

Abstract
Contractile force development of smooth muscle is controlled by balanced kinase and phosphatase activities toward the myosin regulatory light chain (RLC). Numerous biochemical and pharmacological studies have investigated the specificity and regulatory activity of smooth muscle myosin light-chain phosphatase (MLCP) bound to myosin filaments and comprised of the regulatory myosin phosphatase target subunit 1 (MYPT1) and catalytic protein phosphatase 1cβ (PP1cβ) subunits. Recent physiological and biochemical evidence obtained with smooth muscle tissues from a conditional MYPT1 knockout suggests that a soluble, MYPT1-unbound form of PP1cβ may additionally contribute to myosin RLC dephosphorylation and relaxation of smooth muscle. Using a combination of isoelectric focusing and isoform-specific immunoblotting, we found here that more than 90% of the total PP1c in mouse smooth muscles is the β isoform. Moreover, conditional knockout of PP1cα or PP1cγ in adult smooth muscles did not result in an apparent phenotype in mice up to 6 months of age and did not affect smooth muscle contractions ex vivo. In contrast, smooth muscle–specific conditional PP1cβ knockout decreased contractile force development in bladder, ileal, and aortic tissues and reduced mouse survival. Bladder smooth muscle tissue from WT mice was selectively permeabilized to remove soluble PP1cβ to measure contributions of total (α-toxin treatment) and myosin-bound (Triton X-100 treatment) phosphatase activities toward phosphorylated RLC in myofilaments. Triton X-100 reduced PP1cβ content by 60% and the rate of RLC dephosphorylation by 2-fold. These results are consistent with the selective dephosphorylation of RLC by both MYPT1-bound and -unbound PP1cβ forms in smooth muscle.
Keywords: protein phosphorylation, smooth muscle, myosin, muscle physiology, protein phosphatase, contraction, MYPT1, PP1c
Introduction
Force development of smooth muscle is initiated by phosphorylation of myosin regulatory light chain (RLC),3 where the extent of RLC phosphorylation is determined by the balanced activities of smooth muscle–specific Ca2+/calmodulin–dependent MLCK and MLCP (1–4). Smooth muscle MLCP is defined biochemically as a heterotrimeric protein comprised of a myosin phosphatase target (MYPT1) regulatory subunit, a PP1cβ (also referred to as PP1cδ in the smooth muscle field) catalytic subunit, and a small subunit of unknown function (M20) (2, 3, 5–7). MYPT1 from smooth and nonmuscle cells targets PP1cβ to myosin and potentiates the protein phosphatase activity toward phosphorylated RLC (1, 8). Binding of PP1cβ, but not the PP1cα or PP1cγ isoforms, to MYPT1 has been shown in co-expression studies (7) and detailed in a high-resolution structure (9). MYPT1 protein knockout showed a concomitant reduction in PP1cβ, consistent with the idea that the interaction of the two proteins stabilized the complex in vivo (10–12).
Genetic approaches have provided novel insights that complement and extend biochemical and biophysical studies of the roles of MLCK and MYPT1 in smooth muscle contraction. Although it has been suggested that multiple protein kinases phosphorylate smooth muscle RLC (13–15), conditional knockout of MLCK in smooth muscles of adult mice demonstrated its primary physiological role for smooth muscle RLC phosphorylation. Additionally, transgenic expression of biosensor MLCK showed tight coupling between increasing [Ca2+]i and Ca2+/calmodulin activation followed by feedback autoinhibition of the activation by MLCK phosphorylation (16–19). Thus, MLCK activity is fine-tuned by different cellular mechanisms. Genetic approaches have provided additional insights into the role of mutations in MLCK that cause familial aortic dissections in humans (20–22).
Smooth muscle–specific conditional knockout of MYPT1 and knockin mutations at its phosphorylation sites have also provided novel insights. Conditional knockout of MLCK in smooth muscle cells in adult mice is lethal because of contractile failure (21, 23–25). However, knockout of MYPT1 in smooth muscle cells did not result in smooth muscle failure in intact mice and was associated with modest changes in agonist- and KCl-induced contractile and relaxation responses of isolated ileal (12, 26), mesenteric arterial (11), bladder (10), and tracheal (27) tissues. Knockout of MYPT1 reduced the amount of PP1cβ by about half, which was associated with a 50% reduction in the rate of RLC dephosphorylation and relaxation. Thus, MYPT1 was shown not to be absolutely required for smooth muscle function, suggesting other contributory cell mechanisms for RLC dephosphorylation. Significant phosphatase activity toward phosphorylated RLC bound to soluble heavy meromyosin was found in the supernatant fraction of smooth muscle homogenates where MYPT1 was not present. This activity was inhibited by calyculin A but not a low concentration of okadaic acid, which inhibits PP2A, consistent with PP1cβ being involved (10).
Collectively, these recent reports suggest that protein phosphatase activity directed physiologically to phosphorylated RLC is not restricted to PP1cβ bound to MYPT1. Therefore, we determined the relative abundance of different PP1c isoforms in smooth muscle and their importance in maintaining smooth muscle function. We also investigated the contribution of soluble, non-MYPT1-bound PP1cβ in dephosphorylating myosin in myofilaments by selective permeabilization of smooth muscle fibers. The results support the conclusions that PP1cβ is the main isoform expressed in smooth muscle, that ablation of PP1cβ expression in smooth muscle causes a functional loss and death, and that phosphorylated RLC is dephosphorylated by both myosin-targeted and soluble PP1cβ activities.
Results
Conditional knockout of PP1cβ, but not PP1cα or PP1cγ, was lethal
We had previously developed an experimental strategy to specifically ablate MLCK, MYPT1, and myosin in smooth muscle cells of adult mice to avoid untoward developmental and nonsmooth muscle effects that complicate experimental results (10, 12, 20, 21, 27–29). Mice containing floxed alleles of genes expressed in smooth muscle cells were crossed with a transgenic mouse line expressing a fusion protein of the Cre recombinase with the modified estrogen receptor binding domain (CreERT2) under the control of the smooth muscle myosin heavy chain promoter (30). Cre-mediated recombination occurred robustly and exclusively in smooth muscle cells in tissues, but only after tamoxifen treatment (30).
Using this approach with mice containing floxed alleles for PP1cα, PP1cβ, and PP1cγ (31), we conditionally knocked out the α, β, or γ PP1c isoforms. With knockout of PP1cα or PP1cγ, mice appeared healthy with development of no apparent phenotype for up to 6 months (n > 20). Thus, all mice survived (Fig. 1A). In contrast, knockout of PP1cβ reduced survival, with death starting as early as 12 days and complete lethality by day 43 (n = 16). Apparent hypercontraction of intestines was observed in the PP1cβ knockouts during dissections for experiments (Fig. S1), but veterinary anatomic pathology evaluations, including necropsy and histology, did not reveal obvious abnormalities in organs from the knockout animals that might account for animal death. Thus, smooth muscle–specific knockout of only PP1cβ was lethal, but not knockout of PP1cα or PP1cγ.
Figure 1.

Conditional knockout of specific PP1c isoforms in smooth muscles of adult mice. A, comparison of mouse survival after tamoxifen-induced knockout of PP1cα (dashed line, n > 20), PP1cγ (dotted line, n > 20), or PP1cβ (solid line, n = 16) in smooth muscles. B, representative immunoblot of bladder smooth muscle samples (5 μg/lane) for PP1c isoforms from conditional knockout animals more than 20 days after start of tamoxifen injections. The primary antibodies used in immunoblotting are described under “Experimental procedures.” The band detected below PP1cα is nonspecific (n.s.) based on specific knockdown of a single band in immunoblots of same samples after separation by IEF.
Isoform-specific immunoblotting showed a significant reduction in the respective PP1c proteins as early as 5 days after a 2-week tamoxifen treatment protocol, with a greater reduction after 20 days (Fig. 1B). PP1cγ was reduced by 56% (n = 3), α by 60% (n = 3), and β by 60% (n = 6). There was no significant compensatory increase in nontargeted isoforms. Thus, the conditional knockout treatment reduced the respective contents of PP1cα, PP1cβ, and PP1cγ, but only loss of PP1cβ was lethal.
