compared with the control condition (Fig. 4A, p < 0.02). However, C117S that was pretreated with H2O2 (C117Sox) had no effect on KCa 1.1 channel open probability (p = 0.2413). As a control, no effect on channel activity was observed with the addition of 500 nm H2O2, the concentration that equaled the maximum final concentration of H2O2 in the bath solution after the addition of PKG Iαox constructs.
Discussion
PKG Iα is an essential signaling molecule in the vasculature and can be activated by cGMP or oxidation (10, 35–38). The first study to describe that PKG Iα can be constitutively activated by oxidation used cupric cations as the oxidizing agent (11). This activity was found to be blocked by iodoacetimide, which indicated involvement of cysteine residues in this mechanism. Moreover, three possible disulfide bonds were identified by nonreducing SDS-PAGE and Edmann degradation: an interprotomer disulfide bond between Cys-42 and Cys-42′, an intraprotomer disulfide bond between Cys-117 and Cys-195, and an intraprotomer disulfide bond between Cys-312 and Cys-518. Of the three, the last disulfide bond could not be definitively confirmed, and no other functions have been ascribed to the remaining cysteine residues.
Previous analyses of PKG I isoforms determined that PKG Iα, but not PKG Iβ, exhibits sensitivity to oxidation (10, 12). Thus, it was concluded that Cys-42, the only cysteine residue unique to PKG Iα, must be the primary regulator of oxidation-dependent activation (10). Moreover, formation of this interprotomer disulfide bond between Cys-42 and Cys-42′ of the opposing protomer was found to correlate with a vasodilatory response in isolated perfused rat hearts (10). These observations helped to cement oxidation as an additional mechanism by which PKG Iα can be activated. Despite an abundance of data ascribing physiological importance to the formation of the interprotomer disulfide bond between Cys-42 and Cys-42′, the structural and biochemical underpinnings of how this disulfide bond would activate PKG Iα remain unclear. Thus, we sought to expand upon these initial observations to more thoroughly understand the mechanism of oxidation-dependent activation in PKG Iα. Signaling by PKG I isoforms is mediated by both substrate targeting and activation (6, 23, 24, 26, 36, 39–42). More importantly, they can be considered independent because disruption of either facet alters PKG I signaling pathways (23–26). In this study, we sought to separate these aspects of PKG I signaling to better understand how oxidation may regulate each of these processes. The use of a synthetic peptide substrate allowed for a thorough examination of the effect of oxidation that was independent from substrate targeting through the dimerization domain.
Under reducing conditions, the cGMP-dependent activation characteristics of PKG Iα WT were consistent with previously reported values (Fig. 2C) (29, 30). Furthermore, its preincubation with H2O2 induced interprotomer disulfide bond formation (Fig. 2D) (10, 11, 43). We observed that exposure of WT enzyme to 500 μm H2O2 for 30 min at 24 °C resulted in the greatest amount of constitutive activation, determined by an increase in basal activity and a corresponding decrease incGMP-dependent activation (Fig. 2B). Although this concentration of H2O2 is significantly higher than what has been implemented in prior studies, we sought to measure the H2O2 concentration dependence on PKG Iα activity in a way similar to that used by Landgraf et al. (10–12, 43).
Characterization of C42S provides insight into rodent models of oxidation-dependent activation of PKG Iα
When cGMP-dependent activation of C42S was examined under reducing conditions, we observed that the presence of the serine mutant significantly altered enzyme activity (Fig. 3A). This overall shift in efficacy and potency (3-fold) culminated in a 90% reduction in activity for C42S under cGMP concentrations near the Ka for PKG Iα WT. Even under maximum cGMP concentrations typically observed in smooth muscle cells (1–2 μm), C42S exhibited only about 30% of the maximal activity of PKG Iα WT (Fig. 3A) (44). Based upon these results, we conclude that the C42S mutant retains sensitivity to oxidation, and, unlike the WT enzyme, the reduced form of C42S displays a significantly altered kinetic profile toward the synthetic peptide substrate used in the in vitro phosphotransferase assays. In agreement with previous results, C42S did not form an interprotomer disulfide bond in the presence of H2O2 (Fig. 3E) (10, 43). These results are consistent with a recent study that found that the reduced form of C42S exhibited a 5-fold shift in its activation constant for cGMP. Taken together, these results suggest that the interprotomer disulfide bond does not mediate the oxidative phenotype but does offer an explanation of the detrimental physiological phenotypes associated with this mutation.
Previous studies investigating the physiological effects of the interprotomer disulfide bond used a PKG Iα C42S knock-in mouse model (14). These mice were found to be hypertensive compared with littermate controls. Based upon our kinetic analyses of PKG Iα C42S, we suggest that this phenotype may result from the compromised kinetic profile of the enzyme rather than an inability to be activated by oxidation. This conclusion is further substantiated by the effects of PKG Iα constructs on KCa 1.1 channel activity (Fig. 4A). The similarities in channel responses between the oxidizing and reducing conditions suggest that substrate targeting of PKG through its dimerization domain to KCa 1.1 is not compromised by formation of the interprotomer disulfide bond between Cys-42 and Cys-42′ in the dimerization domain.
A comparison of KCa 1.1 channel characteristics subjected to either C42S or WT uncovered the presence of channel “flickers” for patches treated with C42S and an overall increase in the number of channel opening events over an equivalent time period (Fig. 4Bb and Figs. S4 and S5). Moreover, the KCa 1.1 channel flickers were observed for all patches exposed to C42S (with or without cGMP and under oxidizing conditions) (Fig. 4Bb and Figs. S4 and S5). Within this context, a study examining pressure-induced constriction of third order mesentery arteries expressing C42S observed that application of the KCa 1.1 channel blocker, paxilline, did not induce vessel constriction (13). In light of the modified KCa 1.1 channel characteristics that were observed herein with C42S, the mutation may possess hitherto unknown properties, such as unique interactions with the channel pore or altered targeting to other PKG Iα substrates.
