

Abstract
Introduction
We investigated the effect of antihypertensive (aHTN) medications and cholinesterase inhibitors (ChEIs) on the cognitive decline in patients with Alzheimer's disease (AD) and analyzed synergism by chemogenomics systems pharmacology mapping.
Methods
We compared the effect of aHTN drugs on Mini-Mental State Examination scores in 617 AD patients with hypertension, and studied the synergistic effects.
Results
The combination of diuretics, calcium channel blockers, and renin-angiotensin-aldosterone system blockers showed slower cognitive decline compared with other aHTN groups (Δβ = +1.46, P < .0001). aHTN medications slow down cognitive decline in ChEI users (Δβ = +0.56, P = .006), but not in non-ChEI users (Δβ = −0.31, P = .53).
Discussion
aHTN and ChEI drugs showed synergistic effects. A combination of diuretics, renin-angiotensin-aldosterone system blockers, and calcium channel blockers had the slowest cognitive decline. The chemogenomics systems pharmacology–identified molecular targets provide system pharmacology interpretation of the synergism of the drugs in clinics. The results suggest that improving vascular health is essential for AD treatment and provide a novel direction for AD drug development.
Keywords: Alzheimer's disease, Cognitive decline, Cholinesterase inhibitors, Antihypertensive medications, Combination therapy, Systems pharmacology, Clinical data mining
Highlights
-
•
Combination of three antihypertensive drug classes slowed down cognitive decline.
-
•
Antihypertensives and cholinesterase inhibitor showed synergistic cognitive benefit.
-
•
Computational systems pharmacology revealed molecular mechanisms for synergism.
1. Introduction
Alzheimer's disease (AD) has loomed as a major health challenge worldwide. To date, there is no cure for AD, and the currently available treatments, such as cholinesterase inhibitors (ChEIs), achieve very limited therapeutic advantage [1]. Because of the complexity in the pathology and etiology of AD, it has been proposed that combination therapies may be more advantageous compared with monotherapies, and there is a great need for studies on combination therapies in AD [2].
Hypertension is one of the most prevalent coexisting diseases in patients with AD, comprising 42% of the AD population [3]. In addition to the fact that both diseases are age-related, it has also been proposed that vascular abnormalities can etiologically contribute to the onset and progression of AD [4]. Therefore, targeting the vascular system is a potential strategy for treating AD.
Numerous epidemiologic studies have investigated the relationship among hypertension, antihypertensive (aHTN) medications, and AD, but the results were mixed [5]. Several longitudinal studies have consistently reported that mid-life hypertension is an important risk factor for developing AD and dementia in late age [6]. In older subjects, however, the role of blood pressure in relation to AD seems more obscure and intricate, whereas high incidences of AD or dementia have been reported to have an association with low blood pressure in elderly patients [7], [8]; other studies also reported no association [9], [10], association with high blood pressure [11], or association with both high and low blood pressure [12]. On the other hand, aHTN drugs have been associated with reduced risk of dementia in observational studies [13], [14] and randomized controlled trials (RCTs) [15], [16]. Particularly, the use of diuretics, renin-angiotensin-aldosterone system (RAAS) inhibitors, and β-adrenergic blockers was related to a slower rate of cognitive decline and lower risk of dementia in elderly patients [17]. Importantly, however, the study population of all the aforementioned studies was elderly individuals rather than patients already diagnosed with AD. Therefore, these results do not necessarily translate to a therapeutic effect in patients with AD. In one study, the use of diuretics was associated with a slower decline in cognitive function among patients diagnosed with dementia [18]. However, this study did not differentiate between AD and other types of dementia including vascular dementia, which is pathologically distinct from AD and may interact with hypertension via different pathways. Therefore, direct evidence of the therapeutic effect of aHTN medications on patients with AD is still lacking. Moreover, none of the studies done in patients diagnosed with AD have examined the effect of combinations of different classes of aHTN drugs, or the combination of aHTN drugs and currently available treatments for AD.
In this observational study, we applied clinical data-mining analyses to investigate the effect of aHTN medications on the cognitive decline in patients with clinically diagnosed AD. We then assessed the effect of the combined use of multiple hypertensive drug classes and the combination of aHTN drugs with ChEIs. Furthermore, we applied systems pharmacology drug-target network analyses to better understand, from the molecular level, the effect of anti-AD and aHTN medications on AD and their synergism in clinical contexts. Systems pharmacology applies the principles of systems biology to study pharmacology, and it seeks to understand how medicines work on various molecular targets from complex systems of the body. Then, the drug-target network studies can serve as predictors on new indication(s) for approved drugs and to guide combinational therapy in clinics. Xie and Wang have developed chemogenomics systems pharmacology target (CSP-target) mapping technique [19] by computational analyses on interaction networks of multiple drugs and multiple targets, from a systems pharmacology perspective, using high-throughput docking (HTDocking) [20] and TargetHunter algorithm [21] based on the AD [22] and cardiovascular disease (CVD) [23] domain-specific chemogenomics knowledgebases. Such integrated computational methodologies enable the classification of drugs according to their chemical structures and to which proteins they bind, and the CSP-target analyses make predictions about the therapeutic effects of drugs for complex diseases and possible off-target effects. The outcomes from such integrated CSP-target analyses of AD and HTN medications used in patients led to new understanding of drug action synergy by correlating molecular pharmacology with clinical observation for complex diseases. By investigating the molecular targets involved in HTN and AD, the CSP-target map analysis will provide a mechanistic insight into the effect of aHTN medications on AD in clinical contexts. We also correlated such analyses with the reported AD pathology and the mechanism of drug actions in the literature and clinical trials for AD treatment. The outcomes of such studies could be used to guide rationale clinical therapy and even to new drug design and discovery for AD.
2. Methods
2.1. Participants
The subjects of this study were patients examined at the University of Pittsburgh Alzheimer's Disease Research Center from April 1983 to March 2015. The protocols for patient diagnosis and information collection were published previously [24]. Briefly, the patients received a series of clinical examinations evaluating their physical, cognitive, and neurologic status, and a diagnosis was made by a neurologist and a psychiatrist and then reviewed by a committee. Follow-up surveys and cognitive evaluations using the Mini-Mental State Examination (MMSE) were conducted at annual clinic visits and semiannual phone interviews regarding their current status and history of disease and medication.