PP1cβ was found to be the most abundant PP1c expressed in smooth muscle
Because PP1cβ knockout alone affected smooth muscle contraction and mouse survival, the relative content of the different PP1c isoforms was assessed. We optimized conditions to separate the different PP1c isoforms extracted from bladder smooth muscle by preparative IEF (Fig. 2A). The relative positions of the distinct isoforms were determined by specific antibodies and were consistent with pI predictions from amino acid content, with PP1cβ the most acidic, followed by PP1cα and PP1cγ. A pan-antibody that was raised to sequences that are identical among the three PP1c isoforms showed cross-reactivity with PP1cβ and PP1cα, but the abundance of PP1cγ was too low to detect unless the gels were overloaded with protein. A nonspecific band did not co-migrate with any of the PP1c isoforms identified with specific antibodies. Quantification of the different PP1c isoforms immunoblotted with the pan-antibody showed that 97% of the total PP1c was PP1cβ, and 3% was PP1cα with PP1cγ undetectable (Fig. 2B). Thus, even if PP1cα had activity toward phosphorylated RLC, the low amount expressed in smooth muscle cells would limit its physiological contribution to RLC dephosphorylation and relaxation.
Figure 2.
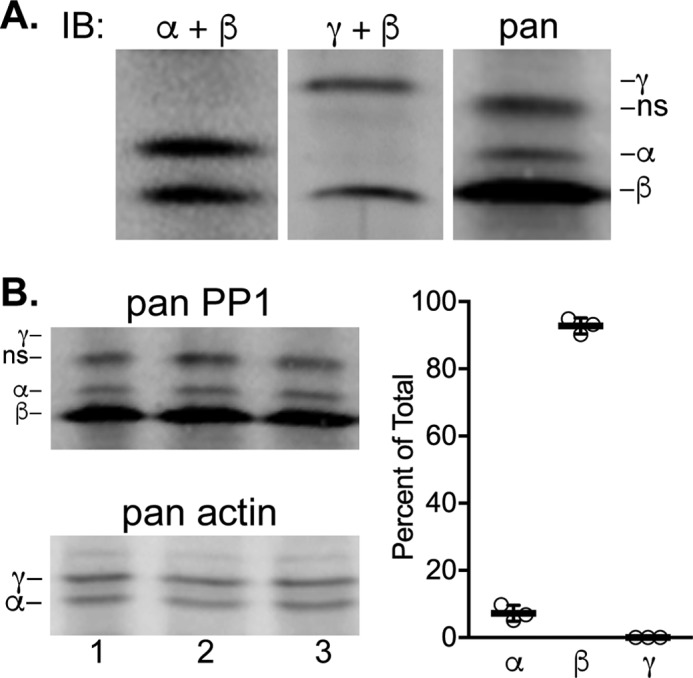
Quantification of PP1c isoforms expressed in bladder smooth muscle by immunoblotting after preparative IEF. A, identification of the positions of PP1c isoform-specific bands by immunoblot (IB) analysis. Mixtures of antibodies used to detect isoforms are indicated. ns, nonspecific. B, total PP1 protein in bladder samples (10 μg/lane) from three WT mice immunoblotted after separation by preparative IEF. Actin isoforms detected from the same membrane are shown as loading controls. Relative amounts of the isoforms are shown in the graph.
PP1cβ knockout resulted in reduction of MYPT1 content
H&E-stained histological sections of bladder tissue from PP1cβ knockout mice did not reveal significant changes in tissue structure (Fig. 3A). PP1cβ and smooth muscle actin expression, detected by immunofluorescence of bladder tissue sections, shows that PP1cβ expression is reduced uniformly in smooth muscles (Fig. 3A). Coomassie-stained SDS-PAGE showed a normal presence of smooth muscle proteins, including myosin heavy chain and actin, in bladder tissue from PP1cβ knockout mice (Fig. 3B). Thus, the reduced maximal force development responses were not due to significant pathological deterioration of the smooth muscle myofilament. Immunoblot of samples subjected to IEF confirmed the specific knockout of most of the PP1cβ protein (Fig. 3C). Comparison of immunoblots of MLCK and MYPT1 in tissue from PP1cβ knockout mice showed increased expression of MLCK by 33% ± 14%, a reduction of MYPT1 and PP1cβ to 20% ± 8%, and 40% ± 18% of the respective average WT contents (n = 5 or 6; mean ± S.D.) (Fig. 3D). This reduction in MYPT1 with knockout of PP1cβ mirrors the reduction in PP1cβ observed with knockout of MYPT1 (10). We permeabilized intact PP1cβ knockout bladder smooth muscle strips with α-toxin to introduce small pores for buffer transfer, or Triton X-100, which removes the membrane to allow washout of soluble proteins. MYPT1 was not removed with either treatment (Figs. S3 and S4). Comparable ratios of PP1cβ/MYPT1 in α-toxin– and Triton X-100–treated PP1cβ knockout bladder smooth muscle strips indicate that the remaining 40% of PP1cβ is stabilized in part by binding to the remaining MYPT1 (Fig. 3E). To determine whether smooth muscle RLC phosphorylation was increased in vivo, mice were anesthetized, and empty bladders were snap-frozen in situ with clamps prechilled in liquid nitrogen. Tissue extracts from PP1cβ knockout mice had higher amounts of phosphorylated RLC than the WT (Fig. 3, F and G).
Figure 3.
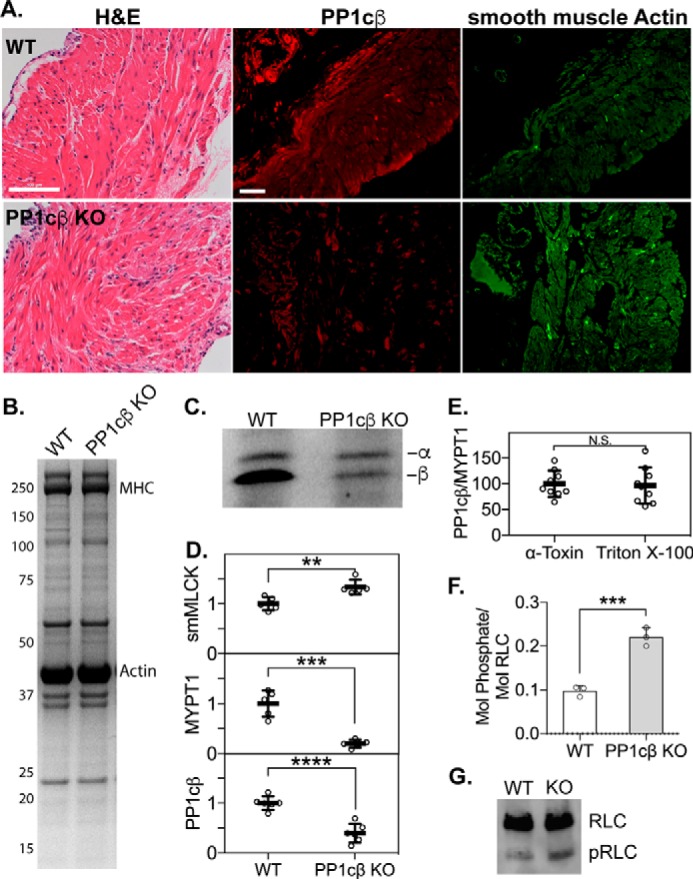
Effects of conditional PP1cβ knockout on smooth muscle proteins. A, H&E-stained sections of bladder tissue from WT and PP1cβ knockout (KO) mice (left) and immunofluorescence images of PP1cβ (center, red), and smooth muscle actin (right, green). Scale bars = 100 μm. B, Coomassie-stained gel following SDS-PAGE, showing the smooth muscle proteins myosin heavy chain (MHC) and actin in 5 μg of total protein. C, representative immunoblot of PP1cα and PP1cβ proteins in bladder homogenates after separation by preparative IEF. A rabbit monoclonal pan-PP1c antibody was used (Abcam). D, quantitation of smooth muscle MLCK, MYPT1, and PP1cβ relative to RLC. n = 5 or 6. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by t test using GraphPad Prism. E, ratio of PP1cβ/MYPT1 in 5 μg of protein extracts from PP1cβ knockout intact bladder smooth muscles after permeabilization with α-toxin or Triton X-100. N.S., not significant. F, comparison of the extent of RLC phosphorylation in WT and PP1cβ knockout empty bladders that were snap-frozen in situ. n = 3. ***, p < 0.001 by t test using GraphPad Prism. G, representative RLC immunoblot after separation of phosphorylated RLC (pRLC) from nonphosphorylated RLC in 2 μg of total bladder extract.
