

ABSTRACT
Efforts to reduce immunosuppression in the solid tumor microenvironment by blocking the recruitment or polarization of tumor associated macrophages (TAM), or myeloid derived suppressor cells (MDSCs), have gained momentum in recent years. Expanding our knowledge of the immune cell types, cytokines, or recruitment factors that are associated with high-grade disease, both within the tumor and in circulation, is critical to identifying novel targets for immunotherapy. Furthermore, a better understanding of how therapeutic regimens, such as Dexamethasone (Dex), chemotherapy, and radiation, impact these factors will facilitate the design of therapies that can be targeted to the appropriate populations and retain efficacy when administered in combination with standard of care regimens. Here we perform quantitative analysis of tissue microarrays made of samples taken from grades I-III astrocytoma and glioblastoma (GBM, grade IV astrocytoma) to evaluate infiltration of myeloid markers CD163, CD68, CD33, and S100A9. Serum, flow cytometric, and Nanostring analysis allowed us to further elucidate the impact of Dex treatment on systemic biomarkers, circulating cells, and functional markers within tumor tissue. We found that common myeloid markers were elevated in Dex-treated grade I astrocytoma and GBM compared to non-neoplastic brain tissue and grade II-III astrocytomas. Cell frequencies in these samples differed significantly from those in Dex-naïve patients in a pattern that depended on tumor grade. In contrast, observed changes in serum chemokines or circulating monocytes were independent of disease state and were due to Dex treatment alone. Furthermore, these changes seen in blood were often not reflected within the tumor tissue.
Conclusions: Our findings highlight the importance of considering perioperative treatment as well as disease grade when assessing novel therapeutic targets or biomarkers of disease.
KEYWORDS: Glioblastoma, tumor associated macrophage, Dexamethasone, immunotherapy, cancer immunology
Introduction
Glioblastoma (GBM), a WHO grade IV astrocytoma, is a primary brain tumor which has an extremely poor prognosis in adults, with a 5-year overall survival (OS) of 9.8% following surgery, temozolomide treatment, and radiation.1 The infiltrative nature of tumor cells into surrounding brain tissue and distant sites makes complete microscopic surgical resection improbable and recurrence inevitable, necessitating an immune therapy that can effectively eliminate transformed cells while leaving surrounding neural and glial structures intact.
As with most solid tumors, immunotherapy for GBM has proven challenging, in large part due to the immunosuppressive tumor microenvironment (TME). Tumor associated macrophages (TAMs), accounting for ~ 20–30% of the cells in the GBM tumor mass,2 express soluble factors and surface molecules that prevent immune surveillance by endogenous T and NK cells, and shut down crosstalk between the adaptive and innate immune systems.3-5 Novel therapies that circumvent this suppressive milieu by blocking the recruitment or polarization of TAMs6,7 are complicated by the paucity of markers that discriminate dysfunctional cells8 and a poor understanding of the tumor-derived factors that influence their phenotype or trafficking.9,10 Furthermore, gene expression analysis suggests that TAMs differ across grades of glioma11,12 and regions within heterogeneous GBM tumors,13 although it is not clear how these factors impact TAMs phenotypically and functionally.8 Finally, the search for systemic biomarkers in myeloid cell populations necessitates an improved delineation of the cells and proteins in circulation as they reflect the conditions of the tumor microenvironment.
Complicating efforts to generate effective myeloid-targeted experimental therapies is the treatment of glioma patients with the immunosuppressive drug Dexamethasone (Dex). Although the relationship between Dex treatment and circulating myeloid cell frequency or phenotype has been reported,14 this variable is often overlooked in the development of novel therapeutic targets. Furthermore, though the impact of Dex treatment on immune suppression15,16 and survival14 in GBM patients has been studied, it is yet unclear if these effects are systemic or restricted to the tumor microenvironment in patients with low and high grade astrocytomas.
In this study, we sought to identify myeloid markers that distinguish low and high-grade astrocytomas and determine if they are altered by Dex treatment. Quantitative analysis of tissue microarrays (TMAs) made of samples taken from grades I-III astrocytomas and GBM were used to assess the frequency of the common myeloid markers CD163, CD68, CD33, and S100A9. We performed serum, flow cytometric, and Nanostring analysis to better understand the impact of Dex treatment on systemic biomarkers, circulating cells, and functional proteins within tumor tissue. We found that that CD163, CD68, and S100A9 frequencies were elevated in Dex-treated grade I astrocytoma and GBM compared to non-neoplastic tissue and grade II-III tumors. Cell frequencies in these samples differed significantly from those in Dex-naïve patients in a tumor grade-dependent pattern. In contrast, changes in serum chemokines or circulating monocytes were often independent of disease state and were due to Dex treatment alone. Furthermore, changes in circulation were often not reflected within the tumor tissue, indicating that additional methods may be required to identify cells and proteins associated with prognosis using minimally invasive procedures. Collectively, our data suggest that the complexity of the interaction between circulating cells and those in the tumor microenvironment, as well as perioperative steroid treatment, may confound the identification of novel systemic biomarkers that are associated with prognosis and tumor burden. This may be of particular interest when prospectively or retrospectively stratifying patients participating in immunotherapy clinical trials.
Materials and methods
TMA construction
Study subjects were identified from the University of Washington medical records and Pathology database after approval from the Seattle Children’s Research Institutional (SCRI) Review Board #14412. All research subjects included in the study underwent surgery at University of Washington Medical Center between 2009–2015. Human tissue was obtained at the time of surgery; neoplastic tissue from tumor resections and non-neoplastic tissue from anterior temporal lobectomies performed in chronic epilepsy patients. Following surgery formalin-fixed, paraffin-embedded tissue blocks (FFPE) were made during gross dissection following tissue processing. Hematoxylin and eosin (H&E) stained slides were reviewed by a Neuropathologist at the time of initial diagnosis and again in order confirm the diagnosis before inclusion into the study. 25 glioblastomas, 25 grade II-III astroctyomas, 25 grade I pilocytic astrocytomas, and 25 non-neoplastic anterior temporal lobe tissue were identified and sampled in accordance with institutional regulatory agencies. Triplicate 1 mm tissue cores per case were harvested (two from an intratumoral region and one from either a peritumoral region in grade I astrocytomas or from the infiltrating edge in grade II-III astrocytomas) in neoplastic cases and from cortical parenchyma in non-neoplastic cases. The TMA was designed and coded by a research scientist in a randomized fashion to reduce the effect that staining artifacts and regional tissue spot loss may have on analysis. These tissue cores were arrayed into a new recipient paraffin block with a manual tissue arrayer (MTA-1; Beecher Instruments, Sun Prairie, WI To account for potential heterogeneity within the tumor, two 1.5 mm cores from each biopsy were taken to create the tissue microarray. Consecutive sections at 4 microns were generated for immunohistochemistry analyses.
IHC
Immunostaining was performed manually. PBS (pH 7.4) was used as a wash between all steps. The TMA slides were deparaffinized, and pretreated in citrate buffer (Invitrogen #005000) placed in a steamer for 20 minutes. A solution of 5% goat serum (Jackson Immuno #005–000-121) + 0.3% Triton X (“Tx”, Thermo Fisher #BP151100) in PBS + Avidin/Biotin solution (Vector #SP-2001) was applied to block nonspecific protein, endogenous biotin and avidin binding sites. Antibodies were diluted in the goat serum + Tx solution, then biotin was added. The solution was applied to the slides and incubated for 1 hour at room temperature. A biotinylated anti-mouse secondary antibody (Jackson Immuno #115–067-003) was applied for 30 minutes. To visualize antibodies, the Vectastain ABC HRP kit was used (Vector #PK-4000), followed by a DAB solution (Vector #SK-4105) applied for 5 minutes. The slides were counterstained in hematoxylin, dehydrated in alcohol, cleared in xylene substitute, and cover slipped. For antibody information see Supplemental Table 1.
Image analysis
TMA cores were imaged on the Nuance multispectral imaging system (Perkin Elmer) on a Nikon Eclipse Ci with a 10x Plan Apo lens (NA 0.45). Multispectral images were analyzed using InForm Tissue Finder Analysis software v2.3 (Perkin Elmer). Multispectral images were first spectrally unmixed into DAB and Hematoxylin components using a spectral library generated from positive control images. Images were then manually segmented for tissue area vs. blank area to determine the number of pixels in the image that represent the TMA core area. The image was further segmented into cells using the software’s cell segmentation algorithm with a threshold set using the hematoxylin spectrum. These software-identified cells were then scored using a threshold set using the DAB spectrum.
For whole sections, all slides were stained, imaged and quantified together. For each case immunostaining for HLA-DR, DP, DQ and CD86 was quantified in 5 random sections of tumor, at 20X magnification. Immunostained fields were visualized and captured with a digital camera mounted on a Nikon Eclipse 80i microscope and analyzed using NIS-Elements Advanced Research Software v4.13 (Nikon Instruments Inc., Melville, NY); consistent settings were maintained between images. No image processing was carried out prior to intensity analysis. Resulting immunostained area for HLA-DR, DP, DQ or CD86 was expressed as a percentage of the total tumor area analyzed.
Blood/serum processing
Blood was collected under SCRI Institutional Review Board (IRB) approved protocols #14982 and #14412. During surgery, 15 total milliliters (mL) of peripheral blood was drawn into BD Vacutainer Sodium Citrate Tubes (whole blood) or BD red top serum tubes for serum analysis through the intravenous lines of patients. Tubes were centrifuged at 750 xg for 5 minutes at room temperature; plasma and serum were then removed and frozen at −80C. The remaining blood product was placed in 1x Red Blood Cell (RBC) Lysis Buffer (eBioscience) at 10 mL RBC Lysis Buffer per 1 mL blood product for 10 minutes at room temperature. Lysing was stopped with 20 mL 1x Phosphate Buffered Saline (PBS) and centrifuged at 750xg for 5 minutes. The supernatant was removed and remaining peripheral blood mononuclear cell (PBMC) pellet was resuspended in PBS plus human Fc block and immunostained immediately.
Multiplex assays
Serum collected in BD Serum Tubes was thawed on ice, diluted 1:5, and used in the Bio-Rad 40-plex Human Chemokine assay according to manufacturer’s instructions.
Nanostring gene expression analysis
RNA was isolated from FFPE GBM samples using High Pure FFPET RNA Isolation Kit (Roche, Indianapolis, IN). Five μL of RNA was used directly in the Human PanCancer Immune Profiling Panel (Nanostring, Seattle, WA) per the manufacturer’s protocols. Nanostring nSolver software was used to normalize gene expression across samples, calculate the geometric means, fold change, and p values.
PBMC Flow Cytometry
Live cells were immunostained for flow cytometry immediately following blood processing, fixed using 100 μL of 2% paraformaldehyde for 5 minutes at room temperature. Cells were spun at 553 xg for 5 minutes, washed 3x in 200 μL PBS, and run in 100 μL volume of PBS on a five laser BD Fortessa. Data was analyzed using FlowJo 10.0 (Treestar). For antibody information see Supplemental Table 1.