Probable AD cases with concomitant hypertension (n = 617) were selected from a total of 4364 participants in clinics. These patients will be referred to as set 1. Among these AD cases, 399 had records of aHTN drug use. The aHTN drugs were categorized into four drug classes: diuretics; calcium channel blockers (CCBs); RAAS inhibitors; and others (β-adrenergic blockers, α-adrenergic blockers, arterial vasodilators, and miscellaneous aHTN agents). On the basis of their use of aHTN drugs or their combinations, the set 1 patients were divided into nine groups (Table 1). Later, we contrasted the particular treatment group with the slowest cognitive decline (combination of diuretics + CCB + RAAS) against all other groups. Their baseline characteristics are shown in Table 2. Apolipoprotein E (APOE) genotyping was performed on isolated DNA from blood as described previously or by using TaqMan genotyping assays [25].
Table 1.
Baseline characteristics for set 1 patients (number of subjects under different aHTN drug classes in set 1)
| Group | No. of years since the first record of aHTN drug use |
|||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | |
| No aHTN drug | 218 | 182 | 99 | 53 | 36 | 34 |
| Diuretics only | 29 | 27 | 18 | 11 | 6 | 5 |
| CCB only | 25 | 22 | 13 | 12 | 7 | 1 |
| RAAS only | 79 | 72 | 47 | 29 | 17 | 8 |
| Diuretics + RAAS | 44 | 38 | 28 | 16 | 10 | 8 |
| CCB + RAAS | 31 | 29 | 23 | 17 | 11 | 6 |
| Diuretics + CCB | 7 | 5 | 4 | 3 | 4 | 2 |
| Diuretics + CCB + RAAS | 24 | 20 | 17 | 13 | 12 | 9 |
| Other groups | 160 | 142 | 108 | 63 | 42 | 30 |
| Total | 617 | 537 | 357 | 217 | 145 | 103 |
Abbreviations: aHTN, antihypertensive; CCB, calcium channel blocker; RAAS, renin-angiotensin-aldosterone system.
Table 2.
Baseline characteristics for set 1 (diuretics + CCB + RAAS vs. all other groups)
| Group | Diuretics + CCB + RAAS | Other groups | χ2/t | P value |
|---|---|---|---|---|
| N | 24 | 593 | ||
| Age | 77.3 (7.2) | 76.3 (−7.6) | −0.54 | 0.54 |
| Education | 13.7 (3.2) | 13.2 (3.3) | −0.7 | 0.48 |
| Baseline MMSE (SD) | 21.9 (4.7) | 20.4 (5.2) | −1.39 | 0.16 |
| Gender: female (%) | 20 (83%) | 397 (67%) | 2.83 | 0.09 |
| APOEε4 carrier (%) | 11 (46%) | 280 (47%) | 0.02 | 0.89 |
Abbreviations: CCB, calcium channel blocker; MMSE, Mini-Mental State Examination; RAAS, renin-angiotensin-aldosterone system.
To investigate the potential synergistic effect of ChEI and aHTN drugs on the cognitive decline during the first 2 years after being diagnosed with probable AD, we selected the patients who did not switch treatment (i.e., from ChEI user to nonuser and vice versa) from set 1. These patients will be later referred to as set 2 (N = 419). On the basis of their usage of ChEI and aHTN drugs, the patients in set 2 were divided into four groups, and their baseline characteristics are shown in Table 3.
Table 3.
Baseline characteristics for set 2 patients by ChEI and aHTN drug use
| aHTN drug use | No ChEI |
ChEI |
χ2F | P value | ||
|---|---|---|---|---|---|---|
| No | Yes | No | Yes | |||
| N | 40 | 46 | 169 | 174 | ||
| Education (SD) | 13.2 (2.8) | 13.1 (2.6) | 14.8 (3.1) | 13.6 (3.0) | 21.93 | <.0001 |
| Age (SD) | 75.7 (9.8) | 77.6 (6.8) | 72.7 (9.4) | 75.9 (8.0) | 18.25 | <.0001 |
| Baseline MMSE (SD) | 19.4 (4.2) | 22.2 (3.4) | 21.5 (4.3) | 21.2 (4.1) | 10.59 | <.0001 |
| Gender: female (%) | 30 (75.0) | 36 (78.7) | 90 (53.3) | 108 (61.9) | 2.926 | .09 |
| APOEε4 carrier∗ (%) | 23 (57.5) | 23 (50.0) | 105 (62.1) | 100 (57.5) | 1.546 | .238 |
Abbreviations: aHTN, antihypertensive; ChEI, cholinesterase inhibitor; MMSE, Mini-Mental State Examination.
Missing data for APOEε4 genotype: n = 2, both in the ChEI−/aHTN+ group.
2.2. Statistical analysis
The cognitive function, as measured by MMSE, was the primary outcome of interest in this study. Because of skewness, the MMSE scores were power transformed before conducting the mixed-effect linear model analysis. Student t test, analysis of variance, or χ2 tests compared the baseline characteristics of the two sets of patients.
The study data were maintained and managed using SPSS for Windows (v12–v15); the analyses were carried out using SPSS and SAS (SAS Institute, Cary, NC). α = 0.05 was used as the threshold for statistical significance for all analyses.
2.3. Effect of aHTN drugs on cognitive decline
To test the effects of different classes and combinations of aHTN drugs on the rate of cognitive decline in AD patients with hypertension (set 1), we implemented mixed-effect regression analysis with random intercept and trend. The nine aHTN treatment groups (see Table 1) and time after first record of aHTN drug use were used as the primary predictors for the power-transformed MMSE score. We also assessed the interaction term between the treatment group and time to test whether the treatment groups differed in their rates of cognitive decline. The analysis was conducted using an autoregressive variance-covariance matrix, and controlled for covariates including age at baseline, sex, years of education, and APOEε4 carrier status.
