

ABSTRACT
EGFR signaling has a critical role in oncogenic KRAS-driven tumorigenesis of the pancreas, whereas it is dispensable in other organs. The complex signaling network engaged by oncogenic KRAS and its modulation by EGFR signaling, remains incompletely understood. In order to study early signaling events activated by oncogenic KRAS in the pancreas, we recently developed a novel model system based on murine primary pancreatic epithelial cells enabling the time-specific expression of mutant KrasG12D from its endogenous promoter. Here, we discuss our findings of a KrasG12D-induced autocrine EGFR loop, how this loop is integrated by the MYC oncogene, and point to possible translational implications.
KEYWORDS: autocrine signaling, EGFR, kras, MYC, pancreatic cancer
KRAS in pancreatic ductal adenocarcinoma (PDAC)
KRAS belongs to the RAS family of small GTPases, which are controlled by a molecular switch between an active GTP-bound and an inactive GDP-bound state, a process controlled by guanine nucleotide exchange factors (GEFs), which promote RAS activation, and GTPase activating proteins (GAPs), which stimulate the intrinsic GTPase activity and RAS inactivation.1 Oncogenic mutations of KRAS occur in over 90% of pancreatic ductal adenocarcinomas (PDAC)2 and according to recent sequencing studies.3,4 activating mutations of KRAS found in PDAC patients affected codon 12 in 93% of all patients (G12D: 38%; G12V: 38%; G12R: 14%; G12C: 2%; G12S: 1%), codon 61 in 6% of the patients (Q61H: 3.5%; Q61R: 1.5%; Q61K: 1%), and codon 13 or codon 146 in 0.5% of all patients. PDAC is a malignancy with an uniquely poor prognosis and a 5-year-survival rate of less than 8%. Despite its low incidence compared with other solid cancers, it is currently the fourth leading cause of cancer-related death in the western world and is projected to be the second most common cause in the near future.
Mutant KRAS is refractory to GAP-induced GTP hydrolysis, favoring an active GTP-bound state.5 For a detailed description of the highly complex signaling induced by oncogenic KRAS in the pancreas, we like to refer the reader to recent reviews.6-8 Compelling experimental evidence demonstrates that oncogenic KRAS initiates the development of PDAC via pre-neoplastic lesions, including pancreatic intraepithelial neoplasia (PanIN) lesions.9-11 Furthermore, oncogenic KRAS is also required to maintain PDAC growth.12,13 Therefore, analysis of signaling pathways controlled by mutated KRAS is of prominent importance to define opportunities to interfere with cancer initiation, progression, and maintenance.
Primary pancreatic ductal epithelial cells (PDECs): A relevant model to study KRAS signaling in the context of the pancreas
Current knowledge argues that KRAS engages several major pathways, including the RAF/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK)-, and the phosphoinositide-3-kinase (PI3K)/AKT-pathway. However, the signaling network regulated by oncogenic KRAS is certainly by far more complex and controlled by feed forward and backward loops, which are incompletely defined. In an attempt to establish a cell-based model to study early oncogenic KRAS signaling in the pancreas, we used genetically engineered murine primary pancreatic ductal epithelial cells (PDECs).14 Together with acinar cells, these cells contribute to the exocrine compartment of the pancreas.15 Expression of a tamoxifen-activatable Cre recombinase expressed from the ubiquitous Rosa26 or the ductal-specific Hnf1b promoter (R26CreERT2 or Hnf1b-CreER mouse lines) allows activation of the KrasG12D oncogene via a Lox-Stop-Lox (LSL) strategy (LSL-KrasG12D knock-in allele) in