The effects of disrupted substrate targeting have been observed previously for a mutant leucine zipper knock-in mouse model in which the leucine residues in the dimerization domain were mutated to alanine residues. This mutant was unable to phosphorylate the myosin-binding subunit and the Ras homolog A (RhoA), despite retaining its sensitivity to cGMP and ability to phosphorylate the synthetic substrate, BPDEtide (23). Furthermore, these mice, like the C42S knock-in model, were also hypertensive. Since substrate targeting through the dimerization domain and activation are both required for phosphorylation of substrates by PKG Iα, we suggest that either explanation is possible.
C117S drives constitutive activation of PKG Iα by oxidation
To test our hypothesis that the oxidative phenotype is driven by residues that lie outside of the N terminus, we used the PKG Iα truncation construct, Δ53 (30). Similar to C42S, Δ53 does not form an interprotomer disulfide bond (Fig. 3E). However, oxidation had the most pronounced effect on this construct, as indicated by the complete insensitivity to cGMP. These results corroborate the C42S data and overwhelmingly demonstrate that the interprotomer disulfide bond is not required for oxidation-induced activation.
Finally, we tested the contribution of the intraprotomer disulfide bond that has been shown to form between Cys-117 and Cys-195 in the cGMP-A domain (11, 27). We observed that oxidized C117S exhibited only a modest increase in basal activity and no reduction in maximal velocity compared with its reduced form and that of WT (Fig. 3C). We also consistently observed robust activation by cGMP under oxidizing conditions, indicating that the C117S mutation nearly abolishes the oxidative phenotype; these results are further corroborated by the inside-out patching experiments. Finally, with the C195S mutant, we observed an activation profile similar to that of WT under both reducing and oxidizing conditions. Thus, we conclude that Cys-117 plays a unique role in mediating oxidative activation of PKG Iα in vitro.
The initial observation that PKG Iα and Iβ exhibit differing responses to H2O2 has predominantly focused the field on Cys-42, the cysteine residue specific to the Iα isoform. However, the results described herein indicate that the oxidative phenotype is controlled by Cys-117 in the cyclic nucleotide binding domain-A (CBD-A). PKG Iα and Iβ are 100% identical in this region, and the residue analogous to Cys-117 in PKG Iβ is Cys-132. Thus, we examined PKG Iβ and found that oxidation had a significant impact on its maximal velocity that was similar to the effect on PKG Iα. However, only a slight increase in basal activity was observed. Our results are consistent with a previous study that focused solely on activation of PKG Iβ with H2O2 in the absence of cGMP (10) and indicate that examination of the full cGMP-dependent activation profile is necessary to observe the full effects of oxidation on PKG Iβ activity. Differences in PKG Iα and Iβ (cGMP-dependent activation profiles and targeting to substrates) are attributable to their distinct N termini (35, 45, 46). We propose that their N termini also dictate their oxidation-dependent phenotypes and suggest a model by which this could occur.
A model for oxidation-driven activation in PKG Iα and considerations for pathophysiological conditions
The results obtained using the PKG Iα mutant constructs suggest that Cys-117 is necessary and sufficient to stimulate constitutive activation of PKG Iα. Under mild oxidizing conditions, Cys-117 and Cys-195 would form sulfenic acid intermediates, which can be reduced to the free thiol or condensed to a disulfide form (Fig. 5A). The fact that no discernible structural changes have been observed between the reduced and disulfide-bonded forms of CBD-A suggests that the site maintains an inactive conformation in both states and that disulfide bond formation between Cys-117 and Cys-195 likely mitigates oxidative activation (19, 27, 47). As determined by this study and Landgraf et al. (11), increasing the concentrations of oxidant causes the Vmax of the kinase to decay, resulting in a rise in constitutive activity. The reversibility of the oxidized phenotype measured previously was determined at concentrations that did not affect the Vmax (10, 11). This suggests that formation of sulfinic and sulfonic acid species at Cys-117 may drive the cGMP insensitivity that is observed at high concentrations of H2O2.
Figure 5.
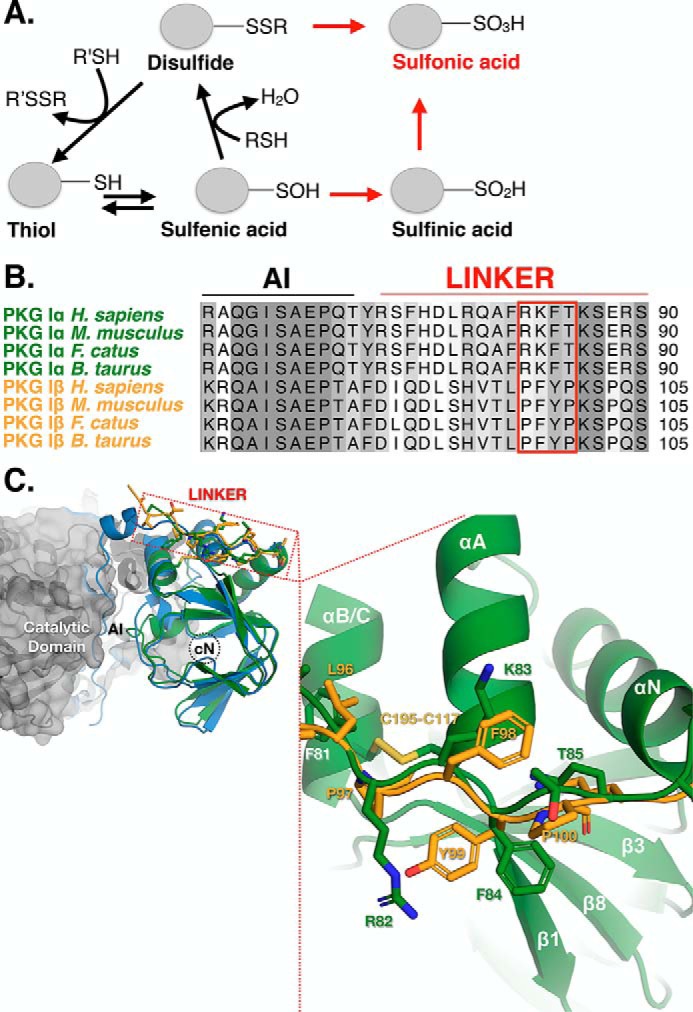
Model of the oxidation-dependent activation of PKG Iα. A, cysteine residues can be oxidized to multiple states, which proceed through a sulfenic acid intermediate. When in proximity to a free thiol, sulfenic acids can be condensed to a disulfide. Further oxidation of sulfenic acids results in the irreversible sulfinic and sulfonic acid forms. The sulfonic acid form can also be reached via a thiosulfinate intermediate. B, sequence alignment of mammalian PKG Iα (green) and Iβ (gold) isoforms denoting the autoinhibitory (AI) domain and linker region. The segment within the linker region that is in close proximity to Cys-117 is boxed in red. C, structural alignment of the PKG Iα (green; PDB code 3SHR) and the Iβ (yellow; PDB code 3OD0) A-sites superimposed with the RIα:C holoenzyme of PKA (RIα (blue) and C (gray); PDB code 2QCS). The boxed region indicates the location of the linker region of PKG Iα and Iβ with their corresponding residues within close proximity to Cys-117.