Dexamethasone flow cytometry
GM-CSF-differentiated macrophages were plated at 500K per well and treated with 0, 0.1, 1, or 10 uM dexamethasone in RPMI-1640 (Gibco) containing 10% FBS (Hyclone) for 24 hours, detached with Versene (Gibco), then analyzed immediately by flow cytometry. The following antibodies were used: CD40-BUV395, CD80-BV786, and HLA-DR/DP/DQ-FITC (BD Biosciences), HLA-ABC-Pacific blue, CD86-BV605, CD163-PerCPCy5.5, PD-L1-PE, and CCR2-PECy7 (BioLegend). For washout experiments, macrophages were treated with 0.1 uM dexamethasone for 24 hours, then standard RPMI-1640 with 10% FBS added for an additional 6 days prior to flow cytometry.
Cortisol ELISA
Cortisol was detected in serum samples from healthy donors and grade I-IV astrocytoma patients being treated or not with Dexamethasone at the time of sample collection. Cortisol ELISA kit was purchased from Eagle Bioscience and manufacturer’s protocols followed. Data was analyzed in Graphpad.
Human subjects
Blood and tumor tissue was collected from brain tumor patients who signed our Brain Tumor Bank acquisition consent form. Data is only correlated with pathology of tumor diagnosis, and steroid treatment, and identifying data was not collected for these experiments. All use of human subject material has been approved by the University of Washington Institutional Review Board and Committee on Human Research. In addition, all investigators have completed and passed the NIH computer-based training course on the Protections of Human Research Subjects.
This study has been performed in accordance with the principles of the Declaration of Helsinki (1964) as revised in Tokyo (1975) Venice (1983), Hong Kong (1989), Somerset West (1996), and Edinburgh (2000) and with the Note for Guidance (ICH Harmonized Tripartite Guideline) Good Clinical Practice for Trials on Medicinal Products in the European Community as well as adopted by the US Food and Drug Administration.
The study was also based on the following ethical and formal considerations:
Informed consent of the subject.
Declaration of Helsinki.
Laws and regulations in the United States and State of Washin
Statistics
Unless otherwise stated, results were analyzed with Prism software (GraphPad), using student’s T test, or one-way ANOVA followed by Tukey’s multiple comparisons test comparing differences across all samples. For non-parametric TMA data we used the Mann-Whitney test (for two comparisons) or Kruskal-Wallace ANOVA (for multiple comparisons) followed by Dunn’s multiple comparisons test comparing differences across all samples. Statistical significance is denoted with an asterisk where * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001.
Results
CD163 is elevated in grades i and IV astrocytoma
CD163, a common maker of tumor associated myeloid cells (TAMs),17 functions as a hemoglobin-haptoglobin scavenger receptor, and is up-regulated on the surface of macrophages, monocytes, and microglia in response to anti-inflammatory cytokines such as IL-10,18,19 and glucocorticoids such as Dex.20-22 A recent study evaluating grades II-IV astrocytomas demonstrates that CD163 mRNA expression increases with increasing grade of astrocytoma, and that CD163+ cells produce IL-10 in the GBM tumor microenvironment.23 Furthermore, this report suggests that CD163 could be used as a prognostic indicator for grade III astroctyomas. Given the described import of CD163 positive cells as a source of immune-modulatory factors and a potential prognostic marker, we sought to determine whether the observed mRNA expression was reflected at the protein level or if this trend extended to grade I astrocytoma. We immunostained sections from four TMAs comprised of cores taken from non-neoplastic brain tissue, grade I, grade II-III astrocytoma (20% isocitrate dehydrogenase (IDH-1 (R132H) wild-type, 46% IDH-1 (R132H) mutant, 34% not otherwise specified), and IDH-1 (R132H) mutant, 34% not otherwise specified), and IDH wild type GBM. Sections for these analyses were serial, with cores selected from two locations per patient- one from peritumoral and one from a central region based on neuropathology review. Regions were selected for areas with limited necrosis and high cellularity. Unless otherwise stated, tumors were resected from patients who were treated with Dexamethasone (Dex) prior to surgery. Quantitative image analysis was performed to determine the percent of total cells staining positive for CD163, and number of CD163+ cells per square millimeter using InForm software (Figure 1A). Of note, CD163 staining was restricted to perivascular cells in non-neoplastic tissue (Figure 1A), but was distributed throughout the tissue of astrocytomas. When compared to non-neoplastic tissue, we found that CD163 staining was elevated in grade I and GBM when calculated as either percent of total cells (median = 30.9% and 15.6% respectively) or cells per square millimeter (Figure 1B-C). To our surprise, samples in the grade II-III TMA were not significantly different than those in the non-neoplastic tissue TMA, but had significantly fewer CD163+ cells than either grade I or GBM (Figure 1B, C). Compared to GBM, which has an overall 5-year survival of 9.8%,1 patients diagnosed with grade I pilocytic astrocytoma have a favorable outcome: 96.5% 5-year survival in patients 5–19, and 52.9% in patients over 60.24 While future studies with larger cohorts will be needed to confirm these findings, this data indicates that although accumulation of CD163 expressing cells may predict survival within grade III patient populations,23 it does not correlate with prognosis across all grades. Further, these data suggest that CD163 expression itself is not indicative of TAM functions in astrocytoma patients.
Figure 1.
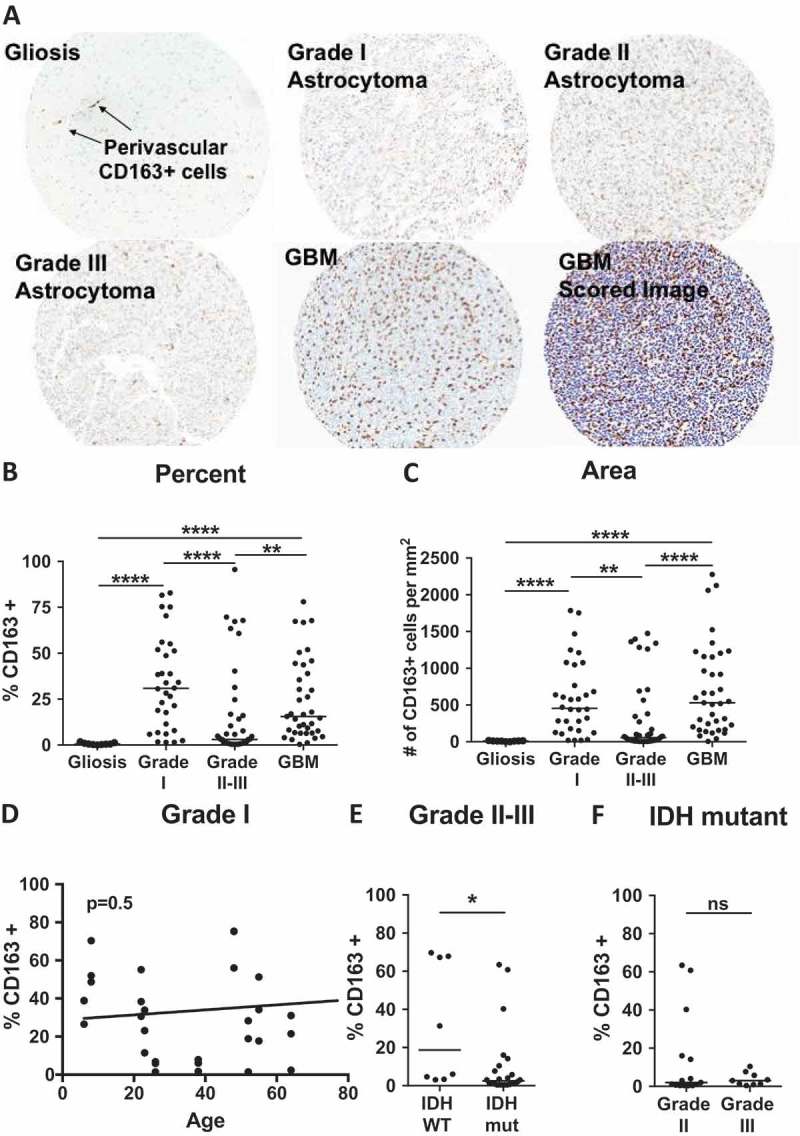
CD163 is elevated in grades I and IV astrocytoma. (A) Representative cores from gliosis, grade I, II, III and GBM TMAs stained for CD163. Bottom right image is an example image scored by InForm software, where blue indicates nuclei, and brown indicates CD163+ cells. (B-C) Quantitative analysis of TMAs represented as (B) % of total cells that are CD163+, or (C) number of CD163+ cells per square millimeter. P-values calculated with the Kruskal–Wallis one-way ANOVA followed by Dunn’s multiple comparisons test. (D) Linear regression analysis of patient age versus CD163 positivity in grade I pilocytic astroctyomas. P-value 0.5 indicates the slope does not significantly deviate from zero. (E) Comparison CD163 infiltration in IDH wild-type versus mutant samples from grade II-III astrocytomas. (F) Comparison CD163 infiltration in grade II versus III astrocytomas among IDH mutant samples. P-values were calculated with the Mann-Whitney test. Statistical significance is denoted with an asterisk where * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Line indicates median.
Unlike the grade II-III and GBM TMAs that are comprised of tumors from adult patients, the grade I pilocytic astrocytoma TMA represents patients age 6 to 77. To determine whether immune system maturity influenced the distribution of CD163 infiltration seen in Figure 1B, or if this marker correlated with the lower survival rates seen in adult patients,24 we represented CD163 positivity as a function of age, but found no relationship between the two factors (Figure 1D).
One of the greatest prognostic indicators among grades II-III astrocytomas is IDH1/2 mutation status. Though IDH wild-type grade II-III astrocytomas are rare, making up only 21.6% of patients,25 their prognosis is poor with an overall survival of 31.8 vs 63.8 months for those with IDH mutant tumors (comparison made among TERT promoter wild type individuals).26 The mutation status of gliomas is also associated with effects on the immune landscape of the tumor microenvironment, suggesting an impact on TAM protein expression in patient tumors.27 A comparison of cores from IDH wild type and mutant tumors from our grade II-III TMA showed a slight increase in percentage of CD163 expressing cells in the IDH wild-type population (Figure 1E). Although it is tempting to speculate that CD163 infiltration may be associated with a poorer prognosis among these patients, the small sample size of wild type tumors (N = 3) precludes this conclusion and will be addressed in future studies evaluating a larger sample size. Recent WHO guidelines suggest that among IDH mutant astrocytomas, there may be little prognostic difference between grades II and III,28 a finding supported by the consistency of CD163 expressing cells in IDH mutant grade II and III astrocytomas (Figure 1F).
