

Abstract
Cyclic α-maltosyl-(1→6)-maltose (CMM, cyclo-{→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→})is a cyclic glucotetrasaccharide with alternating α-1,4 and α-1,6 linkages. CMM is composed of two maltose units and is one of the smallest cyclic glucooligosaccharides. Although CMM is resistant to usual amylases, it is efficiently hydrolyzed by CMM hydrolase (CMMase), belonging to subfamily 20 of glycoside hydrolase family 13 (GH13_20). Here, we determined the ligand-free crystal structure of CMMase from the soil-associated bacterium Arthrobacter globiformis and its structures in complex with maltose, panose, and CMM to elucidate the structural basis of substrate recognition by CMMase. The structures disclosed that although the monomer structure consists of three domains commonly adopted by GH13 and other α-amylase–related enzymes, CMMase forms a unique wing-like dimer structure. The complex structure with CMM revealed four specific subsites, namely −3′, −2, −1, and +1′. We also observed that the bound CMM molecule adopts a low-energy conformer compared with the X-ray structure of a single CMM crystal, also determined here. Comparison of the CMMase active site with those in other enzymes of the GH13_20 family revealed that three regions forming the wall of the cleft, denoted PYF (Pro-203/Tyr-204/Phe-205), CS (Cys-163/Ser-164), and Y (Tyr-168), are present only in CMMase and are involved in CMM recognition. Combinations of multiple substitutions in these regions markedly decreased the activity toward CMM, indicating that the specificity for this cyclic tetrasaccharide is supported by the entire shape of the pocket. In summary, our work uncovers the mechanistic basis for the highly specific interactions of CMMase with its substrate CMM.
Keywords: glycoside hydrolase, crystal structure, X-ray crystallography, carbohydrate metabolism, oligosaccharide, amylase, carbohydrate degradation, cyclic oligosaccharides, cyclodextrins, neopullulanase, CMM catabolism, hydrolytic enzyme
Introduction
Various types of cyclic oligosaccharides with unique properties such as cyclodextrins (CDs)2 (1, 2), cyclodextran (3, 4), cyclomaltopentaose cyclized by an α-1,6-linkage (ICG5) (5–7), and cyclic α-nigerosyl-(1→6)-nigerose (CNN, also denoted as cycloalternan) (8–14) have been studied. Cyclic oligosaccharides potentially have applications in various fields, such as food, cosmetic, pharmaceutical, chemical, textile, and agricultural industries, due to their ability to increase solubility and stability, to mask odors (sequestration), and to alter reactivity and physical properties (e.g. viscosity) of guest molecules (2). In relation to the functional cyclic oligosaccharides, enzymes that produce or hydrolyze CDs have been extensively studied. Cyclo(malto)dextrin glucanotransferase (EC 2.4.1.19) acts on α-1,4-glucan and generates CDs, which consist of α-1,4-linked 6 (α-), 7 (β-), or 8 (γ-) glucopyranose units by intramolecular transglycosylation (15–17). In the Carbohydrate-Active enZymes (CAZy) database (http://www.cazy.org/)3 (18), cyclo(malto)dextrin glucanotransferases are categorized in the subfamily 2 of the glycoside hydrolase (GH) family 13 (GH13_2) (19). Although general α-amylases cannot hydrolyze CDs, several GH13 enzymes belonging to GH13_20 subfamilyexhibit CD-hydrolyzing activity (e.g. cyclomaltodextrinase, EC 3.2.1.54). For example, Thermoactinomyces vulgaris R-47 α-amylase II (TVAII) (20), Geobacillus stearothermophilus neopullulanase (GsNPL) (21), Thermus sp. IM6501 maltogenic α-amylase (ThMAA) (22), and Bacillus. sp. I-5 cyclomaltodextrinase (22) have been demonstrated to hydrolyze CDs. In particular, the molecular basis of the recognition and hydrolysis of CDs by TVAII has been extensively studied (23–27). The CD-hydrolyzing GH13_20 enzymes generally have broad-substrate selectivity. GsNPL is a unique enzyme that exhibits hydrolysis and glucosyltransferase activities for both α-1,4 and α-1,6 linkages (neopullulanase activity, EC 3.2.1.135) (28).
We have previously identified a novel enzymatically produced cyclic tetrasaccharide, cyclic α-maltosyl-(1→6)-maltose (CMM, cyclo-{→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→}) (29). CMM consists of two maltose molecules cyclized with two α-1,6 linkages and thus has alternating α-1,4 and α-1,6 linkages (Fig. 1). CMM was initially obtained by reacting the culture supernatant of a soil-isolated bacterium, Arthrobacter globiformis M6, on starch. Further studies showed that the action of a single enzyme contained in the culture supernatant, 6-α-maltosyltransferase (6MT, EC 3.2.1.-), was responsible for the synthesis of CMM (30). It was also shown that 6MT acts on maltooligosaccharides of a degree of polymerization = 3 or higher and produces CMM by a two-stage catalysis of inter- and intramolecular α-1,6-transglucosylation. A. globiformis M6 can grow on CMM as the sole carbon source, but no CMM decomposition product was detected in the culture supernatant. A further study revealed that an intracellular enzyme, CMM hydrolase (CMMase, EC 3.2.1.-), plays a key role in the CMM catabolism (31–33). CMMase degrades CMM into two maltose molecules by a two-stage hydrolysis of the α-1,6 linkages via maltosyl-maltose (MM, Fig. 1). CMMase belongs to GH13_20, whereas 6MT is unclassified in the currently established 40 subfamilies of GH13. Interestingly, CMMase is highly specific to CMM and MM, although other GH13_20 enzymes generally do not hydrolyze CMM and exhibit wide substrate specificities toward CD, α-1,4, and α-1,6 linkages.
Figure 1.
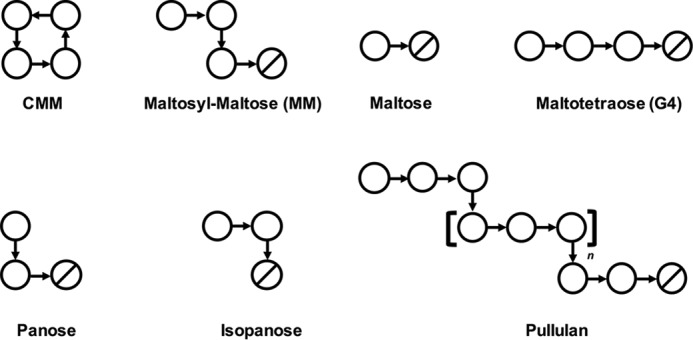
Schematics of ligands used in this study. Circles and slashed circles indicate glucose and reducing end glucose, respectively. α-1,4 and α-1,6 linkages are shown as horizontal and vertical arrows, respectively.
Recently, crystal structures of enzymes and proteins related to the metabolism of CNN, which is the other type of cyclic glucotetrasaccharide linked by alternating α-1,3 and 1,6 linkages, by Listeria monocytogenes have been reported (34, 35). However, the structural basis for the CMM metabolism has not yet been elucidated.
In this study, we determined the crystal structures of CMMase in a ligand-free form and in complex forms with three glucooligosaccharide ligands, including CMM. A comparison of the active site of CMMase with the GH13_20 enzymes revealed significant differences responsible for the substrate specificity. The X-ray structure of a single CMM crystal was also determined, and its conformation was compared with the CMMase-bound CMM molecule. Furthermore, a site-directed mutational analysis of the key residues in the substrate-binding site confirmed their importance in the recognition of CMM.
Results
Overall structure of CMMase
The crystal structure of CMMase was solved by molecular replacement using the GsNPL structure (33.8% sequence identity by EMBOSS Needle Pairwise Alignment, PDB code 1J0H) as a search model. Structures in a ligand-free form and the complex forms with maltose (α-d-Glcp-(1→4)-α-d-Glcp), panose (α-d-Glcp-(1→6)-α-d-Glcp-(1→4)-α-d-Glcp), and CMM were determined at 1.6–2.4 Å resolutions (Table 1). The monomer enzyme is composed of three domains: a catalytic (β/α)8-barrel domain A (residues 1–119 and 175–377); domain B (residues 120–174), which protrudes from domain A; and domain C (residues 378–450) (Fig. 2B). The three-domain architecture is generally present in GH13 and α-amylase–related clan GH-H enzymes (e.g. GH70 and GH77) (36). In the GH13_20 subfamily, crystal structures of six bacterial and three archaeal enzymes, which have activities of α-amylase (EC 3.2.1.1) and cyclomaltodextrinase (EC 3.2.1.54) (37), debranching enzyme or amylo-α-1,6-glucosidase (EC 3.2.1.33) (38), amylopullulanase (EC 3.2.1.41) (39), cyclomaltodextrinase (EC 3.2.1.54) and maltogenic α-amylase (EC 3.2.1.133) (22, 40), and neopullulanase (EC 3.2.1.135) (41, 42), have been reported. A database search using the DALI server (43) revealed that CMMase shows a high structural similarity to GsNPL (Z-score, 53.3; RMSD, 1.5 Å for 435 Cα atoms), TVAII (Z-score, 53.3; RMSD, 1.3 Å for 435 Cα atoms), ThMAA (Z-score, 52.3; RMSD, 1.6 Å for 432 Cα atoms), and a debranching enzyme from Nostoc punctiforme (NpDBE, Z-score, 46.6; RMSD, 2.0 Å for 420 Cα atoms). Among them, GsNPL, TVAII, and ThMAA share an additional domain at the N terminus (domain N), whereas NpDBE has a typical three-domain monomer structure such as CMMase.
Table 1.
