

ABSTRACT
The role of T cell receptor (TCR) signaling for adaptive immune responses is essential. The ability to respond to a broad spectrum of tumor antigens requires an adaptive selection of various TCR. So far, little is known about the role of TCR richness and clonality in the cellular immune response to head and neck cancer (HNC), though the Endothelial Growth Factor Receptor (EGFR)-specific CD8+ T cell response can be enhanced by cetuximab therapy. Therefore, we investigated differences in TCR sequences between human papillomavirus (HPV)+ and HPV− HNC patients, as well as differences in TCR sequence characteristics between T cells of peripheral blood mononuclear cells (PBMC) and tumor infiltrating lymphocytes (TIL). Additionally, we were able to investigate the TCR richness and clonality in samples pre- and post- treatment in a prospective clinical trial of neoadjuvant cetuximab. Interestingly, HPV+ and HPV− HNSCC did not significantly differ in the extent of TCR clonality and richness in PBMC or TIL. However, neoadjuvant cetuximab treatment increased the number of unique TCR sequences in PBMC (p = 0.0003), which was more prominent in the clinical responder patients compared to non-responders (p = 0.04). A trend toward TCR gene focusing was observed in TIL (p = 0.1) post-treatment. Thus, an increase in richness of TCR sequences in the periphery with a focusing at the tumor site is associated with an improved treatment response, suggesting an influence of peripheral quantity and intratumoral quality on adaptive immunity in cetuximab treated patients.
KEYWORDS: TCR, immunosequencing, cetuximab, EGFR, cytotoxic T cells, head and neck cancer, immunotherapy
Introduction
Antigen recognition through the T cell receptor (TCR) is one of the primary determinants regulating adaptive immune responses. The HLA-associated presentation of tumor antigen (TA), together with costimulatory pathways and cytokines, is responsible for T cell activation. These TA are often specific neoantigens resulting from tumor specific mutations,1 and thereby introduce a high TCR and patient specificity. Additionally, many TA are overexpressed, shared (wild type) proteins, such as epidermal growth factor receptor (EGFR). Thus, immunotherapeutic approaches targeting the adaptive immune response highly depend on the recognition of these neoantigens or other shared overexpressed TA. Recent developments of checkpoint inhibitors have shown profound results in various tumor entities for many patients in advanced stages.2 Yet, only a fraction of the treated patients demonstrate clinical response.3 In part, therapeutic failure can be caused by insufficient recognition of TA-specific T cells dependent on novel protein sequences. HNC presents a useful tumor type to profile TA-specificity and TCR genotypes with clinical response, given the presence of neoantigens vs. viral sequences encoded by HPV− or HPV+ HNC, respectively. Indeed, there have been recent attempts to increase T cell specificity against shared TA and neoantigens.4
Physiologically, the variety of antigens is immense for foreign pathogens and, in the case of cancer, self-derived cellular components. Therefore, the requirements for diversity and yet specificity of antigen presentation and antigen detection are met through a specific recombination and selection process. In the case of TCR, the diversity is achieved through variation of the involved components of the TCR complex. The TCR α and β chains consist of three hypervariable complementarity-determining regions (CDR) that influence HLA antigen binding. The variable TCR regions of each somatic T cell undergo a genetic recombination of the DNA encoding segments, generating high diversity of the antigen recognizing components. The combination of high diversity in TCR and a clonal expansion of activated antigen specific cells allows for adaptive immune responses to a broad spectrum of TA. Recently, the technological advances allowed the introduction of “immunosequencing” in order to analyze specimens from peripheral blood or (tumor) tissue for specific quantification of TCR diversity.5 In particular, antigen specificity of T cells is mainly determined by the CDR region coding for the variable antigen binding pocket, CDR3. Hence, by analyzing PCR-based amplified CDR3 sequences of a patient sample by high throughput sequencing, the TCR immune receptor profile and clonal diversity can be determined.6 This allows for analysis of clonality and richness of patient TCR and more importantly, observation of intra-individual changes after immunotherapy as well as differences between the circulation and the tumor site. In tumor infiltrating lymphocytes (TIL), TA-specific T cells have been shown to express activation markers such as programmed death-1 (PD-1) or cytotoxic T lymphocyte associated protein-4 (CTLA-4).7 Immunotherapeutic approaches targeting these pathways have demonstrated good clinical responses in a variety of tumor types.8–11 Still, in the case of TA-specific immunity, the role of different treatment modalities on TCR properties and their therapeutic consequences is poorly understood.
In recent years, several TA-specific monoclonal antibodies (mAb) were approved for the treatment of various malignancies. Cetuximab is FDA approved for the treatment of patients with squamous cell carcinoma of the head and neck (HNC). We previously demonstrated that cetuximab is able to significantly enhance antigen presentation and NK:DC priming of adaptive immune responses in TIL.12 However, direct evidence for actual changes in adaptive immunity in relation to clinical response is lacking. Therefore, in this study we investigated the effect of cetuximab on the impact of TCR receptor and clinical implications utilizing a prospective neoadjuvant “window of opportunity” clinical trial of neoadjuvant cetuximab therapy (UPCI 08–013, NCT01218048), with a focus on changes in TCR diversity and clonality.