Impact of dexamethasone on CD163 infiltration is tumor grade-dependent
Corticosteroids such as Dexamethasone (Dex) are commonly used pre- and post-operatively to control cerebral edema.14 Although glucocorticoid signaling increases expression of CD163 on myeloid cells,22,29 the effects on accumulation of CD163+ cells in tumor tissue are less well understood. Recent work suggests that variation in the expression of macrophage immune modulatory proteins is more critical to disease progression than macrophage accumulation within the tumor.30 Supporting this study, we found that only grade I astrocytoma patients treated with Dex had increased accumulation of CD163 expressing cells when comparing Dex-treated samples with those from untreated non-neoplastic tissue or low-grade astrocytoma patients (Figure 2A-C). Although there exists an association, to date it is not clear whether CD163+ cells infiltrate tumor tissue, necessitating Dex administration, or if Dex administration increases CD163+ cell accumulation. Because surgical resection of tumor from GBM patients without pre-operative Dex treatment is rare, our TMAs were constructed exclusively from tumors isolated from GBM patients recently (within 7 days) treated with steroids. To evaluate patients who had not been treated with steroids, we acquired freshly resected tumors from newly diagnosed GBM patients either treated with Dex or not (N = 8 each). Tumors were formalin fixed, paraffin embedded, sectioned and stained for CD163 according to the methods used for TMA analysis. We imaged four random 10x fields from each section, avoiding areas of necrosis, and found a significant increase in the percent of CD163+ cells in the steroid naïve patient samples (median = 12.8% Dex-treated vs 18.9% untreated, p < 0.05) (Figure 2D), suggesting that the impact of Dex on CD163 accumulation in the tumor is dependent on tumor grade. Although the TMA samples in Figure 2A-C and the whole GBM tissue sections in Figure 2D cannot be directly compared, it is important to point out that among Dex-naïve samples, the infiltration of CD163+ cells increases with increasing tumor grade (medians: non-neoplastic tissue = 0.9%, grade I = 1.1%, grade II-III = 3.8%, and GBM = 18.9%) (Figure 2A-D).
Figure 2.
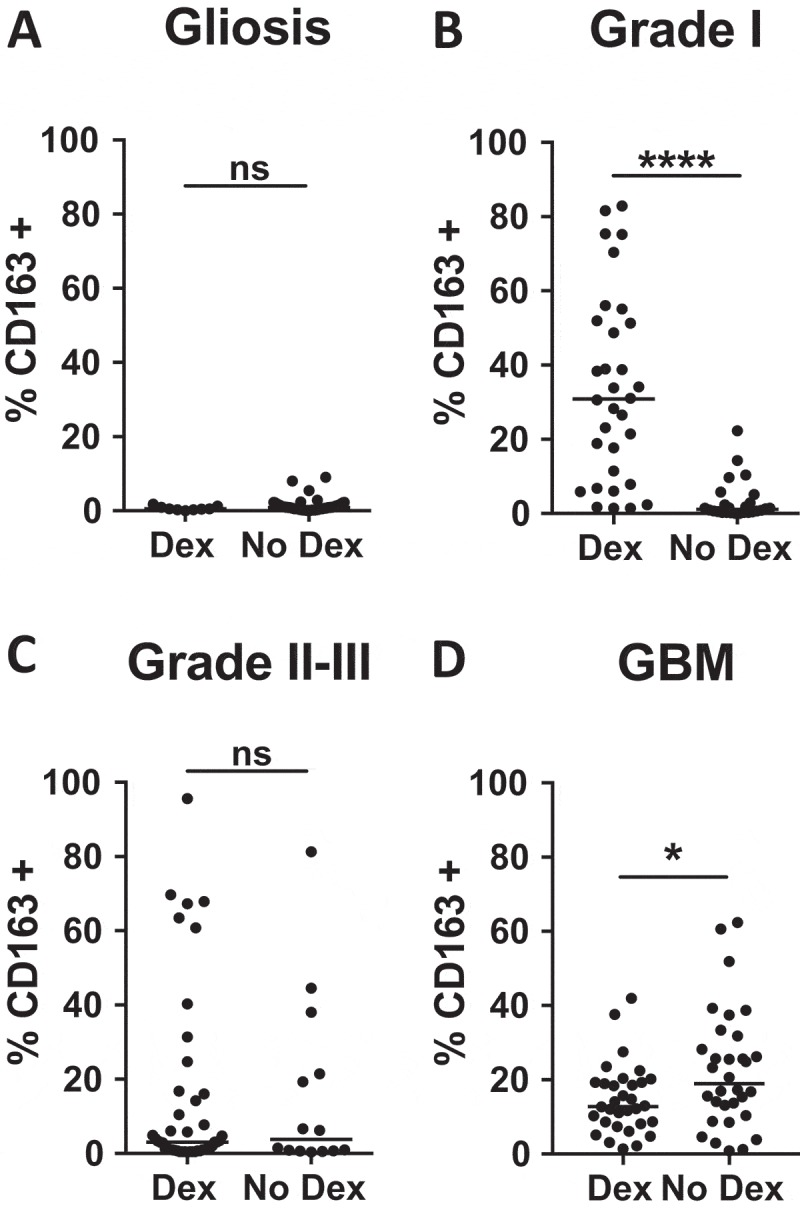
Impact of Dexamethasone on CD163 infiltration is tumor grade-dependent. Comparison of CD163 positivity in Dex-treated and Dex-naïve samples from (A) gliosis, (B) grade I astrocytoma, (C) grade II-III astrocytoma, and (D) GBM patients. P-values were calculated with the Mann-Whitney test. Statistical significance is denoted with an asterisk where * = p < 0.05, **** = p < 0.0001. Line indicates median.
CD68, CD33, S100A9 are not associated with high grade disease
CD68, a marker of both pro- and anti-inflammatory macrophages,17 exhibited a similar pattern to CD163 in that it was also elevated in grade I and GBM tumors when compared to non-neoplastic brain tissue and grade II-III astrocytoma (Figure 3A).
Figure 3.
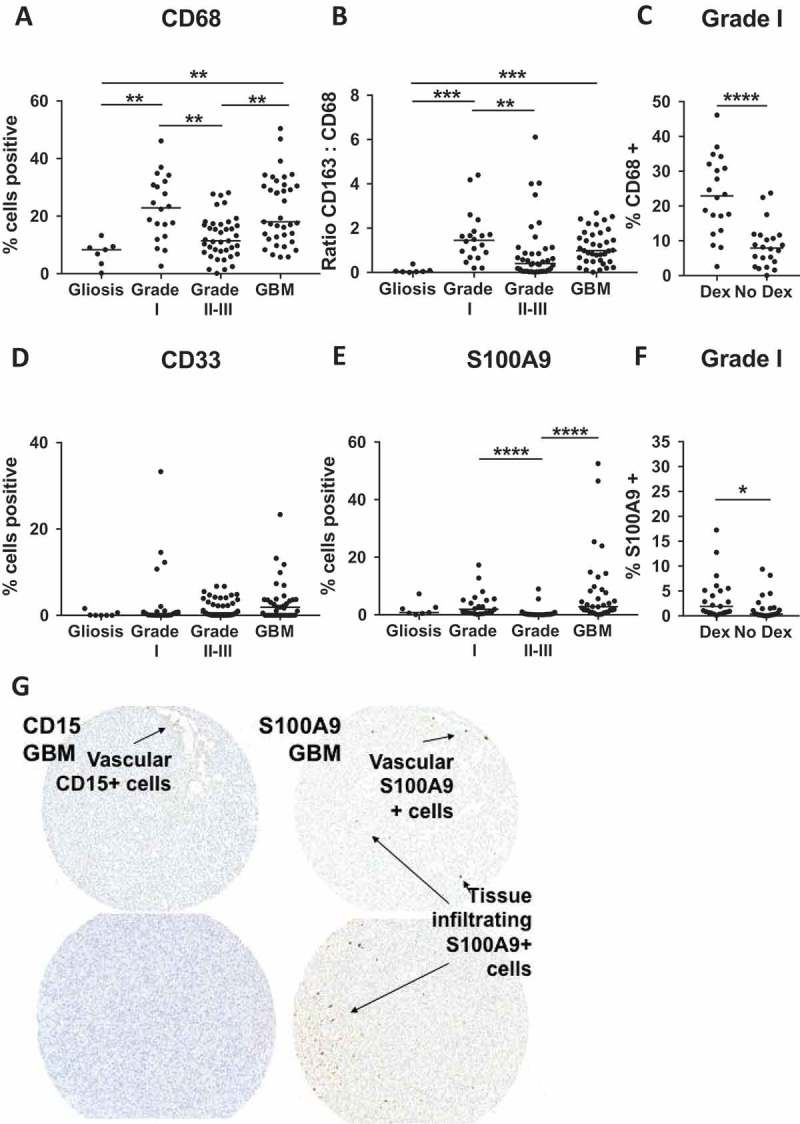
CD68, CD33, S100A9 are not associated with high grade disease. (A) Quantitative analysis of TMAs represented as % of total nuclei that are CD68 + . (B) Ratio of CD163 to CD68 positivity calculated from adjacent TMA sections. (C) Comparison of CD68 positivity in Dex-treated and Dex-naïve samples from grade I astrocytoma patients. Quantitative analysis of TMAs represented as % of total cells that are (D) CD33+ and (E) S100A9 + . (F) Comparison of S100A9 positivity in Dex-treated and Dex-naïve samples from grade I astrocytoma patients. (G) CD15 (left images) S100A9 (right images) showing vascular CD15+ neutrophils, and both vascular and tumor infiltrating S100A9+ cells. Images from left to right were taken from serial sections. Statistical significance is denoted with an asterisk where * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Line indicates median.
Although the levels of CD68 and CD163 staining were comparable in GBM tissue (18.0% vs 15.6% respectively), in non-neoplastic tissue, the median for CD68 was 16.6 times higher than that for CD163 (8.3% CD68+ vs 0.5% CD163+), suggesting that the markers are non-redundant, and that CD68 may have stained CD163 negative tissue resident myeloid cells. A higher ratio of CD68 to CD163 (representing pro-inflammatory “M1” functions) has been associated with a favorable outcome in response to ipilimumab treatment in melanoma patients.31 Here we calculated the ratio of CD163 to CD68 as a measure of skewing toward an anti-inflammatory, “M2” phenotype. Unexpectedly, we see that this ratio is highest in grade I astrocytoma (CD163:CD68 median = 1.4) (Figure 3B) indicating that these two markers together cannot predict prognosis when comparing across grades. Consistent with the pattern observed for CD163 (Figure 2B), CD68 frequency also increased in Dex-treated grade I tumors compared to those from Dex-naïve patients (Figure 3C). This suggests that Dex-treatment may not simply up-regulate CD163 on cells infiltrating tumor tissue, but instead either promotes myeloid cell accumulation in grade I tumors or is required to reduce edema in patients grade I tumors that accumulate CD163+ cells through other, as yet defined, mechanisms. This effect was not found in non-neoplastic tissue tissue or grade II-III tumors where Dex had no impact on either CD163 (Figure 2A, C) or CD68 (Supp Figure 1A, B).
Myeloid derived suppressor cells (MDSCs) are a heterogeneous population of immature cells from the myeloid lineage that are associated with immune suppression and tumor progression32-36 In blood, these cells are described as polymorphonuclear (PMN-MDSC) CD14-/CD11b+/CD15+/CD33dim (separated from PMNs in the low density fraction of a centrifugation gradient), or monocytic (M-MDSC) CD11b+/CD14+/HLA-DR low/-/CD15-37. In tissue, it is difficult to distinguish between PMNs, or monocytes, and MDSCs. CD33 does not discriminate between MDSCs, macrophages, dendritic cells or other myeloid cells.38 S100A9, a protein expressed in non-terminally differentiated myeloid cells,39 may exclude some dendritic cells and macrophages, but is present on subsets of monocytes, neutrophils, as well as MDSCs.37 With these limitations in mind, we stained TMAs for CD33 and S100A9 expression to determine if these markers were associated with tumor grade. We found that the percentage of cells staining for CD33 was not significantly elevated in any grade of astrocytoma compared to non-neoplastic tissue (Figure 3D), nor was this marker influenced by Dex-treatment (Supp Figure 1C-E). S100A9 staining was elevated in grade I and GBM tumors relative to grade II-III astrocytoma, but not when compared to non-neoplastic tissue (Figure 3E). Steroid treated grade I astrocytoma patients also had increased S100A9 staining (Figure 3F), which was not found in patients with either non-neoplastic tissue or grade II-III (Supp Figure 1F, G). Although this marker stains both neutrophils and cells of the monocytic lineage, we found that very few CD15+ neutrophils infiltrate viable GBM tissue, and were instead primarily restricted to blood vessels (Figure 3G, left panels). In contrast, S100A9 marked both cells contained within the vasculature and tumor infiltrating cells (Figure 3G, right panels), suggesting that within GBM, S100A9 staining is identifying cells of the monocyte lineage.