2.4. Synergistic effects of aHTN drugs and ChEIs on cognitive decline
To test for the possible synergistic effects of aHTN drugs and ChEIs on the rate of cognitive decline, we divided the set 2 patients into two layers: ChEI users and nonusers. We first compared the time trends of MMSE decline in these two layers, and then investigated the effect of aHTN drug use on the MMSE decline in both layers using a mixed-effect regression model. Baseline characteristics including age, sex, years of education, APOEε4 genotype, and MMSE score were adjusted for in the analyses, and an autoregressive variance-covariance matrix was used in the analyses.
2.5. Data collection for CVD and AD
The information on approved drugs, drugs in clinical trials, and protein targets associated with AD and CVD were gathered from various databases, including the DrugBank, ClinicalTrials.gov, BindingDB, AlzGene, PubChem, ChEMBL, Therapeutic Target Database, and SciFinder. The information from different sources was standardized with the same format, including protein full name, gene name, UniProt Entry ID, and Entry name. All the information was double checked by a second person, according to our data collection protocols [22]. To be noticed, CVDs included coronary artery diseases such as angina and myocardial infarction, stroke, hypertension, rheumatic heart disease, cardiomyopathy, heart arrhythmia, congenital heart disease, valvular heart disease, carditis, aortic aneurysms, peripheral artery disease, and venous thrombosis. The tables listing the disease-specific targets can be found on our website (http://cbligand.org/AD/target_list.php for AD and http://cbligand.org/CVD/target_list.php for CVD) [20], [21].
2.6. Construction of disease-target network for AD and hypertension
To find the shared multiple drug targets in the overlapping pathways between AD and hypertension, which may point to the combinational treatment for these two coexisting medical conditions, we used our established AD (http://www.cbligand.org/AD) and CVD (http://www.cbligand.org/CVD) databases for the analysis. From the CVD database, we only extracted the drug targets specifically for hypertension treatment to simplify the analysis. Most of the targets we included were the proteins with one or more drugs in the market or under investigation for the treatment of hypertension. We mainly focused on targets with confirmed associations with the disease validated by at least two different sources. Then we combined the target information from two disease-specific databases. The UniProt Entry name and the corresponding disease (AD or hypertension) were further used to map out a disease-target network (DTN) by using Cytoscape 3.1.2, an open-source program for visualizing complex networks.
2.7. Mechanism study of diuretics + CCB + RAAS combination using CSP-target mapping
To understand the molecular mechanism of the synergistic effect among the diuretics + CCB + RAAS combination on the cognitive decline in patients with AD, we carefully examined drug (most frequently used) from each of the aHTN drug classes. The representative drugs are hydrochlorothiazide (HCTZ) for diuretics, amlodipine for CCB, and losartan for RAAS inhibitor. Then, we predicted the AD-related protein targets that each of the drugs potentially acts on by docking the drug molecule against the AlzPlatform target library [22] using our HTDocking program, a web-based software package for high-throughput docking (http://www.cbligand.org/HTDocking/). A docking score of ≥6 was used as a threshold for potential interactions. Then, we ranked the potential protein targets for each drug based on the docking scores, and searched in the literature for experimental validation results for the top 10 interactions. To visualize the results, a CSP-target mapping was constructed using SpiderPLot to construct DTN. The docking poses and detailed interactions were visualized using PyMOL.
3. Results
3.1. Effect of aHTN medications on cognitive decline
Among the 617 patients who had been diagnosed with AD and hypertension (set 1), 399 (64.7%) patients had at least one record for using an aHTN agent. The number of patients under different drug classes over time is shown in Table 1. Time zero is defined as the time for the first record of aHTN drug use in the database. For patients without hypertension and hypertensive patients who did not have any record of aHTN drug use, time zero is defined as the date of the first record in the database. The rate of cognitive decline in probable AD patients without HTN (n = 627) was not significantly different from that of those who had probable AD and HTN but were not taking aHTN medications (P = .14, see Fig. S1). A significant difference (P = .02) was found among different treatment groups in their trajectories of cognitive decline (see Fig. 1). The patient group under the combination of CCB, diuretics, and RAAS showed the lowest rate of cognitive decline, with virtually no decrease in the first 3 years.
Fig. 1.

The cognitive decline in set 1 patients by different aHTN treatment groups was analyzed using a mixed-effect regression model. Combination of diuretics, calcium channel blockers, and renin-angiotensin-aldosterone system inhibitors (diuretics + CCB + RAAS) was associated with the slowest rate of cognitive decline among all groups tested (Δβ = +1.46, P < .0001). This effect was most prominent in the first 3 years of aHTN treatment. Abbreviations: aHTN, antihypertensive; MMSE, Mini-Mental State Examination.
To further confirm this observation, we compared the MMSE trajectories of patients with the CCB + diuretics + RAAS combination group against patients in all other groups in the first 3 years using the mixed-effect regression model with random intercept and slope. There was no significant difference between the CCB + diuretics + RAAS group and other groups in terms of the baseline characteristics (Table 2). However, the estimated slope of cognitive decline of all other groups was −2.15, whereas the slope of the CCB + diuretics + RAAS group was −0.69 (Δβ = +1.46, P < .0001), indicating a significantly slower rate of decline.
3.2. Synergistic effect of aHTN medications and ChEI on cognitive decline
Table 3 shows the baseline characteristics of set 2 patients. In general, ChEI users received more education than nonusers. aHTN drug users were older compared with nonusers. Those who never used either ChEI or aHTN drugs also had the lowest MMSE score at baseline.
We first compared the rate of cognitive decline over the 24-month follow-up between the ChEI group and the non-ChEI group, controlling for baseline characteristics including age, sex, years of education, APOEε4 genotype, and MMSE score. The result showed that ChEI use had a trend of slowing MMSE decline (P = .068). Next, we compared the effect of aHTN medications in the ChEI group and non-ChEI group in set 2, respectively (see Fig. 2). In the non-ChEI stratum, the ChEI−/aHTN− patients had a slope of −2.09 (P < .0001), and the ChEI−/aHTN+ patients also had a similar trajectory (Δβ = −0.31, P = .53). On the other hand, ChEI+/aHTN− patients had a similar slope to that of ChEI−/aHTN− patients (β = −2.13, P < .0001). Interestingly, however, ChEI+/aHTN+ patients had a slope for cognitive decline of −1.57, indicating a significantly reduced rate of cognitive decline compared with the ChEI+/aHTN− patients (slope difference Δβ = +0.56, P < .01).