Results
Antigen specific T cells from HNC patients express exhaustion markers
In order to investigate the levels of activation and antigen experienced status in TA-specific CD8+ T cells,13,14 we utilized EGFR (853–861)-, MAGE(271–279)- and HPV-E7(11–20)- specific multimers and counter-stained for PD-1 and CTLA-4 to indicate antigen experienced status (HPV+ n = 18, HPV− n = 18). PD-1+ HLA-A2:EGFR(853–861)- and HLA-A2:MAGE(271–279)- specific T cells from both HPV+ and HPV− patients were significantly higher than PD-1 and CTLA-4 double positive cells or CTLA-4 single positive cells (Figure 1(a) and B, p < 0.05). Interestingly, differences in circulating, shared TA-specific T cells were not detected between HPV+ and HPV− HNC patients, suggesting the induction of polyclonal antigen specific immunity regardless of HPV status. While HPV− HNC patients showed no detectable staining for HPV-specific multimers (not shown), a subset of HPV-16 HLA-A2:E7(11–20)-specific T cells detected in the circulation of HPV+ patients were found to express PD-1 (8.5%, Figure 1(c)) as a marker of antigen experience and exhaustion. As PD-1+ T cells appear to be clonal expansions of anti-tumor effector cells in the tumor microenvironment,7 the relationship between TCR and anti-tumor efficacy was further investigated, using a broader method than multi-color flow cytometry.
Figure 1.
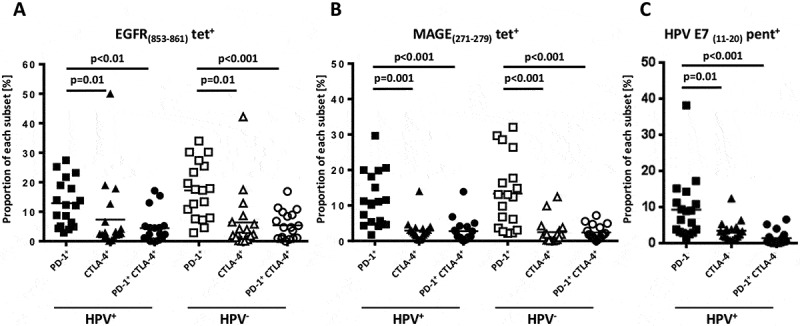
Antigen specific T cells of HNC patients express exhaustion markers. Antigen specific CD8+T cells were identified using EGFR (853–861)-, MAGE(271–279)- and HPV-E7(11–20)-specific tetramers/ pentamers and counter-stained with PD-1 and CTLA-4 (t test, HPV+ n = 18, HPV− n = 18).
Peripheral blood TCR characteristics among HNC patients
Pre- and post- treatment PBMC samples from 14 patients were analyzed for changes in TCR sequences, with a median of over one million sequences and over 70000 unique sequences generated per sample. Most samples were diverse, with some expanded clones, with a median clonality of 0.131. Figure 2(a) compares the similarity of these 28 different samples, based on sequence counts, demonstrating few similarities between individual patient PBMC. Sorted frequencies of clones decreased exponentially and CDR3 length showed Gaussian distribution among all samples (Figure 2(b)). Using a clone tracker tool, we exported the top ten amino acid sequences of each sample and tracked their presence across all 56 samples. Most of the top amino acid sequences (267/359) were shared in less than four samples. The heatmap of frequencies of variable gene families per samples displayed similar distributions across all 28 PBMC samples (Figure 2(c)).
Figure 2.
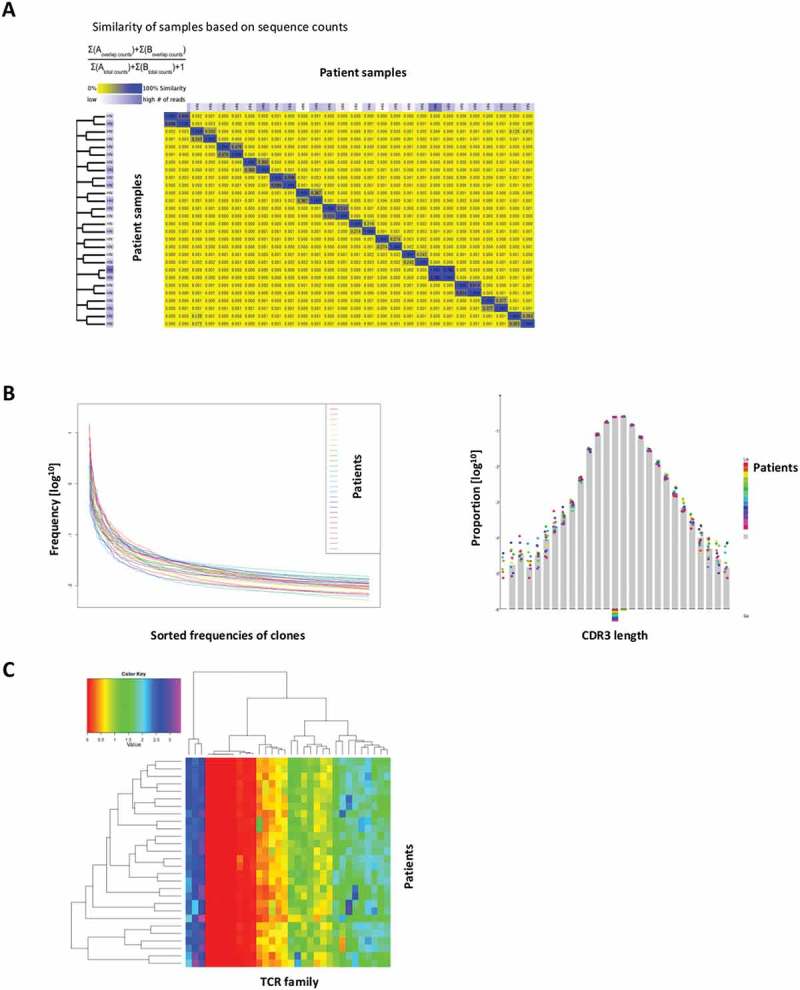
TCR characteristics among trial patients. (a) Sample similarity based on sequence counts per sample. (b) Sorted frequencies of clones per sample, CDR3 length distribution per sample. (c) Heatmap of frequencies of v families per samples.