Chemokine levels are influenced by dex-treatment
Given the accumulation of myeloid cells in GBM tumor tissue, we hypothesized that myeloid recruitment factors secreted by tumor cells 40–43 would be elevated in patient serum. We assayed serum from 14 healthy donors, 17 Dex-treated GBM patients, and 8 steroid naïve GBM patients using the BioRad Bioplex human chemokine array, which quantifies 40 soluble analytes. Surprisingly, we found that the only significant differences in concentrations of serum analytes were attributable to steroid treatment. As reported previously, we saw Dex-dependent suppression of IP-10 (CXCL10) and MCP-1 (CCL2),16 in addition to 6Ckine, CTACK, MDC, and SDF1a/b (Figure 4A, Supp Figure 2A). The only Dex-elevated protein was MPIF-1 (CCL23) (Figure 4A).
Figure 4.
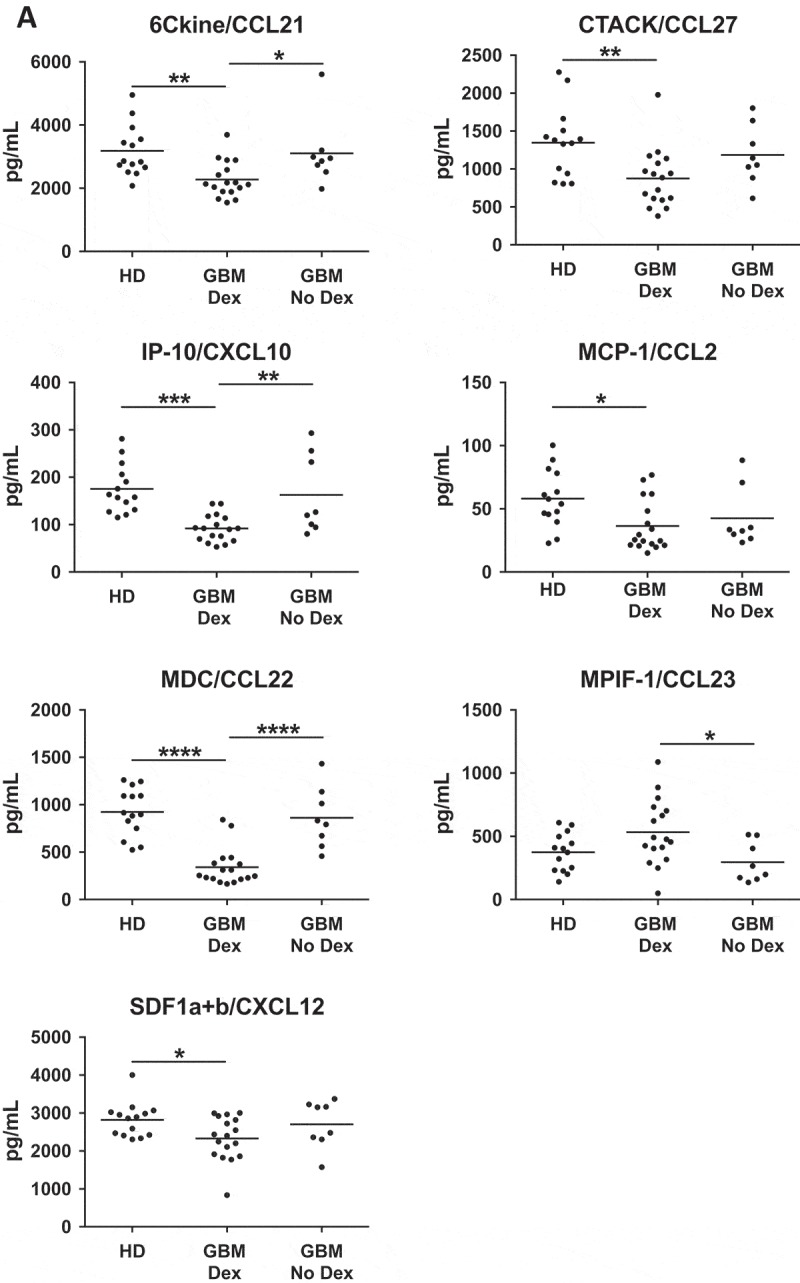
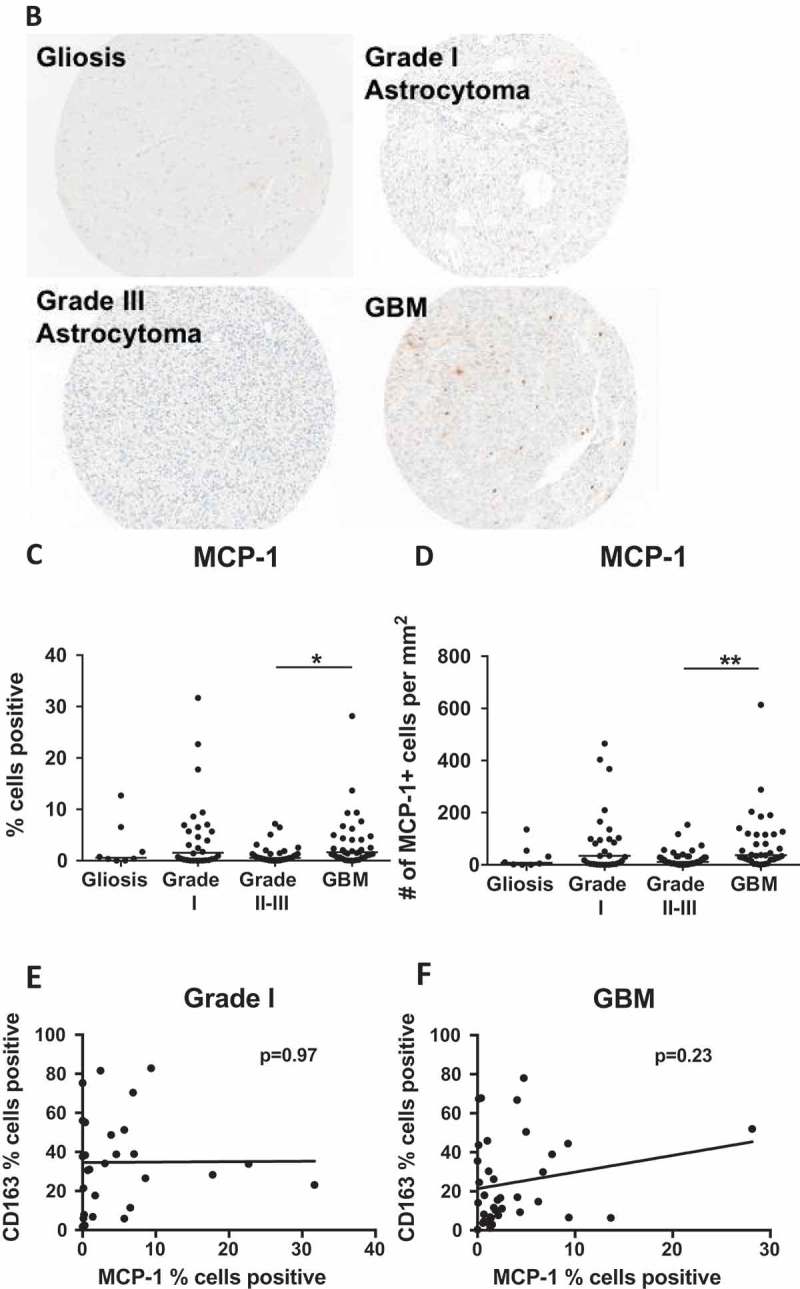
Chemokine levels are influenced by Dex-treatment. (A) Serum samples from healthy donors (HD) or Dex-treated and naïve GBM patients were analysed using the Biorad Human Chemokine Panel 40-plex. Dex-dependent differences were found in 6Ckine (CCL21), CTACK (CCL27), IP-10 (CXCL10), MCP-1 (CCL2), MDC (CCL22), MPIF-1 (CCL23), and SDF-1 (CXCL12). P-values calculated using one-way ANOVA followed by Tukey’s multiple comparisons test. Line indicates mean. (B) Representative cores from gliosis, grade I, III and GBM TMAs stained for MCP-1. (C-D) Quantitative analysis of TMAs represented as (C) % of total cells that are MCP-1+, or (D) number of MCP-1+ cells per square millimeter. P-values calculated with the Kruskal–Wallis one-way ANOVA followed by Dunn’s multiple comparisons test. Line indicates median. Linear regression analysis of MCP-1 versus CD163 positivity in (E) grade I pilocytic astroctyoma and (F) GBM. P-values indicate the slope does not significantly deviate from zero. Comparison of MCP-1 positivity in Dex treated and Dex naïve samples from (G) gliosis (H) grade I, and (I) grade II-III TMAs.
The Dex-dependent decrease, and similarity of healthy donor and GBM serum levels of MCP-1 have been seen before,16 but the relationship between serum levels and tumor tissue expression is not clear. MCP-1 is often reported as a key tumor-derived factor responsible for myeloid cell accumulation.40,44,45 These studies have led to the proposal of MCP-1 blockade as a treatment for GBM.46,47 A detailed examination of human gliomas demonstrated that MCP-3, not MCP-1 correlates with myeloid cell accumulation in glioblastoma,48 calling into question the utility of MCP-1 inhibition in preventing TAM infiltration.
To determine if MCP-1 is elevated in GBM, potentially generating a chemokine gradient for myeloid cell recruitment, or if its expression relates to myeloid infiltration in astrocytoma, we performed quantitative analysis of Dex-treated samples and found that MCP-1 expression was highest in grade I and GBM, but only statistically different between grades II-III and GBM (Figure 4B-D). Consistent with other quantitative analyses of human glioma samples,48 we saw no correlation between the percentage of cells staining for CD163 and MCP-1 in either grade I or GBM tumors (Figure 4E, F).
Figure 4.
(Continued).
Figure 4.
(Continued).