In support of this model, we observed cysteine modifications on tryptic peptides of the oxidized form of PKG Iα by MS and found that Cys-117 can adopt disulfide and sulfonic acid species, whereas Cys-195 can only adopt a disulfide-bound form (Table 2). This suggests that the higher-order sulfur acids occur preferentially on Cys-117 over Cys-195. Consequently, it is equally probable for the oxidation of Cys-117 to higher-order sulfur acids to proceed with or without the disulfide-bound intermediary form. We also found that Cys-42 can adopt disulfide, sulfinic, and sulfonic acid forms in the oxidized state, which further reinforce that, despite the presence of higher-order sulfur acid species in the N terminus, their effects on enzymatic activity are limited. Based upon the available crystal structures and these data, we propose a model of constitutive, cGMP-independent activation of PKG Iα that is driven through oxidation of Cys-117 to either the sulfenic, sulfinic, or sulfonic acid intermediates. However, due to its exposure to solvent, it is likely that the interprotomer disulfide bond at Cys-42 may act as a buffering mechanism to preclude the formation of sulfur acid species at Cys-117.
Table 2.
Identification of cysteine-containing peptides from oxidized PKG Iα
| Cysteine | Sequence with modificationsa | z+ | Theoretical m/z (monoisotopic) | Observed m/z (monoisotopic) | Concentration | Retention time |
|---|---|---|---|---|---|---|
| ppm | min | |||||
| Cys-42 | K↓C@QSVLPVPSTHIGPR↓T | 2 | 819.917 | 819.917 | 0.00 | 37.88 |
| K↓C#QSVLPVPSTHIGPR↓T | 2 | 811.920 | 811.921 | 1.23 | 37.96 | |
| K↓C⋀QSVLPVPSTHIGPR↓T | 2 | 824.435 | 824.437 | 2.42 | 34.34 | |
| K↓C∼QSVLPVPSTHIGPR↓T | 2 | 844.433 | 844.435 | 2.37 | 40.13 | |
| Cys-117 | K↓NLELSQIQEIVDC@M*YPVEYGK↓D | 3 | 845.726 | 845.724 | −2.36 | 58.92 |
| K↓NLELSQIQEIVDC⋀M*YPVEYGK↓D | 3 | 848.738 | 848.739 | 1.17 | 58.25 | |
| K↓NLELSQIQEIVDC∼M*YPVEYGK↓D | 3 | 862.070 | 862.069 | 1.15 | 58.89 | |
| Cys-195 | R↓QC∼FQTIMMR↓T | 2 | 627.775 | 627.776 | 1.59 | 24.48 |
| R↓QC∼FQTIM*MR↓T | 2 | 635.773 | 635.772 | 1.57 | 30.43 | |
| R↓QC∼FQTIM*M*R↓T | 2 | 643.770 | 643.771 | 1.55 | 34.03 |
a Modifications were as follows: *, oxidation; #, sulfinic acid; @, sulfonic acid; ⋀, iodoacetamide (free thiol, previously disulfide-bound); ∼, maleimide (free thiol).
A sequence alignment of PKG Iα and Iβ paired with a structural alignment of the PKG I CBD-A and the PKA RIα:C holoenzyme shows that Cys-117 is oriented proximal to the linker region that connects the autoinhibitory domain and CBD-A. This region contains very low sequence identity (∼6%) between Iα and Iβ (Fig. 5B). We propose that the presence of the basic residues (Arg-82 and Lys-83) within the linker region of PKG Iα may form hydrogen bonds or a salt bridge with the sulfur acid forms of Cys-117 (Fig. 5C). In comparison, this region in PKG Iβ contains hydrophobic residues (Pro-97 and Phe-98) at the equivalent positions. In this model, we propose that an interaction between the acid form of Cys-117 and the linker region in PKG Iα could alter the conformation of the linker to release the autoinhibitory segment from the catalytic domain, resulting in constitutive activation. We further propose that our model may provide an explanation of the differing responses to oxidation between the two type I isoforms.
PKG Iα has been reported to be activated by cGMP-independent mechanisms in both physiological and pathophysiological states (10, 11, 12, 15, 48). Oxidizing agents, particularly H2O2, have been suggested to play important roles in a number of conditions associated with vascular remodeling, including angiogenesis, atherosclerosis, and hypertension (16–22, 49). Typically, H2O2 concentrations in mammalian cells are estimated to average between 1 and 700 nm (50). However, local levels caused by bursts in H2O2 production are suggested to greatly exceed the average value and depend on the distance from the source and local concentrations of antioxidants, such as catalase, GSH peroxidases, and thioredoxin reductases. A recently developed H2O2 sensor (HyPer-Tau) coupled with superresolution microscopy suggested that local bursts of hydrogen peroxide can reach 50–100 μm (49). In addition, the presence of peroxiredoxin hubs has been suggested to neutralize ∼90% of the H2O2 produced in cells, which would further limit the effect of H2O2 to specific microdomains (51). The data presented herein indicate that constitutive PKG Iα activation via oxidation does not occur until 500 μm and involves long-lived exposure to the oxidant. Thus, this mechanism would be specifically relevant to PKG Iα anchored near hydrogen peroxide sources or under pathophysiological conditions associated with high global intracellular concentrations of hydrogen peroxide.