X-ray data collection and refinement statistics of CMMase crystals
| Wild-type |
D201N, CMM | |||
|---|---|---|---|---|
| Ligand-free | Maltose | Panose | ||
| Data collection | ||||
| Beamline | SPring-8 | KEK-PF | KEK-PF | KEK-PF |
| BL26B1 | BL1A | BL17A | NW12A | |
| Wavelength (Å) | 1.0000 | 1.0000 | 0.9800 | 1.0000 |
| Space group | P21 | C2221 | P21 | P41212 |
| Unit cell (Å) | a = 48.3 | a = 47.0 | a = 48.3 | a = b = 72.7 |
| b = 179.8 | b = 115.7 | b = 180.2 | c = 209.2 | |
| c = 63.4 | c = 182.2 | c = 62.9 | ||
| β (°) = 113.6 | β (°) = 111.6 | |||
| Resolution (Å)a | 50.0–2.40 (2.44–2.40) | 50.0–2.10 (2.14–2.10) | 50.0–1.94 (1.97–1.94) | 50.0–1.60 (1.63–1.60) |
| Total reflections | 114,848 | 106,382 | 243,620 | 1,000,344 |
| Unique reflectionsa | 35,608 | 26,693 | 72,961 | 73,605 |
| Completeness (%)a | 92.2 (92.1) | 88.6 (96.7) | 99.5 (93.0) | 98.2 (98.5) |
| Redundancya | 3.2 (3.1) | 4.0 (4.6) | 3.3 (3.0) | 13.6 (14.0) |
| Mean I/σ (I)a | 23.2 (13.1) | 11.5 (2.2) | 13.7 (2.4) | 29.5 (6.0) |
| Rmerge (%)a | 4.8 (9.6) | 14.1 (55.2) | 10.1 (35.2) | 8.0 (43.3) |
| CC1/2 (%) | (98.4) | (83.9) | (79.1) | (95.9) |
| Refinement | ||||
| Resolution (Å) | 89.9–2.40 | 91.1–2.05 | 90.1–1.94 | 68.7–1.60 |
| No. of reflections | 33,636 | 25,316 | 69,198 | 69,714 |
| Rwork/Rfree (%) | 18.8/24.8 | 25.2/32.3 | 18.8/22.3 | 16.2/19.0 |
| RMSD from ideal values | ||||
| Bond lengths (Å) | 0.015 | 0.015 | 0.020 | 0.027 |
| Bond angles (°) | 1.855 | 1.777 | 1.983 | 2.337 |
| Ramachandran plot (%) | ||||
| Favored | 96.4 | 95.2 | 96.4 | 96.8 |
| Allowed | 3.2 | 4.4 | 3.0 | 3.0 |
| Outlier | 0.3 | 0.5 | 0.6 | 0.2 |
| PDB code | 5ZXG | 6A0L | 6A0K | 6A0J |
a Values in parentheses are for the highest-resolution shell.
Figure 2.

Overall structure of CMMase and electron density maps of ligands. A, dimer structure in the crystals. One monomer is colored by domain (catalytic domain A in cyan, domain B in red, and domain C in yellow), and the other monomer is shown in gray. B, monomer structure of CMMase–CMM complex. The catalytically important residues (Asp-201 as nucleophile, Glu-230 as acid/base, and Asp-297 as fixer), bound CMM, and a calcium ion are shown as cyan sticks, gray sticks, and a green sphere, respectively. C–E, mFo − Fc omit electron density maps of CMM (C, contoured at 4.0 σ), maltose (D, 2.0 σ), and panose (E, 2.0 σ), in the complex structures.
The molecular masses of CMMase, as deduced from the amino acid sequence, estimated by SDS-PAGE and calibrated by size-exclusion chromatography (in 150 mm NaCl and 20 mm Tris-HCl (pH 8.0)), were 51.6, 53.4, and 105.2 kDa, respectively, suggesting that it is dimeric in solution. In the ligand-free structure and those of two complex forms (maltose and panose), a wing-like dimer structure was observed in the crystal packing in space group P21 (two molecules in the asymmetric unit) or C2221 (one molecule in the asymmetric unit) (Fig. 2A). The dimer interface comprises two β-sheets in domain C. A molecular interface analysis using PISA (44) indicates that the interface area is 1,060 Å2, with 19 hydrogen bonds, 11 salt bridges, and an estimated ΔiG value of −3.2 kcal/mol, and it is implied to be the most likely dimer interface among crystal packing interfaces (complexation significance score = 1). However, this dimer structure is not present in the CMM complex structure of the P41212 space group. This is probably because the CMM complex crystal was grown under alkaline conditions with a high-salt concentration (0.1 m glycine NaOH (pH 9.3), 0.2 m lithium sulfate, and 0.8 m sodium/potassium tartrate), and the salt bridges and the hydrogen bonds in the interface may have been broken during the crystal growth. The two active sites are well-separated in the dimer structure, and the dimerization apparently does not affect the enzymatic function.
To the best of our knowledge, the C domain–mediated wing-like dimer structure of CMMase (Fig. 3A) is unique among GH13 enzymes, and no similar type of dimer structures have been reported. In GH13_20 enzymes, GsNPL forms a dimer with the characteristic domain N (Fig. 3B) (42), and the dimer interface is substantially involved in the formation of the active site. Similar dimer formation has also been observed in other GH13_20 enzymes containing domain N, such as TVAII and ThMAA (22, 41). As shown in Fig. 3C, NpDBE exhibits a unique boat-shaped dimer structure mediated by domains A and B (38). A neighboring molecule is only slightly involved in the formation of the active site, and it has been shown that dissociation into monomers does not affect the activity (45).
Figure 3.

Surface models of dimer structure of GH13_20 enzymes. A, CMMase (ligand-free form). B, GsNPL (PDB code 1J0H). C, NpDBE (PDB code 2WC7). Domains N, A, B, and C of chain A are shown in green, cyan, red, and yellow, respectively. The catalytic residues of chain B are shown in red.
A calcium-binding site is observed in the CMM complex structure (Fig. 2B). The same calcium ion was observed in chain A of the ligand-free and panose complex structures but was not found in chain B of these structures or in either chain of the maltose complex. The calcium ion is coordinated by the side-chain oxygens of Asn-23, Asp-29, and Asp-49, the main chain carbonyl of Asp-25 and Gly-47, and one water molecule. The calcium-binding site in CMMase is different from the conserved site in other GH13 subfamilies, e.g. that of GH13_5 α-amylase from Bacillus licheniformis (46) but is similar to that of GsNPL (42). The calcium ion of CMMase is not required for activity; we previously reported that EDTA treatment or the addition of calcium ion did not change the activity (31).
Active site of CMMase complexed with substrate and products
In addition to the natural substrate (CMM), a partial structure of the intermediate product/substrate MM (panose) and the final degradation product (maltose) of CMMase (Fig. 1) were located in the active site of the complex structures. The electron densities of these ligands were clearly observed(Fig. 2, C–E) near the catalytic residues (Asp-201 as the nucleophile and Glu-230 as the acid/base catalyst), which are conserved in GH13. To avoid the hydrolysis of CMM, a nucleophile residue variant (D201N) was made and used for the co-crystallization with CMM.
In the maltose complex structure, one maltose molecule is bound at subsites −1 and −2 (Fig. 4). The maltose molecule is extensively recognized by protein side chains through hydrogen bonds (Tyr-168, Asp-341, and Arg-345 at subsite −2, and Glu-230, His-296, Asp-297, and His-121 at subsite −1) and stacking interactions (His-79 and Tyr-81 at subsites −2and −1, respectively) (Fig. 5A). These interactions are well-conserved among the GH13_20 enzymes.
Figure 4.
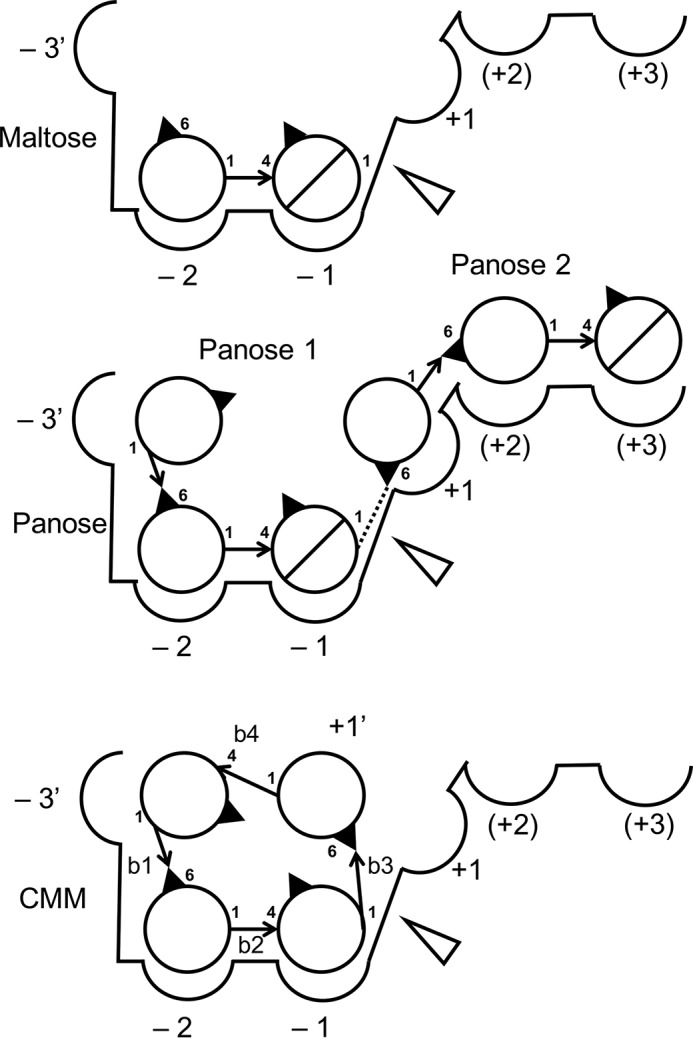
Schematic model of ligand binding to the active site of CMMase crystal structures. Circles and slashed circles indicate glucose and reducing end glucose, respectively. The cleavage site is shown as an open triangle. Arrows indicate glycoside linkages. Identifiers of the four glycosidic bonds in CMM (b1–b4) are labeled.
Figure 5.

Stereoviews of the active site of CMMase. A, maltose complex (pink). B, panose complex (yellow). C, CMM complex (gray). Protein residues, hydrogen bonds, and water molecules are shown as green sticks, yellow dashed lines, and red spheres, respectively.