PP1cβ knockout reduced the maximal developed force of phasic and tonic smooth muscles
Phasic smooth muscles are characteristic of the gastrointestinal and urogenital systems and display rhythmic contractile activity. Tonic smooth muscles are found in large blood vessels and airways and exhibit sustained contractions (32). We compared the contractile responses of the mouse bladder, ileum, and aorta to KCl to induce smooth muscle cell depolarization or to agonist for receptor-mediated signaling. Vehicle-treated floxed littermates were used as WT controls. PP1cα or PP1cγ knockout did not significantly affect the maximal developed forces of the different smooth muscles over the WT (Fig. 4A). In contrast, PP1cβ knockout caused significant maximal force reductions in the bladder, ileum, and aorta (Fig. 4, A and B). The ileal smooth muscle contractile force was reduced to the greatest extent, which predicts that failure of the gastrointestinal system might be a primary contributor to death. Measurements of maximum developed forces at earlier times post-tamoxifen treatment show a time-dependent decrease in muscle contraction (Fig. 4C). The magnitudes of reductions in force are similar to the decreases in PP1cβ and MYPT1 proteins, which were, respectively, reduced to 44% ± 7% and 25% ± 6% at less than 20 days and 36% ± 4% and 15% ± 3% at more than 20 days after tamoxifen treatment (Fig. 4D). Resting RLC phosphorylations in ileal smooth muscles were elevated, indicating hyperphosphorylation of the smooth muscle RLC in response to decreased phosphatase activity (Fig. 4E). The reduction in maximal developed forces (Fig. 4, A and C), which are calculated from the difference in peak and resting forces, are coincident with increased resting RLC phosphorylations (Fig. 4, C and E).
Figure 4.

Effects of reduction in specific PP1c isoforms on smooth muscle contraction. A, maximal developed forces by smooth muscles of the bladder (B), ileum (I) and aorta (A) from conditional knockout animals in response to KCl (top panel) or agonist (bottom panel). Specific isoforms knocked out are indicated. Agonists included carbachol for the bladder and ileum and phenylephrine for the aorta. Tissues were harvested at least 20 days after the start of tamoxifen injections. Dots represent forces relative to average responses for tissues from matched WT mice (vehicle-treated floxed littermates). Error bars represent averaged values with S.D.; scattered dots represent n ≥ 6 from at least 3 animals. *, p < 0.05 by ANOVA (GraphPad Prism). B, representative force traces for bladder, ileum, and aorta tissues from WT (gray line) or PP1cβ knockout (black line) mice. C, maximal developed forces in response to KCl or carbachol (CCh) by ileal smooth muscles from PP1cβ knockout animals, measured at less than 20 days (gray columns, 11–18 days) and at more than 20 days (dark gray columns, 21–24 days) post-tamoxifen treatment to induce gene ablation. Error bars represent averaged values with S.D.; scattered dots represent n ≥ 11 from at least 4 animals. ***, p < 0.001; ****, p < 0.0001 by 2-way ANOVA, followed by Dunnett's multiple comparisons test (GraphPad Prism). N.S., not significant. D, comparison of PP1cβ and MYPT1 protein amounts, arbitrary units (AU), in WT and PP1cβ knockout animals at less than 20 days (light gray) and more than 20 days (dark gray) post-tamoxifen treatment. E, extent of RLC phosphorylation in ileal smooth muscle samples snap-frozen at rest (R) and 10 s post-carbachol treatment, measured by immunoblotting with a smooth muscle RLC antibody after separation of phosphorylated RLCs from nonphosphorylated RLCs by urea/glycerol-PAGE. Comparisons were made between WT and PP1cβ knockout ilea. Error bars represent averaged values with S.D.; scattered dots represent n ≥ 4 from at least 4 animals. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by 2-way ANOVA, followed by Dunnett's multiple comparisons test (GraphPad Prism).
RLC dephosphorylation was slower in Triton X-100–treated compared with α-toxin–treated smooth muscle strips
To directly measure the potential contribution of soluble PP1cβ toward smooth muscle function, we optimized conditions to permeabilize smooth muscle fibers to initiate RLC phosphorylation by endogenous MLCK in myofilaments and then measured the rate of dephosphorylation under controlled conditions. Treatment with α-toxin forms pores that are permeable to small molecules such as ions but not larger protein molecules (33, 34). However, Triton X-100 effectively removes the sarcolemma, allowing passage of large soluble protein molecules (35).
As expected, α-toxin–treated bladder smooth muscle strips contained filamin, myosin heavy chain, and actin when analyzed by SDS-PAGE followed by Coomassie blue staining (Fig. S4). Triton X-100 treatment did not result in the removal of these myofilament proteins, but other unidentified proteins, particularly lower-molecular-mass proteins, were removed compared with untreated strips (Fig. S3) and results obtained with α-toxin (Fig. S4).
We compared the amount of PP1cβ in intact WT and PP1cβ knockout bladder smooth muscle after treatment with α-toxin or Triton X-100 (Fig. 5A). Quantification showed that, compared with α-toxin–treated smooth muscles, PP1cβ/MYPT1 was reduced to 44% ± 16% (n > 20; mean ± S.D.) following Triton X-100 treatment (Fig. 5A). Thus, PP1cβ appears to consist of two pools, one bound to MYPT1 and the other soluble.
Figure 5.
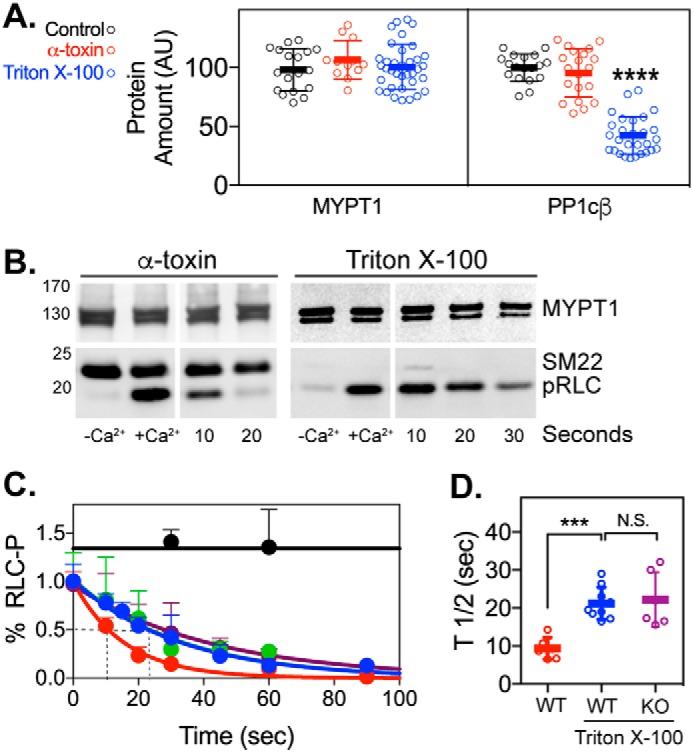
Effects of α-toxin and Triton X-100 treatment on smooth muscle protein content and RLC dephosphorylation in bladder tissue. A, quantitation of MYPT1 and PP1cβ protein content (arbitrary units (AU)) in untreated control-treated (black), α-toxin–treated (red), and Triton X-100–treated (blue) bladder strips from WT mice. The amounts of MYPT1 and PP1cβ were normalized to the Coomassie-stained myosin heavy chain protein amount and are relative to the average MYPT1 content in the control bladder strips. n > 8 for all groups. ****, p < 0.0001 by ANOVA (GraphPad Prism). B, representative immunoblots of MYPT1, SM22, and phosphorylated RLC (pRLC) in treated bladder strips from WT mice. Left panels, effects of presence of Ca2+ in phosphorylation buffer. Right panels, immunoblots at different times after starting dephosphorylation. C, after permeabilization with α-toxin (red) or Triton X-100 (blue), bladder strips were incubated in Ca2+ buffer to induce phosphorylation of myosin RLC by endogenous MLCK. The extent of phosphorylation at the indicated seconds after switching to Ca2+-free buffer is shown relative to the amount after 5 min in Ca2+ buffer. The effects of 1 μm calyculin A (black) and 1 nm okadaic acid (green) added to Ca2+ on RLC phosphorylation in Triton X-100–treated strips are shown as controls. Dephosphorylation of RLC in PP1cβ knockout bladder strips after Triton X-100 treatment (purple), is also shown. Each point in the dephosphorylation curve represents mean ± S.D. RLC phosphorylation was normalized to the MYPT1 immunoblot or the MHC Coomassie stain loading control. Data were fit to a nonlinear regression one-phase decay curve using GraphPad Prism. D, t½ values from individually fitted curves are shown as mean ± S.D. (n ≥ 6). ***, p < 0.001 by ANOVA using GraphPad Prism.