HPV+ and HPV− patients demonstrate similar TCR richness and clonality
Reasons for the prognostic differences between HPV+ and HPV− HNC patients have been under investigation.15 Therefore, we investigated differences of TCR clonality and richness in these patients, as well as a comparison between pre- and post-cetuximab TCR’s from peripheral blood and TIL (n = 14 patients, 56 samples). Interestingly, HPV+ and HPV− HNC patients demonstrated comparable clonality frequencies for PBMC at baseline (p = 0.75) or post-treatment (p = 0.67), as well as in TIL (p = 0.75 and 0.28 for pre- and post-cetuximab treatment, Figure 3(a)). In PBMC, the HPV+ and HPV− patients also manifested no significant difference in TCR richness at both baseline and post-treatment (p = 0.66, Figure 3(b). Similar results were seen for TCR richness in TIL at both baseline (p = 0.11) and post-treatment (p = 0.85).
Figure 3.
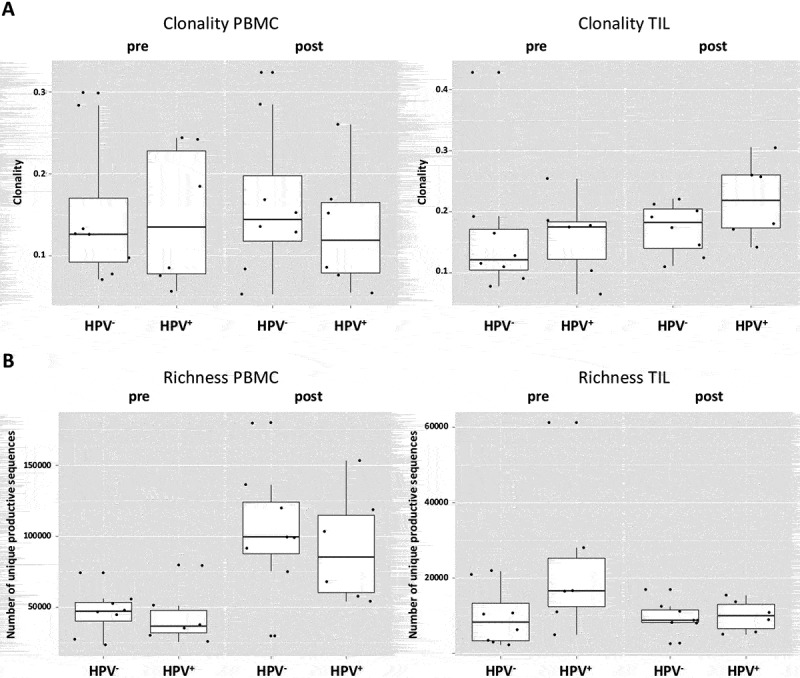
HPV+ and HPV− patients demonstrate similar TCR richness and clonality. (a) HPV+ and HPV− demonstrate comparable clonality frequencies for PBMC at baseline (p = 0.75) or post-treatment (p = 0.67), as well as in TIL (p = 0.75 and 0.28 for pre- and post-treatment). (b) In PBMC, the HPV+ and HPV− patients manifest no significant difference in TCR richness at both baseline and post-treatment (p = 0.66), similar results account for the richness In TIL at both baseline (p = 0.11) and post-treatment (p = 0.85).
Cetuximab treatment increases TCR richness in PBMC and is associated with clinical response
Since the induction of adaptive immunity has been observed in cetuximab-treated HNC patients,13 the effect of cetuximab treatment on TCR clonality in PBMC and TIL was analyzed. Changes in clonality of the PBMC samples before and after cetuximab treatment were moderate (Figure 4(a)). Neoadjuvant treatment with cetuximab significantly increased the richness of TCR in PBMC (p = 0.0003), with a more modest effect in the TCR richness in TIL (p = 0.15, Figure 4(b)). In TIL, there was a trend toward increased clonality after cetuximab treatment (p = 0.1). PBMC as well as TIL clonality and richness of TCR were investigated in cetuximab clinical responders and nonresponders. Responders and nonresponders did not differ in clonality either at baseline (p = 0.87) or post-treatment (p = 0.76) in PBMC as well as in TIL (p = 0.76 baseline; p = 0.64 post-treatment, Figure 5(a)). The richness in TIL did not differ between responders and nonresponders at either baseline or post-treatment time points (p = 0.85 and p = 0.76). However, significantly higher TCR richness in PBMC was observed in cetuximab responders at both baseline and post-treatment (p = 0.03, Figure 5(b)). Five out of 7 nonresponders and 4 out of 5 responders demonstrated an increase in TIL clonality post-treatment (Figure 6(a)). In cetuximab responders, the increase of TCR richness in the periphery is higher than in the nonresponder group (p = 0.04, Figure 6(b)).
Figure 4.