Experimental procedures
Site-directed mutagenesis of WT PKG Iα
WT PKG Iα and PKG Iβ from Bos taurus (NCBI entries NP_776861.1 and CAA70155.1) were cloned into pFAST Bac HTA as described previously (34, 49). The PKG Iα construct was used as a template for site-directed mutagenesis to produce the following mutants: PKG Iα C42S, PKG Iα C42A, PKG Iα C117S, and PKG Iα C195S. The forward and reverse primers used to generate these constructs were synthesized as follows: C42S, 5′-AAG AGG AAA CTC CAT AAA AGC CAG TCA GTG-3′ (sense) and 5′-GGG CAG CAC TGA CTG GCT TTT ATG GAG-3′ (antisense); C42A, 5′-AAG AGG AAA CTC CAT AAA GCC CAG TCA GTG-3′ (sense) and 5′-GGC AGC ACT GAC TGG GCT TTA TGG-3′ (antisense); C117S, 5′-ATC CAA GAG ATT GTG GAT AGT ATG TAC CCA GTG-3′ (sense) and 5′-GCC GTA CTC CAC TGG GTA CAT ACA ATC CAC AAT-3′ (antisense); C195S, 5′-GCC ATT GAC CGA CAA TCT TTT CAG ACG-3′ (sense) and 5′-CCT CAT TAT CGT CTG AAA AGA TTG TCG-3′ (antisense). All site-directed mutagenesis experiments were carried out using the QuikChange (Stratagene) method.
Expression and purification of PKG I constructs
PKG Iα and Iβ WT, PKG Iα mutant constructs, and PKG Iα Δ53 were expressed in Sf9 cells using the Bac-to-Bac Baculovirus Expression System as described previously (29, 30). Constructs were purified from Sf9 cell pellets by nickel immobilized metal ion affinity chromatography using either prepacked (5 ml; Bio-Rad) or hand-packed nickel-nitrilotriacetic acid columns. Protein purified for use in inside-out patch clamp experiments was prepared without the N-terminal hexahistidine tag, by incubation with His-tagged tobacco etch virus (TEV) protease at a 1:20 mass ratio (TEV/PKG) during the first dialysis step (50 mm MES, 300 mm NaCl, 1 mm TCEP, pH 6.9, 4 °C, 16 h). TEV and the histidine tag were subtracted from the cleaved protein by flowing over a nickel immobilized metal ion affinity chromatography column. The resulting flow-through containing PKG I was concentrated to 1 mg/ml and dialyzed against 1 liter of 50 mm MES, 150 mm NaCl, 1 mm TCEP, 10% glycerol, pH 6.9, at 4 °C. The resulting protein solution was divided into 20–100-μl aliquots, flash-frozen in liquid nitrogen, and stored at −80 °C.
Determination of H2O2 concentration
The primary H2O2 stock (stored at 4 °C) was measured by absorption spectroscopy at λ = 240 nm (43.6 m−1 cm−1) to determine its concentration (31, 32). Fresh dilutions were prepared daily from the stock for use in experiments.
Measurement of interprotomer disulfide bond formation by SDS-PAGE
PKG I constructs were buffer-exchanged (1–4 times) into 50 mm MES, 150 mm NaCl, 1 mm TCEP, pH 6.9, using 3000 MWCO ultracentrifugal filter units (Amicon) to remove glycerol (to <1%) in the storage buffer. PKG concentration was determined by measuring the absorbance at 280 nm (equal to 81,250 m−1 cm−1 as calculated by ProtParam (51)). All constructs were adjusted to 1 mg/ml (13.1 μm of monomer), and PKG (3.6 μl) was added to 1.4 μl of H2O2 (freshly prepared 10× stocks that ranged from 0 to 50 mm) and 9 μl of 50 mm MES, 150 mm NaCl, pH 6.9 (in the absence of reducing agent) (14-μl reactions). Reactions proceeded for 30 min at 24 °C and were subsequently quenched by the addition of 2 μl of 1 m maleimide (10% DMSO stock) and 4 μl of 5× SDS-loading buffer without reducing agent. Samples were boiled for 2 min at 95 °C and separated by SDS-PAGE using 4–20% TGX gradient gels (Bio-Rad). The resulting gels were stained with InstantBlue Protein Stain (Expedeon) and imaged using an Odyssey IR Scanner (LI-COR) at 700 nm. Bands were selected and quantified in the Image Studio Software (LI-COR) using intensity analysis relative to background. The background was subtracted by implementing the mean intensity background subtraction method. Images were exported as 600 DPI gray-scale TIFF files for use as figures. Data analysis was achieved using Prism version 7 (GraphPad).
In vitro phosphotransferase assays
Activation of reduced PKG I constructs by cGMP (BioLog) was assessed by measuring phosphorylation of a synthetic peptide substrate (W15, TQAKRKKSLAMA) with [γ-32P]ATP as described previously (35). To measure the cGMP-dependent activation under oxidized conditions, PKG I constructs were preincubated with 500 μm H2O2 in 50 mm MES, pH 6.9, for 30 min at 24 °C. Under these conditions, the 1 mm TCEP in the PKG storage buffer was diluted to a final concentration of 500 nm. Oxidized PKG I was then added to the other reaction components (in the absence of reducing agent) to initiate the reactions. All subsequent steps were performed following the same procedure as the reduced PKG conditions.