Permeable smooth muscle fibers retain contractile proteins in myofilaments, thus preserving a functional structure. Permeable tissue strips were incubated in Ca2+ buffer with calmodulin to induce phosphorylation of RLC in smooth muscle myosin by endogenous MLCK also bound in myofilaments. After 5-min incubation, myosin phosphorylation increased to 0.45 ± 0.01 mol of phosphate/mol RLC in both α-toxin– and Triton X–100 treated strips (Fig. 5, B and C). Dephosphorylation rates were obtained by measuring the normalized extent of RLC phosphorylation at various times after removal of Ca2+ and fitting the data to a nonlinear regression (Fig. 5, C and D). Averaged t½ values for RLC dephosphorylation in α-toxin– and Triton X-100–treated WT bladder strips were 9 ± 3 (n = 6) and 21 ± 4 s (n = 11), respectively (p < 0.001) (Fig. 5D). RLC dephosphorylation was not affected by 1 nm okadaic acid (t½ = 23 ± 2 s), confirming that the phosphatase activity was not from PP2A. Thus, the 60% reduction in PP1cβ content was associated with a 2-fold decrease in the rate of RLC dephosphorylation in Triton X-100–treated strips. The t½ value for RLC dephosphorylation in Triton X-100–treated PP1cβ knockout bladder strips was 22 ± 7 s (n = 6) (Fig. 5D) and not significantly different from Triton X-100–treated WT bladder strips. These results are consistent with a model where PP1cβ tightly bound to MYPT1 as well as PP1cβ in a soluble form both dephosphorylate RLC in smooth muscle myofilaments.
Discussion
Protein phosphatases constitute a diverse set of superfamilies of enzymes that have distinct 3D structures and differing active sites with different mechanisms of hydrolysis (36–38). Regulation and specificity are achieved through assembly of multisubunit holoenzymes, subunit phosphorylation, and the action of inhibitor proteins (36, 39, 40). Biochemical identification of PP1c as a primary RLC phosphatase focused on the protein phosphatase activity in myofilament fractions from skeletal and smooth muscles, where MYPT2 and MYPT1 were identified as targeting subunits, respectively, along with an accessory protein, M20, of unknown function (3, 5, 8, 41–45). Binding of MYPT2 and MYPT1 to myosin as well as PP1cβ identified a tethering or scaffolding role. Reconstitution of individually expressed MYPT1 and PP1c proteins showed many of the properties of the native complex, with activity enhanced toward RLC (46–49). Thus, smooth muscle MLCP was defined as the holoenzyme complex consisting of MYPT1, PP1cβ, and M20.
Based on the central importance of MLCP in the regulatory scheme for RLC phosphorylation/dephosphorylation in smoothmuscle, it was surprising to find that smooth muscle–specific conditional knockout of MYPT1 in adult mice had only modest effects on RLC dephosphorylation and relaxation (10–12, 26, 27). There was a coincident 60% decrease in the amount of PP1cβ, with no changes in PP1cα or PP1cγ. The decrease in the amount of PP1cβ could be related to the loss of the stable complex between MYPT1 and PP1cβ. However, it is important to note that smooth muscle RLC was rapidly dephosphorylated in MYPT1-deficient contracting isolated smooth muscle tissues when KCl or agonists were washed out. Also, the amounts of other members of the MYPT1 family were not significantly increased to compensate for the loss of MYPT1. Thus, a possible conclusion was that another protein phosphatase activity complements MLCP to dephosphorylate RLC. This idea was supported by biochemical assays of smooth muscle fractions (10).
Based on these observations, we focused on the role of PP1c in smooth muscle using our genetic approach of smooth muscle–specific conditional knockout in adult mice. Quantitation of PP1c isoforms in smooth muscle show very high levels of PP1cβ compared with PP1cα or PP1cγ. Knockout of PP1cβ, but not PP1cα or PP1cγ, was lethal in mice, probably because of smooth muscle failure, as shown by compromised contractile function of isolated tissues. Knockout of PP1cβ, but not PP1cα or PP1cγ, specifically in cardiac myocytes resulted in concentric remodeling, interstitial fibrosis, and contractile dysregulation with enhanced contractility of isolated myocytes and phosphorylation of RLC and myosin binding protein C (50). Cardiac RLC phosphorylation is not essential for contraction but does play an important role in enhancing contractile force to optimize cardiac performance (51–53)
The severe phenotype with smooth muscle–specific knockout of PP1cβ could be due to hyperphosphorylation of RLC and smooth muscle contracture, as expected for the loss of protein phosphatase activity directed to phosphorylated RLC. We measured increased RLC phosphorylation in untreated bladder tissues from PP1cβ knockout mice snap-frozen in situ. We have no direct evidence of RLC hyperphosphorylation, potentially because of compensation by the autonomic nervous system to maintain RLC phosphorylation in a normal range, as PP1cβ is diminished until a threshold is reached for RLC hyperphosphorylation, leading to smooth muscle cell failure. The significant reduction in PP1cβ would be expected to lead to RLC phosphorylation by other protein kinase activities revealed previously with chemical inhibitors of MLCP in addition to Ca2+/calmodulin-activated MLCK, with a hyperphosphorylated state associated with vasospasm and other pathological states (21, 54), similar to our observation regarding the hypercontraction of small intestines. This period of RLC hyperphosphorylation would probably be short before failure, so our attempts to identify it were not successful with tissues isolated on different days after starting tamoxifen injection, along with significant variation in animal responses.
Alternatively, the loss of PP1cβ may affect other essential smooth muscle cell functions. In addition to regulation of smooth muscle contraction via interaction with MYPT1, PP1 catalyzes a major fraction of all protein dephosphorylation events in eukaryotic cells and regulates a wide array of processes (36, 39, 40, 55–57). Consistent with its pleiotropic action, PP1 displays broad substrate specificity and has more than 200 validated interacting partners that both localize PP1 to distinct regions of the cell and modulate its substrate specificity. Although roles of PP1cβ in smooth muscle cell function besides RLC dephosphorylation have not been identified, it is expected that it may be important for other essential smooth muscle cell processes (5, 36, 39, 40, 55–57).
RLC may be dephosphorylated by both PP1cβ bound to MYPT1 as well as a soluble form. Only about half of the smooth muscle cell PP1cβ appears to be insoluble and bound MYPT1 with myosin in the myofilaments. Interestingly, there are no structural insights into how PP1cβ tightly bound to MYPT1 on myosin filaments can rapidly dephosphorylate RLC, considering that the molar amount of myosin is 20- to 30-fold greater than MYPT1. Although MLCK content is similar to MYPT1 and is also bound tightly to myofilaments, structural and biochemical studies show that it is sufficiently elongated with binding at its N terminus tail to extend its C-terminal catalytic core to multiple myosin heads in thick filaments to phosphorylate RLC (58–62). Sequence truncation studies and a crystal structure of a fragment of MYPT1 and PP1cβ have identified the interacting sequences to be at the N terminus of MYPT1, near its myosin-interacting site (9, 39, 56). Thus, it is not clear how a catalytic subunit that is bound tightly to this specific site in MYPT1 can dephosphorylate most of the RLC in myosin thick filaments.