In the panose complex structure, two panose molecules are bound at subsites −3′ to −1 (panose 1) and at subsites +1 to +3 (panose 2) (Fig. 4). Notably, the nonreducing end Glc moiety of panose 1 was significantly deviated from the normal subsite −3 position of GH13_20 enzymes (discussed below); thus, we designated this subsite as −3′. The α-1,6 linkages of panose 1 and 2 are located between the subsites −3′ and −2 and subsites +1 and +2, respectively. Therefore, panose 1 mimics the binding of MM, whereas panose 2 appears to represent binding of linear α-glucans such as pullulan. The maltose moiety of panose 1 is bound very similarly to the maltose complex structure, but subsite −3′ of CMMase is located in a vertical position (Fig. 5B). The subsite −3′ is not extensively recognized by the protein, but the side chains of Ser-164 and Cys-163 make a hydrogen bond to the O6 hydroxyl and a hydrophobic interaction, respectively. Panose 2 is loosely recognized compared with panose 1. The side chains of Tyr-204 and Glu-231 form direct hydrogen bonds and a stacking interaction at subsites +2 and +3, and several water-mediated hydrogen bonds additionally support the recognition. The reducing end Glc of panose 2 was observed as the β-anomer (Fig. 2E), probably due to the loose recognition at this subsite (+3). The observed oxygen–oxygen distances between the O1 atom of Glc at subsite −1 and the O6 atom of Glc at subsite +1 are too close (1.2 Å, dotted line in Fig. 4); thus, we set the occupancies of the two panose molecules to 0.5. This implies that the two Glc moieties mimic an α-1,6 linkage of isopanose and pullulan, which can be cleaved by CMMase (Fig. 1) (31). The O1 and C1 atoms of Glc at subsite −1 are located near the oxygen atoms of the side chains of the acid/base catalyst (Glu-230, distance = 2.6 Å) and the nucleophile (Asp-201, distance = 2.9 Å), suggesting that the Glc moiety canonically occupies the catalytic subsite.
In the complex structure with CMM, the α-1,6 linkage is positioned near the catalytic residues. The distances between the C1 of Glc at subsite −1 and the side-chain oxygen of Asp(Asn)-201 and the scissile glycosidic bond oxygen and that of Glu-230 are 3.1 and 3.2 Å, respectively (Fig. 5C). Because the Glc moiety connecting the sugars in subsites −3′ and −1 is again significantly deviated from the normal subsite +1 position for linear glucan substrates, we designated this subsite as +1′ (Fig. 4). The Glc at subsite +1′ forms hydrogen bonds with Asp-297 and several water molecules. The binding mode of CMM to subsites −3′ to −1 is similar to that of panose 1. At subsite −3′, there are also interactions with Cys-163 and Ser-164. The maltose moiety at subsites −2 and −1 are extensively recognized with similar interactions as those of the maltose complex, as described above.
In summary, for the structural features of the active site, CMMase binds a Glc moiety at subsite +1 by placing the O6 hydroxyl near the acid/base catalyst (cleavage preference for an α-1,6 bond). In addition, CMMase has strong subsites for the maltose moiety at subsites −2 and −1, whereas the flanking subsites (−3′ and +1′/+1) appear to be relatively weak. The extensive interactions at subsites −2 and −1 suggest that only α-1,4-linked Glc moieties are allowed to bind here. These structural features are consistent with the substrate preference for CMMase (31), with hydrolysis ratios of 99.4% for CMM, 76.2% for MM, 34.6% for pullulan, 28.7% for isopanose, 4.2% for maltotetraose (G4), and 1.8% for maltopentaose. Furthermore, we have measured the kinetic parameters of CMMase to CMM and MM. CMMase exhibited typical Michaelis-Menten–type kinetics (hyperbolic saturation curve) to the both substrates (data not shown). The Km, kcat, and kcat/Km values for CMM were 3.6 ± 0.2 mm, 59.7 ± 2.9 s−1, and 17.8 ± 1.4 s−1 mm−1, respectively (Table 2). The Km, kcat, and kcat/Km values for MM were 51 ± 14 mm, 250 ± 39 s−1, and 5.0 ± 2.8 s−1 mm−1, respectively. Therefore, CMMase preferentially binds CMM for hydrolysis compared with MM. From a structural comparison of the ligand-free and complex structures, it was found that CMMase did not significantly change its structure upon substrate (ligand) binding, as the RMSD values for the Cα atoms between all pairs are within 0.19–0.47 Å. A significant movement was only observed for Arg-233 in the panose complex (Fig. 5B), whose side chain moves into the subsite +3 when it is occupied.
Table 2.
Kinetic parameters of the wild-type CMMase and variants for CMM
The enzymatic reaction was performed in 50 mm sodium acetate buffer (pH 6.0) at 25 °C, and reducing power was measured.
| Enzyme | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | s−1 | s−1 mm−1 | |
| WT | 3.6 ± 0.2 | 59.7 ± 2.9 | 17.8 ± 1.4 |
| Y168A | 45.5 ± 5.3 | 111.1 ± 8.9 | 2.4 ± 0.3 |
| Y204A | 5.0 ± 0.3 | 101.4 ± 2.7 | 20.4 ± 1.5 |
| P203A/Y204N/F205E | 40.8 ± 6.3 | 126.1 ± 12.9 | 3.1 ± 0.6 |
| Y168Q/Y204N | 29.5 ± 2.1 | 146.7 ± 7.0 | 5.0 ± 0.4 |
| Y168Q/P203A/Y204N/F205E | 110.6 ± 56.3 | 45.0 ± 18.6 | 0.4 ± 0.3 |
| C163A | 69.7 ± 5.0 | 74.4 ± 4.4 | 1.1 ± 0.1 |
| C163V | 15.2 ± 0.5 | 32.6 ± 0.6 | 2.1 ± 0.1 |
| C163L | 85.9 ± 14.8 | 97.7 ± 13.3 | 1.1 ± 0.2 |
| C163S | 43.5 ± 5.3 | 33.4 ± 3.0 | 0.8 ± 0.2 |
| S164A | 16.8 ± 0.9 | 127.1 ± 3.9 | 7.9 ± 0.5 |
We also examined inhibition behavior and potency of maltose and panose, whose binding interactions with CMMase were revealed by the crystallography. Both of the compounds exhibited typical competitive inhibition with CMM substrate (data not shown), and the Ki values for maltose and panose were 4.8 ± 0.3 and 2.8 ± 0.4 mm, respectively. The Ki values were comparable with the Km value for CMM (3.6 mm). The relatively small Ki value of panose is consistent with the more extensive interactions of the panose 1 molecule in subsites −3′ to −1 compared with maltose in subsite −1 and −2 (Fig. 5, A and B).
Comparison of the active site and ligand-binding mode with neopullulanase belonging to GH13_20
Fig. 6 shows a comparison of the active sites of the two representative GH13_20 enzymes, CMMase and GsNPL. The complex structures of CMMase with CMM, panose, and maltose and those of GsNPL with isopanose and maltotetraose (G4) are shown by superimposing the ligands. As expected by the moderate sequence homology, these enzymes have similar overall structures, as described above. Accordingly, the active-site residues are basically conserved, especially for those depicted on the left side and the centerline in Fig. 6, B and D. In particular, residues involved in the strong interactions at subsites −2 and −1 are highly conserved. However, residues depicted on the right side in Fig. 6, B and D (circled by dotted lines), are not conserved, and thus significant differences at other subsites (−3/−3′, +1, +2, and +3) are found. As shown in Fig. 6, A and C, by surface models with hydrophobicity from white to red, the active-site cleft of GsNPL is elongated and boat-shaped, with a hydrophobic protrusion above subsite −1, whereas that of CMMase is bowl-shaped with a relatively hydrophobic platform at subsites +2 and +3.
Figure 6.

Comparison of the active sites of CMMase and GsNPL. A and B, molecular surface (A) and stick model (B) of CMMase. Maltose (pink), panose (yellow), and CMM (gray) in the complex structures are superimposed onto the CMMase–CMM complex structure (green in B). C and D, molecular surface (C) and stick model (D) of GsNPL. Maltotetraose (pink, PDB code 1J0J), isopanose (purple, 1J0K), and panose (orange, 1J0I) in the complex structures are superimposed onto the panose complex structure (cyan in D). The surface model is color-coded according to the surface hydrophobicity.
The residues involved in formation of the characteristic walls of the cleft of each enzyme can be grouped as follows: PYF (Pro-203, Tyr-204, and Phe-205), CS (Cys-163 and Ser-164), and Y (Tyr-168) in CMMase, and ANE (Ala-330, Asn-331, and Glu-332), FA (Phe-289 and Ala-290), and Q (Gln-294) in GsNPL (Fig. 6, B and D). The cleft of GsNPL is relatively deeper at subsites +1 and +2, whereas that of CMMase is shallower at subsites +2 and +3. Moreover, the cleft of GsNPL is narrower at subsites +1 and −1 compared with that of CMMase. The changes at the PYF/ANE and CS/FA walls are responsible for these differences. The recognition of Glc at subsite +1 in GsNPL is supported by hydrophobic interactions with Phe-289, Val-329, Trp-359, and Tyr-45* (Fig. 6D, asterisk indicates that this residue is from the neighboring monomer) (42), and this subsite appears to be relatively stronger than that of CMMase. In particular, the unique subsite −3′ of CMMase is formed by the CS wall. Subsite −3 (and also possible subsite −4) of GsNPL is open to the solvent, suggesting that it prefers linear glucan substrates. In contrast, the cleft of CMMase is blocked at the corresponding position by the side chain of Tyr-168 (Fig. 6B), and the subsite −3′ is located at an elevated position. Therefore, the difference at subsite −3/−3′ appears to be caused by the amino acid change of Q/Y.