Measurement of KCa 1.1 activation by inside-out patch clamp of vascular smooth muscle cells
Male C57BL/6 mice (3–6 months old) were euthanized using procedures approved by the institutional animal care and use committee at the University of Vermont and performed in accordance with the National Institutes of Health policy on the care and use of laboratory animals. The anterior and posterior pial and superior cerebellar arteries were isolated and cleaned of connective tissue. The arteries were then incubated at 37 °C in dissociation solution (55 mm NaCl, 80 mm sodium glutamate, 6 mm KCl, 2 mm MgCl2, 10 mm glucose, 10 mm HEPES, pH 7.3) containing 0.5 units/ml papain (Worthington) and 1 mg/ml dithioerythritol for 12 min followed by a 10-min incubation in dissociation solution containing 1 mg/ml collagenase (Worthington Type 4). Arteries were then removed and placed back in the isolation solution without enzymes for an additional 10 min before trituration with a polished Pasteur pipette to yield single smooth muscle cells. Cells were left to stick to the glass coverslip in the experiment chamber for 10 min before use.
Membrane patches were pulled from the isolated vascular smooth muscle cells to achieve the inside-out configuration and contained 2–10 KCa 1.1 channels each. KCa 1.1 single-channel currents were recorded under the following conditions. The pipette solution contained 6 mm KCl, 134 mm NaCl, 1 mm MgCl2, 10 mm HEPES, pH 7.4 (NaOH); the bathing solution contained 135 mm KCl, 2 mm MgCl2, 1.4787 mm CaCl2, 2 mm EGTA, 10 mm HEPES, adjusted to pH 7.2 with 14.85 mm KOH. The concentration of free Ca2+ on the intracellular face was calculated to be 500 nm using the online software WEBMAXC Standard (https://web.stanford.edu/∼cpatton/webmaxcS.htm;4 Stanford University, Stanford, CA). Single KCa 1.1 channel recordings were obtained at room temperature (25 °C) for at least 5 min at 0 mV and using an Axopatch 200 B amplifier coupled to a Digidata 1332A digital-to-analog converter. Currents were filtered at 2 kHz, digitized at 20 kHz using pCLAMP version 9.2 software (Axon Instruments), and then analyzed with Clampfit version 9.2 using the half-amplitude threshold method. The baseline activity was recorded for at least 20 min to ensure patch stability. The experimental condition was measured after the addition of 100 μm ATP and either reduced, inactive PKG Iα (no cGMP added), PKG Iα constructs activated with 5 μm cGMP, or PKG Iα constructs activated with 500 μm H2O2. For the oxidized condition, PKG Iα constructs were first buffer-exchanged into 50 mm MES, 150 mm NaCl, 1 mm TCEP, pH 6.9, using MWCO 3000 ultracentrifugal filter units (Amicon) to remove the glycerol (to <1%) in the storage buffer. Assuming a 1:1 negation of TCEP with H2O2 (31, 32), PKG Iα constructs were oxidized with a final concentration of 500 μm H2O2 at 24 °C for 30 min before use in patch clamp experiments. The addition of PKG Iα constructs to the bath solution (5 ml) resulted in a final concentration of H2O2 that was less than 500 nm. The -fold activation was determined as the open probability of the channels (NPo) during the experimental condition/NPo at baseline. The -fold activations of the experimental conditions were compared with the control condition (reduced PKG Iα, 0 cGMP) using unpaired t tests with Welch's correction in Prism version 7 (GraphPad). KCa 1.1 channel open times within 5-min windows were measured from full-length PKG Iα, C42S, and their respective baseline recordings using Clampfit version 9.2. The mean channel open dwell times (τ) and interquartile ranges were calculated using the Mann–Whitney test, and the differences between the baseline control and experimental dwell times were compared using a nonparametric Hodges–Lehmann analysis in Prism version 7 (GraphPad). Channel recordings examining the effects of C42S on KCa 1.1 were run under symmetrical K+, 5 μm Ca2+ in the presence of reduced C42S (−cGMP) at +40, 0, and −40 mV.
Sequence alignments
Sequences for PKG Iα and Iβ were downloaded from the National Center for Biotechnology Information (NCBI) database, and a multiple-sequence alignment was performed with JalView Desktop using CLUSTAL-O (52, 53). The following accession numbers were used: Homo sapiens, Iα-NP_001091982.1 and Iβ-NP_006249.1; Mus musculus, Iα-NP_001013855.1 and Iβ-NP_035290.1; Felis catus, Iα-XP_023096056.1 and Iβ-XP_023096055.1; B. taurus, Iα-NP_776861.1 and Iβ-CAA70155.1.
Mass spectrometry
For MS analysis to identify the cysteine oxidation states induced by H2O2, His-tagged PKG Iα was thawed on ice and buffer-exchanged into 50 mm MES, 300 mm NaCl, 1 mm TCEP (pH 6.9) (Buffer 1) using a 10,000 MWCO centrifugal concentrator (Millipore). Samples were oxidized with a final concentration of 500 μm H2O2 for 30 min at room temperature and then buffer-exchanged three times into 50 mm MES, 300 mm NaCl (pH 6.9) (Buffer 2). Dimedone (200 mm stock in 10% DMSO) was added to a final concentration of 20 mm to the reduced and oxidized samples and incubated for 20 min at room temperature. Excess dimedone was removed by buffer exchange by centrifugal concentrator into Buffer 2. Maleimide (200 mm in double-distilled H2O) was added to samples at a final concentration of 20 mm, incubated at 24 °C for 20 min, and then removed by buffer exchange as described above. Samples were centrifuged at ∼14,500 × g to remove any protein aggregates, and the supernatants were transferred to fresh tubes. TCEP was added at a final concentration of 1 mm and incubated for 20 min at room temperature. Iodoacetimide (200 mm stock in in double-distilled H2O) was added to the samples at a final concentration of 20 mm and incubated at 24 °C for 20 min. Samples were buffer-exchanged five times in Buffer 1 to ensure removal of the cysteine-modifying agents and digested with trypsin overnight at 37 °C. Tryptic peptides were flash-frozen in liquid nitrogen and stored at −80 °C pending analysis.