Our results with selective permeabilization and quantification of the rates of RLC dephosphorylation support the idea that soluble PP1cβ contributes to protein phosphatase activity toward RLC in myofilaments. This proposal is consistent with the observations that RLC is rapidly dephosphorylated in smooth muscle tissues with knockout of MYPT1 (10–12, 26, 27, 63). These results are also consistent with previously reported biochemical measurements, where the specific activity of PP1cβ in the total smooth muscle homogenate was the same as that found in the supernatant fraction not containing MYPT1 (10). We refer here to a soluble form of PP1cβ, but it should be recognized that this form may be bound to a still-to-be identified regulatory subunit (36, 39, 40, 56).
In summary, PP1cβ was found to be the primary PP1c isoform in smooth muscle, and its smooth muscle–specific conditional knockout led to compromised smooth muscle contraction and death in mice. Evidence was presented that washout of a soluble form of PP1cβ in permeable fibers decreased the rate of RLC dephosphorylation. These observations, along with previous reports regarding the effects of smooth muscle–specific knockout of MYPT1 and identification of robust dephosphorylation of RLC bound to myosin heavy chain in smooth muscle supernatant fraction not containing MYPT1 are consistent with the idea that physiologically selective dephosphorylation of RLC occurs by both MYPT1-bound and -unbound PP1cβ in smooth muscle cells.
Experimental procedures
Ethics approval
Experiments were performed in accordance with the National Institutes of Health and Institutional Animal Care and Use Guidelines. The Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center approved all procedures and protocols. Animals were sacrificed by intraperitoneal administration of a lethal dose of tribromoethanol (250 mg/kg) for tissue collection.
Generation of genetically modified mice
Mice containing floxed alleles for PP1cα, PP1cβ, and PP1cγ (50) were crossed with a transgenic mouse line expressing a fusion protein of Cre recombinase with the modified estrogen receptor binding domain (CreERT2) under the control of the smooth muscle myosin heavy chain promoter (30). Mice were bred and screened as described previously (10, 26, 30). Male mice (8–12 weeks old) were injected intraperitoneally with tamoxifen or vehicle for 5 consecutive days each week for 2 weeks at a dose of 1 mg/day (10). Aorta, ileum, and bladder smooth muscle tissues were harvested between 16 and 40 days after start of tamoxifen or vehicle treatment from mice homozygous for the floxed allele and Cre recombinase. Mice showing visible signs of distress leading to morbidity were sacrificed.
Measurements of mouse smooth muscle contraction
Aortic, ileal, and bladder tissues were dissected in physiological salt solution (118.5 mm NaCl, 4.75 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 24.9 mm NaHCO3, 1.6 mm CaCl2, and 10.0 mm D-glucose, aerated with 95% O2 and 5% CO2 to maintain the pH at 7.4) and mounted onto force transducers in water-jacketed organ baths as described previously (10, 12, 27). A resting force of 0.9 to 1.0 g was applied, and tissues were equilibrated for at least 45 min, followed by precontraction with alternating treatments of 75 mm KCl three times every 10 min to establish viability. At the end of the equilibration period, tissues were stimulated with 10 μm carbachol or 10 μm phenylephrine. Force development was recorded isometrically by a Grass FT03 force transducer connected to a Powerlab 8/SP data acquisition unit (AD Instruments, Colorado Springs, CO). Force measurements were normalized as grams of developed force per tissue wet weight for all tissues.
At the indicated times after specific treatments, tissues were quick-frozen by clamps prechilled in liquid nitrogen and processed as described previously (21). Briefly, frozen tissues were stored at −80 °C until they were added to a frozen slurry of acetone with 10% TCA and 10 mm DTT and slowly thawed at room temperature. Tissues were rinsed with ethyl ether (three times for 10 min each), exposed to air for a few minutes to evaporate the ether, and suspended in urea sample buffer containing 8 m urea, 20 mm Tris (pH 8.6), 23 mm glycine, 10 mm DTT, 4 mm EDTA, and 5% sucrose. Proteins were then solubilized in a Bullet blender with addition of urea crystals to saturation (Next Advance, Inc., Averill Park, NY) (with 2-mm zirconium oxide beads, four spins for 3 min each at setting 9). Protein content was determined by Bradford assay (Bio-Rad) with BSA as the standard. Bromphenol blue was added to 0.004%, and samples were stored at −0 °C.
Western blot analysis
Tissue extracts solubilized in urea sample buffer were added to 0.25 volumes of SDS sample buffer containing 250 mm Tris (pH 6.8), 10% SDS, 50 mm DTT, 40% glycerol, and 0.01% bromphenol blue and boiled for SDS-PAGE (4–12% polyacrylamide gradient). Proteins were transferred onto a nitrocellulose membrane and visualized by immunoblotting with antibodies to pan-MYPTs (LSBio), PP1cα (mouse, Invitrogen), PP1cβ (rabbit, Millipore), PP1cγ (goat, Santa Cruz Biotechnology), pan-actin (mouse, Sigma), MLCK (mouse, K36, Sigma), and glyceraldehyde-3-phosphate dehydrogenase (rabbit, Santa CruzBiotechnology). Total protein stain by Coomassie gel was used to adjust protein loading, and quantification of western blots was determined by quantitative densitometry of the protein of interest to smooth muscle RLC (for bladder tissue extracts) or a Ponceau S–stained membrane total lane (for ileal tissue extracts) using the ImageQuantTL software package (GE Healthcare). The amount of RLC expressed in all experiments was confirmed to not be affected by the genetic (Fig. S2) or biochemical manipulations (Fig. S3).
Separation of PP1 isoforms by preparative IEF
The composition of the preparative IEF mini-gels was as follows: 9.1 m urea, 5% acrylamide (19:1), 10% glycerol, 2% ampholyte (pH 5–7, SERVA), and 2% CHAPS. The gel mixture was degassed for 10 min at room temperature and polymerized for 1 h after addition of 0.05% ammonium persulfate and 0.1% N,N,N',N'-tetramethylethylenediamine. The gel running buffers were 50 mm l-histidine (inner buffer) and 20 mm phosphoric acid (outer buffer). Tissues were weighed and homogenized in a 50× volume of Bio-Rad rehydration solution using a glass homogenizer. Additional urea crystals were added to saturation during continuous shaking at room temperature for at least 30 min. The tissue samples were then clarified by centrifugation at 20,000 × g for 2 min. Following gel polymerization and assembly of the apparatus, wells were prefilled with 25% Bio-Rad rehydration solution, and samples were underlayed to prevent contact with the inner buffer. Samples were focused at 400 V overnight (∼16 h). Following IEF, the gels were removed, washed in 100 ml of gel fixation buffer (50% methanol and 2% SDS) for 1 h (20 min with 3 buffer changes), and transferred to a nitrocellulose membrane in standard wet transfer buffer with 0.1% SDS for 3 h at 10 V (Mini Blot transfer apparatus, Thermo, plate electrodes). PP1c isoforms were detected with a rabbit mAb to the N-terminal region of PP1c, where sequences between the three isoforms are nearly identical (Abcam).
Measurement of RLC phosphorylation
RLC phosphorylation was measured by urea/glycerol-PAGE as described previously (21, 64). Muscle proteins in 8 m urea sample buffer were subjected to urea/glycerol-PAGE to separate nonphosphorylated from monophosphorylated RLC. Following electrophoresis, proteins were transferred to nitrocellulose membranes and probed with a mouse mAb against smooth muscle RLC from Kathleen Trybus (University of Vermont). The ratio of monophosphorylated RLC to total RLC (nonphosphorylated plus monophosphorylated) was determined by quantitative densitometry of developed immunoblots and expressed as mol of phosphate per mol of protein.