Amino acid sequence alignment revealed that, among the GH13_20 enzymes, the key residues of the three walls are unique to CMMase (Fig. 7). The ANE and FA motifs in GsNPL are conserved in other GH13_20 enzymes that exhibit neopullulanase-like activity (TVA II and ThMAA), but the corresponding residues of Gln-294 vary (Ala or His).
Figure 7.

Partial amino acid sequence alignment of the catalytic domain of CMMase and GH13_20 subfamily enzymes. The secondary structures of CMMase are shown above. The four conserved regions (I–IV) of α-amylase family enzymes are underlined. Three key regions for the substrate specificity of CMMase are boxed with green frames.
Identification of important residues for activity by site-directed mutational analysis
To investigate the importance of these residues for the activity of CMMase, we constructed 10 variant enzymes containing single to quadratic site-directed mutations at the three wall positions by mimicking the active site of GsNPL with substitution of PYF, CS, and Y for ANE, FA, and Q, or by substituting them with Ala. Their kinetic parameters for CMM (Table 2) were compared with the WT enzyme. A single Ala substitution in the PYF wall (Y204A) caused a concomitant increase of the kcat and Km values for CMM, resulting in slight increase of the kcat/Km value. A single Ala substitution at the Y wall (Y168A) also caused increases in both the kcat and Km values, but the significant increase of the Km value had a large impact on the kcat/Km value (7.4-fold decrease compared with the WT). The triple substitutions from PYF to ANE (P203A/Y204N/F205E) and the combinational double mutation of Tyr-204 and Tyr-168 (Y168Q/Y204N) also significantly decreased the kcat/Km value due to increases of the Km value. The quadratic variant from PYF-Y to ANE-Q (Y168Q/P203A/Y204N/F205E) exhibited the highest Km and the lowest kcat/Km values among all variants tested. These results indicate that the PYF and Y walls at the −2 and plus subsites concertedly support formation of the bowl-shaped cleft of CMMase that is suitable for the small cyclic substrate. Substitutions at the CS wall also significantly increased the Km value for CMM. The effect of the substitution at Ser-164 (S164A) was milder than those at Cys-163 (C163A, C163V, C163L, and C163S). Among the four Cys-163 variants that we tested, C163V and C163S showed the highest and lowest kcat/Km values, respectively. Therefore, we conclude that the small side chain of Cys-163 mainly contributes to the substrate affinity at the unique −3′ subsite of CMMase with its hydrophobicity.
We also measured activities (hydrolysis ratio) of the variant enzymes toward various substrates and compared themwith those of the WT enzyme (Table 3). Notably, the Y204A and P203A/Y204N/F205E variants exhibited a significantly increased activity toward G4, suggesting that elimination of the large side chain of Tyr-204 at subsite +2 shifted the specificity of CMMase from α-1,6 linkage toward the linear α-1,4-linked substrate. This result is consistent with the conservation pattern of the residue corresponding to Tyr-204 of the PYF motif (Fig. 7). Enzymes with neopullulanase-like activity (GsNPL, TVAII, and ThMAA), which have intermediate specificities to α-1,4 and α-1,6 linkages, have Asn at this position, although the α-1,6-specific debranching enzyme (NpDBE) has an aromatic residue (Phe-213). In the study of TVAII, substitution of Val-326 (the residue corresponding to Val-202) could modulate the preference for α-1,6 and α-1,4 linkages (47). The V326A variant of TVAII favored the α-1,4 linkage, although V326I favored the α-1,6 linkage, suggesting that size and hydrophobicity of the residue at this position modulate the linkage preference. As shown in Fig. 5, Val-202 of CMMase is located close to the PYF wall.
Table 3.
Substrate specificity of the wild-type CMMase and variants toward various substrates (10 mm oligosaccharides or 1% pullulan)
The enzymatic reaction was performed in 50 mm sodium acetate buffer (pH 6.0) with 0.1 mg/ml enzyme at 25 °C for 18 h. The hydrolysis was monitored by TLC and classified by the following marks: −, not hydrolyzed; +, ≦10% hydrolyzed; ++, 10–50% hydrolyzed; and +++ ≧50% hydrolyzed.
| Enzyme | CMM | MM | G4 | Isopanose | Pullulan |
|---|---|---|---|---|---|
| WT | +++ | +++ | + | ++ | +++ |
| Y168A | +++ | ++ | + | − | − |
| Y204A | +++ | +++ | +++ | ++ | +++ |
| P203A/Y204N/F205E | +++ | +++ | +++ | + | ++ |
| Y168Q/Y204N | +++ | ++ | + | + | + |
| Y168Q/P203A/Y204N/F205E | ++ | + | ++ | − | + |
| C163A | +++ | + | − | − | − |
| C163V | +++ | − | − | − | − |
| C163L | +++ | + | − | − | + |
| C163S | +++ | − | − | − | − |
| S164A | +++ | +++ | + | + | ++ |
The substitution at Tyr-168 (Y168A, Y168Q/Y204N, and Y168Q/P203A/Y204N/F205E) significantly decreased the activity toward pullulan and isopanose. Moreover, the substitutions at the CS wall generally decreased the activities toward substrates other than CMM. In particular, the Cys-163 substitutions showed significantly decreased activities toward MM. These results indicate that changes at the CS and Y walls, which constitute the essential part of the pocket of CMMase, are also destructive for binding of the linear substrates.
Crystallography of CMM
Previously, we reported the preparation process and characteristics of pentahydrate crystals of CMM (29). Here, we determined the X-ray crystal structure of a single CMM crystal (Table 4 and supporting information). Fig. 8 shows superimposition of the structure of CMM in the single crystal (Fig. 8, cyan) with the CMM molecule bound to CMMase (white). The two CMM molecules unexpectedly exhibit large conformational differences. We also modeled an energy-minimized CMM molecule in the active site (Fig. 8, thin orange sticks), and we found that it adopts a similar conformation as the CMMase-bound CMM molecule. The CMM in the single crystal shows the largest deviations from the other two at the Glc unit at subsite −3′, swinging to the left side in Fig. 8. The conformational differences arise from the two α-1,6 bonds (b1 between −3′ and −2 and b3 between −1 and +1′) and the two α-1,4 bonds (b2 between −2 and −1 and b4 between +1′ and −3; see Fig. 4 for the designation). Therefore, we measured the torsion angles of the four glycosidic bonds of the three CMM structures (Table 5). Dowd et al. (48) calculated isoenergy surfaces of the α-1,6 bond of isomaltose based on the MM3 force field. The conformations of the b1 and b3 bonds in the CMM single crystal are placed near the second lowest local minimum of the energy map of α-isomaltose (ϕ = −44.3°, ψ = −174.6°, and ω = −50.5°), and those in CMM complexed with CMMase are close to the third minimum (ϕ = −43.5°, ψ = 179.7°, and ω = 60.6°). Therefore, the two α-1,6 bonds are not high-energy conformers even though they adopt distinct conformations (ω ∼ −58° in a single crystal and ω ∼ +40° in complex in CMMase). For the α-1,4 bond of the α-maltose unit, the GlycoMapsDB web tool was used to map the torsion angles with 2,512 PDB entries (black for 1,988 disaccharide fragments and red for 524 exact structures) onto a calculated energy map (Fig. 9) (49). The b2 and b4 bonds in CMMase were mapped in the most frequently observed low-energy conformation area, although those in the single CMM crystal were mapped in a rarely observed high-energy conformer area, whose calculated energy is more than 5 kcal/mol higher than that of the global minimum. Therefore, the single-crystal structure of CMM may be a relatively high-energy conformer trapped during the crystal formation, and the CMM molecule bound in CMMase represents one of the lowest energy conformations in solution.
Table 4.
X-ray data collection and refinement statistics of a CMM crystal.
| Empirical formula | C72H150O75 or C72H120O60·15H2O |
| Formula weight (g mol−1) | 2215.93 |
| Crystal size (mm) | 0.340 × 0.060 × 0.020 |
| Crystal color and shape | Colorless, prism |
| Temperature (K) | 100 |
| Radiation (λ, Å) | 0.71075 |
| Crystal system | Monoclinic |
| Space group | P21 (4) |
| a (Å) | 0.4366 (18) |
| b (Å) | 21.595 (5) |
| c (Å) | 22.159 (3) |
| β (°) | 91.671 (11) |
| V (Å3) | 4992.0 (16) |
| Z | 2 |
| dcalcd (g/cm3) | 1.474 |
| R(int) | 0.1350 |
| No. of observations (all reflections) | 22,371 |
| Reflection/parameter ratio | 16.91 |
| Final R indices (I >2.00σ(I)) | R1 = 0.1463 |
| Final R indices (all data) | R = 0.1957 |
| wR2 = 0.3963 | |
| Goodness of fit | 1.091 |
Figure 8.

Stereographic superimposition of CMM in a single crystal (cyan sticks), CMM bound to CMMase (white sticks), and energy-minimized CMM molecule model (thin orange sticks). The catalytic residues (green sticks) and subsites of CMMase are shown. Energy minimization of the CMM molecule model was performed using PCModel version 9.20 (Serena Software) with MMX force field. The CMM molecules were superimposed using the Glc unit bound at subsite −1.
Table 5.
Torsion angles (°) of the glycosidic bonds of CMM structures
| Bond and anglesa | In CMMase | Energy-minimized | Single crystal |
|---|---|---|---|
| b1 ϕ/ψ/ω | −48.5/−149.8/40.0 | −43.2/163.4/−158.7 | −63.6/−177.6/−58.7 |
| b2 ϕ/ψ | −7.6/13.3 | 2.1/−38.9 | −49.7/158.3 |
| b3 ϕ/ψ/ω | −56.9/−151.1/41.8 | −61.1/−150.1/19.5 | −58.9/177.9/−58.4 |
| b4 ϕ/ψ | −26.9/17.5 | 6.0/30.8 | −56.1/164.0 |
a Designation of the bonds (b1–b4) are shown in Fig. 4. Definitions of the torsion angles are as follows: H1′–C1′–O4–C4 for ϕ, C1′–O4–C4–H4 for ψ, and O1–C6′–C5′–H5′ for ω (α-1,6 bond).
Figure 9.