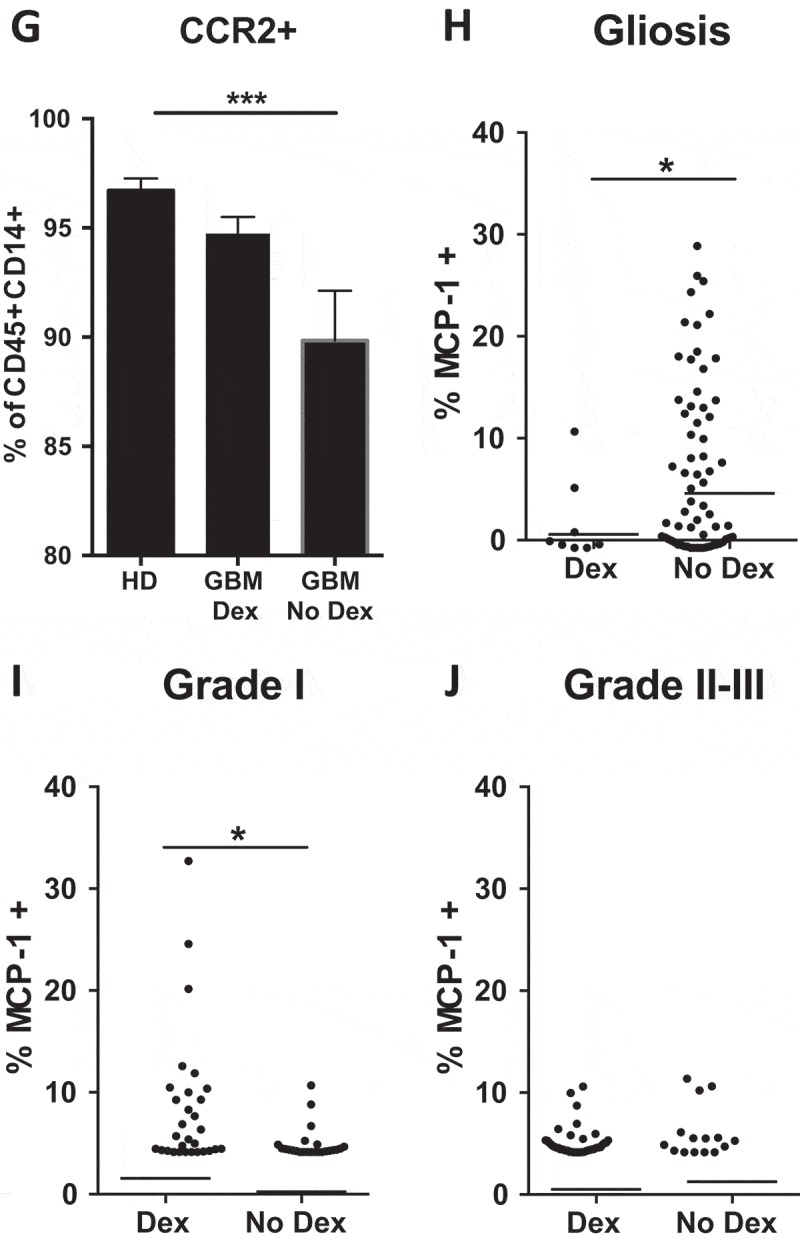
It has been demonstrated in non-neoplastic patients that Dex up-regulates CCR2, a receptor for MCP-1, and increases the migratory response of human monocytes.49,50 This suggests that a Dex-dependent decrease in serum MCP-1 may be compensated for by an increase in CCR2. Consistent with this, flow cytometric analysis of circulating monocytes revealed that Dex-naïve GBM patients have fewer CCR2+ cells, but Dex treatment restored CCR2 frequencies to those seen in healthy donors (Figure 4G). In spite of the observed Dex-induced increase in CCR2+ monocytes, the correlation data in Figure 4F suggests that there exists another, possibly redundant mechanism for myeloid recruitment to human GBM.
Surprisingly, we found that Dex-treatment significantly suppressed MCP-1 expression in non-neoplastic tissue (median = 0.6% vs 4.6%) (Figure 4H), amplified MCP-1 expression in grade I tumors (median = 1.6% vs 0.24%) (Figure 4I), but had no impact on MCP-1 expression in grade II-III astrocytomas (Figure 4J). These observations support the hypothesis that clinical diagnosis or other variables not considered in this study can result in varied effects of Dex-treatment on the tumor microenvironment.51
It is well-established that dexamethasone treatment reduces plasma cortisol levels in healthy individuals, but baseline cortisol levels may be altered in neoplastic patients.51 To determine if the observed variations in Dex-mediated effects between tumor grade can be attributed to cortisol levels, we performed ELISA analysis on serum collected from healthy donors, or from Dex-treated or Dex-naïve glioma patients. While serum from grades I and II Dex-naïve patients trended lower for cortisol levels, there was no significant difference between groups (Supp Figure 2B). As expected, all samples from Dex-treated patients had undetectable levels of cortisol.
Multiple myeloid-related genes are altered by dex-treatment in GBM tumors
Given that Dex treatment altered CD163 infiltration (Figure 2D) and serum chemokine levels (Figure 4A) in GBM patients, we sought to determine the effects of steroid treatment on other immune modulating factors, particularly those related to myeloid cell function in the tumor microenvironment. We performed Nanostring RNA expression analysis using the Pan Cancer Immune panel on FFPE tumor tissue resected from newly diagnosed GBM patients that had been treated with Dex (n = 5) or not (n = 4). To minimize the effects of variance in CD163 positive cell accumulation and cell death on our analysis, we selected non-necrotic tissue with comparable CD163 infiltration (Supp. Figure 3A). Consistent with this image analysis, Nanostring analysis revealed no change in CD163 or CD68 transcript across these samples (Supplemental Table 2). We found that 70 genes were decreased in Dex-treated patient tumor tissue, including genes involved in myeloid cell differentiation and recruitment (MCSF, CSF1R), and antigen presentation (HLA-DR, DP, DQ; CD86) (Table 1). Surprisingly, we saw down-regulation of genes for chemokines Fractalkine (CX3CL1) and CXCL16 (Table 1), that was not reflected in the serum analysis (Supp Figure 2). Conversely, serum proteins determined to be Dex-regulated by Bioplex, MCP-1 (CCL2), SDF-1 (CXCL12), or IP-10 (CXCL10), were unchanged at the mRNA level in tissue (Supplemental Table 2), Whether this was due to the biased sample selection, or the fact that Dex-treatment does not impact expression of these genes within tumor tissue is unclear. Regardless, these findings highlight that changes in key soluble factors in GBM tumors are not necessarily reflected in circulation.
Table 1.
Comparison of Dex-treated versus untreated GBM tumors using the Nanostring Pan-Cancer Immune panel. FFPE tumors from each group were selected based on comparable number of CD163 positive cells. Of 770 genes on the panel, 542 had average raw counts greater than 50. 71 of these genes have a p-value less than 0.05 and fold change greater than 2. Only 1 of the 71 genes, Complement C7, is up-regulated by Dex-treatment.
| Gene Name | Accession # | Fold Change Dex vs No Dex | P value |
|---|---|---|---|
| Anti-inflammatory | |||
| ATF1 | NM_005171.2 | −2.07 | 0.025 |
| CSF1 | NM_000757.4 | −3.96 | 0.024 |
| CSF1R | NM_005211.2 | −12.64 | 0.010 |
| IL13RA1 | NM_001560.2 | −2.04 | 0.044 |
| PDCD1LG2 | NM_025239.3 | −22.19 | 0.006 |
| SMAD3 | NM_005902.3 | −2.94 | 0.043 |
| SOCS1 | NM_003745.1 | −19.48 | 0.014 |
| STAT3 | NM_139276.2 | −2.63 | 0.012 |
| TANK | NM_004180.2 | −2.38 | 0.042 |
| TGFB2 | NM_003238.2 | −4.66 | 0.001 |
| Antigen Presentation | |||
| CD86 | NM_175862.3 | −14.94 | 0.003 |
| HLA-DPA1 | NM_033554.2 | −3.5 | 0.047 |
| HLA-DQA1 | NM_002122.3 | −56.6 | 0.020 |
| HLA-DQB1 | NM_002123.3 | −23.6 | 0.038 |
| HLA-DRA | NM_019111.3 | −4.01 | 0.044 |
| MR1 | NM_001531.2 | −2.19 | 0.042 |
| Complement | |||
| C1R | NM_001733.4 | −4.1 | 0.009 |
| C3 | NM_000064.2 | −3.17 | 0.020 |
| C7 | NM_000587.2 | 39.95 | 0.025 |
| Metabolism | |||
| GPI | NM_000175.2 | −2.45 | 0.041 |
| HPRT1 | NM_000194.1 | −2.56 | 0.033 |
| LGALS3 | NM_001177388.1 | −2.32 | 0.019 |
| OAS3 | NM_006187.2 | −9.77 | 0.048 |
| Migration and Adhesion | |||
| APP | NM_000484.3 | −2.48 | 0.032 |
| BST2 | NM_004335.2 | −4.6 | 0.040 |
| CD37 | NM_001774.2 | −7.96 | 0.037 |
| CD44 | NM_001001392.1 | −2.67 | 0.025 |
| CD99 | NM_002414.3 | −3.23 | 0.003 |
| CX3CL1 | NM_002996.3 | −2.91 | 0.039 |
| CXCL16 | NM_001100812.1 | −2.35 | 0.034 |
| CXCR4 | NM_003467.2 | −3.15 | 0.038 |
| ITGB4 | NM_001005731.1 | −11.07 | 0.049 |
| MUC1 | NM_001018017.1 | −19.27 | 0.018 |
| TMUB2 | NM_024107.2 | −3.32 | 0.033 |
| VCAM1 | NM_001078.3 | −10.18 | 0.017 |
| Phagocytosis | |||
| ANXA1 | NM_000700.1 | −5.01 | 0.003 |
| CD276 | NM_001024736.1 | −3.01 | 0.023 |
| FCGR1A | NM_000566.3 | −8.02 | 0.026 |
| LAMP2 | NM_001122606.1 | −2.76 | 0.026 |
| Pro-inflammatory Response | |||
| CD48 | NM_001778.2 | −7.28 | 0.045 |
| CD58 | NM_001779.2 | −4.79 | 0.031 |
| CFI | NM_000204.3 | −4.12 | 0.003 |
| ELK1 | NM_005229.3 | −2.79 | 0.019 |
| IL15RA | NM_002189.2 | −6.88 | 0.046 |
| IL6ST | NM_002184.2 | −2.94 | 0.016 |
| IRF5 | NM_002200.3 | −6.29 | 0.038 |
| LY96 | NM_015364.2 | −21.64 | 0.008 |
| MICA | NM_000247.1 | −21.43 | 0.045 |
| MYD88 | NM_002468.3 | −2.62 | 0.029 |
| NFKB1 | NM_003998.2 | −3.56 | 0.039 |
| NFKBIA | NM_020529.1 | −2.24 | 0.023 |
| RIPK2 | NM_003821.5 | −3.2 | 0.047 |
| TLR3 | NM_003265.2 | −4.91 | 0.007 |
| TLR4 | NM_138554.2 | −4.05 | 0.047 |
| TLR6 | NM_006068.2 | −2.18 | 0.047 |
| TNFRSF10C | NM_003841.3 | −9.92 | 0.039 |
| TNFRSF1A | NM_001065.2 | −2.66 | 0.031 |
| TNFSF13B | NM_006573.4 | −3.67 | 0.033 |
| TNFSF8 | NM_001244.3 | −7.57 | 0.021 |
| Survival and Differentiation | |||
| BCL10 | NM_003921.2 | −2.09 | 0.033 |
| BCL6 | NM_001706.2 | −2.16 | 0.012 |
| DUSP6 | NM_001946.2 | −2.75 | 0.031 |
| MAP4K2 | NM_004579.2 | −3.16 | 0.048 |
| PTPRC | NM_080921.3 | −3.49 | 0.038 |
| RORA | NM_134261.2 | −4.41 | 0.014 |
| STAT5B | NM_012448.3 | −3.04 | 0.013 |
| ZNF346 | NM_012279.2 | −2.64 | 0.012 |
| Tissue Modeling and Repair | |||
| FOXJ1 | NM_001454.3 | −41.34 | 0.003 |
| LRP1 | NM_002332.2 | −2.26 | 0.019 |
| PDGFC | NM_016205.2 | −3.72 | 0.024 |
| SYT17 | NM_016524.2 | −22.94 | 0.002 |
Few immune-related genes are altered in recurrent GBM tumors
To determine the impact of prior resection, or chemotherapy, and radiation treatment on immune factors in the GBM TME we performed the same Nanostring analysis comparing patients with newly diagnosed disease to those with recurrent GBM who had not been given steroids for at least 2 weeks prior). Despite a slight increase in CD163 staining in recurrent tumors (Supp Figure 3B), we found significant differences in the expression of just 8 genes (Table 2), including HAMP, a protein necessary for iron storage in macrophages.