a temporally controlled fashion in PDECs (Fig. 1). Experimental evidence in mice support the notion that KRASG12D-induced de-differentiation/reprogramming of pancreatic acinar cells to duct-like cells, a process called acinar-to-ductal metaplasia (ADM), plays a prominent role in PDAC formation. Furthermore, murine ductal cells seem to be refractory to KRASG12D-mediated transformation.16,17 To analyze the relevance of PDECs as a potential cell of PDAC origin, we conducted orthotopic transplantation experiments of PDECs with ex vivo activated KRASG12D expression.14 “KRASG12D-on” PDECs transplanted at a relative low number into the pancreas of immunodeficient mice, failed to form preneoplastic lesions. In contrast, increasing the number of cells to 7.5 × 105 cells resulted in the development of ductal/PanIN-like structures, an observation consistent with data published by the Bar-Sagi group.18 Next we activated expression of KRASG12D ex vivo in PDECs and concomitantly deleted the tumor suppressor Cdkn2a, which is frequently inactivated in human PDAC. After orthotopic implantation of these cells into the pancreas of immunodeficient mice, we observed formation of invasive PDACs and metastasis.14 Similarly, concomitant activation of KRASG12D and inactivation of the tumor suppressor p53 by biallelic expression of mutated p53R172H, which is the murine ortholog of the p53R175H hotspot mutation found in human PDAC, resulted in invasive cancers after orthotopic transplantation (unpublished data). In a complementary approach, we could demonstrate that RNAi-mediated silencing of p16INK4, which is encoded by the Cdkn2a locus, or p53 in “KRASG12D-on” PDECs results in PDAC formation in vivo.19 Consistent with these results, an important recent publication by the Leach group demonstrated PDAC formation by activating KrasG12D and mutationally inactivating p53 (via expression of p53R172H from both alleles) in the ductal (HNF1b positive) lineage in vivo.20 In summary, murine PDECs can serve as the cell of origin of murine PDAC after inactivation of important tumor barriers. Thereby, this model mimics human PDAC formation, which depends on conquering the same roadblocks. Considering the exceptional cellular plasticity of the pancreas,16 it is not surprising that several cells of origin exist. Indeed, expressing oncogenic Kras in ductal, acinar, or even endocrine cells of the murine pancreas induces transformation under certain experimental conditions.14,20-23 It will be important for future research, to determine whether PDACs derived from distinct cellular lineages are characterized by a specific mutational landscape or specific molecular alterations, reflecting the unique requirements of the different pancreatic compartments, and whether the cell of origin has an impact on the behavior of the tumor with respect to clinical outcomes as well as therapeutic vulnerabilities. In addition, a PanIN-independent road to cancer originating in the ductal compartment was suggested,20 which needs further investigations. Beyond these considerations for future research directions, the presented data show that our new model is relevant to investigate oncogenic KRAS signaling in the context of the pancreas.
Figure 1.
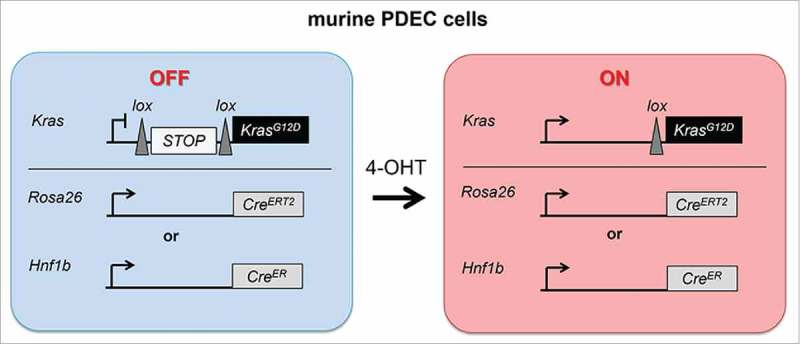
Expression of KRASG12D signaling in PDECs. Depicted is the genetic strategy to activate KRASG12D-expression in primary pancreatic ductal epithelial cells (PDECs) in a temporally controlled manner. Treatment of PDECs with 4-hydroxytamoxifen activates a Cre recombinase, expressed from the Rosa26 or the Hnf1b promoter. This leads to the expression of KRASG12D, driven by the endogenous promoter.