ZipTip pipette tips (Merck Millipore) were used to desalt the tryptic peptide samples by first activating with 100% acetonitrile (MeCN) and equilibrated in 0.1% TFA before introduction of tryptic peptides. Bound peptides were washed in 0.1% TFA and eluted with 0.1% TFA in 50% MeCN and dried in a SpeedVac. Peptides were resuspended in Solvent A (2.5% MeCN, 0.15% formic acid) and separated via HPLC using the Easy n-LC 1200 system before MS/MS analysis on the Q Exactive Plus mass spectrometer fitted with a Nanospray Flex ion source and supplied with Thermo Xcalibur version 4.0 software (Thermo Fisher Scientific). Chromatography columns (15 cm × 100 μm) were packed in-house with 2.7-μm C18 packing material (Bruker, Halo, pore size = 90 Å). Peptides were eluted using a 0–50% gradient of Solvent B (80% MeCN, 0.15% formic acid) over 60 min and electrosprayed into the mass spectrometer. This gradient was followed by 10 min at 100% Solvent B before a 15-min equilibration in 100% Solvent A. The precursor scan (scan range = 360–1700 m/z, resolution = 7.0 × 104, automatic gain control = 1.0 × 106, maximum ion injection time = 100 ms) was followed by 10 collision-induced dissociation tandem mass spectra for a top 10 approach. Collision-induced dissociation spectra were acquired for the top 10 ions in the precursor scan (resolution = 3.5 × 104, automatic gain control = 5.0 × 104, maximum ion injection time = 50 ms, isolation window = ±1.6 m/z, collision energy = 26 eV).
Raw spectra were searched for matches within the forward and reverse sequence of the hexahistidine-tagged PKG Iα using SEQUEST, either requiring tryptic peptides or with no enzyme specified, permitting the denoted modifications with a mass tolerance of 4 ppm. For the SEQUEST search requiring tryptic peptides, cross-correlation (XCorr) scores were filtered to include scores that met the following criteria: for z = +1, XCorr 1.8; for z = +2, XCorr 2.0; for z = +3, XCorr 2.2; for z = +4, XCorr 2.4; for z = +5, XCorr 2.6.
Author contributions
J. L. S., T. M. M., and W. R. D. conceived of and designed the experiments. J. L. S. and A. D. B., and A. M. S. performed experiments. J. L. S., T. M. M., and W. R. D. wrote the manuscript with contributions from A. D. B., M. T. N., A. M. S., and B. A. B.
Supplementary Material
Acknowledgments
We thank Werner Tegge (Helmholtz Centre for Infection Research (HZI), Braunschweig, Germany) for synthesizing the W15 peptide substrate used in the phosphotransferase assays. Mass spectrometry was supported by the Vermont Genetics Network through National Institutes of Health (NIH) Grant 8P20GM103449 from the INBRE program of NIGMS. SDS-PAGE experiments were imaged with support from NIH Grants 5 P30 RR032135 from the COBRE Program of the National Center for Research Resources and 8 P30 GM103498 from NIGMS. We also thank the Hondal Laboratory at the University of Vermont for helpful discussions and use of resources.
This work was supported by National Institutes of Health Grants 5T32 HL007594-30 (to J. L. S.) and R01 HL121706 and R01 HL131181 (to M. T. N.) and additional support from the Totman Trust for Biomedical Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PKG I
- cGMP-dependent protein kinase I
- TCEP
- tris(2-carboxyethyl)phosphine
- PDB
- Protein Data Bank
- TEV
- tobacco etch virus
- MWCO
- molecular weight cut-off
- MeCN
- acetonitrile
- Xcorr
- cross-correlation.
References
- 1. Friebe A., and Koesling D. (2003) Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 93, 96–105 10.1161/01.RES.0000082524.34487.31 [DOI] [PubMed] [Google Scholar]
- 2. Hofmann F., Bernhard D., Lukowski R., and Weinmeister P. (2009) cGMP regulated protein kinases (cgk). Handb. Exp. Pharmacol., 137–162 10.1007/978-3-540-68964-5_8 [DOI] [PubMed] [Google Scholar]
- 3. Fukao M., Mason H. S., Britton F. C., Kenyon J. L., Horowitz B., and Keef K. D. (1999) Cyclic GMP-dependent protein kinase activates cloned bkca channels expressed in mammalian cells by direct phosphorylation at serine 1072. J. Biol. Chem. 274, 10927–10935 10.1074/jbc.274.16.10927 [DOI] [PubMed] [Google Scholar]
- 4. Sausbier M., Arntz C., Bucurenciu I., Zhao H., Zhou X. B., Sausbier U., Feil S., Kamm S., Essin K., Sailer C. A., Abdullah U., Krippeit-Drews P., Feil R., Hofmann F., Knaus H. G., et al. (2005) Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in bk channel-deficient mice. Circulation 112, 60–68 10.1161/01.CIR.0000156448.74296.FE [DOI] [PubMed] [Google Scholar]
- 5. Schubert R., and Nelson M. T. (2001) Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol. Sci. 22, 505–512 10.1016/S0165-6147(00)01775-2 [DOI] [PubMed] [Google Scholar]
- 6. Kato M., Blanton R., Wang G. R., Judson T. J., Abe Y., Myoishi M., Karas R. H., and Mendelsohn M. E. (2012) Direct binding and regulation of rhoa protein by cyclic GMP-dependent protein kinase iα. J. Biol. Chem. 287, 41342–41351 10.1074/jbc.M112.421040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun X., Kaltenbronn K. M., Steinberg T. H., and Blumer K. J. (2005) Rgs2 is a mediator of nitric oxide action on blood pressure and vasoconstrictor signaling. Mol. Pharmacol. 67, 631–639 [DOI] [PubMed] [Google Scholar]
- 8. Yuen S., Ogut O., and Brozovich F. V. (2011) Mypt1 protein isoforms are differentially phosphorylated by protein kinase G. J. Biol. Chem. 286, 37274–37279 10.1074/jbc.M111.282905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuen S. L., Ogut O., and Brozovich F. V. (2014) Differential phosphorylation of lz+/lz- mypt1 isoforms regulates mlc phosphatase activity. Arch. Biochem. Biophys. 562, 37–42 10.1016/j.abb.2014.08.011 [DOI] [PubMed] [Google Scholar]