Table 2.
Comparison of recurrent versus newly diagnosed GBM tumors using the Nanostring Pan-Cancer Immune panel. All patients were Dex-naïve at the time of surgery. Of 770 genes on the panel, 527 had average raw counts greater than 50. Eight of these genes have a p-value less than 0.05 and fold change greater than 2. All 8 show loss of mRNA expression in recurrent tumors.
| Gene Name | Accession # | Fold Change Recurrent vs Newly Diagnosed |
P value | Function |
|---|---|---|---|---|
| CHIT1 | NM_003465.2 | −2.23 | 0.001 | Innate Immune Response |
| TLR2 | NM_003264.3 | −2.3 | 0.028 | Pro-inflammatory |
| MR1 | NM_001531.2 | −2.36 | 0.045 | Antigen Presentation |
| THY1 | NM_006288.2 | −3.95 | 0.026 | Immune Regulation |
| CFB | NM_001710.5 | −4 | 0.029 | Innate Immune Response |
| SAA1 | NM_199161.1 | −6.93 | 0.046 | Innate Immune Response |
| CFI | NM_000204.3 | −7.91 | 0.021 | Pro-inflammatory |
| HAMP | NM_021175.2 | −31.28 | 0.026 | Innate Immune Response |
MHC-II and CD86 protein levels are unchanged by pre-operative dex-treatment
Antigen presentation by macrophages and microglia to T cells is critical for activation of an anti-tumor immune response. Having found decreased expression of proteins critical to this process in our Dex-focused Nanostring analysis (Table 1), we immunostained GBM tissue sections for CD86 and MHC class II. Inconsistent with our Nanostring analysis, we saw no change in the percent area positive for either marker in GBM tissue, regardless of Dex treatment (Figure 5A, B). Explanations for this observation include the insensitivity of IHC staining of fixed tumor tissues that may yield a false negative of variation in protein expression, despite having a functional impact on antigen presentation and T cell activation, as well as port-transcriptional regulation of protein expression that has been demonstrated for other immune activating proteins such as IFNγ.52 Of note, immunofluorescent staining revealed that HLA-DR, DP, DQ expression was restricted to CD163+ cells, and not GFAP+ astrocytes within GBM tumors (Figure 5C).
Figure 5.
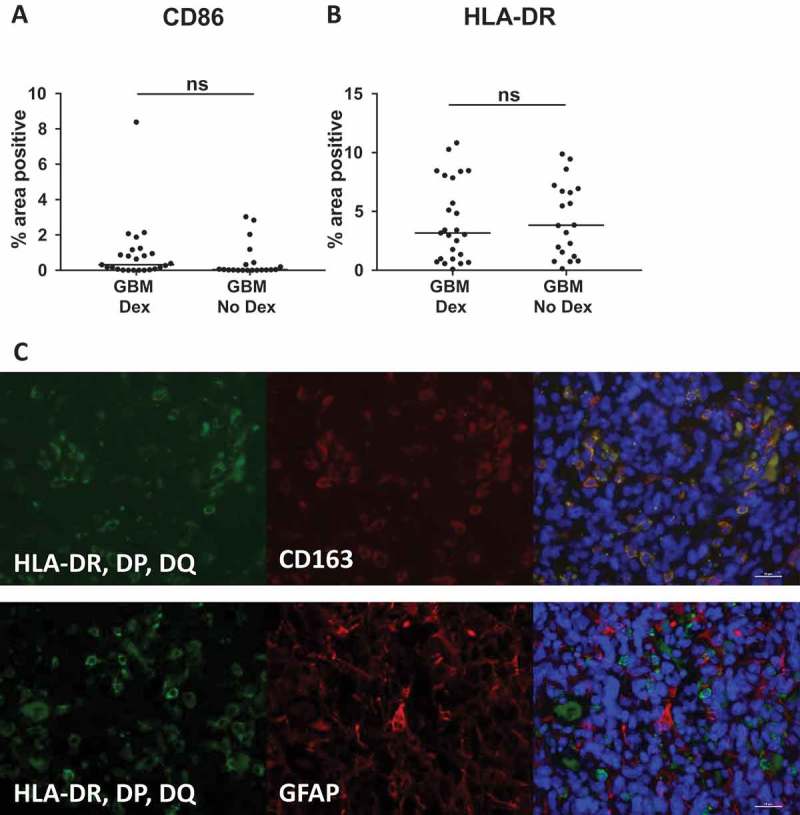
MHC-II and CD86 protein levels are unchanged by pre-operative Dex-treatment. Quantitative image analysis of FFPE samples used in Nanostring analysis (Table 1) reveal no difference in (A) HLA-DR, DP, DQ or (B) CD86 protein expression. Line indicates median. (C) Immunofluorescent staining for HLA-DR, DP, DQ (green) and CD163 (red, top image) or GFAP (red, bottom image) show that MHC-II is restricted to CD163+ cells and is not expressed by GFAP+ tumor cells or astrocytes.
Dex-treatment, but not tumor grade, influences circulating monocyte phenotype
To determine whether steroid treatment impacts protein expression in circulating monocytes, we ran flow cytometry on freshly isolated, RBC-lysed blood from healthy donors (HD), and patients with low or high-grade gliomas. Dex-dependent enrichment of the HLA-DR low/- monocyte population has been shown previously in patients with GBM.16 Here we confirm these findings and show that HLA-DR/DP/DQ loss in Dex-treated patients is independent of disease grade (Figure 6A). Consistent with the fact that the gene for CD163 is glucocorticoid-regulated,18,22,29 we found that CD163 was expressed on a higher percentage of circulating CD14+ monocytes in Dex-treated low and high-grade glioma patients (Figure 6B). In contrast, we see no change in the frequency of the commonly used MDSC marker CD33 (Figure 6C) or the T cell suppressive marker PD-L1 (Figure 6D), indicating that Dex doesn’t universally suppress surface protein expression, but may have specific targets. Collectively, these data suggest that many of the monocytic markers associated with immunosuppressive functions in tumor patients may be independent of disease grade, but instead may be caused by perioperative steroid treatment. For example, patients on concomitant steroid treatment may have artifactual reduction in MHC class II expression, a hallmark for monocytic myeloid derived suppressor cells.
Figure 6.
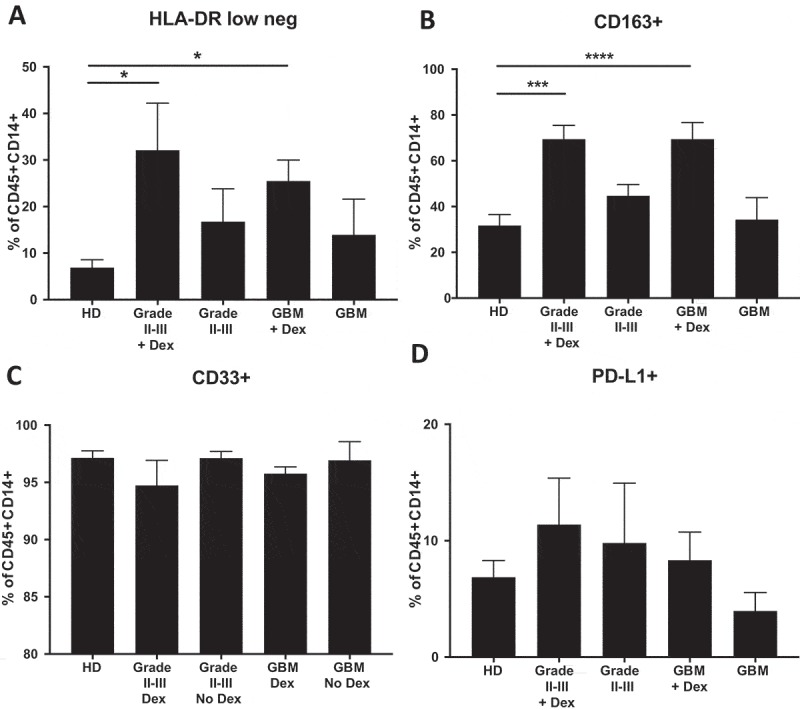
Dex-treatment, but not tumor grade, influences circulating monocyte phenotype. Fresh, RBC-lysed blood from healthy donors (HD), and Dex-treated and naïve grade II-III glioma, and GBM patients was immunostained for myeloid markers (A) HLA-DR, (B) CD163, (C) CD33, and (D) PD-L1 and analyzed by flow cytometry. All frequencies are represented as the percent of CD45+, CD14+ cells. Line and error bars indicate mean and SEM.
Dex-treatment suppresses expression of myeloid markers in vitro
To determine if steroid treatment would universally impact terminally differentiated myeloid cell phenotype independently of disease, we differentiated macrophages from monocytes isolated from 3 healthy donors using previously described methods53 and treated them for 24 hours with 0.1, 1, 10 μM Dex. Dexamethasone concentrations were selected based on previously reported concentrations in serum and tissues of cancer patients using HPLC.16 Flow cytometry analysis revealed that the decreases in MHC-II expression found in Dex-treated GBM tumors (Table 1) and monocytes isolated from Dex-treated patients (Figure 6A) could be reproduced in healthy donor macrophages following steroid treatment (Figure 7A). Similarly, steroid treatment in vitro was sufficient to induce a decrease in other components of antigen presentation including CD86, CD80, CD40, HLA-ABC (MHC-I), as well as the MCP-1 receptor CCR2 (Figure 7B-F). Interestingly, PD-L1 expression was also decreased on in vitro Dex-treated macrophages (Figure 7G). As with circulating monocytes isolated from steroid treated patients, CD163 was up-regulated in macrophages, although this observation was consistent in only 2 of 3 healthy donors (Figure 7H), suggesting that there may be variability in donor-dependent responsiveness to this drug. Taken together these data indicate that in vitro Dex treatment is sufficient to recapitulate our clinical observation.
Figure 7.