KRAS and the EGFR-dependent autocrine feed-forward loop
Activation of KrasG12D expression in PDECs ex vivo induced a proliferative response.14,24-26 To understand the requirements of “KRASG12D-on” PDECs to remain in the cell cycle, we profiled mRNA expression in comparison to “KRASG12D-off” cells. We observed molecular signatures linked to the epidermal growth factor receptor (EGFR) pathway.14 EGFR belongs to the EGFR/ErbB family of receptor tyrosine kinases and can be activated by several ligands, including epidermal growth factor (EGF), amphiregulin (AREG), epiregulin (EREG), transforming growth factor α (TGF-α), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), and epigen (EGN). Indeed, several EGFR ligands, including AREG and EREG, are induced upon expression of KRASG12D in PDECs and EGFR becomes subsequently autophosphorylated and thus activated.14 Furthermore, we could show that the EGFR signal is needed for KRASG12D-dependent cell cycle entry of PDECs.14 These data argue for an EGFR-dependent feed-forward loop engaged by oncogenic KRAS. Several lines of evidence in different model systems document the contribution of an EGFR-dependent autocrine loop toward the KRAS driven transformation in the pancreas. In organoids established from duct-like cells of Pdx1-Cre;LSL-KrasG12D/+ mice, KRASG12D induces expression of EGFR ligands.27 Activation of EGFR signaling by induction of EGFR and EGFR ligand expression occurs early in the KRASG12D-driven carcinogenesis in mice as well as in human low-grade PanINs.28,29 Mechanistically, KRASG12D induces the formation of reactive oxygen species (ROS), which activate the expression of EGFR and some of its ligands via the NFκB pathway.30,31 It is clear that ectopic expression of EGFR ligands accelerates the carcinogenesis in the pancreas.32-34 Inversely, knockout of the Egfr gene in KRASG12D- and KRASG12V-driven mouse models of the disease completely blocked pancreatic tumor formation.28,29 Importantly, the requirement of EGFR for oncogenic KRAS-driven tumor formation seems to be tissue-specific and depends on co-existing mutations. In contrast to the pancreas, KRASG12V-driven tumor formation in the lung and the intestine is EGFR-independent.28 Although tissue specific engagement of oncogenic effector pathways is common in cancer biology, the molecular underpinnings for the tissue-specific requirement of the EGFR remains unclear. In addition, EGFR signaling is essential for tumor formation in a KRASG12V-driven Cdkn2a-deficient PDAC model, whereas it is dispensable in a p53-deficient background.28,29 Interestingly, when we compared transcriptome profiles of “KRASG12D-on” PDECs to profiles of PDECs with additional biallelic expression of the p53R172H mutant, we detected enrichment of metabolic signatures in p53-inactivated PDECs by KEGG pathway analysis (unpublished data). Such observations are compatible with the well-known anti-Warburg function of wild type p53 to impair glycolysis and to promote mitochondrial respiration.35 In PDAC cells, glucose fluxes to anabolic pathways, such as the hexosamine biosynthesis pathway, which is needed for protein glycosylation, and the non-oxidative branch of the pentose phosphate pathways, which is important for ribose synthesis.12 Numerous cancer-relevant proteins, including potent oncogenic drivers, such as the transcription factor MYC,36 are regulated/activated via glycosylation.37 Thus, the metabolic rewiring in KRASG12D-expressing cells linked to an inactivation of p53 might activate potent oncogenic drivers, thereby reducing the requirement of the autocrine EGFR-loop to activate growth and to enter the cell cycle. Therefore, disabling the anti-Warburg effect of p53 might switch the augmentation of oncogenic KRAS signaling from specific extrinsic requirements to intrinsic ones. Such a process might be linked to a specific oncogenic driver, explaining discrepant outcomes of EGFR functions in different cancer types.
However, it is important to note that the autocrine EGFR loop does not stand alone to support oncogenic KRAS-mediated transformation of the pancreas. Autocrine activation of insulin like growth factor 1 receptor (IGF1R) is required for KRASG12D and BRAFV600E induced transformation of Cdnk2a/p53-deficient PDECs. This might point to the possibility that the usage of specific loops is determined by the set of inactivated tumor suppressor genes.25 Furthermore, inflammation is a major risk factor for PDAC and pro-inflammatory molecules may act in an autocrine fashion to augment the signaling threshold of oncogenic KRAS.38 A recent important example is the control of the KRAS-driven carcinogenesis in the pancreas by the YAP and TAZ transcription factors.39,40 In ductal cells, it was demonstrated that KRASG12D engages YAP to induce expression of secreted factors, including pro-inflammatory molecules like IL6 or IL1α, needed for proliferation.40
Integration of the EGFR-loop - connection to licensing nodes
Considering that signaling of oncogenic KRAS is highly complex, with several effector pathways acting in parallel with redundant branches modulated by autocrine signaling loops, a major goal is to define non-redundant signaling nodes. An important requirement of such a node is the authority to license profound cellular decisions, for instance to grow or not to grow, to enter the cell cycle or to stay in quiescence.