Fig. 2.
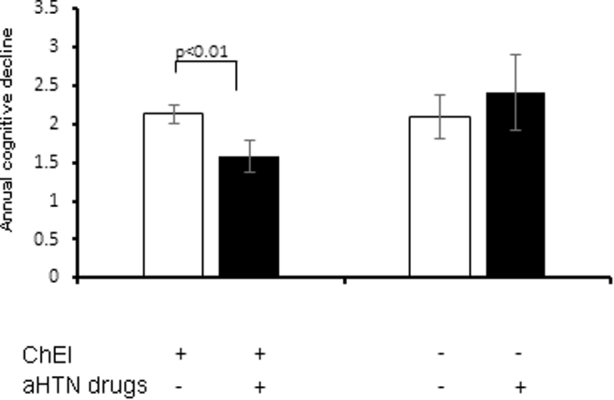
Annual MMSE decline in AD cases with hypertension (set 2) by ChEI and aHTN drug use. Trajectories for cognitive decline among different groups in set 2 were analyzed using a linear mixed-effect regression model. Bars and error bars represent the coefficient β and standard error of the mean for the time variable in each group, respectively. In ChEI+ group, concomitant use of aHTN drugs was associated with significantly slower cognitive decline (slope difference Δβ = +0.56, P < .01). The use of aHTN drug was not associated with a significantly different rate of cognitive decline in the ChEI− group (Δβ = −0.31, P = .53). Note that “+” denotes drug users and “−” denotes nonusers. For example, ChEI+/aHTN− represents patients who used ChEIs but not aHTN drugs. Abbreviations: AD, Alzheimer's disease; aHTN, antihypertensive; ChEI, cholinesterase inhibitor; MMSE, Mini-Mental State Examination.
3.3. Mechanism study using CSP-target mapping
Using our AD and CVD chemogenomics-guided CSP-target mapping, we identified 128 molecular targets for the signaling pathways related to hypertension and 108 molecular targets related to AD (Fig. 3). Twenty-eight targets were found in the intersection between the two diseases, including several targets for RAAS inhibitors (angiotensin-converting enzyme [ACE], angiotensin receptor 1 [AGTR1], and AGTR2) and CCBs (CACNA1A, CACNB2, CACNA2D1, and CACNA2D4). We also identified the drugs targeting two important molecular targets that are related to aHTN treatment, lipoprotein lipase, and ACE (Table S1). These drugs have already been approved by the Food and Drug Administration or are in clinical trials for treating AD.
Fig. 3.

Overlapping protein targets for Alzheimer's disease (red) and hypertension (cyan). Each node represents a protein target name that is associated with the disease(s) linked to it by a straight line. Targets highlighted in solid line circles indicate the drug classes that are shown to have potential synergetic effects by our data-mining analysis in clinical and molecular levels. Abbreviations: ACE, angiotensin-converting enzyme; AGTR1, angiotensin receptor 1; CACNA1A, calcium voltage-gated channel auxiliary subunit alpha1 A; CACNA2D1, calcium voltage-gated channel auxiliary subunit alpha2delta1; CACNA2D4, calcium voltage-gated channel auxiliary subunit alpha2delta4; CACNB2, calcium voltage-gated channel auxiliary subunit beta1; PPARγ, peroxisome proliferator–activated receptor gamma.
We docked the three representative drugs (HCTZ, amlodipine, and losartan) from the three aHTN drug classes against the AD-specific target library. From the docking results, we identified 43 potential targets for HCTZ, 38 potential targets for amlodipine, and 46 potential targets for losartan, whose docking scores with the corresponding aHTN drugs were ≥6. We searched in the literature for the reported drug-target interactions among the top 10 targets for each aHTN drug and constructed a CSP-target mapping analysis as shown in Fig. 4. For each aHTN compound, we selected two predicted protein targets (one experimentally validated and one nonvalidated) as examples; their docking poses and detailed DTN interactions are shown in Fig. 5. The CCB drug amlodipine has been predicted to form hydrogen bonds with its validated target, acetylcholinesterase (AChE) [26], in residues Gln71, Tyr72, Tyr124, Ser125, and Tyr137 (Fig. 5A). Amlodipine has also been predicted to form two hydrogen bonds with its nonvalidated target, the mitogen-activated protein kinase (MAPK), in residues Ala111 and Asp168 (Fig. 5B). Meanwhile, the diuretic drug HCTZ was predicted to form four hydrogen bonds with its validated target phosphodiesterase 4B (PDE4B) [27] in residues Tyr233, Tyr403, Met411, and Met431 (Fig. 5C). Three hydrogen bonds were predicted to form between HCTZ and its nonvalidated target, cyclooxygenase 2 (COX-2) in residues Phe381, Met522, and Ser530 (Fig. 5D). Finally, the RAAS drug losartan was predicted to form four hydrogen bonds with its validated target, the peroxisome proliferator–activated receptor gamma (PPARγ) [28] in residues Cys285, Gln286, Tyr327, and Tyr473, respectively (Fig. 5E). With the nonvalidated target monoamine oxidase B (MAOB), Losartan was predicted to form four hydrogen bonds in residues Ile198, Gln206, Lys296, and Tyr 435 (Fig. 5F). The distances of the hydrogen bonds are all within 2.5 to 4.0 Å range. These results showed that these three representative aHTN medications may potentially act on a number of AD-related protein targets in addition to their primary targets, and a number of these interactions have been experimentally confirmed in the literature.
Fig. 4.

Chemogenomics systems pharmacology (CSP) target mapping analysis for the molecular mechanism of HCTZ, amlodipine, and losartan for AD treatment. Each blue circle represents an aHTN drug, and each of the other nodes represents a predicted protein target either with experimentally validated binding affinities (green) or without experimental validation (magenta). Each edge connecting an aHTN drug and a protein target represents either an unconfirmed (dashed line) or a confirmed (solid line) drug-target interaction predicted by the HTDocking algorithm. The numbers on the edges represent the docking scores (predicted log Ki's) of the drug-target interaction. Abbreviations: AD, Alzheimer's disease; AChE, acetylcholinesterase; ADRB2, β-2 adrenergic receptor; aHTN, antihypertensive; BChE, butyrylcholinesterase; CHRM2, cholinergic receptor muscarinic 2; COX, cyclooxygenase; MAOB, monoamine oxidase B; MAPK, mitogen-activated protein kinase; NOS, nitric oxide synthase; PDE, phosphodiesterase; PPARγ, peroxisome proliferator–activated receptor gamma.