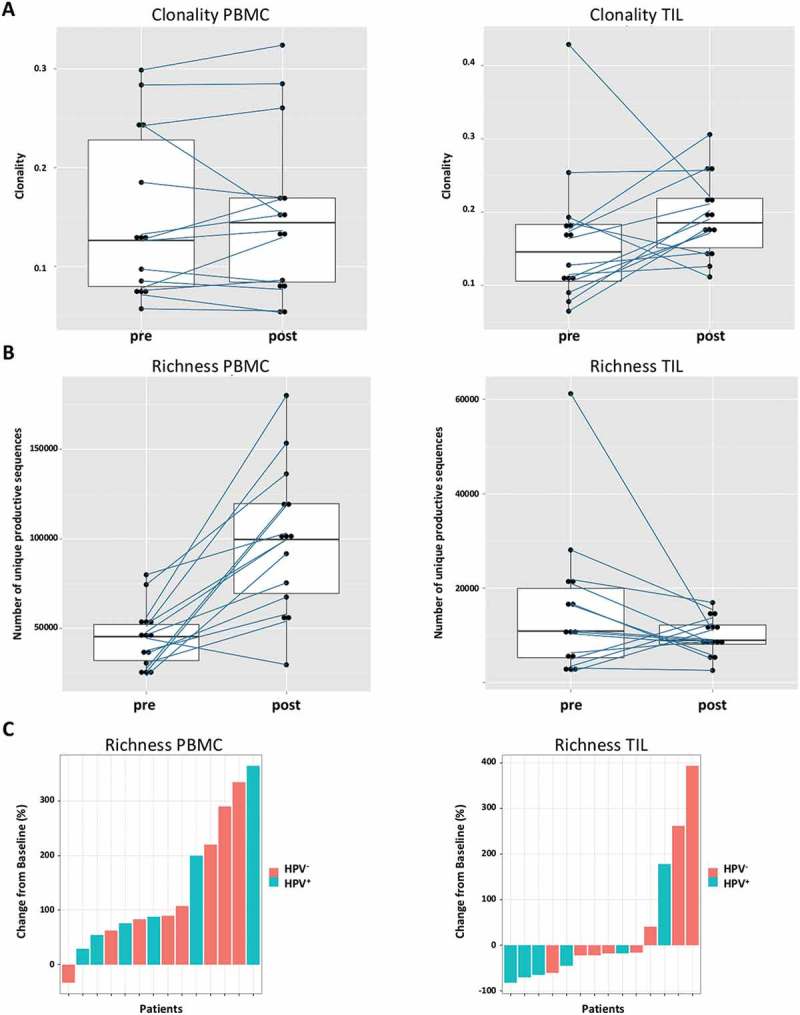
Cetuximab treatment increases TCR richness in PBMC. (a) The changes of clonality in the PBMC samples before and after treatment are moderate. In TIL, there is a trend of increased clonality after treatment (p = 0.1). (b) The treatment with cetuximab increases the richness of TCR in PBMC (p = 0.0003) in contrast to richness in TIL (p = 0.15). (c) Waterfall plots for changes of richness from baseline after treatment.
Figure 5.
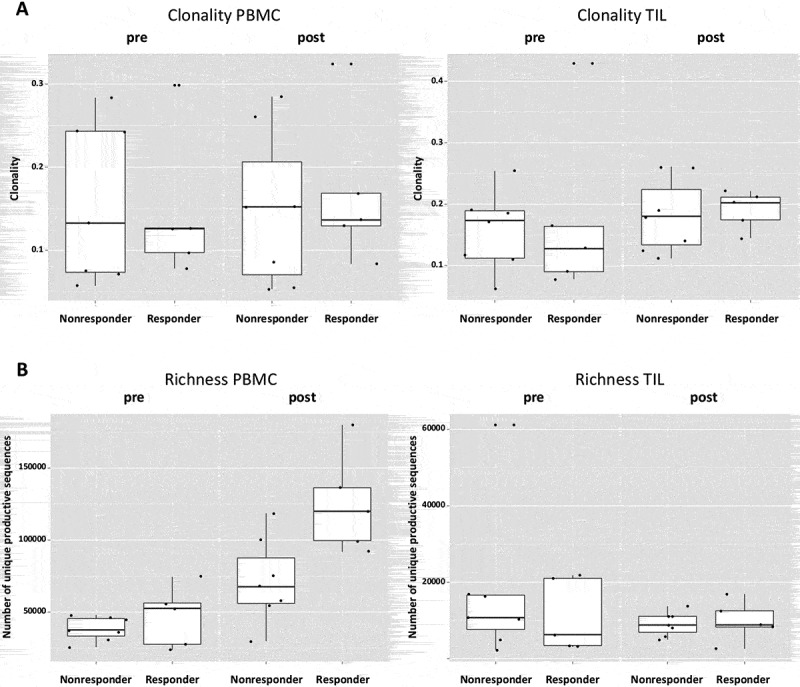
A higher TCR richness in PBMC is associated with treatment response. (a) Both responders and nonresponders do not differ in clonality either at baseline (p = 0.87) or post-treatment (p = 0.76) in PBMC as well as in TIL (p = 0.76 baseline; p = 0.64 post-treatment). (b) Cetuximab responders have higher TCR richness at both time points in PBMC (p = 0.03). The richness in TIL does not differ in responders and nonresponders at both time points (p = 0.85 and p = 0.76).
Figure 6.