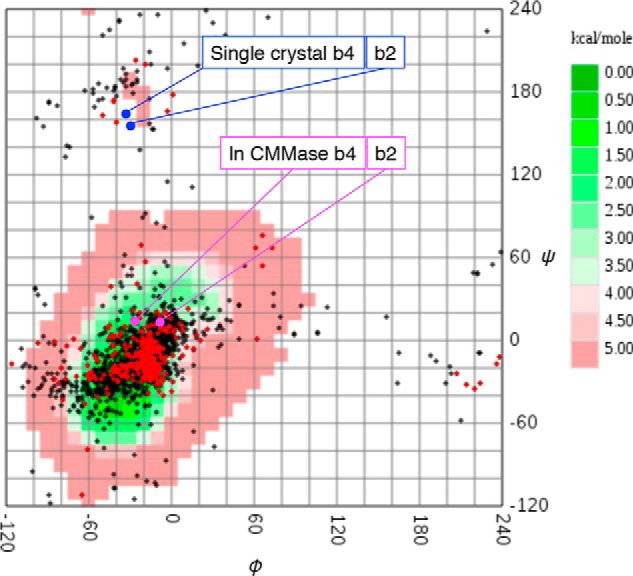
α-1,4-Glycosidic bond torsion angles of α-maltose units in PDB (GlycoMAP ID: 7565) and CMM structures. Exact α-maltose structures (red) and α-maltose-containing glycans (black) in the PDB are plotted on a calculated conformational free energy map (coded green to red for low- to high-energy conformations). The torsion angles of CMM molecules in the single crystal (blue) and in the CMMase complex structure (magenta) are also plotted. The b2 and b4 bonds of CMM are designated in Fig. 4.
Discussion
Fig. 10 illustrates our proposed reaction steps of the two-stage hydrolysis of CMM to maltose by CMMase, according to the results of this study: step 1, CMM binds to the active site; step 2, CMM is cleaved at one of the two α-1,6 linkages, and the reaction product, MM, is tentatively released from the active site. For step 3, MM rebinds so that the remaining α-1,6 linkage is placed near the catalytic residue, and for step 4, the second α-1,6 linkage is hydrolyzed, and the products (two molecules of maltose) are released from the active site. The −2 and −1 subsites strongly recognize the maltose molecule and play a critical role in the substrate binding. In addition, interaction at −3′ subsite, which is mainly supported by the hydrophobic interaction by Cys-163, fixes the cyclic molecule. The PYF and Y walls also support formation of the bowl-shaped cleft, which is complementary for CMM. Comparison between the apo and complex structures suggested that the active site of CMMase does not have an induced-fit–type feature. Our kinetic analysis indicated that the enzyme preferentially binds CMM for hydrolysis compared with MM. This is probably due to the rigid and cyclic structure of the CMM substrate, whose low-energy conformer fits into the static active site.
Figure 10.

Proposed reaction steps of the two-stage hydrolysis of CMM to maltose by CMMase. Circles and arrows indicate glucose and glycoside linkages, respectively. The cleavage site is shown as an open triangle.
Among the several known cyclic oligosaccharides, the metabolic pathways of CDs have been extensively studied for various bacteria and archaea, such as Klebsiella oxytoca, Thermococcus sp., Bacillus subtilis, and T. vulgaris (50–53). The microbial CD metabolism generally consists of three stages: synthesis of the cyclic oligosaccharides (CDs) outside the cells, uptake into the cells via ATP-binding cassette transporters, and degradation by intracellular enzyme(s). It has been suggested that this type of metabolic pathway is advantageous in the competition of carbon source acquisition by transiently changing the molecular form of a digestible glucan (starch) into a special cyclic form (CDs), which is rarely assimilated by other microorganisms. A similar three-stage metabolic pathway for CNN has been studied for L. monocytogenes (35). The CNN metabolic pathway is coded in two operons with 10 genes in total (lmo2446--lmo2444 and lmo0178--lmo0184). The functional and structural study revealed that the CNN metabolic pathway involves at least two extracellular enzymes for CNN formation, a specific ABC transporter, several intracellular enzymes, and a translational regulation system of a ROK family protein. In the CNN metabolic pathway, the crystal structure of GH31 Trueperella pyogenes cycloalternan-degrading enzyme (TpCADE), which is a homolog of the intracellular CNN-hydrolyzing enzyme of L. monocytogenes (Lmo0182), has been reported (34, 35). Fig. 11 shows a comparison of the active sites of CMMase and TpCADE by aligning the structures with the catalytic components for α-glycosidic bond hydrolysis. However, the two enzymes having the hydrolytic activity toward cyclic glucotetrasaccharides adopt completely different substrate-recognition architectures. TpCADE has the cleavage specify toward the α-1,3 bond of CNN and recognizes the substrate by a stacking interaction ranging from subsites −1 to +1, and two direct hydrogen bonds to sugars in subsites −1 and −2.
Figure 11.

Comparison of the active sites of CMMase and TpCADE. A, CMMase (green)–CMM (gray) complex structure. B, TpCADE (cyan)–CNN (black) complex structure (PDB 5I0G). The structures were superimposed with the following atoms and presented side-by-side: side chain oxygen atoms of the catalytic residues (nucleophile and acid/base catalyst), the scissile glycosidic bond oxygen, and the sugar ring atoms in subsite −1. Hydrogen bonds and the nucleophilic attack distances are shown as yellow and red dashed lines, respectively.
The metabolic pathway of A. globiformis M6 for CMM also consists of three-stage components (29–31, 33), but it is relatively simpler than that for CNN. The CMM metabolic pathway consists of a single cluster containing seven genes (cmmA–G). Among them, an extracellular CMM-forming enzyme, 6MT (CmmA), and an intracellular CMM-degrading enzyme, CMMase (CmmF), have been characterized. The cmmB gene encodes an intracellular GH13_30 α-glucosidase (33). The α-glucosidase exhibits high activity toward MM, panose, and maltose, and it has been suggested that CMM is degraded to glucose by the synergistic action of CMMase and the α-glucosidase CmmB (31). The cmmC, -D, and -E genes show 27, 44, and 49% sequence identity (by BLAST) to a solute-binding protein from Streptomyces avermitilis, a putative permease from Deinococcus geothermalis, and a putative permease from Bacillus clausii of the ABC sugar transport system, respectively. Therefore, CmmC, CmmD, and CmmE likely form an ABC importer system specific for CMM. The cmmG gene encodes a putative LacI/PurR family transcriptional regulator.
Our study revealed that the shape and interactions in the active site of CMMase are highly specific for the CMM substrate, and we identified several critical residues for the recognition. CMMase might have emerged through molecular evolution from a GH13_20 family enzyme, which has a wide substrate specificity similar to that of neopullulanase, under pressure from the “selfish” metabolic pathway for the cyclic oligosaccharide, in combination with the molecular evolution of the CMM-forming enzyme, 6MT. To date, the CMMase activity has not been confirmed for enzymes other than CMMase from A. globiformis M6, even though many GH13 enzymes show a certain sequence identity (>40% by BLAST). For example, neopullulanase-like enzymes, cyclomaltodextrinases, general type α-amylases, dextranases (EC 3.2.1.11), isoamylases (EC 3.2.1.63), pullulanases, and debranching enzymes cannot hydrolyze CMM. To investigate putative CMMases from the gene database, we performed a protein BLAST search. Table 6 shows the amino acid sequence alignment of the top 10 homologous proteins from different organisms. Most of the bacterial species belong to the Actinobacteria class, except for Chlamydia. These putative proteins exhibit relatively higher sequence identities (>55%) to CMMase than to the neopullulanase-like enzymes, and the critical residues forming the PYF, CS, and Y walls are basically conserved. Therefore, we assume that these putative homologs also have CMMase activity and that their source organisms likely have a three-stage metabolic pathway for CMM, similar to that of A. globiformis M6. Further studies on 6MT and the putative ABC transporter (CmmC solute-binding protein) will identify key residues for their functions and clarify the whole picture of the three-stage metabolic pathway of CMM, which may be prevalent in Actinobacteria.
Table 6.
Partial amino acid sequence alignment of CMMase and its homologs
The residues corresponding to important residues for CMM specificity in CMMase are shown in bold. The regions conserved in CMMase are underlined.
| Speciesa | Annotation | Id (%)b | GenBank and RefSeq ID | Partial sequence conservation |
|
|---|---|---|---|---|---|
| CS and Y | PYF | ||||
| A. globiformis M6 (Micrococcales) | Cyclic maltosyl–maltose hydrolase | BAI67607 | NYRTCSGCYYLP | RLDVPYFINH | |
| Microbacterium hydrocarbonoxydans (Micrococcales) | GH13 protein | 73 | WP_045259135 | NYRTCSGCYYLP | RLDVPYFINH |
| Corynebacterium-like bacterium B27 (Corynebacteriales) | GH13 protein | 70 | WP_022897950 | NYRTCSGCYYLP | RLDVPYFINH |
| Cellulomonas sp. KRMCY2 (Micrococcales) | GH13 protein | 69 | WP_024285493 | NYRTCSGCHYLP | RLDVPYFINR |
| Actinomyces sp. Marseille-P3257 (Actinomycetales) | GH13 protein | 66 | WP_076464478 | NYRTCSGCEYLP | RLDVPYFINK |
| Actinobacteria bacterium 69-20 (Actinomycetales) | α-Amylase | 63 | OJV25818 | NYQTCSGCYYLP | RLDVPYFVPT |
| T. pyogenes (Actinomycetales) | GH13 protein | 60 | WP_024963853 | NYRTCSGCEYLP | RLDVPYFINM |
| Gardnerella sp. (Bifidobacteriales) | GH13 protein | 57 | WP_004105641 | NYKTCSGCYYLP | RLDVPYFVNK |
| Alloscardovia omnicolens (Bifidobacteriales) | GH13 protein | 57 | WP_049206603 | NYKTCSGCYYLP | RLDVPYFVNK |
| Chlamydia trachomatis (Chlamydiales) | GH protein | 55 | CRH73918 | NYKTCSGCEYLP | RLDVPYFIYP |
| Mobiluncus mulieris (Actinomycetales) | GH13 protein | 55 | WP_004012112 | NYKTCSGCYYLP | RLDVPYYVNM |
a The order of each organism is shown in parentheses.
b Sequence identity was by BLAST.