Fig. 5.

Docking poses of the representative aHTN compounds against the predicted targets. Each of the aHTN compounds was docked against an experimentally validated target (left two columns—A, C, and E) and a nonvalidated target (right two columns—B, D, and F) predicted in our CSP-target mapping study. The first and third columns show the relative positions of the binding pockets (pink surfaces) in the proteins. The second and fourth columns show the detailed interactions between the aHTN compounds and the adjacent residues in the binding pocket of the protein targets. The aHTN compounds were shown as blue sticks, and the interacting residues were shown as orange sticks with residue numbers labeled. The polar interactions between the aHTN compounds and the protein targets were shown as dotted lines, and their bond lengths (Å) were labeled. Abbreviations: AChE, acetylcholinesterase; aHTN, antihypertensive; CCB, calcium channel blockers; COX, cyclooxygenase; CSP, chemogenomics systems pharmacology; HCTZ, hydrochlorothiazide; MAOB, monoamine oxidase B; MAPK, mitogen-activated protein kinase; PDE, phosphodiesterase; PPARγ, peroxisome proliferator–activated receptor gamma; RAAS, renin-angiotensin-aldosterone system.
4. Discussion
We applied clinical data-mining analyses to investigate patients with AD in three decades of clinical observations and evaluated whether/which medication(s) used for HTN, a common coexisting disease of AD, is related to cognitive decline in patients diagnosed with AD. As shown previously, the use of aHTN medications was associated with a reduced rate of cognitive decline only in those patients who also used ChEI. This suggests that aHTN medications and ChEIs may produce a synergistic effect against cognitive decline in AD patients with hypertension. Moreover, patients under one specific combination of three classes of aHTN drugs, namely diuretics, CCB, and RAAS, were associated with the most significant reduction in the rate of cognitive decline compared with all other groups.
Our study provides new and enriched data on the protective effect of aHTN medications against cognitive decline in patients with AD, and the results are congruent with the previous reports of decreased risk of cognitive decline and developing AD in hypertension-medicated patients and elderly people [13], [14], [15], [16], [17], [18]. Our results are also in line with the most recent findings from the National Institutes of Health Systolic Blood Pressure Intervention Trial-Memory and Cognition IN Decreased Hypertension (SPRINT-MIND) [29] presented at the Alzheimer's Association International Conference 2018, which suggested that aggressive blood pressure control reduced the risk of mild cognitive impairment and dementia. Our study provides new evidence on the protective effect of aHTN medications against cognitive decline in patients with AD. This finding bears clinical importance because it suggests that aHTN medications are not only a prophylaxis against developing AD and cognitive decline, but can also be used as an add-on treatment.
Moreover, our study is the first to discover an optimal combination of different aHTN drug classes on the cognitive decline in AD patients with hypertension. According to a recent review article on the protective effect of aHTN medications against AD and cognitive decline [30], most studies have supported a preference for RAAS inhibitors [31], [32], [33] and CCBs [15], [34] in protecting against cognitive decline, whereas some studies also reported no difference among the aHTN classes [35]. It is worth noting that although many studies also involved patients under combinations of aHTN drug classes, most of them tested the effect of different drug classes separately [16], [17]. Few studies have reported different combination therapies that had a more pronounced protective effect against cognitive decline [13], [15], [32], [36], [37], but their patient populations are generally elderly patients or elderly patients identified with high-risk comorbidities, rather than those already diagnosed with AD. According to our results, there seems to be no difference in cognitive decline when each of the drug classes were used alone, but the combination of RAAS + CCB + diuretics was significantly better. This observation does not contradict either side of the previous studies, but further points out a possibility that the preference for RAAS inhibitors and CCBs could be explained by the fact that they can produce a stronger protective effect when used in the specific combination. Our result also highlights the importance of considering specific drug combinations as a distinct group instead of testing each of its components separately.
In addition, our results uncovered that aHTN medications may have a synergistic effect with ChEIs on decreasing the rate of cognitive decline in AD patients with hypertension. First, we analyzed the effect of ChEI on the cognitive decline in AD patients with hypertension over a 24-month follow-up. The result showed that ChEI users had a marginally significant slower rate of cognitive decline (P = .068) compared with the non-ChEI users. We then compared the effect of aHTN medications in the ChEI users and nonusers, respectively (Fig. 2). The rate of cognitive decline in the ChEI monotherapy group and the ChEI−/aHTN+ group was not significantly different from that in the ChEI−/aHTN− group, whereas the cognitive decline rate of ChEI+/aHTN+ group was significantly slower. To the best of our knowledge, our study is the first to discover the interaction between ChEIs and aHTN medications. As the first-line treatment for AD, the ChEIs have been shown by numerous RCTs and observational studies to have a significant effect on slowing cognitive decline in different AD populations [38], [39], [40]. However, it is important to note that most of these RCTs typically have a short time span of 24 to 26 weeks. Studies on the long-term effect (≥24 months) of the ChEIs on cognitive decline are few and reveal little to no clinically significant benefits on MMSE measures [41], whereas some studies did find a significant improvement using other end points such as nursing home admission [42]. The beneficial effect of ChEI monotherapy is also reported to have considerable interindividual variability [43]. A recent study [44] has searched for clinical factors that can predict ChEI response, yet it did not find any correlations between the pattern of ChEI response and any of the factors investigated, except for initial response in the first few months. Their scope of search included ChEI dose, APOE genotype, and CYP2D6 polymorphisms but did not include the common comorbidities and coadministered medications. In our study, we were the first to discover that the therapeutic effect of ChEIs may be dependent on concomitant use of aHTN medications in AD patients with coexisting hypertension. This synergistic effect suggests that aHTN drugs may serve as an add-on therapy for delaying cognitive decline in these patients.