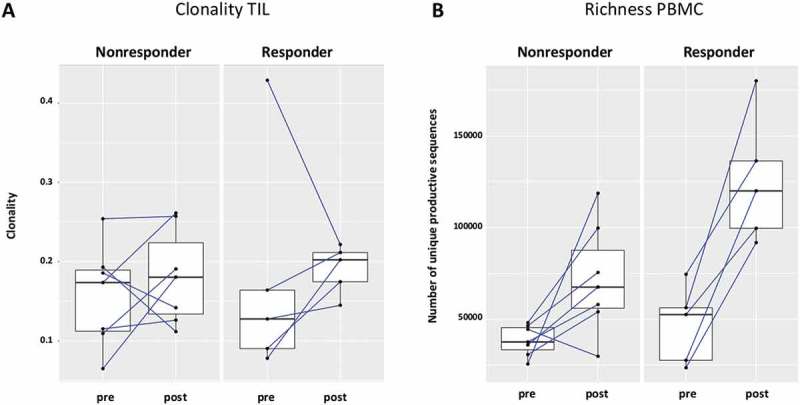
The increase of TCR richness is higher in cetuximab responders. (a) 5 out of 7 nonresponders and 4 out of 5 responders had increased clonality post-cetuximab treatment, the difference between responders and nonresponders did not reach significance (p = 0,76) (b) In the patients responding to cetuximab treatment, the change of TCR richness post-treatment is higher as compared to nonresponders (p = 0.04).
Discussion
The spectrum of TCR epitopes that are responsible for the TA formation is diverse, resulting from each tumor cell’s somatic mutations derived from genetic alterations. As such, even for tumors with similar histological origin, the “genomic landscape” can be highly variable.16 Similar to the response of foreign antigens, it is crucial to develop and maintain a diversified TCR repertoire for TA detection of a highly variable landscape. Therefore, this highly variable genomic landscape of tumor cells is shared by the TCR landscape of TIL,17 accounting for differences in magnitude, location and ratio of individual T cell subsets.18
Immunosequencing of TCR is a sensitive, highly accurate technology that is based on multiplex PCR high throughput sequencing.19 The possibility to quantify and specify the clonal distribution of lymphocytes in patient samples provides new insight in immunologic processes.20 In tumor immunology, it enables new ways of analyzing treatment responses in adaptive immunity, which is of importance as intratumoral T-cell populations have been demonstrated to crucially influence disease course.21,22 TCR analysis can be performed on multiple samples from the same patient simultaneously, providing an overview of distinct T cell compositions, depending on the tissue origin. So far, differences in PBL and in TIL have been observed.23 Still, factors that influence the landscape of TIL are poorly understood.
In this manuscript, we utilize TCR analysis not only for a single “snapshot” but compare TCR diversity and clonality at different time points, in order to observe possible therapeutic influences on the TCR landscape over time. Therapeutic interventions and their effect on the TIL clonality and diversity are of high interest, especially regarding recent developments of specific T cell modulations through immune checkpoint inhibitor therapy. Therefore, we investigated TCR characteristics of peripheral blood and TIL before and after systemic treatment with anti-EGFR mAb cetuximab, in particular to evaluate the suitability of cetuximab as an agent for combination with immunotherapeutic agents, such as checkpoint inhibitors.
In previous investigations, our group has demonstrated enhanced dendritic cell (DC)- mediated cross priming of CD8+ T cells, during treatment with cetuximab.12,24 Intriguingly, cetuximab therapy can induce clinical responses in the absence of mutations in EGFR,25 suggesting additional antitumor mechanisms besides downstream signaling blockade, such as antibody dependant cellular cytotoxicity (ADCC). Correale and colleagues demonstrated in vitro that cetuximab ameliorates DC-mediated phagocytosis of colon cancer cells, which thereby enhances CTL dependent anti-tumor response.26 Our group has demonstrated substantial cross-presentation through enhanced NK:DC cross talk,27 Cetuximab associated NK-cell induced DC maturation therefore influences TA cross-presentation.13,27,28 This effect on adaptive immunity was also observed by Lou and colleagues, who reported effects on T cell diversity after a combination of cetuximab treatment with chemotherapy in a subgroup of patients.29
Immune checkpoint receptors such as PD-1 and CTLA-4, whose blockade through mAbs recently showed significant response rates in otherwise pretreated patients,11,30 are upregulated on activated T cells. In the tumor microenvironment, T cells expressing PD-1 have been identified as clonal expansion of tumor reactive T cell populations, thus representing functionally important components in order to eliminate tumor cells.7 Furthermore, Gros and colleagues were able to recently identify PD1+ CD8+ T cells in circulation of three melanoma patients that targeted unique patient-specific neoantigens.31 Our findings, that tumor antigen specific T-cells (including EGFR, MAGE and HPV) in the periphery express PD-1 and CTLA-4, support these observations. Thus, circulating TA-specific T cells can be identified in peripheral blood with important consequences for the development of individualized TCR analyses and therefore personalized therapies. Additionally, these findings support the importance of circulating T cells and investigations of factors that influence their TA-specific characteristics, especially focusing on their TCR properties.
The importance of TCR diversity for immunologic competence has been demonstrated for various aspects of immune responses. In patients following hematopoietic stem cell transplantation, T cell repertoire recovery was observed through TCR sequencing. Van Heijst and colleagues report consistent findings of patient groups with increased TCR diversity and decreased risk of infection and cancer relapse.32 In patients following septic shock, TCR diversity is reduced and normalization of TCR diversity and PD-1 expression was reported to positively correlate with survival.33 In patients with hepatitis C virus (HCV) infection, Miles and colleagues observed that TCR repertoire diversity is associated with HCV clearance.34 Hence, TCR diversity is associated with a greater likelihood of a competent immune recognition, which is not restricted to foreign pathogens, with important consequences for cancer immunotherapy.