Experimental procedures
Protein preparation
The CMMase-encoding gene was amplified from pRSET–CMMase (33) by PCR to express as an N-terminally His-tagged (His6) protein, and inserted between the NdeI and BamHI sites of pET-28b(+) vector (Novagen, Madison, WI). The following primers were used: 5′-GGAATTCCATATGACCGCTCCCGACTGG-3′; 5′-CGCGGATCCTTACGCAGAGCTCCCGGG-3′ (restriction enzyme sites are underlined). This plasmid is designated pET28b_CMMase. Escherichia coli Rosetta2 (DE3) (Novagen) transformed by this plasmid was cultured in Luria-Bertani medium containing antibiotics (50 mg/liter kanamycin and 34 mg/liter chloramphenicol) at 37 °C until A600 nm = 0.6. To induce protein expression of the transformant, 0.1 mm (final concentration) of isopropyl 1-thio-β-d-galactopyranoside (FUJIFILM Wako Pure Chemical Co., Osaka, Japan) was added to the medium. The medium was then continuously cultured at 15 °C for 24 h. The cultured cells were harvested by centrifugation at 8,000 × g for 15 min and suspended in 50 mm Tris-HCl (pH 8.0) and 500 mm NaCl. To obtain cell-free extracts, the suspended solution was sonicated and centrifuged at 18,000 × g for 45 min. First, CMMase was purified by nickel affinity column chromatography using cOmplete His-Tag Purification Resin (Sigma). CMMase was then further purified by column chromatography with a Mono Q 10/100 GL and a Superdex 200 pg 16/60 using the ÄKTA system (GE Healthcare, Buckinghamshire, UK). The concentration of CMMase was determined by a NanoDrop ND-1000 spectrophotometer (ThermoFisher Scientific, Waltham, MA) using the extinction coefficient ϵ280 nm = 90,090 m−1 cm−1, which was estimated from the amino acid sequence.
Site-directed mutagenesis
Site-directed mutants were constructed using a PrimeSTAR mutagenesis basal kit (TaKaRa Bio, Ohtsu, Japan) and a QuikChange multisite–directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The following primer pairs were used for the PrimeSTAR mutagenesis basal kit: 5′-CGCACGGTTTCGGGCTGCTACTACCTG-3′ and 5′-GCCCGAAACCGTGCGGTAGTTCGGCGT-3 for C163V; 5′-CGCACGCTGTCGGGCTGCTACTACCTG-3′ and 5′-GCCCGACAGCGTGCGGTAGTTCGGCGT-3′ for C163L; 5′-CGCACGTCGTCGGGCTGCTACTACCTG-3′ and 5′-GCCCGACGACGTGCGGTAGTTCGGCGT-3′ for C163S; 5′-ACGTGTGCGGGCTGCTACTACCTGCCG-3′ and 5′-GCAGCCCGCACACGTGCGGTAGTTCGG-3′ for S164A; 5′-TGCTACGCACTGCCGAAGTGGAACGCG-3′ and 5′-CGGCAGTGCGTAGCAGCCCGAACACGT-3′ for Y168A; 5′-TGCTACCAACTGCCGAAGTGGAACGCG-3′ and 5′-CGGCAGTTGGTAGCAGCCCGAACACGT-3′ for Y168Q; 5′-CGGGTGGCGGCTCAATGTGCCGTACTT-3′ and 5′-AAGTACGGCACATTGAGCCGCCACCCG-3′ for D201N; 5′-GTGCCGGCATTCATCAACCACACGTTC-3′ and 5′-GATGAATGCCGGCACATCGAGCCGCCA-3′ for Y204A; and 5′-GTGCCGAACTTCATCAACCACACGTTC-3′ and 5′-GATGAAGTTCGGCACATCGAGCCGCCA-3′ for Y204N. The following primers and complementary strands were used for the QuikChange multisite–directed mutagenesis kit: 5′-CCGAACTACCGCACGGCTTCGGGCTGCTACTA-3′ for C163A, and 5′-GGGTGGCGGCTCGATGTGGCGAACGAGATCAACCACACGTTCTGG-3′ for P203A/Y204N/F205E. D201N, which was used for crystallization, was expressed and purified using the same procedure as for the WT. Other mutant proteins for the activity assay were purified using the His tag and Mono Q columns.
Protein crystallography of CMMase
Protein crystallization was performed by the sitting drop vapor diffusion method at 20 °C. WT crystals were obtained by mixing 0.5 μl of protein solution containing 20 mg/ml CMMase in 20 mm Tris-HCl (pH 8.0), and an equal volume of reservoir solution containing 0.1 m sodium citrate (pH 5.6), 0.22 m ammonium sulfate, and 30% (w/v) PEG4000. The crystals were cryoprotected in the reservoir solutions supplemented with 30% (w/v) PEG400 or 30% maltose (for maltose complex). To obtain the complex structure with panose, 50 mm panose was added to the cryoprotectant solution. D201N–CMM complex crystals were obtained by co-crystallization by mixing 0.5 μl of protein solution containing 13 mg/ml CMMase in 20 mm Tris-HCl (pH 8.0) and 5 mm CMM with an equal volume of reservoir solution containing 0.1 m glycine-NaOH (pH 9.3), 0.2 m lithium sulfate, and 0.8 m sodium/potassium tartrate. The D201N–CMM complex crystals were cryoprotected in the reservoir solution supplemented with 30% (w/v) PEG400 and 10 mm CMM. X-ray diffraction data were collected at BL26B1 at SPring-8 (Hyogo, Japan) and at BL1A, BL17A, and NW12A at the Photon Factory of the High Energy Accelerator Research Organization (KEK-PF, Tsukuba, Japan). The diffraction images were processed with HKL2000 (54). Molecular replacement was performed with MOLREP (55). The model wasfurther built manually with COOT (56) and refined with REFMAC5 (57). Molecular graphic images were prepared using PyMOL (Schrödinger LLC, New York).
Crystallography of CMM
Crystals of CMM were prepared as described previously (29). Details of the X-ray data collection, data reduction, structure solution, and crystallographic refinement are described in Table 4 and the supporting information.
Enzyme assay and kinetic analysis
For the kinetic analysis using CMM as substrate, the enzymatic reaction was initiated by mixing enzyme solution (40 μl), containing 2–10 μg/ml CMMase (or its variant), in 50 mm sodium acetate (pH 6.0) and substrate solution (160 μl), containing 0.625–62.5 mm CMM in 50 mm sodium acetate (pH 6.0). The mixture was incubated at 25 °C for 6 min. After stopping the reaction by heat treatment at 100 °C for 10 min, the reducing power of each mixture was measured by the bicinchoninic acid assay according to the method of Utsumi et al. (58). For the kinetic analysis for MM, substrate solutions containing 1.25–75 mm MM were used. After stopping the reaction by heat treatment at 100 °C for 10 min, a decreased amount of MM was measured by the HPLC system (Shimadzu Corp., Kyoto, Japan) by using two YMC-Triart C18 columns (YMC Co., Ltd., Kyoto, Japan) in tandem. The kinetic parameters and their fitting errors were calculated by nonlinear fitting of the experimental data to the Michaelis-Menten equation using KaleidaGraph 3.6 (Synergy Software, Reading, PA).
For the inhibition kinetics assay for maltose and panose, reaction mixtures containing 1, 4, or 12 mm CMM and 0, 5, 10, or 30 mm inhibitor (maltose or panose) at final concentrations were used, and an increased amount of MM after 6 min of incubation at 25 °C was measured by HPLC. The kinetic parameters were calculated by nonlinear global curve-fitting the experimental data to the theoretical equation for competitive inhibition using R 3.51 (R Foundation for Statistical Computing, Vienna, Austria).
For the substrate specificity assay, the enzymatic reaction was initiated by mixing enzyme solution (1 μl), containing 1 mg/ml CMMase (or its variant), in 20 mm Tris-HCl (pH 8.0), and substrate solution (9 μl), containing 10.0 mm CMM, 10.0 mm MM, 10.0 mm maltotetraose, 10.0 mm isopanose, or 1.00% pullulan, in 50 mm sodium acetate (pH 6.0). The mixture was incubated at 25 °C for 18 h. After stopping the reaction by heat treatment at 100 °C for 10 min, 2 μl of the mixture was spotted onto a TLC Silica Gel 60 F254 plate (Merck, Darmstadt, Germany). Subsequently, the TLC plate was developed by a solution consisting of 55% (v/v) 1-butanol, 36% (v/v) pyridine, and 9% (v/v) water. Sugar spots were detected by spraying 20% (v/v) sulfuric acid/methanol and heating in an oven for several minutes. The contrast of the spot was then visually judged.
Author contributions
M. K., T. A., and H. O. formal analysis; M. K. and H. O. investigation; M. K. and S. F. visualization; M. K. and S. F. writing-original draft; M. K., T. A., H. O., T. M., T. N., and S. F. writing-review and editing; T. A. and S. F. validation; T. A., T. N., and S. F. project administration; T. M., T. N., and S. F. conceptualization; T. M. resources; T. N. and S. F. supervision; S.F. data curation.
Supplementary Material
Acknowledgments
We thank the staff of the Photon Factory and SPring-8 for the X-ray data collection; Dr. Chihaya Yamada for technical help; Dr. Takashi Shibuya, Akiko Miyake, and Michie Ito (R&D Division, Hayashibara Co., Ltd.) for X-ray crystal structure determination of CMM; and Drs. Carly Huitema and Geoff P Horsman for introducing scripts of R software for enzyme inhibition kinetic analysis on bioRxiv (doi: 10.1101/316588) (59).
This work was supported in part by the Platform Project for Support in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5ZXG, 6A0L, 6A0K, and 6A0J) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Crystallographic data for the structure of CMM have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 1865912.