To understand the molecular mechanisms behind the synergistic effects between AD and HTN medications observed in clinical settings, we applied our developed AD and CVD database-guided CSP-target mapping methodology techniques to map out DTNs for AD and HTN [20], [21], [22], [23]. As shown in Fig. 3, we identified certain protein targets associated with two diseases, which indicated that aHTN drug(s) targeting these proteins could also have a direct effect on AD pathologic pathways. Such systems pharmacology DTN mapping analyses also suggested a molecular level synergism in accordance with the clinical level synergistic treatment of patients with AD with combinations of ChEI and aHTN drugs.
There have been some studies investigating the relationship between aHTN medication use and cognitive improvement. However, these articles only reported a reduced risk of AD in the population with hypertension treatment [15], [45], [46]. Because many previous studies have shown that elevated blood pressure is one of the major risk factors for AD, researchers make an assumption that aHTN medications may reduce the incidence of AD by controlling the blood pressure. Nevertheless, others have suggested that the aHTN drugs belonging to different drug classes may have specific protective effects in reducing AD risk [47]. In addition, some reports also found that controlling changes in blood pressure did not significantly alter the risk of AD dementia [46]. Thus, it is suggested that the aHTN drugs have a beneficial role in reducing the incidence of AD that is in addition to or independent from their benefit on blood pressure control.
The mechanism for the protective effect of diuretics against AD has not been widely studied. Although diuretics are a general class of aHTN medications with different mechanisms of action, further analysis of the medication history of the patients in diuretics + CCB + RAAS group in set 1 patients indicated that potassium-sparing diuretics and thiazides are the prevalent diuretics used in this combination. Some studies indicated that potassium-sparing diuretics had a potential to decrease AD risk because of a protective role of high potassium levels related to reduced vasoconstriction and chronic inflammation [45], [46], [47], presumably via inhibiting their primary therapeutic target mineralocorticoid receptor (NR3C2) [48]. However, some other studies found no significant differences between potassium-sparing diuretics and other nonsparing diuretics in decreasing AD risks [46]. On the other hand, the thiazide diuretics have been reported to inhibit carbonic anhydrases (CA1, CA2, and CA4) [49] in addition to their primary target SLC12A3 [50]. Although there has not been any study showing a connection between SLC12A3 and AD risk, inhibition of carbonic anhydrases has been reported to lead to a decreased release of cytochrome c from mitochondria to the cytoplasm, and hence reduce the amyloid beta (Aβ)-induced neurotoxicity [51], which could be a potential mechanism for the protective effect of thiazide diuretics against AD.
The effect of CCBs in reducing AD incidence is controversial. Some epidemiologic studies showed that the use of CCB is related to a reduced risk of dementia [15], [52]. Some others found no significant improvement in primary outcome measures [53], [54], [55]. Many CCBs were tested in clinical trial for AD treatment. Nimodipine and nilvadipine were shown to prevent cognitive decline in some trials, whereas other drugs within the same family failed [55]. Calcium homeostasis has been implicated in a role in AD. Aβ neurotoxicity results in an intracellular calcium influx via CACNA1C channels, which further leads to hyperphosphorylated tau and autophagy dysfunction [53], [56]. In addition, L-type voltage-gated calcium channel (CACNA1C, CACNA1D, CACNA1S, and CACNA1F) blockers prevent neurotoxicity with the potential to reduce Aβ formation and maintain calcium homeostasis [53].
The aHTN therapies targeting RAAS [57], including ACE inhibitors (ACEI), angiotensin II receptor blocker (ARB) [58], and renin inhibitor, have been indicated to play a complicated role in AD pathogenesis. The beneficial effect of RAAS drugs to improve brain function was implicated in many studies. It was thought that the main mechanism of this improvement is to increase cerebral blood flow (CBF) by reducing vasoconstriction [47], [59].
As for ACEIs, the effect is conflicted. On one hand, ACEIs have been reported to slow down the cognitive decline and dementia process [59], [60]. On the other hand, ACE may be involved in the degradation of Aβ, thus ACEIs might contribute to Aβ pathology and induce both the incidence [45] as well as the mortality [61] of AD. In addition, ACEIs augment levels of substance P, a substance degraded by ACE, which leads to an increased activity of another Aβ degrading enzyme, neprilysin, thus having indirect beneficial effects for Aβ clearance [59], [62]. It makes the effect of ACEIs on AD more complicated [59].
ARBs also block angiotensin II signaling by acting on AGTR1 and AGTR2, and hence reduce vasoconstriction. This can result in increased CBF and improved cognitive function. Yet, recent reports suggested that the level of angiotensin II in the brain had a more important role in cognitive function. If ARB blocks the interaction between angiotensin II and the angiotensin II receptor 1, more angiotensin II will be converted to angiotensin III and then to angiotensin IV [47], [63]. Angiotensin IV acts on c-Met receptor, which is associated with increases in long-term potentiation, synaptic plasticity, and CBF [64]. Angiotensin IV can also inhibit receptor insulin-regulated aminopeptidase, thus reducing the catalytic activity on vasopressin and oxytocin, both of which are related to memory consolidation [47], [65], [66]. The interaction between angiotensin IV and insulin-regulated aminopeptidase has also been reported to increase the uptake of glucose, further supporting its cognitive enhancing effects [47], [66]. Another aspect of angiotensin II that is related to AD is the regulation of glycogen synthase kinase 3β (GSK3β). Angiotensin II can enhance the GSK3β level, which is thought to contribute to tau phosphorylation [67], inhibition of acetylcholine (ACh) release [68], and induced oxidative stress [58], [59]. ARBs can reduce angiotensin II–mediated GSK3β elevation, and its contribution to these cognitive-impairing factors, thus enhancing brain function [59].