Our TCR analysis showed very low TCR similarities in between the samples based on sequence count and a normal distribution regarding CDR3 length, similar to reports about TCR analysis of different cancer types.35 This suggests that immunogenic epitopes are not frequently shared in between HNC patients, which is even more interesting regarding our observations and that of others that between HPV+ and HPV− patients, no differences could be observed for TCR richness and TCR clonality in both peripheral blood and TIL.36 Therefore, mutational variability accounts for both HPV+ and HPV− HNC patients, revealing no specific TCR clonotype for HPV+ patients that could explain improved therapeutic treatment responses in HPV+ patients so far.
Comparing TCR properties between TIL and PBMC, Jia and colleagues have shown that coding degenerate clonotypes in tumor tissue are significantly enriched.35 In primary liver carcinoma, TCR analysis was performed by Shi and colleagues in five different tumor regions of the same patient, revealing a higher frequency of the most abundant TIL clones in comparison to peripheral blood.37 At the same time, Shi and colleagues observed that intratumoral TCR clonality was distributed heterogeneously. In our results, we were able to observe an increased clonality in TIL in the responder group after cetuximab treatment, supporting the above described induction of an adoptive immune response through cetuximab treatment. Most importantly, TCR richness was significantly increased in the periphery after cetuximab treatment, with the highest increase in the responder group, supporting the hypothesis that an improved local tumor response is associated to an increase in peripheral TCR diversity. This might impose important therapeutic consequences, especially for possible combinational therapies. Park and colleagues observed in a combination of chemotherapy and the use of trastuzumab, an anti-Her2/neu mAb, that the induced immunity reaction through trastuzumab is reduced by the administration of chemotherapeutics.38
Additionally, CTLA-4 blockade itself was also shown to induce T cell repertoire evolution.39 Recently, effects of CTLA-4 blockade on the TCR repertoire in peripheral blood have been investigated.40 Robert and colleagues reported a diversification of the peripheral T cell pool after treatment with CTLA-4 blocking antibody (tremelimumab) in contrast to healthy donor PBMC. Similar to anti-CTLA-4 therapy, treatment with cetuximab enhances anti-tumor immunity in a subset of patients. Although therapeutic mechanisms of these two mAb are quite different, T cell responses against tumor cells are induced and/or activated.13 However, it is not yet fully understood how these mAb evoke adaptive immune responses beyond enhanced cross-presentation of tumor antigens through intratumoral DC. Increased T cell diversity in the peripheral circulation after cetuximab treatment and a higher amount of unique productive TCR sequences was observed, but expansion of specific TCR clones was not seen in predominance over the others, suggesting that this mode of therapy does not result in the specific detectable expansion of clonal T cells in the circulation. Most intriguingly, an increase in richness of TCR sequences in the periphery (PBMC) but not in TIL was associated with an improved treatment response to cetuximab, suggesting an influence of mAb induced peripheral quantity on intratumoral quality.
Our data are consistent with an immunological effect of cetuximab mediated by a broad T cell proliferation in the circulation. Prior findings suggest the induction of proliferation of T lymphocytes in lymphoid organs and to some extent at the tumor site, which may contribute to the observation of increased TCR richness in blood after treatment with cetuximab. The significant increase in diversity of TCR in peripheral blood may reflect a pharmacodynamic effect of the mAb therapy that is related to a general ability of that patient’s immune system to be activated, rather than a sole correlation with anti tumor immune responses, given the lack of association of TIL changes with clinical response. This finding may be related to insufficient power to detect such an effect. However, the correlation of TCR diversity in PBMC with clinical response could provide a novel biomarker for monitoring cetuximab treated patients in the clinic. Combinations with checkpoint receptor specific mAb would often appear promising. In conclusion, we report an increased post-treatment richness of T cells in the peripheral blood, without detectable expansion of specific clonal populations in the periphery, correlating with treatment response. This increased T cell diversity, together with their increased T cell activation state, may be mechanistically linked to the development of tumor responses in these cetuximab-treated patients.
Materials and methods
Patients and specimens
Patients with stage III/IV, previously untreated HNSCC who were candidates for surgical resection and had no prior history of head and neck cancer were recruited to this single-center, prospective phase II “window of opportunity” clinical trial (UPCI 08–013, NCT01218048). The study was approved by the Institutional Review Board (IRB) at the University of Pittsburgh and performed in accordance with Good Clinical Practice guidelines and the ethical principles outlined in the Declaration of Helsinki. All subjects provided written, informed consent before enrollment. Treatment consisted of 3–4 weekly doses of neoadjuvant cetuximab, followed by surgical resection within 36–48 hours of the last cetuximab dose received. Tumor and peripheral blood specimens were collected at screening and again at the time of surgery under an IRB-approved tissue banking protocol (UPCI 99–069).