The integration of the EGFR-loop and the oncogenic KRAS signaling network in the pancreas remains unclear. EGFR signaling has been connected to canonical ERK-, AKT-, STAT3-, NFAT/AP-1, p70 S6 kinase-, or GSK3-signaling.28,29,32,34,41 Using the EGFR inhibitors erlotinib and gefitinib, we detected no significant influence of the EGFR-loop on KRASG12D-induced ERK- and AKT-signaling.14 To identify licensing nodes connected to the definite pro-proliferative decision transmitted via the EGFR, we performed unbiased transcriptome profiling. Focusing on transcription factors, we detected that the majority of signatures connected to the EGFR-loop belong to MYC- or E2F-pathways.14 Both, MYC as well as E2F transcription factors can be considered as decision-making switch factors that allow cellular growth/cell cycle entry or S-phase entrance, respectively.
Due to MYC's important role in the carcinogenesis in the pancreas and our finding that E2F is downstream of MYC in a subset PDAC cells, we further investigated the link of EGFR and MYC.42,43 Multiple layers control and fine tune MYC transcriptional activity. In PDECs, we detected N-terminal phosphorylation of MYC in response to KRASG12D expression.14 It is well established that threonine 58 (T58) and serine 62 (S62) residues at the N-terminal MYC homology box I control MYC activity (Fig. 2). ERK is a kinase that can phosphorylate S62 of MYC, leading to its stabilization and activation.44 In a hierarchical fashion, phosphorylation of S62 can be followed by a glycogen synthase kinase-3β (GSK3β)-dependent phosphorylation of T58, to limit MYC stability.44 In KRASG12D-expressing PDECs, the EGFR-loop has no effect on the activating phosphorylation of ERK or the inactivating phosphorylation of GSK3β. However, we detected markedly reduced expression of both MYC protein and mRNA upon EGFR inhibition.14 Since we detected no significant changes in oncogenic KRAS-activated canonical (ERK) and non-canonical (e.g. PI3K-AKT-GSK3) signaling pathways, it is likely that the EGFR-loop controls MYC mRNA abundance. To that end, the mechanism connecting EGFR to MYC mRNA expression in PDAC remains unclear and awaits further investigations. In sum, in addition to activation of a MYC stabilization signal and the inactivation of a degradation signal by oncogenic KRAS, the EGFR loop directs a further layer of control upon acute activation of the Kras oncogene in the ductal pancreatic context (Fig. 2). Hereby, the KRASG12D-induced autocrine EGFR-loop allows MYC to license cellular growth and cell cycle entry.14 Beyond licensing of growth and proliferation, another molecular MYC function might be noteworthy. An ongoing current debate includes how MYC is regulating gene expression programs on a global scale45-52 In 2012, a role for MYC to increase merely the output of an active and pre-existing cellular transcriptome was postulated, called the amplifier model.45-47 Other models include the existence of a specific set of direct MYC target genes and it was demonstrated that MYC controls (activates/represses) a specific set of target genes,51,52 that can secondarily feed back to global mRNA expression levels.49,52 It will be important for further work to determine whether the relevant KRASG12D-induced signaling branches discussed in this article synergize to induce MYC expression levels able to amplify - directly or indirectly - the transcriptome induced by oncogenic Kras signaling (Fig. 2). Such a scenario is attractive since it allows augmentation of the KRASG12D-signal at a very distal position, without the need to directly manipulate the activity of the canonical (KRAS-MEK-ERK) or the non-canonical (e.g. PI3K-AKT) branch of the pathway. However, such a possibility and the contribution of such a mechanism to the transformation in the pancreas clearly demand further analysis of the MYC function on a global scale.
Figure 2.