This article contains Tables S1–S14.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CD
- cyclodextrin
- CMM
- cyclic α-maltosyl-(1→6)-maltose
- CNN
- cyclic α-nigerosyl-(1→6)-nigerose
- MM
- maltosyl-maltose
- G4
- maltotetraose
- Glc
- glucose
- GH
- glycoside hydrolase
- GsNPL
- G. stearothermophilus neopullulanase
- TVAII
- T. vulgaris R-47 α-amylase II
- ThMAA
- Thermus sp. IM6501 maltogenic α-amylase
- NpDBE
- N. punctiforme debranching enzyme
- RMSD
- root mean square deviation
- CMMase
- CMM hydrolase
- PDB
- Protein Data Bank
- 6MT
- 6-α-maltosyltransferase.
References
- 1. Schardinger F. (1903) Über Thermophile bakterien aus verschiedenen Speisen und Milch, sowie über einige Umsetzungsprodukte derselben in kohlenhydrathaltigen Nährlö sungen, darunter krystallisierte polysaccharide (dextrine) aus Stärke. Z. Unters. Nahr. Genussm. 6, 865–880 10.1007/BF02067497 [DOI] [Google Scholar]
- 2. Biwer A., Antranikian G., and Heinzle E. (2002) Enzymatic production of cyclodextrins. Appl. Microbiol. Biotechnol. 59, 609–617 10.1007/s00253-002-1057-x [DOI] [PubMed] [Google Scholar]
- 3. Oguma T., Horiuchi T., and Kobayashi M. (1993) Novel cyclic dextrins, cycloisomaltooligosaccharides, from Bacillus sp. T-3040 culture. Biosci. Biotechnol. Biochem. 57, 1225–1227 10.1271/bbb.57.1225 [DOI] [PubMed] [Google Scholar]
- 4. Oguma T., Tobe K., and Kobayashi M. (1994) Purification and properties of a novel enzyme from Bacillus spp. T-3040, which catalyzes the conversion of dextran to cyclic isomaltooligosaccharides. FEBS Lett. 345, 135–138 10.1016/0014-5793(94)00418-8 [DOI] [PubMed] [Google Scholar]
- 5. Watanabe H., Nishimoto T., Sonoda T., Kubota M., Chaen H., and Fukuda S. (2006) An enzymatically produced novel cyclomaltopentaose cyclized from amylose by an α-(1→6)-linkage, cyclo-{→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→4)-α-d-Glcp-(1→4)-α-d-Glcp-(1→4)-α-d-Glcp-(1→}. Carbohydr. Res. 341, 957–963 10.1016/j.carres.2006.02.028 [DOI] [PubMed] [Google Scholar]
- 6. Watanabe H., Nishimoto T., Mukai K., Kubota M., Chaen H., and Fukuda S. (2006) A novel glucanotransferase from a Bacillus circulans strain that produces a cyclomaltopentaose cyclized by an α-1,6-linkage. Biosci. Biotechnol. Biochem. 70, 1954–1960 10.1271/bbb.60131 [DOI] [PubMed] [Google Scholar]
- 7. Watanabe H., Nishimoto T., Kubota M., Chaen H., and Fukuda S. (2006) Cloning, sequencing, and expression of the genes encoding an isocyclomaltooligosaccharide glucanotransferase and an α-amylase from a Bacillus circulans strain. Biosci. Biotechnol. Biochem. 70, 2690–2702 10.1271/bbb.60294 [DOI] [PubMed] [Google Scholar]
- 8. Côté G. L., and Biely P. (1994) Enzymically produced cyclic α-1,3-linked and α-1,6-linked oligosaccharides of d-glucose. Eur. J. Biochem. 226, 641–648 10.1111/j.1432-1033.1994.tb20091.x [DOI] [PubMed] [Google Scholar]
- 9. Bradbrook G. M., Gessler K., Coté G. L., Momany F., Biely P., Bordet P., Pérez S., and Imberty A. (2000) X-ray structure determination and modeling of the cyclic tetrasaccharide cyclo-{→6)-α-d-Glcp-(1→3)-α-d-Glcp-(1→6)-α-d-Glcp-(1→3)-α-d-Glcp-(1→}. Carbohydr. Res. 329, 655–665 10.1016/S0008-6215(00)00212-3 [DOI] [PubMed] [Google Scholar]
- 10. Nishimoto T., Aga H., Mukai K., Hashimoto T., Watanabe H., Kubota M., Fukuda S., Kurimoto M., and Tsujisaka Y. (2002) Purification and characterization of glucosyltransferase and glucanotransferase involved in the production of cyclic tetrasaccharide in Bacillus globisporus C11. Biosci. Biotechnol. Biochem. 66, 1806–1818 10.1271/bbb.66.1806 [DOI] [PubMed] [Google Scholar]
- 11. Aga H., Maruta K., Yamamoto T., Kubota M., Fukuda S., Kurimoto M., and Tsujisaka Y. (2002) Cloning and sequencing of the genes encoding cyclic tetrasaccharide-synthesizing enzymes from Bacillus globisporus C11. Biosci. Biotechnol. Biochem. 66, 1057–1068 10.1271/bbb.66.1057 [DOI] [PubMed] [Google Scholar]
- 12. Aga H., Higashiyama T., Watanabe H., Sonoda T., Nishimoto T., Kubota M., Fukuda S., Kurimoto M., and Tsujisaka Y. (2002) Production of cyclic tetrasaccharide from starch using a novel enzyme system from Bacillus globisporus C11. J. Biosci. Bioeng. 94, 336–342 10.1016/S1389-1723(02)80174-8 [DOI] [PubMed] [Google Scholar]
- 13. Kim Y. K., Kitaoka M., Hayashi K., Kim C. H., and Côté G. L. (2004) Purification and characterization of an intracellular cycloalternan-degrading enzyme from Bacillus sp. NRRL B-21195. Carbohydr. Res. 339, 1179–1184 10.1016/j.carres.2004.02.008 [DOI] [PubMed] [Google Scholar]
- 14. Tagami T., Miyano E., Sadahiro J., Okuyama M., Iwasaki T., and Kimura A. (2016) Two novel glycoside hydrolases responsible for the catabolism of cyclobis-(1→6)-α-nigerosyl. J. Biol. Chem. 291, 16438–16447 10.1074/jbc.M116.727305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paloheimo M., Haglund D., Aho S., and Korhola M. (1992) Production of cyclomaltodextrin glucanotransferase of Bacillus circulans var. Alkalophilus ATCC21783 in B. subtilis. Appl. Microbiol. Biotechnol. 36, 584–591 [DOI] [PubMed] [Google Scholar]
- 16. Lima H. O., De Moraes F. F., and Zanin G. M. (1998) β-Cyclodextrin production by simultaneous fermentation and cyclization. Appl. Microbiol. Biotechnol. 70–72, 789–804 [DOI] [PubMed] [Google Scholar]
- 17. Uitdehaag J. C., van der Veen B. A., Dijkhuizen L., Elber R., and Dijkstra B. W. (2001) Enzymatic circularization of a malto-octaose linear chain studied by stochastic reaction path calculations on cyclodextrin glycosyltransferase. Proteins 43, 327–335 10.1002/prot.1044 [DOI] [PubMed] [Google Scholar]
- 18. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stam M. R., Danchin E. G., Rancurel C., Coutinho P. M., and Henrissat B. (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase–related proteins. Protein Eng. Des. Sel. 19, 555–562 10.1093/protein/gzl044 [DOI] [PubMed] [Google Scholar]
- 20. Tonozuka T., Mogi S., Shimura Y., Ibuka A., Sakai H., Matsuzawa H., Sakano Y., and Ohta T. (1995) Comparison of primary structures and substrate specificities of two pullulan-hydrolyzing α-amylases, TVA I and TVA II, from Thermoactinomyces vulgaris R-47. Biochim. Biophys. Acta 1252, 35–42 10.1016/0167-4838(95)00101-Y [DOI] [PubMed] [Google Scholar]
- 21. Kamasaka H., Sugimoto K., Takata H., Nishimura T., and Kuriki T. (2002) Bacillus stearothermophilus neopullulanase selective hydrolysis of amylose to maltose in the presence of amylopectin. Appl. Environ. Microbiol. 68, 1658–1664 10.1128/AEM.68.4.1658-1664.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee H. S., Kim M. S., Cho H. S., Kim J. I., Kim T. J., Choi J. H., Park C., Lee H. S., Oh B. H., and Park K. H. (2002) Cyclomaltodextrinase, neopullulanase, and maltogenic amylase are nearly indistinguishable from each other. J. Biol. Chem. 277, 21891–21897 10.1074/jbc.M201623200 [DOI] [PubMed] [Google Scholar]
- 23. Kondo S., Ohtaki A., Tonozuka T., Sakano Y., and Kamitori S. (2001) Studies on the hydrolyzing mechanism for cyclodextrins of Thermoactinomyces vulgaris R-47 α-amylase 2 (TVAII). X-ray structure of the mutant E354A complexed with β-cyclodextrin, and kinetic analyses on cyclodextrins. J. Biochem. 129, 423–428 10.1093/oxfordjournals.jbchem.a002873 [DOI] [PubMed] [Google Scholar]
- 24. Ohtaki A., Kondo S., Shimura Y., Tonozuka T., Sakano Y., and Kamitori S. (2001) Role of Phe286 in the recognition mechanism of cyclomaltooligosaccharides (cyclodextrins) by Thermoactinomyces vulgaris R-47 α-amylase 2 (TVAII). X-ray structures of the mutant TVAIIs, F286A and F286Y, and kinetic analyses of the Phe286-replaced mutant TVAIIs. Carbohydr. Res. 334, 309–313 10.1016/S0008-6215(01)00190-2 [DOI] [PubMed] [Google Scholar]
- 25. Kamitori S., Abe A., Ohtaki A., Kaji A., Tonozuka T., and Sakano Y. (2002) Crystal structures and structural comparison of Thermoactinomyces vulgaris R-47 α-amylase 1 (TVAI) at 1.6 A resolution and α-amylase 2 (TVAII) at 2.3 A resolution. J. Mol. Biol. 318, 443–453 10.1016/S0022-2836(02)00111-0 [DOI] [PubMed] [Google Scholar]
- 26. Ohtaki A., Mizuno M., Tonozuka T., Sakano Y., and Kamitori S. (2004) Complex structures of Thermoactinomyces vulgaris R-47 α-amylase 2 with acarbose and cyclodextrins demonstrate the multiple substrate recognition mechanism. J. Biol. Chem. 279, 31033–31040 10.1074/jbc.M404311200 [DOI] [PubMed] [Google Scholar]