- 10. Burgoyne J. R., Madhani M., Cuello F., Charles R. L., Brennan J. P., Schröder E., Browning D. D., and Eaton P. (2007) Cysteine redox sensor in pkgia enables oxidant-induced activation. Science 317, 1393–1397 10.1126/science.1144318 [DOI] [PubMed] [Google Scholar]
- 11. Landgraf W., Regulla S., Meyer H. E., and Hofmann F. (1991) Oxidation of cysteines activates cGMP-dependent protein kinase. J. Biol. Chem. 266, 16305–16311 [PubMed] [Google Scholar]
- 12. Müller P. M., Gnügge R., Dhayade S., Thunemann M., Krippeit-Drews P., Drews G., and Feil R. (2012) H2O2 lowers the cytosolic Ca2+ concentration via activation of cGMP-dependent protein kinase iα. Free Radic. Biol. Med. 53, 1574–1583 10.1016/j.freeradbiomed.2012.08.011 [DOI] [PubMed] [Google Scholar]
- 13. Khavandi K., Baylie R. A., Sugden S. A., Ahmed M., Csato V., Eaton P., Hill-Eubanks D. C., Bonev A. D., Nelson M. T., and Greenstein A. S. (2016) Pressure-induced oxidative activation of PKG enables vasoregulation by Ca2+ sparks and BK channels. Sci. Signal. 9, ra100 10.1126/scisignal.aaf6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prysyazhna O., Rudyk O., and Eaton P. (2012) Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 18, 286–290 10.1038/nm.2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rudyk O., Prysyazhna O., Burgoyne J. R., and Eaton P. (2012) Nitroglycerin fails to lower blood pressure in redox-dead Cys42Ser PKG1α knock-in mouse. Circulation 126, 287–295 10.1161/CIRCULATIONAHA.112.101287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chan S. L., and Baumbach G. L. (2013) Deficiency of Nox2 prevents angiotensin II-induced inward remodeling in cerebral arterioles. Front. Physiol. 4, 133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan S. L., and Baumbach G. L. (2013) Nox2 deficiency prevents hypertension-induced vascular dysfunction and hypertrophy in cerebral arterioles. Int. J. Hypertens. 2013, 793630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gray S. P., Di Marco E., Kennedy K., Chew P., Okabe J., El-Osta A., Calkin A. C., Biessen E. A., Touyz R. M., Cooper M. E., Schmidt H. H., and Jandeleit-Dahm K. A. (2016) Reactive oxygen species can provide atheroprotection via Nox4-dependent inhibition of inflammation and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 36, 295–307 10.1161/ATVBAHA.115.307012 [DOI] [PubMed] [Google Scholar]
- 19. Kim Y. M., Kim S. J., Tatsunami R., Yamamura H., Fukai T., and Ushio-Fukai M. (2017) ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 312, C749–C764 10.1152/ajpcell.00346.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patel D., Lakhkar A., and Wolin M. S. (2017) Redox mechanisms influencing cGMP signaling in pulmonary vascular physiology and pathophysiology. Adv. Exp. Med. Biol. 967, 227–240 10.1007/978-3-319-63245-2_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rashdan N. A., and Lloyd P. G. (2015) Fluid shear stress upregulates placental growth factor in the vessel wall via NADPH oxidase 4. Am. J. Physiol. Heart Circ. Physiol. 309, H1655–H1666 10.1152/ajpheart.00408.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schiffrin E. L. (2012) Vascular remodeling in hypertension: mechanisms and treatment. Hypertension 59, 367–374 10.1161/HYPERTENSIONAHA.111.187021 [DOI] [PubMed] [Google Scholar]
- 23. Blanton R. M., Takimoto E., Aronovitz M., Thoonen R., Kass D. A., Karas R. H., and Mendelsohn M. E. (2013) Mutation of the protein kinase I α leucine zipper domain produces hypertension and progressive left ventricular hypertrophy: a novel mouse model of age-dependent hypertensive heart disease. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1351–1355 10.1093/gerona/glt042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casteel D. E., Boss G. R., and Pilz R. B. (2005) Identification of the interface between cGMP-dependent protein kinase iβ and its interaction partners TFII-I and IRAG reveals a common interaction motif. J. Biol. Chem. 280, 38211–38218 10.1074/jbc.M507021200 [DOI] [PubMed] [Google Scholar]
- 25. Feil R., Kellermann J., and Hofmann F. (1995) Functional cGMP-dependent protein kinase is phosphorylated in its catalytic domain at threonine-516. Biochemistry 34, 13152–13158 10.1021/bi00040a029 [DOI] [PubMed] [Google Scholar]
- 26. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., and Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase iα. Science 286, 1583–1587 10.1126/science.286.5444.1583 [DOI] [PubMed] [Google Scholar]
- 27. Osborne B. W., Wu J., McFarland C. J., Nickl C. K., Sankaran B., Casteel D. E., Woods V. L. Jr., Kornev A. P., Taylor S. S., and Dostmann W. R. (2011) Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure 19, 1317–1327 10.1016/j.str.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin L., Reger A. S., Guo E., Yang M. P., Zwart P., Casteel D. E., and Kim C. (2015) Structures of cGMP-dependent protein kinase (PKG) iα leucine zippers reveal an interchain disulfide bond important for dimer stability. Biochemistry 54, 4419–4422 10.1021/acs.biochem.5b00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moon T. M., Tykocki N. R., Sheehe J. L., Osborne B. W., Tegge W., Brayden J. E., and Dostmann W. R. (2015) Synthetic peptides as cGMP-independent activators of cGMP-dependent protein kinase iα. Chem. Biol. 22, 1653–1661 10.1016/j.chembiol.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moon T. M., Sheehe J. L., Nukareddy P., Nausch L. W., Wohlfahrt J., Matthews D. E., Blumenthal D. K., and Dostmann W. R. (2018) An N-terminally truncated form of cyclic GMP-dependent protein kinase iα (PKG Iα) is monomeric, autoinhibited, and provides a model for activation. J. Biol. Chem. 293, 7916–7929 10.1074/jbc.RA117.000647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han J., Yen S., Han G., and Han P. (1996) Quantitation of hydrogen peroxide using tris(2-carboxyethyl)phosphine. Anal. Biochem. 234, 107–109 10.1006/abio.1996.0059 [DOI] [PubMed] [Google Scholar]