Integration of oncogenic KRAS signaling by MYC in PDECs. GTP-bound oncogenic RAS induces several effector pathways. Depicted are the pathways with clear and definite impact on the carcinogenesis in the pancreas. Canonical signaling leads to activation of the RAF - (mitogen-activated kinase/ERK kinase) MEK - extracellular regulated kinase (ERK) protein kinase signaling pathway. Non-canonical signaling initiates the phosphoinositide 3-kinase (PI3Ks)-AKT-glycogen synthase kinase 3 (GSK3) cascade. ERK can phosphorylate MYC at serine 62 (S62), contributing to its stabilization and activity. AKT-mediated inhibitory phosphorylation of GSK3 impairs MYC threonine 58 (T58) phosphorylation and degradation. The KRASmut induced EGFR-loop increases MYC mRNA expression. The function of MYC to license growth and cell cycle entry is depicted.
Conclusion
Considering I) the complexity and the difficulties to analyze endogenous mouse models of PDAC, II) the need for in vitro systems to analyze signaling pathways mechanistically, III) the cellular plasticity in the pancreas, and IV) several different cells of PDAC origin, we aimed at developing a novel cellular PDEC-based system that addresses these issues allowing functional studies of early events of oncogenic KRAS signaling in the ductal compartment of the pancreas. We validated the new system, which is highly flexible, versatile and fast in vitro as well as in vivo and provide functional evidence for an EGFR signaling loop that controls MYC activity in pancreatic carcinogenesis.
The importance of cooperation between RAS and MYC for transformation has been demonstrated more than 3 decades ago.53 Our findings connect these 2 pathways in an in vitro model of PDAC and illustrate that EGFR is needed for this link, thus contributing to explain the critical role of EGFR for the tumorigenesis in the pancreas.
Despite intensive efforts and recent progress, oncogenic KRAS clinically still remains a challenging target.54 Therefore, it will be important to see whether molecular circuits discussed in this article are relevant at the stage of tumor maintenance and whether such molecular knowledge can be exploited to develop novel therapeutic concepts. Only a subpopulation of PDAC patients benefit from EGFR inhibition.55 The pre-clinical data discussed in this commentary may argue that especially p53 wild type PDACs depend on an extrinsic augmentation of oncogenic KRAS signaling, such as the activation of the EGFR-loop. Indeed in a recent study, regular p53 expression, indicative for wild type p53, was linked to an improved progression free survival of EGFR inhibitor treated PDAC patients.56 Although such observations point to the power of a detailed molecular understanding of the disease for clinical study design and patient stratification, the study needs definite further prospective validation. Considering MYC as an important integrator of the KRAS signal may pave the way to novel therapeutic efforts to target MYC in PDAC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to apologize for not citing any relevant reports due to the need to selectively choose examples, lack of space, or an oversight on our part.
Funding
This work was supported by: Deutsche Krebshilfe [110908 to G.S. and 111273 to M.R.], Wilhelm-Sander Stiftung [2016.004.1 to G.S.], Else Kröner-Fresenius-Stiftung (2016_A43 to M.W.), Deutsche Forschungsgemeinschaft (DFG) [SFB824/C9 to G.S. and D.S. / SCHN 959/3–1 to G.S.], and DKTK Joint Funding [D.S., and G.S.].