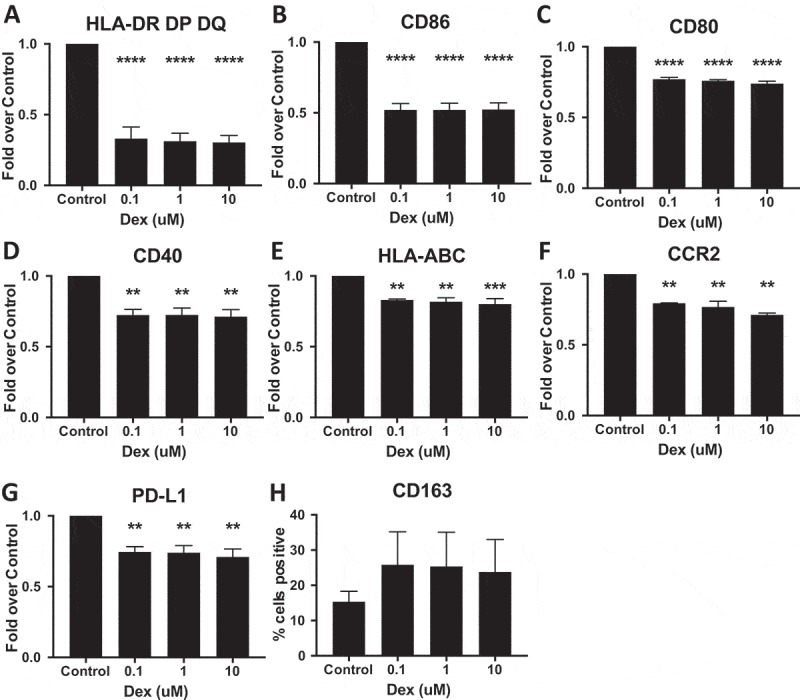
Dex-treatment suppresses expression of myeloid markers in vitro. Monocyte derived macrophages were treated with 0.1, 1, 10 μM Dex for 24 hours, then detached and immunostained for (A) HLA-DR, DP, DQ (MHC-II), (B) CD86, (C) CD80, (D) CD40, (E) HLA-ABC (MHC-I), (F) CCR2 (G) PD-L1, and (H) CD163 and analyzed by flow cytometry. The background mean fluorescence intensity (MFI) of unstained samples was subtracted from experimental samples. Data was then expressed as the fold of treated MFI over untreated (control) MFI. P-values calculated with a one-way ANOVA followed by Dunnett’s multiple comparisons test comparing treated samples to control. Line indicates mean, error bars indicate SEM.
To determine if the impact of steroids on myeloid cell phenotype is permanent or reversible, we performed a washout experiment where GM-CSF-differentiated macrophages were treated for 24 hours with dexamethasone, then allowed to recover in growth media for six days. Cells were then analyzed by flow cytometry for the surface markers above. We found that HLA-DR DP DQ, CD86, CD80, CD40, CCR2, PD-L1 all returned to pre-treatment levels following wash out, whereas CD163 expression remained elevated (Figure 8). Collectively our findings support the need to stratify patients participating in immune modulating therapies, as well as to improve methods to delineate myeloid cell subsets that are linked to immunosuppression and disease progression when seeking therapeutic targets.
Figure 8.
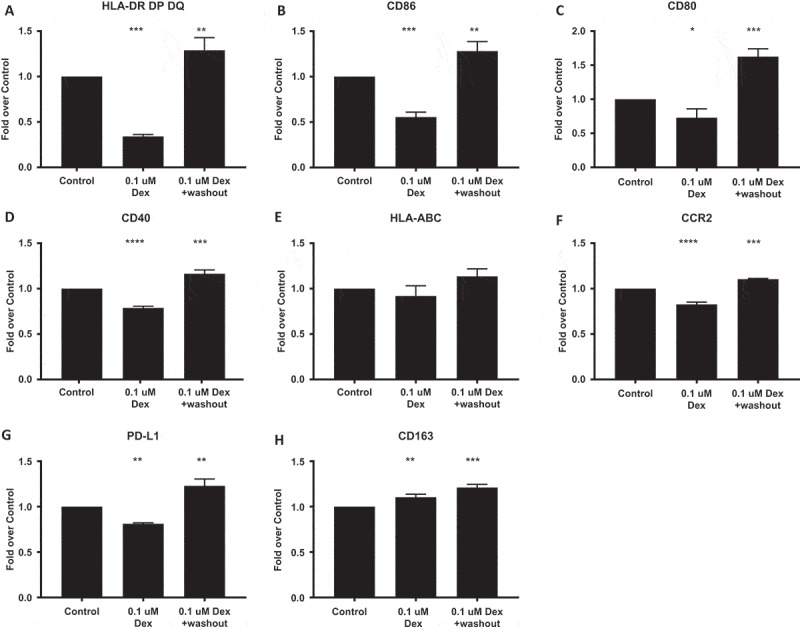
GM-CSF-differentiated macrophages were treated for 24 hours with dexamethasone, then allowed to recover in growth media for six days. Cells were then detached and immunostained for (A) HLA-DR, DP, DQ (MHC-II), (B) CD86, (C) CD80, (D) CD40, (E) HLA-ABC (MHC-I), (F) CCR2 (G) PD-L1, and (H) CD163 and analyzed by flow cytometry. The background mean fluorescence intensity (MFI) of unstained samples was subtracted from experimental samples. Data was then expressed as the fold of treated MFI over untreated (control) MFI. P-values calculated with a one-way ANOVA followed by Dunnett’s multiple comparisons test comparing treated samples to control. Line and error bars indicate mean and SEM.
Discussion
The plasticity of monocytes and macrophages in the circulation and tumor microenvironment of solid tumor patients has been described in recent years, following several successes in immunotherapy clinical trials that indicate the importance of myeloid cell activation in supporting anti-tumor immune responses.54-61 Assessment of myeloid cell phenotypic surface markers may have utility in the fields of novel immunotherapy target identification,6 as well as biomarker discovery to reflect therapeutic efficacy, tumor burden, or treatment failure. Recent literature characterizing cells of the myeloid lineage in cancer patients and animal models highlights their phenotypic and functional variability,6 including those characterized as myeloid derived suppressor cells33 and differentiated macrophages.62,63 To our surprise, we didn’t find significant differences in the expression of the canonical MDSC marker CD33 either in circulation or in the tumor microenvironment. Although typically used to define immunosuppressive TAMs, we find that the number of cells expressing CD163 in glioma tissue is significantly increased in patients with both grade I astrocytoma and GBM. This is supported by previous findings showing that the number of tumor associated macrophages may be less significant than their gene expression and corresponding functions,64,65 and suggests that identification of additional markers is required to define a phenotype associated with immunosuppressive functions. This model is supported by the various redundant mechanisms of immunosuppression in the TME, including soluble GBM tumor cell derived factors such as TGFβ,66 LDH5,5 IL-10,67 and IDO.68 Surface proteins including PD-L1,69 and decreased MHC Class I expression on tumor cells70 that likely influence TAM phenotypes and functions independently of CD163 expression are found in GBM, but not in the tumors of patients with low-grade gliomas.
In this study, we also describe the effects of the frequently administered steroid, dexamethasone, on circulating monocytes and tumor infiltrating macrophages in patients with glioma. In the interest of identifying clinical trial candidates most likely to benefit from immunotherapies, we hypothesized that dexamethasone, which is used to reduce swelling caused by local inflammation or edema prior to surgical resection of gliomas, likely affected circulating and infiltrating myeloid cell functions. While future studies with larger patient groups will be needed to confirm these findings, we show that Dex treatment suppressed the expression of MHC class II (HLA-DR) at the transcriptional and protein level. Given that this is a surface marker whose absence and low expression is typically used to define MDSCs,37 this suggests that myeloid plasticity, and transient suppression of MHC class II in response to steroids, could result in a false positive in quantification of MDSCs that are not functionally suppressive. We also found that several serum proteins and monocyte surface markers that may be candidates for biomarkers in circulation were affected by steroid treatment as opposed to tumor grade in glioma patients, suggesting that steroids may impact responsiveness to cutting edge immunotherapy approaches. Wehave shown that the impact of dexamethasone on myeloid cell phenotype is reversible suggesting any compromise on immunotherapeutic interventions may be avoided by limiting steroid treatment to the peri-operative window. Additionally, alternative approaches to treating cerebral edema, such as the use of VEGF antagonists, are being explored and may improve survival in glioblastoma patients when co-administered with other therapies14
The CCR2-MCP-1 axis has long been defined as the mechanism for monocyte recruitment from the bone marrow to the tumor microenvironment.40 In glioma patients receiving steroid treatment, however, it seems likely that additional mechanisms for recruitment exist, as tumor samples analyzed by IHC did not show increased CCL2/MCP-1 expression, nor did serum analysis reveal elevated CCL2/MCP-1, although the short half -life and rapid internalization of CCL-2 may make it difficult to readily detect in patient sera. In patients with GBM, it is also possible that recruitment to the tumor site is in part the result of tissue resident myeloid cells, such as microglia, endothelial cells, or other infiltrating leukocytes that promote the accumulation of functionally suppressive macrophages in the microenvironment of patients with GBM. Finally, it is not clear whether monocyte and macrophage accumulation is continuous. Several studies seeking to eliminate tumor-associated macrophages suggest that the process is dynamic, in which tumor associated macrophages are rapidly replaced by continuously infiltrating monocytes that differentiate in situ. The longevity of tissue resident macrophages, however, may suggest a different model in which tumor associated macrophage accumulation is dependent on filling a niche early in tumorigenesis. Following establishment of a resident population sufficient to support tumor growth, vascularization, and diffusion, active recruitment wanes until additional resources are needed to support tumor growth.
It is important to note that despite the impact of steroids on many of the canonical phenotypic markers associated with suppressive functions of macrophages, including molecules important for activating T cells, such as HLA-DR and activating costimulatory proteins, we do not propose that dexamethasone treatment alone suppresses pro-inflammatory macrophage functions in GBM patients. As shown in Figure 1E, and described previously,71 the IDH mutation status is a known regulator of gene expression in the tumor microenvironment of glioma patients. Additionally, a defining pathological feature of GBM tumors is necrosis,72 which is associated with neutrophil accumulation.73 As short-lived polymorphonuclear cells, neutrophils are well-established modulators of macrophage functions in non-oncologic immune responses. Specifically, ingestion of neutrophils in areas of necrosis delivers feedback to local macrophages that immune responses have caused tissue damage that needs repair. Given their role in promoting macrophage wound healing phenotypes and functions, we hypothesize that necrotic areas in GBM tumors and the associated signaling cascades that they activate also contribute to the complex interactions between tumor, immune, and stromal cells in gliomas.
The impact of dexamethasone on macrophage functions is not entirely surprising given the known mechanisms of steroid activity, including chromatin remodeling, gene specificity, and duration of effects.20,21,74 The effects described in this study are timely, and have a potential impact on GBM treatment, given the recent development of several immunotherapy approaches predicated on activation of innate immune cell functions, including macrophages that reside within the tumor microenvironment. As a result of the transient nature and reversibility of steroid mediated immune suppression, as well as the plasticity of macrophages found in the tumor microenvironment, patients on steroids are likely good candidates for immunotherapy approaches if weaning below immunomodulatory doses are included in the clinical protocols to restore the antigen presentation and immune activating functions of monocytes and macrophages in patients with gliomas. The involvement of macrophages in supporting immune cell activation and functions in the tumor microenvironment suggest that the consideration of peri-operative treatments and concomitant therapies of patients may therefore enhance efficacy of existing experimental immunotherapies, including checkpoint blockades, recombinant protein immune cell activators, and adoptively transferred, genetically modified NK and T cells.
Funding Statement
This work was supported by the Sponsored Research Agreement Bristol Myers Squibb; Steven Higgins Brain Tumor Research Fund.