Collection of PBMC and TIL
Peripheral blood (30–40 ml) was drawn into heparinized tubes and centrifuged on Ficoll–Hypaque gradients (#17144003, GE Healthcare Bioscience). Peripheral blood mononuclear cells (PBMC) were recovered, washed in RPMI-1640 (#R8758, Sigma) and immediately used for experiments or stored at −80 ºC until further analysis. For TIL isolation, freshly isolated tumors from HNC patients were minced into small pieces in RPMI-1640, which then were transferred to a 70-μm cell strainer and mechanically separated using a syringe plunger. The cells passing through the cell strainer were collected and subjected to Ficoll–Hypaque gradient centrifugation. After centrifugation, mononuclear cells were recovered and stored at −80 ºC until flow cytometry analysis.
Flow cytometry
For cell surface staining, PBMCs were washed twice in staining buffer (2% w/v fetal bovine serum, Corning) and stained for cell surface markers. Cells were incubated with relevant antibodies for 30 min at room temperature (RT) in the dark, washed twice and re-suspended in staining buffer. Flow cytometry was performed using a CyAn flow cytometer (Dako) and LSR Fortessa (Becton Dickinson) and data analyzed using FlowJo software (TreeStar, Inc.). The acquisition and analysis gates were restricted to the lymphocyte gate based on characteristic forward and side scatter properties of the cells.
Antibodies and reagents
The following anti-human monoclonal antibodies (mAb) were used for staining: CD3-Alexa Fluor 405 (#CD0326, Invitrogen), CD4-AF700 (#560836, BD Biosciences), PD-1-APC (#17–9969-41, eBioscience), CTLA-4 PE/Cy7 (#349914, Biolegend), and their respective isotype controls. PE-conjugated EGFR:853–861 tetramer and MAGE:271–279 tetramer were synthesized by the NIH Tetramer Facility (Emory University), and the HPV E7:11–20 Pentamer was commercially obtained (#095, ProImmune). Antibodies and tetramers were pre-titrated using activated as well as non-activated PBMC to determine optimal staining dilutions.
TCR genotyping
TCR genotyping was performed and quantified by Adaptive Biotechnologies, Inc. The maximum available number of samples from the same patient was 4, containing pre- and post-treatment PBMC and pre- and post-treatment TIL.
Statistics
ImmunoSEQ Analyzer (http://www.adaptivebiotech.com/immunoseq/analyzer) was used to retrieve, process and track the TCR sequence data. Pre- and post-treatment richness (S, i.e., the total number of unique productive sequences), Shannon Index (H = -log∑pi(1-pi), pi = the proportion of sequence i), clonality (1-H/log(S), range 0–1, 0 means polyclonal, 1 means monoclonal) were compared by the Wilcoxon Signed Rank tests. Comparisons between two independent groups (i.e. HPV+ vs. HPV−, or responders vs. non-responders) were accomplished with Wilcoxon Rank Sum tests. Frequencies of different T cells subpopulations within the same patient were compared using Wilcox Signed Rank. Data analyses were carried out using the R version 3.2.0.41 All tests are two-sided and a p-value of 0.05 or less is considered statistically significant.
Funding Statement
This work was supported by the HHS | National Institutes of Health (NIH) [R01 DE019727]; IFORES Program University Hospital Essen; University of Pittsburgh Cancer Center Support [P30 CA047904]; HHS | National Institutes of Health (NIH) [P50 CA097190].
Acknowledgments
We would like to thank Bratislav Janjic and Michael Meyer from the Hillman Cytometry Facility for excellent technical assistance. This work is supported by the IFORES program of University of Duisburg-Essen (BAK), NIH Grants R01 DE019727 (RLF) and P50 CA01970 (RLF). This work used the Hillman Biostatistics Facility and Cytometry Facility that are supported in part by award P30CA047904.
Disclosure statement
Robert L. Ferris has received research grants from Bristol Myers Squibb and AZ/Medimmune and has been a consultant/advisory board member for Celgene, Merck, BMS and AZ/Medimmune. The other authors declare no conflict of interest.
References
- 1.Schumacher TN, Schreiber RD.. Neoantigens in cancer immunotherapy. Science (New York, NY). 2015;348:69–74. DOI: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 2.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. DOI: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 3.Mooradian MJ, Sullivan RJ. Immunomodulatory effects of current cancer treatment and the consequences for follow-up immunotherapeutics. Future Oncol. 2017. DOI: 10.2217/fon-2017-0117. [DOI] [PubMed] [Google Scholar]
- 4.Ochi T, Nakatsugawa M, Chamoto K, Tanaka S, Yamashita Y, Guo T, Fujiwara H, Yasukawa M, Butler MO, Hirano N.. Optimization of T-cell reactivity by exploiting TCR chain centricity for the purpose of safe and effective antitumor TCR gene therapy. Cancer Immunology Research. 2015;3:1070–1081. DOI: 10.1158/2326-6066.CIR-14-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Mol Oncol. 2015. DOI: 10.1016/j.molonc.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calis JJ, Rosenberg BR. Characterizing immune repertoires by high throughput sequencing: strategies and applications. Trends Immunol. 2014;35:581–590. DOI: 10.1016/j.it.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. DOI: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. DOI: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. DOI: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, Hodi FS, Joshua AM, Kefford R, Hersey P, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34:4102–4109. DOI: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. DOI: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ming Lim C, Stephenson R, Salazar AM, Ferris RL. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR head and neck cancer cells. Oncoimmunology. 2013;2:e24677 DOI: 10.4161/onci.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, Lopez-Albaitero A, Gibson SP, Gooding WE, Ferrone S, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–1872. DOI: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH.. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res. 2013;73:1764–1776. DOI: 10.1158/0008-5472.CAN-12-3507. [DOI] [PubMed] [Google Scholar]