In addition to the aforementioned pharmacologic effects, our CSP-target map (Fig. 4) also suggests that the diuretics + CCB + RAAS combination may directly act on several AD-related protein targets, with significant overlap. These targets include important proteins from several distinct pathologic pathways of AD, such as the enzymes involved in the clearance of neurotransmitters (AChE and butyrylcholinesterase [BChE]), enzymes related to oxidative stress (MAOB, NOS1, and NOS2), enzymes mediating cellular signaling pathways (PDE4B, PDE4D, PDE5A, PDE7A, MAPK, and COX-2), neurotransmitter receptors (cholinergic receptor muscarinic 2 [CHRM2] and β-2 adrenergic receptor [ADRB2]), a microtubule-associated protein (TAU), and a nuclear receptor related to neuroinflammation (PPARγ). Subsequent literature study has indicated that a number of these predicted interactions have already been reported previously. Fig. 5 demonstrates the binding poses and detailed interactions between the three aHTN drugs and some of their predicted targets. These results suggest that the synergistic effect of the three aHTN medications on cognitive decline may be a net result of all these drug-target interactions. The role of each predicted target in AD as well as its therapeutic effects is addressed subsequently.
The AChE has been predicted to interact with all three representative drugs from the three aHTN medication classes. The AChE catalyzes the breakdown of the neurotransmitter ACh at the synaptic clefts, and the inhibitors of AChE (ChEIs) are the first-line treatment for AD because of their ability to reverse the deficit of ACh in patients with AD. Among the three aHTN drugs, amlodipine has been reported to bind to AChE with a Ki of 0.19 μM [26]. The same study also confirmed that amlodipine may interact with the BChE with a Ki of 0.11 μM. BChE is another subtype of cholinesterase inhibitor, which shares ∼65% amino acid sequence identity with that of AChE and has also played an important role in AD [69].
Our CSP-target map analysis has also predicted that the MAOB, one of the important proteins involved in the oxidative stress mechanism [70], is a potential target for both losartan and HTCZ, which is congruent with the experiments reported in the literature. Oxidative stress is characterized by an imbalance in the redox state of the cells, which occurs as a result of the overproduction of reactive oxygen species or the defects in the antioxidant system [71]. Oxidative stress has been considered to be a major part of the pathophysiology of AD, causing significant tissue damage and neuronal death [72]. Localized in the outer mitochondrial membrane, MAOB catalyzes the oxidative deamination of multiple neurotransmitters including dopamine, serotonin, and norepinephrine, as well as exogenous amine species [73], during which it produces hydrogen peroxide (H2O2) as a side product, which is a potential source of oxidative stress [74]. It is possible that these two drugs together alleviate the cognitive symptoms of AD via reducing the oxidative stress generated by MAOB. The two isoforms of nitric oxide synthase, NOS1 and NOS2, also known as neuronal NOS and inducible NOS, have been reported to contribute to neuronal oxidative stress by producing nitric oxide (NO) [75], which is a free radical itself and can also form peroxynitrite when combined with superoxide anions [76]. In our CSP-target mapping analysis, all three aHTN drugs were predicted to target on NOS2, whereas losartan and HCTZ were predicted to act on NOS1, which may also partially explain the synergistic effect of the three aHTN medications against the cognitive decline in AD.
In addition, several isoforms of the phosphodiesterases (PDE4B, PDE4D, PDE5A, and PDE7A) have also been predicted to be the targets of the aHTN medications, and the inhibition of different PDE isoforms by losartan and HCTZ has been experimentally validated [27], [77]. It has been previously shown in many AD animal models that specific PDE inhibitors improved memory performances by elevating the levels of cyclic adenosine monophosphate and/or cyclic guanosine monophosphate, and hence promoting the gene expression regulated by cyclic adenosine monophosphate response element-binding [78], [79]. These genes are crucial for long-term memory formation and potentiation [80]. Therefore, the inhibition of PDEs provides another possible mechanism underlying the cognitive benefits of the diuretics, CCB, and RAAS inhibitor combination. The p38 MAPK is another cell signaling modulator that has been proposed as a target for treating AD [81]. Activation of the p38MAPK leads to the phosphorylation of serine and threonine residues in various kinases and transcription factors, and upregulates the inflammatory response to cellular stress [82]. Previous studies suggested that elevated MAPK activity is a significant contributing factor for the AD-related neuroinflammation in the brain, particularly in the microglia and astrocytes [83], [84]. Furthermore, the p38MAPK has also been shown to have a direct role in the hyperphosphorylation of the tau protein, which is one of the hallmarks of AD pathology [85]. Therefore, the predicted interaction between MAPK and HCTZ and amlodipine may implicate a role of neuroinflammation in the mechanism underlying the protective effect of the diuretics + CCB + RAAS combination against cognitive decline in patients with AD. Finally, our CSP-target map analysis has also indicated a potential interaction between HCTZ and the COX-2 enzyme. The COX enzyme, also known as prostaglandin H synthase, catalyzes the synthesis of prostanoids such as prostaglandins, prostacyclin, and thromboxane, each of which plays an important role in the inflammation pathway. Because of its role in inflammatory reactions, the COX enzymes have been the primary targets for the nonsteroidal anti-inflammatory drugs [86], [87]. Two selective COX-2 inhibitors, rofecoxib and naproxen, have been accessed in a phase 3 clinical trial for their potential to dampen the cognitive decline in patients with mild to moderate AD, but did not show significant benefit [88]. Given the importance of neuroinflammation in the pathology of AD, however, the author also commented in the article that additional trials using other nonsteroidal anti-inflammatory drugs may still be warranted.
Furthermore, the CHRM2 has been predicted to be the target of both amlodipine and HCTZ by the HTDocking program. The choline deficit is a major symptom of AD, and elevating the choline level by the use of ChEIs has long been the mainstream treatment strategy for AD. The CHRM2 receptor has been shown to regulate the release of ACh from cholinergic neurons, and CHRM2 selective inhibitors have been proposed as a promising strategy to treat AD by elevating the ACh levels [89]. On the other hand, the ADRB2 has been predicted to be a potential target of losartan. Activation of the ADRB2 receptor has been reported to enhance the activity of γ-secretase, and hence accelerate the production of amyloid plaque [90]. Therefore, as a potential inhibitor of ADRB2, losartan may also slow down the AD progression by modulating Aβ production.