- 27. Ohtaki A., Mizuno M., Yoshida H., Tonozuka T., Sakano Y., and Kamitori S. (2006) Structure of a complex of Thermoactinomyces vulgaris R-47 α-amylase 2 with maltohexaose demonstrates the important role of aromatic residues at the reducing end of the substrate binding cleft. Carbohydr. Res. 341, 1041–1046 10.1016/j.carres.2006.01.029 [DOI] [PubMed] [Google Scholar]
- 28. Takata H., Kuriki T., Okada S., Takesada Y., Iizuka M., Minamiura N., and Imanaka T. (1992) Action of neopullulanase. Neopullulanase catalyzes both hydrolysis and transglycosylation at α-(1→4)- and α-(1→6)-glucosidic linkages. J. Biol. Chem. 267, 18447–18452 [PubMed] [Google Scholar]
- 29. Mukai K., Watanabe H., Oku K., Nishimoto T., Kubota M., Chaen H., Fukuda S., and Kurimoto M. (2005) An enzymatically produced novel cyclic tetrasaccharide, cyclo-{→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→6)-α-d-Glcp-(1→4)-α-d-Glcp-(1→} (cyclic maltosyl-(1→6)-maltose), from starch. Carbohydr. Res. 340, 1469–1474 10.1016/j.carres.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 30. Mukai K., Watanabe H., Kubota M., Chaen H., Fukuda S., and Kurimoto M. (2006) Purification, characterization, and gene cloning of a novel maltosyltransferase from an Arthrobacter globiformis strain that produces an alternating α-1,4- and α-1,6-cyclic tetrasaccharide from starch. Appl. Environ. Microbiol. 72, 1065–1071 10.1128/AEM.72.2.1065-1071.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mori T., Nishimoto T., Okura T., Chaen H., and Fukuda S. (2008) Purification and characterization of cyclic maltosyl-(1→6)-maltose hydrolase and α-glucosidase from an Arthrobacter globiformis strain. Biosci. Biotechnol. Biochem. 72, 1673–1681 10.1271/bbb.70759 [DOI] [PubMed] [Google Scholar]
- 32. Mori T., Nishimoto T., Mukai K., Watanabe H., Okura T., Chaen H., and Fukuda S. (2009) Enzymes involved in the biosynthesis and degradation of cyclic maltosyl-maltose in Arthrobacter globiformis M6. J. Appl. Glycosci. 56, 127–136 10.5458/jag.56.127 [DOI] [Google Scholar]
- 33. Mori T., Nishimoto T., Okura T., Chaen H., and Fukuda S. (2011) Cloning, sequencing and expression of the genes encoding cyclic α-maltosyl-(1→6)-maltose hydrolase and α-glucosidase from an Arthrobacter globiformis strain. J. Appl. Glycosci. 58, 39–46 10.5458/jag.jag.JAG-2010_011 [DOI] [Google Scholar]
- 34. Light S. H., Cahoon L. A., Halavaty A. S., Freitag N. E., and Anderson W. F. (2016) Structure to function of an α-glucan metabolic pathway that promotes Listeria monocytogenes pathogenesis. Nat. Microbiol. 2, 16202 [DOI] [PubMed] [Google Scholar]
- 35. Light S. H., Cahoon L. A., Mahasenan K. V., Lee M., Boggess B., Halavaty A. S., Mobashery S., Freitag N. E., and Anderson W. F. (2017) Transferase versus hydrolase: the role of conformational flexibility in reaction specificity. Structure 25, 295–304 10.1016/j.str.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Janecek S., Svensson B., and Henrissat B. (1997) Domain evolution in the α-amylase family. J. Mol. Evol. 45, 322–331 10.1007/PL00006236 [DOI] [PubMed] [Google Scholar]
- 37. Park J. T., Song H. N., Jung T. Y., Lee M. H., Park S. G., Woo E. J., and Park K. H. (2013) A novel domain arrangement in a monomeric cyclodextrin-hydrolyzing enzyme from the hyperthermophile Pyrococcus furiosus. Biochim. Biophys. Acta 1834, 380–386 10.1016/j.bbapap.2012.08.001 [DOI] [PubMed] [Google Scholar]
- 38. Dumbrepatil A. B., Choi J. H., Park J. T., Kim M. J., Kim T. J., Woo E. J., and Park K. H. (2010) Structural features of the Nostoc punctiforme debranching enzyme reveal the basis of its mechanism and substrate specificity. Proteins 78, 348–356 10.1002/prot.22548 [DOI] [PubMed] [Google Scholar]
- 39. Guo J., Coker A. R., Wood S. P., Cooper J. B., Keegan R. M., Ahmad N., Muhammad M. A., Rashid N., and Akhtar M. (2018) Structure and function of the type III pullulan hydrolase from Thermococcus kodakarensis. Acta Crystallogr. D Struct. Biol. 74, 305–314 10.1107/S2059798318001754 [DOI] [PubMed] [Google Scholar]
- 40. Jung T. Y., Li D., Park J. T., Yoon S. M., Tran P. L., Oh B. H., Janeček Š., Park S. G., Woo E. J., and Park K. H. (2012) Association of novel domain in active site of archaic hyperthermophilic maltogenic amylase from Staphylothermus marinus. J. Biol. Chem. 287, 7979–7989 10.1074/jbc.M111.304774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kamitori S., Kondo S., Okuyama K., Yokota T., Shimura Y., Tonozuka T., and Sakano Y. (1999) Crystal structure of Thermoactinomyces vulgaris R-47 α-amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 A resolution. J. Mol. Biol. 287, 907–921 10.1006/jmbi.1999.2647 [DOI] [PubMed] [Google Scholar]
- 42. Hondoh H., Kuriki T., and Matsuura Y. (2003) Three-dimensional structure and substrate binding of Bacillus stearothermophilus neopullulanase. J. Mol. Biol. 326, 177–188 10.1016/S0022-2836(02)01402-X [DOI] [PubMed] [Google Scholar]
- 43. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 45. Choi J. H., Lee H., Kim Y. W., Park J. T., Woo E. J., Kim M. J., Lee B. H., and Park K. H. (2009) Characterization of a novel debranching enzyme from Nostoc punctiforme possessing a high specificity for long branched chains. Biochem. Biophys. Res. Commun. 378, 224–229 10.1016/j.bbrc.2008.11.020 [DOI] [PubMed] [Google Scholar]
- 46. Machius M., Declerck N., Huber R., and Wiegand G. (1998) Activation of Bacillus licheniformis α-amylase through a disorder → order transition of the substrate-binding site mediated by a calcium-sodium-calcium metal triad. Structure 6, 281–292 10.1016/S0969-2126(98)00032-X [DOI] [PubMed] [Google Scholar]
- 47. Ito K., Ito S., Ishino K., Shimizu-Ibuka A., and Sakai H. (2007) Val326 of Thermoactinomyces vulgaris R-47 amylase II modulates the preference for α-(1,4)- and α-(1,6)-glycosidic linkages. Biochim. Biophys. Acta 1774, 443–449 10.1016/j.bbapap.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 48. Dowd M. K., Reilly P. J., and French A. D. (1994) Relaxed-residue conformational mapping of the three linkage bonds of isomaltose and gentiobiose with MM3 (92). Biopolymers 34, 625–638 10.1002/bip.360340505 [DOI] [PubMed] [Google Scholar]
- 49. Frank M., Lütteke T., and von der Lieth C. W. (2007) GlycoMapsDB: a database of the accessible conformational space of glycosidic linkages. Nucleic Acids Res. 35, 287–290 10.1093/nar/gkl907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pajatsch M., Gerhart M., Peist R., Horlacher R., Boos W., and Böck A. (1998) The periplasmic cyclodextrin binding protein CymE from Klebsiella oxytoca and its role in maltodextrin and cyclodextrin transport. J. Bacteriol. 180, 2630–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hashimoto Y., Yamamoto T., Fujiwara S., Takagi M., and Imanaka T. (2001) Extracellular synthesis, specific recognition, and intracellular degradation of cyclomaltodextrins by the hyperthermophilic archaeon Thermococcus sp. strain B1001. J. Bacteriol. 183, 5050–5057 10.1128/JB.183.17.5050-5057.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamionka A., and Dahl M. K. (2001) Bacillus subtilis contains a cyclodextrin-binding protein which is part of a putative ABC-transporter. FEMS Microbiol. Lett. 204, 55–60 10.1111/j.1574-6968.2001.tb10862.x [DOI] [PubMed] [Google Scholar]
- 53. Yopi Tonozuka T., Sakai H., and Sakano Y. (2002) Cloning of a gene cluster for dextrin utilization from Thermoactinomyces vulgaris R-47 and characterization of the cyclodextrin-binding protein. J. Appl. Glycosci. 49, 107–114 10.5458/jag.49.107 [DOI] [Google Scholar]
- 54. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 55. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 56. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 58. Utsumi Y., Yoshida M., Francisco J. P. B., Sawada T., Kitamura S., and Nakamura Y. (2009) Quantitative assay method for starch branching enzyme with bicinchoninic acid by measuring the reducing terminals of glucans. J. Appl. Glycosci. 56, 215–222 10.5458/jag.56.215 [DOI] [Google Scholar]
- 59. Huitema C., and Horsman G. P. (2018) Analyzing enzyme kinetic data using the powerful statistical capabilities of R. bioRxiv 10.1101/316588 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.