- 15.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. DOI: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science (New York, NY). 2013;339:1546–1558. DOI: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon P, Omokoko TA, Breitkreuz A, Hebich L, Kreiter S, Attig S, Konur A, Britten CM, Paret C, Dhaene K, et al. Functional TCR retrieval from single antigen-specific human T cells reveals multiple novel epitopes. Cancer Immunology Research. 2014;2:1230–1244. DOI: 10.1158/2326-6066.CIR-14-0108. [DOI] [PubMed] [Google Scholar]
- 18.Linnemann C, Mezzadra R, Schumacher TN. TCR repertoires of intratumoral T-cell subsets. Immunol Rev. 2014;257:72–82. DOI: 10.1111/imr.12140. [DOI] [PubMed] [Google Scholar]
- 19.Kirsch I. Immune monitoring technology primer: immunosequencing. Journal for Immunotherapy of Cancer. 2015;3:29 DOI: 10.1186/s40425-015-0076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nature Reviews Cancer. 2012;12:298–306. DOI: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 21.Balermpas P, Michel Y, Wagenblast J, Seitz O, Weiss C, Rodel F, Rodel C, Fokas E.. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br J Cancer. 2014;110:501–509. DOI: 10.1038/bjc.2013.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T, Kane LP, Ferris RL.. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015;75:508–518. DOI: 10.1158/0008-5472.CAN-14-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, Drescher CW, Tewari M, Bielas JH, Robins HS.. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol. 2013;231:433–440. DOI: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephenson RM, Lim CM, Matthews M, Dietsch G, Hershberg R, Ferris RL. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother. 2013;62:1347–1357. DOI: 10.1007/s00262-013-1437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA. Responsiveness to cetuximab without mutations in EGFR. N Engl J Med. 2005;353:208–209. DOI: 10.1056/NEJM200507143530218. [DOI] [PubMed] [Google Scholar]
- 26.Correale P, Botta C, Cusi MG, Del Vecchio MT, De Santi MM, Gori Savellini G, Bestoso E, Apollinari S, Mannucci S, Marra M, et al. Cetuximab ± chemotherapy enhances dendritic cell-mediated phagocytosis of colon cancer cells and ignites a highly efficient colon cancer antigen-specific cytotoxic T-cell response in vitro. Int J Cancer. 2012;130:1577–1589. DOI: 10.1002/ijc.26181. [DOI] [PubMed] [Google Scholar]
- 27.Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50:248–254. DOI: 10.1007/s12026-011-8231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, Ferris RL.. CD137 stimulation enhances cetuximab-induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin Cancer Res. 2017;23:707–716. DOI: 10.1158/1078-0432.CCR-16-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo W, He WT, Wen Q, Chen S, Wu J, Chen XP, Ma L.. Changes of TCR repertoire diversity in colorectal cancer after Erbitux (cetuximab) in combination with chemotherapy. Am J Cancer Res. 2014;4:924–933. [PMC free article] [PubMed] [Google Scholar]
- 30.Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jager D, Pietanza MC, Le DT, de Braud F, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–895. DOI: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 31.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nature Medicine. 2016;22:433–438. DOI: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Heijst JW, Ceberio I, Lipuma LB, Samilo DW, Wasilewski GD, Gonzales AM, Nieves JL, van den Brink MR, Perales MA, Pamer EG.. Quantitative assessment of T cell repertoire recovery after hematopoietic stem cell transplantation. Nature Medicine. 2013;19:372–377. DOI: 10.1038/nm.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomino A, Tsuda M, Aoki R, Kajita Y, Hashiba M, Terajima T, Kano H, Takeyama N.. Increased PD-1 expression and altered T cell repertoire diversity predict mortality in patients with septic shock: a preliminary study. PLoS One. 2017;12:e0169653 DOI: 10.1371/journal.pone.0169653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miles JJ, Thammanichanond D, Moneer S, Nivarthi UK, Kjer-Nielsen L, Tracy SL, Aitken CK, Brennan RM, Zeng W, Marquart L, et al. Antigen-driven patterns of TCR bias are shared across diverse outcomes of human hepatitis C virus infection. J Immunol. 2011;186:901–912. DOI: 10.4049/jimmunol.1003167. [DOI] [PubMed] [Google Scholar]
- 35.Jia Q, Zhou J, Chen G, Shi Y, Yu H, Guan P, Lin R, Jiang N, Yu P, Li QJ, et al. Diversity index of mucosal resident T lymphocyte repertoire predicts clinical prognosis in gastric cancer. Oncoimmunology. 2015:4:e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poropatich K, Fontanarosa J, Swaminathan S, Dittmann D, Chen S, Samant S, Zhang B.. Comprehensive T cell immunophenotyping and next generation sequencing from HPV-positive and -negative head and neck squamous cell carcinomas. J Pathol. 2017. DOI: 10.1002/path.4953. [DOI] [PubMed] [Google Scholar]
- 37.Shi L, Zhang Y, Feng L, Wang L, Rong W, Wu F, Wu J, Zhang K, Cheng S.. Multi-omics study revealing the complexity and spatial heterogeneity of tumor-infiltrating lymphocytes in primary liver carcinoma. Oncotarget. 2017;8:34844–34857. DOI: 10.18632/oncotarget.16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK Greene M, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010;18:160–170. DOI: 10.1016/j.ccr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, Fong L.. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6:238ra70 DOI: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20:2424–2432. DOI: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Team RDC R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]