Supplementary material
Supplemental data for this article can be accessd here.
References
- 1.Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Glass R, Synowitz M.. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathologica. 2014;128:347–362. doi: 10.1007/s00401-014-1274-2. [DOI] [PubMed] [Google Scholar]
- 3.Campesato LF, Silva APM, Cordeiro L, Correa BR, Navarro FCP, Zanin RF, Marçola M, Inoue LT, Duarte ML, Molgora M, et al. High IL-1R8 expression in breast tumors promotes tumor growth and contributes to impaired antitumor immunity. Oncotarget. 2017;8:49470–49483. doi: 10.18632/oncotarget.17713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol. 2012;14:584–595. doi: 10.1093/neuonc/nos014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, Fong L, Campbell MJ, Cooper S, Oakes SA, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:12823–12828. doi: 10.1073/pnas.1413933111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, Moretta L, Mantovani A, Moretta A, Bottino C TLR activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of NK cells. Eur J Immunol. 2014;44:1814–1822. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chimal-Ramirez GK, Espinoza-Sanchez NA, Chavez-Sanchez L, Arriaga-Pizano L, Fuentes-Panana EM. Monocyte differentiation towards protumor activity does not correlate with M1 or M2 phenotypes. J immun res. 2016;2016:6031486. doi: 10.1155/2016/6031486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yarchoan M, Xing D, Luan L, Xu H, Sharma RB, Popovic A, Pawlik TM, Kim AK, Zhu Q, Jaffee EM, et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res. 2017;23:7333–7339. doi: 10.1158/1078-0432.CCR-17-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge H, Mu L, Jin L, Yang C, Chang YE, Long Y, DeLeon G, Deleyrolle L, Mitchell DA, Kubilis PS, et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int J Cancer. 2017;141:1434–1444. doi: 10.1002/ijc.30830. [DOI] [PubMed] [Google Scholar]
- 11.Zhu C, Kros JM, Van Der Weiden M, Zheng P, Cheng C, Mustafa DA. Expression site of P2RY12 in residential microglial cells in astrocytomas correlates with M1 and M2 marker expression and tumor grade. Acta Neuropathologica Communications. 2017;5:4. doi: 10.1186/s40478-016-0405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu C, Mustafa D, Zheng -P-P, Van Der Weiden M, Sacchetti A, Brandt M, Chrifi I, Tempel D, Leenen PJM, Duncker DJ, et al. Activation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression. Neuro Oncol. 2017;19:648–659. doi: 10.1093/neuonc/now251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, Watchmaker PB, Yagnik G, Di Lullo E, Malatesta M, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M, Chan TA, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139:1458–1471. doi: 10.1093/brain/aww046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitadze G, Flüh C, Quabius ES, Freitag-Wolf S, Peters C, Lettau M, Bhat J, Wesch D, Oberg -H-H, Luecke S, et al. In-depth immunophenotyping of patients with glioblastoma multiforme: impact of steroid treatment. Oncoimmunology. 2017;6:e1358839. doi: 10.1080/2162402X.2017.1358839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, Dietz AB. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12:631–644. doi: 10.1093/neuonc/noq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buechler C, Ritter M, Orsó E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. 2000;67:97–103. [PubMed] [Google Scholar]
- 19.Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170–182. doi: 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. 2011;286:38703–38713. doi: 10.1074/jbc.M111.275370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immun. 2017;17:233–247. doi: 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heideveld E, Hampton-O’Neil LA, Cross SJ, van Alphen FPJ, van den Biggelaar M, Toye AM, van den Akker E Glucocorticoids induce differentiation of monocytes towards macrophages that share functional and phenotypical aspects with erythroblastic island macrophages. Haematologica. 2018;103(3):395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, Heber-Katz E, Curtis MT, Cotzia P, Hooper DC. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013;19:3776–3786. doi: 10.1158/1078-0432.CCR-12-1940. [DOI] [PubMed] [Google Scholar]
- 24.Johnson DR, Brown PD, Galanis E, Hammack JE. Pilocytic astrocytoma survival in adults: analysis of the surveillance, epidemiology, and end results program of the national cancer institute. J Neurooncol. 2012;108:187–193. doi: 10.1007/s11060-012-0829-0. [DOI] [PubMed] [Google Scholar]
- 25.Killela PJ, Pirozzi CJ, Reitman ZJ, Jones S, Rasheed BA, Lipp E, Friedman H, Friedman AH, He Y, McLendon RE, et al. The genetic landscape of anaplastic astrocytoma. Oncotarget. 2014;5:1452–1457. doi: 10.18632/oncotarget.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Im JH, Hong JB, Kim SH, Choi J, Chang JH, Cho J, Suh C-O. Recurrence patterns after maximal surgical resection and postoperative radiotherapy in anaplastic gliomas according to the new 2016 WHO classification. Sci Rep. 2018;8:777. doi: 10.1038/s41598-017-19014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(42–56):e46. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathologica. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 29.Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, Mack M, Roth J, Sunderkoetter C. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. 2008;84:644–650. doi: 10.1189/jlb.1107768. [DOI] [PubMed] [Google Scholar]
- 30.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raychaudhuri B, Rayman P, Ireland J, et al Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13:591–599. doi: 10.1093/neuonc/nor042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, Loeb M, Bramson JL, Bowdish DME. Blood CD33(+)HLA-DR(-) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol. 2013;93:633–637. doi: 10.1189/jlb.0912461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J immun. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafson MP, Lin Y, Maas ML, Van Keulen VP, Johnston PB, Peikert T, Gastineau DA, Dietz AB, Unutmaz D. A method for identification and analysis of non-overlapping myeloid immunophenotypes in humans. PloS one. 2015;10:e0121546. doi: 10.1371/journal.pone.0121546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, Greten TF, Korangy F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology. 2012;136:176–183. doi: 10.1111/j.1365-2567.2012.03566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathologica. 1997;93:518–527. [DOI] [PubMed] [Google Scholar]
- 41.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, Zhang J, Xu S, Li T, Zhang P, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget. 2016;7:80521–80542. doi: 10.18632/oncotarget.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, Brown JM. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18:797–806. doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuratsu J, Yoshizato K, Yoshimura T, Leonard EJ, Takeshima H, Ushio Y. Quantitative study of monocyte chemoattractant protein-1 (MCP-1) in cerebrospinal fluid and cyst fluid from patients with malignant glioma. J Natl Cancer Inst. 1993;85:1836–1839. [DOI] [PubMed] [Google Scholar]
- 45.Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salacz ME, Kast RE, Saki N, Bruning A, Karpel-Massler G, Halatsch ME. Toward a noncytotoxic glioblastoma therapy: blocking MCP-1 with the MTZ Regimen. Onco Targets Ther. 2016;9:2535–2545. doi: 10.2147/OTT.S100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S, Iwama T, Takami T Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol. 2009;34:1621–1627. [DOI] [PubMed] [Google Scholar]
- 49.Penton-Rol G, Cota M, Polentarutti N, Luini W, Bernasconi S, Borsatti A, Sica A, LaRosa GJ, Sozzani S, Poli G, et al Up-regulation of CCR2 chemokine receptor expression and increased susceptibility to the multitropic HIV strain 89.6 in monocytes exposed to glucocorticoid hormones. J immun. 1999;163:3524–3529. [PubMed] [Google Scholar]
- 50.Yeager MP, Pioli PA, Collins J, Barr F, Metzler S, Sites BD, Guyre PM. Glucocorticoids enhance the in vivo migratory response of human monocytes. Brain Behav Immun. 2016;54:86–94. doi: 10.1016/j.bbi.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Findling JW, Raff H. DIAGNOSIS OF ENDOCRINE DISEASE: differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur J Endocrinol. 2017;176:R205–R216. doi: 10.1530/EJE-16-0946. [DOI] [PubMed] [Google Scholar]
- 52.Villarino AV, Katzman SD, Gallo E, Miller O, Jiang S, McManus MT, Abbas AK. Posttranscriptional silencing of effector cytokine mRNA underlies the anergic phenotype of self-reactive T cells. Immunity. 2011;34:50–60. doi: 10.1016/j.immuni.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moyes KW, Lieberman NA, Kreuser SA, Chinn H, Winter C, Deutsch G, Hoglund V, Watson R, Crane CA Genetically engineered macrophages: a potential platform for cancer immunotherapy. Hum Gene Ther. 2017;28(2):200–215. [DOI] [PubMed] [Google Scholar]
- 54.Lawler SE, Chiocca EA. Oncolytic virus-mediated immunotherapy: a combinatorial approach for cancer treatment. J clin oncol. 2015;33:2812–2814. doi: 10.1200/JCO.2015.62.5244. [DOI] [PubMed] [Google Scholar]
- 55.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- 56.Liao JB, Swensen RE, Ovenell KJ, Hitchcock-Bernhardt KM, Reichow JL, Apodaca MC, D’Amico L, Childs JS, Higgins DM, Buening BJ, et al. Phase II trial of albumin-bound paclitaxel and granulocyte macrophage colony-stimulating factor as an immune modulator in recurrent platinum resistant ovarian cancer. Gynecol Oncol. 2017;144:480–485. doi: 10.1016/j.ygyno.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 57.Mitchell DA, Batich KA, Gunn MD, Huang M-N, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–369. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Penas-Prado M, Hess KR, Fisch MJ, Lagrone LW, Groves MD, Levin VA, De Groot JF, Puduvalli VK, Colman H, Volas-Redd G, et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro Oncol. 2015;17:266–273. doi: 10.1093/neuonc/nou155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27:190–199. doi: 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- 60.Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, Volkmer AK, Volkmer J-P, Liu J, Lim JS, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126:2610–2620. doi: 10.1172/JCI81603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, D’Ambrosio A, Bruce JN, Parsa AT. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. 2017;23:3575–3584. doi: 10.1158/1078-0432.CCR-16-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang W, Li X, Zheng D, Zhang D, Peng X, Zhang X, Ai F, Wang X, Ma J, Xiong W, et al. Dynamic changes and functions of macrophages and M1/M2 subpopulations during ulcerative colitis-associated carcinogenesis in an AOM/DSS mouse model. Mol Med Rep. 2015;11:2397–2406. doi: 10.3892/mmr.2014.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016:1.(2). pii: e85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng W, Ren X, Zhang C, Cai J, Liu Y, Han S, Wu A. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology. 2016;86:2226–2234. doi: 10.1212/WNL.0000000000002770. [DOI] [PubMed] [Google Scholar]
- 66.Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I, Fontana A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J immun. 1989;143:3222–3229. [PubMed] [Google Scholar]
- 67.Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997;17:3217–3224. [PubMed] [Google Scholar]
- 68.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon K-S, Auffinger B, Tobias AL, Han Y, Lesniak MS. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18:6110–6121. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 70.Palesch D, Wagner J, Meid A, Molenda N, Sienczyk M, Burkhardt J, Münch J, Prokop L, Stevanovic S, Westhoff M-A, et al. Cathepsin G-mediated proteolytic degradation of MHC class I molecules to facilitate immune detection of human glioblastoma cells. Cancer Immun Immunother. 2016;65:283–291. doi: 10.1007/s00262-016-1798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31:774–786. doi: 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529–539. [DOI] [PubMed] [Google Scholar]
- 73.Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathologica. 1999;98:349–354. [DOI] [PubMed] [Google Scholar]
- 74.Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.