References
- [1].Fey D, Matallanas D, Rauch J, Rukhlenko OS, Kholodenko BN. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin Cell Dev Biol 2016; 58:96-107. [DOI] [PubMed] [Google Scholar]
- [2].Knudsen ES, O'Reilly EM, Brody JR, Witkiewicz AK. Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology 2016; 150:48-63; http://dx.doi.org/ 10.1053/j.gastro.2015.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al.. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012; 491:399-405; PMID:23103869; http://dx.doi.org/ 10.1038/nature11547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al.. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015; 6:6744; PMID:25855536; http://dx.doi.org/ 10.1038/ncomms7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016; 129:1287-92; PMID:26985062; http://dx.doi.org/ 10.1242/jcs.182873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Logsdon CD, Lu W. The Significance of Ras Activity in Pancreatic Cancer Initiation. Int J Biol Sci 2016; 12:338-46; PMID:26929740; http://dx.doi.org/ 10.7150/ijbs.15020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013; 144:1220-9; http://dx.doi.org/ 10.1053/j.gastro.2013.01.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014; 111:817-22; PMID:24755884; http://dx.doi.org/ 10.1038/bjc.2014.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al.. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4:437-50; PMID:14706336; http://dx.doi.org/ 10.1016/S1535-6108(03)00309-X [DOI] [PubMed] [Google Scholar]
- [10].Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003; 17:3112-26; PMID:14681207; http://dx.doi.org/ 10.1101/gad.1158703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007; 11:291-302; http://dx.doi.org/ 10.1016/j.ccr.2007.01.012 [DOI] [PubMed] [Google Scholar]
- [12].Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al.. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435; http://dx.doi.org/ 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122:639-53; PMID:22232209; http://dx.doi.org/ 10.1172/JCI59227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Diersch S, Wirth M, Schneeweis C, Jors S, Geisler F, Siveke JT, Rad R, Schmid RM, Saur D, Rustgi AK, et al.. Kras(G12D) induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene 2016; 35:3880-6; PMID:26592448; http://dx.doi.org/ 10.1038/onc.2015.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest 2011; 121:4572-8; PMID:22133881; http://dx.doi.org/ 10.1172/JCI57131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Puri S, Folias AE, Hebrok M. Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell 2015; 16:18-31; http://dx.doi.org/ 10.1016/j.stem.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, Pan FC, Akiyama H, Wright CV, Jensen K, et al.. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 22:737-50; PMID:23201164; http://dx.doi.org/ 10.1016/j.ccr.2012.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012; 21:836-47; PMID:22698407; http://dx.doi.org/ 10.1016/j.ccr.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].von Burstin J, Diersch S, Schneider G, Reichert M, Rustgi AK, Schmid RM. Detection of Tumor Suppressor Genes in Cancer Development by a Novel shRNA-Based Method. Mol Cancer Res 2015; 13:863-9; PMID:25724428; http://dx.doi.org/ 10.1158/1541-7786.MCR-14-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bailey JM, Hendley AM, Lafaro KJ, Pruski MA, Jones NC, Alsina J, Younes M, Maitra A, McAllister F, Iacobuzio-Donahue CA, et al.. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016; 35:4282-8; PMID:26592447; http://dx.doi.org/ 10.1038/onc.2015.441 [DOI] [PubMed] [Google Scholar]
- [21].Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011; 19:728-39; PMID:21665147; http://dx.doi.org/ 10.1016/j.ccr.2011.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, et al.. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 2008; 105:18913-8; PMID:19028870; http://dx.doi.org/ 10.1073/pnas.0810097105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009; 16:379-89; PMID:19878870; http://dx.doi.org/ 10.1016/j.ccr.2009.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 2010; 18:448-58; http://dx.doi.org/ 10.1016/j.ccr.2010.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Appleman VA, Ahronian LG, Cai J, Klimstra DS, Lewis BC. KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling. Mol Cancer Res 2012; 10:1228-39; PMID:22871572; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0340-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, Wright CV, Hebrok M, Lewis BC. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci U S A 2007; 104:5103-8; PMID:17372229; http://dx.doi.org/ 10.1073/pnas.0701158104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al.. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015; 160:324-38; PMID:25557080; http://dx.doi.org/ 10.1016/j.cell.2014.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012; 22:318-30; PMID:22975375; http://dx.doi.org/ 10.1016/j.ccr.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al.. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012; 22:304-17; PMID:22975374; http://dx.doi.org/ 10.1016/j.ccr.