- 32. Tan Z., Ihnat P. M., Nayak V. S., and Russell R. J. (2012) Quantitative analysis of tris(2-carboxyethyl)phosphine by anion-exchange chromatography and evaporative light-scattering detection. J. Pharm. Biomed. Anal. 59, 167–172 10.1016/j.jpba.2011.09.034 [DOI] [PubMed] [Google Scholar]
- 33. Alioua A., Huggins J. P., and Rousseau E. (1995) PKG-I α phosphorylates the α-subunit and upregulates reconstituted GKCa channels from tracheal smooth muscle. Am. J. Physiol. 268, L1057–L1063 [DOI] [PubMed] [Google Scholar]
- 34. Kyle B. D., Hurst S., Swayze R. D., Sheng J., and Braun A. P. (2013) Specific phosphorylation sites underlie the stimulation of a large conductance, Ca2+-activated K+ channel by cGMP-dependent protein kinase. FASEB J. 27, 2027–2038 10.1096/fj.12-223669 [DOI] [PubMed] [Google Scholar]
- 35. Dostmann W. R., Taylor M. S., Nickl C. K., Brayden J. E., Frank R., and Tegge W. J. (2000) Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Iα inhibit no-induced cerebral dilation. Proc. Natl. Acad. Sci. U.S.A. 97, 14772–14777 10.1073/pnas.97.26.14772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Francis S. H., Smith J. A., Colbran J. L., Grimes K., Walsh K. A., Kumar S., and Corbin J. D. (1996) Arginine 75 in the pseudosubstrate sequence of type Iβ cGMP-dependent protein kinase is critical for autoinhibition, although autophosphorylated serine 63 is outside this sequence. J. Biol. Chem. 271, 20748–20755 10.1074/jbc.271.34.20748 [DOI] [PubMed] [Google Scholar]
- 37. Heil W. G., Landgraf W., and Hofmann F. (1987) A catalytically active fragment of cGMP-dependent protein kinase: occupation of its cGMP-binding sites does not affect its phosphotransferase activity. Eur. J. Biochem. 168, 117–121 10.1111/j.1432-1033.1987.tb13395.x [DOI] [PubMed] [Google Scholar]
- 38. Ruth P., Landgraf W., Keilbach A., May B., Egleme C., and Hofmann F. (1991) The activation of expressed cGMP-dependent protein kinase isozymes Iα and Iβ is determined by the different amino-termini. Eur. J. Biochem. 202, 1339–1344 10.1111/j.1432-1033.1991.tb16509.x [DOI] [PubMed] [Google Scholar]
- 39. Aitken A., Hemmings B. A., and Hofmann F. (1984) Identification of the residues on cyclic GMP-dependent protein kinase that are autophosphorylated in the presence of cyclic amp and cyclic GMP. Biochim. Biophys. Acta 790, 219–225 10.1016/0167-4838(84)90025-6 [DOI] [PubMed] [Google Scholar]
- 40. Alverdi V., Mazon H., Versluis C., Hemrika W., Esposito G., van den Heuvel R., Scholten A., and Heck A. J. (2008) cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. J. Mol. Biol. 375, 1380–1393 10.1016/j.jmb.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 41. Lee E., Hayes D. B., Langsetmo K., Sundberg E. J., and Tao T. C. (2007) Interactions between the leucine-zipper motif of cGMP-dependent protein kinase and the C-terminal region of the targeting subunit of myosin light chain phosphatase. J. Mol. Biol. 373, 1198–1212 10.1016/j.jmb.2007.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou G. P. (2011) The structural determinations of the leucine zipper coiled-coil domains of the cGMP-dependent protein kinase Iα and its interaction with the myosin binding subunit of the myosin light chains phosphase. Protein Pept. Lett. 18, 966–978 10.2174/0929866511107010966 [DOI] [PubMed] [Google Scholar]
- 43. Kalyanaraman H., Zhuang S., Pilz R. B., and Casteel D. E. (2017) The activity of cGMP-dependent protein kinase Iα is not directly regulated by oxidation-induced disulfide formation at cysteine 43. J. Biol. Chem. 292, 8262–8268 10.1074/jbc.C117.787358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nausch L. W., Ledoux J., Bonev A. D., Nelson M. T., and Dostmann W. R. (2008) Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc. Natl. Acad. Sci. U.S.A. 105, 365–370 10.1073/pnas.0710387105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlossmann J., and Desch M. (2009) cGK substrates. Handb. Exp. Pharmacol., 163–193 10.1007/978-3-540-68964-5_9 [DOI] [PubMed] [Google Scholar]
- 46. Ruth P., Pfeifer A., Kamm S., Klatt P., Dostmann W. R., and Hofmann F. (1997) Identification of the amino acid sequences responsible for high affinity activation of cGMP kinase Iα. J. Biol. Chem. 272, 10522–10528 10.1074/jbc.272.16.10522 [DOI] [PubMed] [Google Scholar]
- 47. Kim J. J., Lorenz R., Arold S. T., Reger A. S., Sankaran B., Casteel D. E., Herberg F. W., and Kim C. (2016) Crystal structure of PKG I:cGMP complex reveals a cGMP-mediated dimeric interface that facilitates cGMP-induced activation. Structure 24, 710–720 10.1016/j.str.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakamura T., Ranek M. J., Lee D. I., Shalkey Hahn V., Kim C., Eaton P., and Kass D. A. (2015) Prevention of PKG1α oxidation augments cardioprotection in the stressed heart. J. Clin. Invest. 125, 2468–2472 10.1172/JCI80275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Warren E. A., Netterfield T. S., Sarkar S., Kemp M. L., and Payne C. K. (2015) Spatially-resolved intracellular sensing of hydrogen peroxide in living cells Sci. Rep. 5, 16929 10.1038/srep16929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brewer T. F., Garcia F. J., Onak C. S., Carroll K. S., and Chang C. J. (2015) Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 84, 765–790 10.1146/annurev-biochem-060614-034018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karplus P. A. (2015) A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 80, 183–190 10.1016/j.freeradbiomed.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.