Intact bladder strip dephosphorylation
The smooth muscle layer of freshly isolated mouse bladders was cut into 200-μm strips in Ca2+-free physiological salt solution (118.5 mm NaCl, 4.75 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 24.9 mm NaHCO3, and 10.0 mm D-glucose, aerated with 95% O2 and 5% CO2 to maintain the pH at 7.4) and pinned down with 0.10 mm Minutiens (Austerlitz) on top of a silicone matrix (Sylgard 184) in a 12-well culture dish. Dissected strip lengths were measured, stretched by 50%, and permeabilized with 20 μg/ml α-toxin or 0.1% Triton X-100 in relaxing buffer as described previously (65) with modifications. The relaxing buffer (60.1 mm potassium methanesulfonate, 10 mm magnesium methanesulfonate, 4 mm Na2ATP, 3 mm EGTA, 30 mm PIPES, 1 mm DTT, 10 mm NaN3, 20 mm phosphocreatine, 0.1% fatty acid–free BSA, and protease inhibitor mixture (Halt, Thermo)) was freshly prepared on the day of assay, and the pH was adjusted to 7.1 with KOH. Permeabilizations were performed at 30 °C. After Triton X-100 treatment, bladder strips were washed in relaxing buffer three times for 15 min each at room temperature to remove Triton X-100 and equilibrated for 1 h in relaxing buffer with 1 μm calmodulin to allow diffusion of soluble proteins and replacement of calmodulin (66), which is necessary for activation of myofilament-bound smooth muscle MLCK. After α-toxin treatment, bladder strips were used directly for dephosphorylation studies. Phosphorylation of RLC in permeable bladder strips was initiated with addition of Ca2+ buffer (3.3 mm Ca(Ms)2, 1 μm calmodulin, and 1 mg/ml creatine kinase in relaxing buffer (pH 7.1)). To measure dephosphorylation, bladder strips were incubated in Ca2+ buffer for 5 min and then switched to Ca2+-free dephosphorylation buffer (70.1 mm KMs, 10 mm EGTA, 30 mm PIPES, 10 mm EDTA, 1 mm DTT, and protease inhibitor mixture (Halt, Thermo (pH 7.1)) for 10–120 s. At the indicated times, the dephosphorylation reaction was stopped with 10% TCA/10 mm DTT to acid-precipitate all proteins. Precipitated bladder strips were dabbed dry and then heated directly in 50 μl of 2× Laemmli buffer for 1 min at 95 °C. RLC phosphorylation was measured by immunoblotting with phospho-RLC antibody (Abcam). The extent of RLC phosphorylation (moles of phosphate per mole of RLC) was calculated from an immunoblot of total RLC in phosphorylated strips after separation by Phos-tag SDS-PAGE (10).
Histology and immunohistochemistry
For histological analyses, isolated smooth muscles from tamoxifen- and vehicle-treated mice were fixed in 4% (w/v) paraformaldehyde overnight, embedded in paraffin, and transversely sectioned at a thickness of 6 μm. After dewaxing and hydration, the sections were stained with standard hematoxylin and eosin (H&E). For immunohistochemistry, fresh tissues were embedded with OCT, cryosectioned, and blocked with PBS containing 0.1% Triton X-100, 0.1% Tween 20, 1% BSA, and 5% nonimmune goat serum for 1 h at room temperature and then incubated with primary antibody (anti-rabbit PP1cβ (Millipore) or anti-mouse smooth muscle α-actin (Sigma)) overnight and incubated with secondary antibodies conjugated with either Alexa Fluor 488 (Life Technologies, Gaithersburg, MD) for smooth muscle α-actin or 569 (Life Technologies) for PP1cβ. H&E-stained sections were imaged using a Zeiss Axioplan2 microscope with a ×10 objective (numerical aperture, 0.3). Fluorescence images were acquired on a Zeiss LSM880 confocal microscope with a ×20 objective (numerical aperture, 0.8).
Statistical analysis
All data are presented as mean ± S.D. Statistical comparisons were performed by Student's t test for comparisons between control and treatment groups. For multiple comparisons, one-way or two-way ANOVA followed by Dunnett's post hoc test was used. Data analyses were performed with statistical software (GraphPad Prism). p < 0.05 was considered statistically significant.
Author contributions
A. N. C., K. E. K., and J. T. S. conceptualization; A. N. C., N. G., Z. L., and J. H. data curation; A. N. C., N. G., Z. L., and J. H. formal analysis; A. N. C. funding acquisition; A. N. C. validation; A. N. C., N. G., K. E. K., and J. T. S. investigation; A. N. C. and A. C. N. methodology; A. N. C., K. E. K., and J. T. S. writing-original draft; A. N. C., K. E. K., and J. T. S. project administration; A. N. C., A. C. N., K. E. K., and J. T. S. writing-review and editing; A. C. N. and J. T. S. resources.
Supplementary Material
Acknowledgments
We thank Kathleen Trybus (University of Vermont) for the mAb to smooth muscle RLC.
This work was supported by an American Heart Association Scientist Development Grant (to A. N. C.), National Institutes of Health Grants DA10044 (to A. C. N) and R01 HL112778 and R01 HL080536 (to J. T. S.), the Moss Heart Fund (to J. T. S.), and the Fouad A. and Val Imm Bashour Distinguished Chair in Physiology (to J. T. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- RLC
- regulatory light chain
- MLCK
- myosin light chain kinase
- MLCP
- myosin light chain phosphatase
- IEF
- isoelectric focusing
- H&E
- hematoxylin and eosin
- ANOVA
- analysis of variance.
References
- 1. Hartshorne D. J. (1998) Myosin phosphatase: subunits and interactions. Acta Physiol. Scand. 164, 483–493 10.1046/j.1365-201X.1998.00447.x [DOI] [PubMed] [Google Scholar]
- 2. Somlyo A. P., and Somlyo A. V. (2003) Ca2+-sensitivity of smooth and non-muscle myosin II: modulation by G proteins, kinases and myosin phosphatase. Physiol. Rev. 83, 1325–1358 10.1152/physrev.00023.2003 [DOI] [PubMed] [Google Scholar]
- 3. Grassie M. E., Moffat L. D., Walsh M. P., and MacDonald J. A. (2011) The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1δ. Arch. Biochem. Biophys. 510, 147–159 10.1016/j.abb.2011.01.018 [DOI] [PubMed] [Google Scholar]
- 4. Kamm K. E., and Stull J. T. (2001) Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530 10.1074/jbc.R000028200 [DOI] [PubMed] [Google Scholar]
- 5. Matsumura F., and Hartshorne D. J. (2008) Myosin phosphatase target subunit: many roles in cell function. Biochem. Biophys. Res. Commun. 369, 149–156 10.1016/j.bbrc.2007.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dippold R. P., and Fisher S. A. (2014) A bioinformatic and computational study of myosin phosphatase subunit diversity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R256–R270 10.1152/ajpregu.00145.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scotto-Lavino E., Garcia-Diaz M., Du G., and Frohman M. A. (2010) Basis for the isoform-specific interaction of myosin phosphatase subunits protein phosphatase 1c β and myosin phosphatase targeting subunit 1. J. Biol. Chem. 285, 6419–6424 10.1074/jbc.M109.074773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimizu H., Ito M., Miyahara M., Ichikawa K., Okubo S., Konishi T., Naka M., Tanaka T., Hirano K., and Hartshorne D. J. (1994) Characterization of the myosin-binding subunit of smooth muscle myosin phosphatase. J. Biol. Chem. 269, 30407–30411 [PubMed] [Google Scholar]
- 9. Terrak M., Kerff F., Langsetmo K., Tao T., and Dominguez R. (2004) Structural basis of protein phosphatase 1 regulation. Nature 429, 780–784 10.1038/nature02582 [DOI] [PubMed] [Google Scholar]