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Storz P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2016:1-5; PMID:27215184; http://dx.doi.org/ 10.1080/21541248.2016.1192714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liou GY, Doppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep 2016; 14:2325-36; PMID:26947075; http://dx.doi.org/ 10.1016/j.celrep.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ray KC, Moss ME, Franklin JL, Weaver CJ, Higginbotham J, Song Y, Revetta FL, Blaine SA, Bridges LR, Guess KE, et al.. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene 2014; 33:823-31; PMID:23376846; http://dx.doi.org/ 10.1038/onc.2013.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sandgren EP, Luetteke NC, Qiu TH, Palmiter RD, Brinster RL, Lee DC. Transforming growth factor alpha dramatically enhances oncogene-induced carcinogenesis in transgenic mouse pancreas and liver. Mol Cell Biol 1993; 13:320-30; PMID:8417334; http://dx.doi.org/ 10.1128/MCB.13.1.320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Siveke JT, Einwachter H, Sipos B, Lubeseder-Martellato C, Kloppel G, Schmid RM. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell 2007; 12:266-79; http://dx.doi.org/ 10.1016/j.ccr.2007.08.002 [DOI] [PubMed] [Google Scholar]
- [35].Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 2015; 16:393-405; PMID:26122615; http://dx.doi.org/ 10.1038/nrm4007 [DOI] [PubMed] [Google Scholar]
- [36].Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, Cantrell DA. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol 2016; 17:712-20; PMID:27111141; http://dx.doi.org/ 10.1038/ni.3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol 2016; 428:3282-94; http://dx.doi.org/ 10.1016/j.jmb.2016.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, Cruz-Monserrate Z, Wang H, Ji B, Logsdon CD. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 2012; 122:1519-28; PMID:22406536; http://dx.doi.org/ 10.1172/JCI59743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gruber R, Panayiotou R, Nye E, Spencer-Dene B, Stamp G, Behrens A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016; 151:526-39; PMID:27215660; http://dx.doi.org/ 10.1053/j.gastro.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, et al.. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 2014; 7:ra42; PMID:24803537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, Regul L, Nagarajan S, Kuhnemuth B, Johnsen SA, et al.. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015; 148:1024-34 e9; PMID:25623042; http://dx.doi.org/ 10.1053/j.gastro.2015.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wirth M, Mahboobi S, Kramer OH, Schneider G. Concepts to Target MYC in Pancreatic Cancer. Mol Cancer Ther 2016; 15:1792-8; PMID:27406986; http://dx.doi.org/ 10.1158/1535-7163.MCT-16-0050 [DOI] [PubMed] [Google Scholar]
- [43].Schild C, Wirth M, Reichert M, Schmid RM, Saur D, Schneider G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol Carcinog 2009; 48:1149-58; PMID:19603422; http://dx.doi.org/ 10.1002/mc.20569 [DOI] [PubMed] [Google Scholar]
- [44].Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635-45; PMID:16064138; http://dx.doi.org/ 10.1038/nrm1703 [DOI] [PubMed] [Google Scholar]
- [45].Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151:56-67; http://dx.doi.org/ 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al.. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012; 151:68-79; PMID:23021216; http://dx.doi.org/ 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rahl PB, Young RA. MYC and transcription elongation. Cold Spring Harb Perspect Med 2014; 4:a020990; PMID:24384817; http://dx.doi.org/ 10.1101/cshperspect.a020990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wolf E, Lin CY, Eilers M, Levens DL. Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol 2015; 25:241-8; PMID:25475704; http://dx.doi.org/ 10.1016/j.tcb.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 2015; 15:593-607; PMID:26383138; http://dx.doi.org/ 10.1038/nrc3984 [DOI] [PubMed] [Google Scholar]
- [50].Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, Wolf E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016; 5:e15161; PMID:27460974; http://dx.doi.org/ 10.7554/eLife.15161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al.. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014; 511:483-7; PMID:25043018; http://dx.doi.org/ 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al.. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014; 511:488-92; PMID:25043028; http://dx.doi.org/ 10.1038/nature13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983; 304:596-602; PMID:6308472; http://dx.doi.org/ 10.1038/304596a0 [DOI] [PubMed] [Google Scholar]
- [54].Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 2016; 15:771-785; PMID:27469033 [DOI] [PubMed] [Google Scholar]
- [55].Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al.. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25:1960-6; PMID:17452677; http://dx.doi.org/ 10.1200/JCO.2006.07.9525 [DOI] [PubMed] [Google Scholar]
- [56].Ormanns S, Siveke JT, Heinemann V, Haas M, Sipos B, Schlitter AM, Esposito I, Jung A, Laubender RP, Kruger S, et al.. pERK, pAKT and p53 as tissue biomarkers in erlotinib-treated patients with advanced pancreatic cancer: a translational subgroup analysis from AIO-PK0104. BMC Cancer 2014; 14:624; PMID:25164437; http://dx.doi.org/ 10.1186/1471-2407-14-624 [DOI] [PMC free article] [PubMed] [Google Scholar]