- 10. Tsai M. H., Chang A. N., Huang J., He W., Sweeney H. L., Zhu M., Kamm K. E., and Stull J. T. (2014) Constitutive phosphorylation of myosin phosphatase targeting subunit-1 in smooth muscle. J. Physiol. 592, 3031–3051 10.1113/jphysiol.2014.273011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qiao Y. N., He W. Q., Chen C. P., Zhang C. H., Zhao W., Wang P., Zhang L., Wu Y. Z., Yang X., Peng Y. J., Gao J. M., Kamm K. E., Stull J. T., and Zhu M. S. (2014) Myosin phosphatase target subunit 1 (MYPT1) regulates the contraction and relaxation of vascular smooth muscle and maintains blood pressure. J. Biol. Chem. 289, 22512–22523 10.1074/jbc.M113.525444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao N., Chang A. N., He W., Chen C. P., Qiao Y. N., Zhu M., Kamm K. E., and Stull J. T. (2016) Physiological signalling to myosin phosphatase targeting subunit-1 phosphorylation in ileal smooth muscle. J. Physiol. 594, 3209–3225 10.1113/JP271703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puetz S., Lubomirov L. T., and Pfitzer G. (2009) Regulation of smooth muscle contraction by small GTPases. Physiology 24, 342–356 10.1152/physiol.00023.2009 [DOI] [PubMed] [Google Scholar]
- 14. Wilson D. P., Sutherland C., Borman M. A., Deng J. T., Macdonald J. A., and Walsh M. P. (2005) Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem. J. 392, 641–648 10.1042/BJ20051173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moffat L. D., Brown S. B., Grassie M. E., Ulke-Lemée A., Williamson L. M., Walsh M. P., and MacDonald J. A. (2011) Chemical genetics of zipper-interacting protein kinase reveal myosin light chain as a bona fide substrate in permeabilized arterial smooth muscle. J. Biol. Chem. 286, 36978–36991 10.1074/jbc.M111.257949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Isotani E., Zhi G., Lau K. S., Huang J., Mizuno Y., Persechini A., Geguchadze R., Kamm K. E., and Stull J. T. (2004) Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc. Natl. Acad. Sci. U.S.A. 101, 6279–6284 10.1073/pnas.0308742101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizuno Y., Isotani E., Huang J., Ding H., Stull J. T., and Kamm K. E. (2008) Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am. J. Physiol. Cell Physiol. 295, C358–C364 10.1152/ajpcell.90645.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding H. L., Ryder J. W., Stull J. T., and Kamm K. E. (2009) Signaling processes for initiating smooth muscle contraction upon neural stimulation. J. Biol. Chem. 284, 15541–15548 10.1074/jbc.M900888200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai M. H., Kamm K. E., and Stull J. T. (2012) Signalling to contractile proteins by muscarinic and purinergic pathways in neurally stimulated bladder smooth muscle. J. Physiol. 590, 5107–5121 10.1113/jphysiol.2012.235424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L., Guo D. C., Cao J., Gong L., Kamm K. E., Regalado E., Li L., Shete S., He W. Q., Zhu M. S., Offermanns S., Gilchrist D., Elefteriades J., Stull J. T., and Milewicz D. M. (2010) Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 87, 701–707 10.1016/j.ajhg.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao N., Huang J., He W., Zhu M., Kamm K. E., and Stull J. T. (2013) Signaling through myosin light chain kinase in smooth muscles. J. Biol. Chem. 288, 7596–7605 10.1074/jbc.M112.427112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milewicz D. M., Trybus K. M., Guo D. C., Sweeney H. L., Regalado E., Kamm K., and Stull J. T. (2017) Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler. Thromb. Vasc. Biol. 37, 26–34 10.1161/ATVBAHA.116.303229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He W. Q., Peng Y. J., Zhang W. C., Lv N., Tang J., Chen C., Zhang C. H., Gao S., Chen H. Q., Zhi G., Feil R., Kamm K. E., Stull J. T., Gao X., and Zhu M. S. (2008) Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135, 610–620 10.1053/j.gastro.2008.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang W. C., Peng Y. J., Zhang G. S., He W. Q., Qiao Y. N., Dong Y. Y., Gao Y. Q., Chen C., Zhang C. H., Li W., Shen H. H., Ning W., Kamm K. E., Stull J. T., Gao X., and Zhu M. S. (2010) Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J. Biol. Chem. 285, 5522–5531 10.1074/jbc.M109.062836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He W. Q., Qiao Y. N., Zhang C. H., Peng Y. J., Chen C., Wang P., Gao Y. Q., Chen C., Chen X., Tao T., Su X. H., Li C. J., Kamm K. E., Stull J. T., and Zhu M. S. (2011) Role of myosin light chain kinase in regulation of basal blood pressure and maintenance of salt-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 301, H584–H591 10.1152/ajpheart.01212.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He W. Q., Qiao Y. N., Peng Y. J., Zha J. M., Zhang C. H., Chen C., Chen C. P., Wang P., Yang X., Li C. J., Kamm K. E., Stull J. T., and Zhu M. S. (2013) Altered contractile phenotypes of intestinal smooth muscle in mice deficient in myosin phosphatase target subunit 1. Gastroenterology 144, 1456–1465, 1465.e1–5 10.1053/j.gastro.2013.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao N., Tsai M. H., Chang A. N., He W., Chen C. P., Zhu M., Kamm K. E., and Stull J. T. (2017) Physiological vs. pharmacological signalling to myosin phosphorylation in airway smooth muscle. J. Physiol. 595, 6231–6247 10.1113/JP274715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuang S. Q., Kwartler C. S., Byanova K. L., Pham J., Gong L., Prakash S. K., Huang J., Kamm K. E., Stull J. T., Sweeney H. L., and Milewicz D. M. (2012) Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ. Res. 110, 1411–1422 10.1161/CIRCRESAHA.111.261743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J., Gao N., Wang S., Milewicz D. M., Kamm K. E., and Stull J. T. (2018) Genetic approaches to identify pathological limitations in aortic smooth muscle contraction. PLoS ONE 13, e0193769 10.1371/journal.pone.0193769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E., Lemmer B., Schütz G., Gutkind J. S., and Offermanns S. (2008) G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14, 64–68 10.1038/nm1666 [DOI] [PubMed] [Google Scholar]
- 31. Braz J. C., Gregory K., Pathak A., Zhao W., Sahin B., Klevitsky R., Kimball T. F., Lorenz J. N., Nairn A. C., Liggett S. B., Bodi I., Wang S., Schwartz A., Lakatta E. G., DePaoli-Roach A. A., et al. (2004) PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10, 248–254 10.1038/nm1000 [DOI] [PubMed] [Google Scholar]
- 32. Fisher S. A. (2010) Vascular smooth muscle phenotypic diversity and function. Physiol. Genomics 42A, 169–187 10.1152/physiolgenomics.00111.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Füssle R., Bhakdi S., Sziegoleit A., Tranum-Jensen J., Kranz T., and Wellensiek H. J. (1981) On the mechanism of membrane damage by Staphylococcus aureus α-toxin. J. Cell Biol. 91, 83–94 10.1083/jcb.91.1.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cassidy P., Hoar P. E., and Kerrick W. G. (1979) Irreversible thiophosphorylation and activation of tension in functionally skinned rabbit ileum strips by [35S]ATP γS. J. Biol. Chem. 254, 11148–11153 [PubMed] [Google Scholar]
- 35. Iizuka K., Ikebe M., Somlyo A. V., and Somlyo A. P. (1994) Introduction of high molecular weight (IgG) proteins into receptor coupled, permeabilized smooth muscle. Cell Calcium 16, 431–445 10.1016/0143-4160(94)90073-6 [DOI] [PubMed] [Google Scholar]
- 36. Brautigan D. L. (2013) Protein Ser/Thr phosphatases: the ugly ducklings of cell signalling. FEBS J. 280, 324–345 10.1111/j.1742-4658.2012.08609.x [DOI] [PubMed] [Google Scholar]
- 37. Cohen P. (1989) The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 58, 453–508 10.1146/annurev.bi.58.070189.002321 [DOI] [PubMed] [Google Scholar]
- 38. Chen M. J., Dixon J. E., and Manning G. (2017) Genomics and evolution of protein phosphatases. Sci. Signal. 10 [DOI] [PubMed] [Google Scholar]
- 39. Heroes E., Lesage B., Görnemann J., Beullens M., Van Meervelt L., and Bollen M. (2013) The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J. 280, 584–595 10.1111/j.1742-4658.2012.08547.x [DOI] [PubMed] [Google Scholar]
- 40. Verbinnen I., Ferreira M., and Bollen M. (2017) Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans. 45, 89–99 10.1042/BST20160154 [DOI] [PubMed] [Google Scholar]
- 41. Chen Y. H., Chen M. X., Alessi D. R., Campbell D. G., Shanahan C., Cohen P., and Cohen P. T. (1994) Molecular cloning of cDNA encoding the 110 kDa and 21 kDa regulatory subunits of smooth muscle protein phosphatase 1M. FEBS Lett. 356, 51–55 10.1016/0014-5793(94)01231-8 [DOI] [PubMed] [Google Scholar]
- 42. Alessi D., MacDougall L. K., Sola M. M., Ikebe M., and Cohen P. (1992) The control of protein phosphatase-1 by targeting subunits: the major myosin phosphatase in avian smooth-muscle is a novel form of protein phosphatase-1. Eur. J. Biochem. 210, 1023–1035 10.1111/j.1432-1033.1992.tb17508.x [DOI] [PubMed] [Google Scholar]
- 43. Shirazi A., Iizuka K., Fadden P., Mosse C., Somlyo A. P., Somlyo A. V., and Haystead T. A. (1994) Purification and characterization of the mammalian myosin light chain phosphatase holoenzyme: the differential effects of the holoenzyme and its subunits on smooth muscle. J. Biol. Chem. 269, 31598–31606 [PubMed] [Google Scholar]
- 44. Pato M. D., and Adelstein R. S. (1983) Purification and characterization of a multisubunit phosphatase from turkey gizzard smooth muscle. The effect of calmodulin binding to myosin light chain kinase on dephosphorylation. J. Biol. Chem. 258, 7047–7054 [PubMed] [Google Scholar]
- 45. Pato M. D., Adelstein R. S., Crouch D., Safer B., Ingebritsen T. S., and Cohen P. (1983) The protein phosphatases involved in cellular regulation: 4: classification of two homogeneous myosin light chain phosphatases from smooth muscle as protein phosphatase-2A1 and 2C, and a homogeneous protein phosphatase from reticulocytes active on protein synthesis initiation factor eIF-2 as protein phosphatase-2A2. Eur. J. Biochem. 132, 283–287 10.1111/j.1432-1033.1983.tb07360.x [DOI] [PubMed] [Google Scholar]
- 46. Feng J., Ito M., Ichikawa K., Isaka N., Nishikawa M., Hartshorne D. J., and Nakano T. (1999) Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 274, 37385–37390 10.1074/jbc.274.52.37385 [DOI] [PubMed] [Google Scholar]
- 47. Zhang A. J., Bai G., Deans-Zirattu S., Browner M. F., and Lee E. Y. (1992) Expression of the catalytic subunit of phosphorylase phosphatase (protein phosphatase-1) in Escherichia coli. J. Biol. Chem. 267, 1484–1490 [PubMed] [Google Scholar]
- 48. Watanabe T., da Cruz e Silva E. F., Huang H. B., Starkova N., Kwon Y. G., Horiuchi A., Greengard P., and Nairn A. C. (2003) Preparation and characterization of recombinant protein phosphatase 1. Methods Enzymol. 366, 321–338 [DOI] [PubMed] [Google Scholar]
- 49. Khasnis M., Nakatomi A., Gumpper K., and Eto M. (2014) Reconstituted human myosin light chain phosphatase reveals distinct roles of two inhibitory phosphorylation sites of the regulatory subunit, MYPT1. Biochemistry 53, 2701–2709 10.1021/bi5001728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu R., Correll R. N., Davis J., Vagnozzi R. J., York A. J., Sargent M. A., Nairn A. C., and Molkentin J. D. (2015) Cardiac-specific deletion of protein phosphatase 1β promotes increased myofilament protein phosphorylation and contractile alterations. J. Mol. Cell. Cardiol. 87, 204–213 10.1016/j.yjmcc.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang A. N., Kamm K. E., and Stull J. T. (2016) Role of myosin light chain phosphatase in cardiac physiology and pathophysiology. J. Mol. Cell. Cardiol. 101, 35–43 10.1016/j.yjmcc.2016.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chang A. N., Battiprolu P. K., Cowley P. M., Chen G., Gerard R. D., Pinto J. R., Hill J. A., Baker A. J., Kamm K. E., and Stull J. T. (2015) Constitutive phosphorylation of cardiac Myosin regulatory light chain in vivo. J. Biol. Chem. 290, 10703–10716 10.1074/jbc.M115.642165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chang A. N., Mahajan P., Knapp S., Barton H., Sweeney H. L., Kamm K. E., and Stull J. T. (2016) Cardiac myosin light chain is phosphorylated by Ca2+/calmodulin-dependent and -independent kinase activities. Proc. Natl. Acad. Sci. U.S.A. 113, E3824–E3833 10.1073/pnas.1600633113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walsh M. P. (2011) Vascular smooth muscle myosin light chain diphosphorylation: mechanism, function, and pathological implications. IUBMB Life 63, 987–1000 10.1002/iub.527 [DOI] [PubMed] [Google Scholar]
- 55. Cohen P. T. (2002) Protein phosphatase 1: targeted in many directions. J. Cell Sci. 115, 241–256 [DOI] [PubMed] [Google Scholar]
- 56. Hendrickx A., Beullens M., Ceulemans H., Den Abt T., Van Eynde A., Nicolaescu E., Lesage B., and Bollen M. (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol. 16, 365–371 10.1016/j.chembiol.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 57. Peti W., Nairn A. C., and Page R. (2013) Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 280, 596–611 10.1111/j.1742-4658.2012.08509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin P., Luby-Phelps K., and Stull J. T. (1999) Properties of filament-bound myosin light chain kinase. J. Biol. Chem. 274, 5987–5994 10.1074/jbc.274.9.5987 [DOI] [PubMed] [Google Scholar]
- 59. Smith L., and Stull J. T. (2000) Myosin light chain kinase binding to actin filaments. FEBS Lett. 480, 298–300 10.1016/S0014-5793(00)01931-1 [DOI] [PubMed] [Google Scholar]
- 60. Hatch V., Zhi G., Smith L., Stull J. T., Craig R., and Lehman W. (2001) Myosin light chain kinase binding to a unique site on F-actin revealed by 3-D image reconstruction. J. Cell Biol. 154, 611–617 10.1083/jcb.200105079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hong F., Haldeman B. D., Jackson D., Carter M., Baker J. E., and Cremo C. R. (2011) Biochemistry of smooth muscle myosin light chain kinase. Arch. Biochem. Biophys. 510, 135–146 10.1016/j.abb.2011.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mabuchi Y., Mabuchi K., Stafford W. F., and Grabarek Z. (2010) Modular structure of smooth muscle myosin light chain kinase: hydrodynamic modeling and functional implications. Biochemistry 49, 2903–2917 10.1021/bi901963e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen C. P., Chen X., Qiao Y. N., Wang P., He W. Q., Zhang C. H., Zhao W., Gao Y. Q., Chen C., Tao T., Sun J., Wang Y., Gao N., Kamm K. E., Stull J. T., and Zhu M. S. (2015) In vivo roles for myosin phosphatase targeting subunit-1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J. Physiol. 593, 681–700 10.1113/jphysiol.2014.283853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kamm K. E., Hsu L. C., Kubota Y., and Stull J. T. (1989) Phosphorylation of smooth muscle myosin heavy and light chains: effects of phorbol dibutyrate and agonists. J. Biol. Chem. 264, 21223–21229 [PubMed] [Google Scholar]
- 65. Kitazawa T., Takizawa N., Ikebe M., and Eto M. (1999) Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in Triton X-100-demembranated rabbit arterial smooth muscle. J. Physiol. 520, 139–152 10.1111/j.1469-7793.1999.00139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gardner J. P., Stout M. A., and Harris S. R. (1989) Calmodulin loss in vascular smooth muscle following Triton X-100 or saponin skinning. Pflugers Arch. 414, 484–491 10.1007/BF00585061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.