The tau phosphorylation pathway is another promising target for the treatment of AD [91], [92]. As a microtubule-associated protein, tau participates in the assembly of tubulin into microtubules in the brain [93]. The hyperphosphorylation of tau protein has been found to be the major cause of the breakdown of microtubules and the formation of neurofibrillary tangles, which is known to be one of the hallmarks of AD [94]. Current strategies for blocking tau hyperphosphorylation mainly include targeting the upstream enzymes of tau phosphorylation, such as (1) inhibiting the kinases catalyzing tau phosphorylation, such as GSK3β, cyclin-dependent kinase 5 (CDK-5), and other kinases; (2) enhancing the activity of tau phosphatases (PP2A); and (3) enhancing the glycosylation of tau by the β-N-acetylglucosamine (O-GlcNAcylation) group [91]. Direct inhibition of tau protein has not yet been reported. In our CSP-target mapping analysis, HCTZ and amlodipine were both predicted to bind to the tau protein, which may potentially block its phosphorylation, and the consequent formation of neurofibrillary tangles.
Importantly, in our docking study, both losartan and amlodipine have been predicted to target the PPARγ, and it has been found that a metabolite of losartan, EXP3179, has a partial agonist activity on PPARγ [28]. PPARγ is another protein target that has been extensively studied for its implications in AD. The PPARs are a family of three nuclear receptors (α, γ, and δ), each of which regulates a set of genes involved in lipid and energy metabolisms [95]. Because of the ability of PPARγ to regulate both lipid and carbohydrate metabolisms, particularly the serum glucose levels and insulin sensitivity, the PPARγ agonists (pioglitazone and rosiglitazone) have been developed into medications for treating type II diabetes mellitus [96]. Apart from that, the PPARγ agonists have also been found to have a therapeutic potential for AD by targeting multiple aspects of AD pathology, including Aβ homeostasis, neuroinflammation, insulin sensitivity, energy metabolism, and lipid metabolism [97]. As a matter of fact, the PPARγ agonist pioglitazone has entered phase III clinical trials for its effect in slowing the cognitive decline in patients with AD. Our CSP-target mapping revealed that these results may also explain the effect of the diuretics + CCB + RAAS combination against AD-related cognitive decline.
AD is a multifactorial disease. The limitations of current treatments are that they target specific downstream neurochemical pathology whereas an upstream underlying mechanism remains to be unveiled. A combination medication that acts on a number of molecular and cellular pathologic pathways in AD, including Aβ accumulation, tau phosphorylation, chronic inflammation, oxidative stress, and impaired CBF, might have more beneficial effects compared with a single medication to treat AD [46]. Using these drugs in combination may help to achieve such effects.
We also point out that the present study has limitations. First, the patient number in set 2 was not large enough to allow further analysis for different aHTN drug classes. Therefore, interpretation of the molecular mechanisms underlying the possible synergism between hypertensive drugs and ChEIs is difficult. Second, we did not measure the blood pressure levels, which might have been an intermediate factor for the effect of aHTN drugs on cognitive decline. Although some studies reported that high blood pressure is a contributing factor to cognitive decline [98], [99], other studies also reported a U-shaped relationship between blood pressure and cognitive impairment [100], [101], suggesting that mildly elevated blood pressure may be beneficial in patients with AD, particularly in APOEε4 carriers [35]. Without blood pressure measurements, our study does not reveal the role of blood pressure in the therapeutic effect of aHTN medications on cognitive decline. Third, one recent study has pointed out that the cognitive benefits of ACEIs in patients with AD are dependent on rs1800764 and rs4291 genotypes [102]. Because we did not have the information regarding the rs1800764 and rs4291 variants, the genetic influence on the effectiveness of the combination therapy remains to be tested. Fourth, one potential factor that may contribute to the better cognitive outcome in patients treated with the three aHTN medications is that these patients might have visited their doctors more frequently, and consequently, they have better control of hypertension and other disorders that we did not examine here. Finally, the prediction of drug-target interactions using the CSP-target mapping analysis was carried out in silico using the HTDocking platform. The actual binding affinities for these interactions need to be experimentally validated in future studies.
5. Conclusions
Our clinical outcome analyses supported the protective effect of aHTN medications against cognitive decline in patients who have already been diagnosed with AD. The combined use of aHTN medications and ChEIs was associated with a significantly slower rate of cognitive decline compared with each of the drugs alone in patients with AD, suggesting a potential synergistic effect. These findings indicate that improving the vascular health of patients with AD can produce a cognitive benefit, and also suggest that the mechanisms of actions of aHTN medications may provide a novel approach for developing therapies against AD. Well-controlled clinical trials that test the effect of the combined use of ChEIs and different classes of aHTN drugs, especially the combination of diuretics, CCBs, and RAAS inhibitors, could be valuable for determining the specific mechanisms of the synergism.
Research in Context.
-
1.
Systematic review: Hypertension is an important risk factor for developing Alzheimer's disease (AD) in later stages of life; hypertension also etiologically contributes to the pathologies of AD.
-
2.
Interpretation: Some antihypertensive (aHTN) drug classes had been shown to prevent or slow cognitive decline in elderly patients. In this study, we confirmed the effect of aHTN medications on slowing cognitive decline in AD cases and examined combinations of aHTN drugs that achieved synergistic effects against cognitive decline. This study also provides a molecular level interpretation for the synergism using a chemogenomics systems pharmacology approach and identified the protein targets of the aHTN drugs that may explain their anti-AD activities.
-
3.
Future directions: The effect of aHTN medications on slowing cognitive decline needs further confirmation by randomized controlled trials. Further studies could also investigate whether aHTN medications have an indirect effect on cognitive decline through lowering blood pressure.
Acknowledgments
This study was supported by NIH grants P30 DA035778, W81XWH-16-1-0490, P50 AG005133, P01 AG025204, R01 DA025612, RF1 AG025516, U01 AG051406, R01 AG027224, R01 AG030653, and R01 AG041718. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health, the National Institutes of Health, the Department of Veterans Affairs, or the United States Government.
Footnotes
Conflict of interest: Dr Oscar L. Lopez has served as a consultant for Grifols, Inc, and Lundbeck. Dr William E. Klunk receives royalty payments from GE Healthcare (indirect through a license agreement with the University of Pittsburgh). Dr X.-Q Xie serves as a consultant for Oxis Biotech under an agreement with the University of Pittsburgh.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.trci.2018.09.001.
Contributor Information
Oscar L. Lopez, Email: LopezOL@upmc.edu.
Xiang-Qun Xie, Email: xix15@pitt.edu.
Supplementary data
Fig. S1.
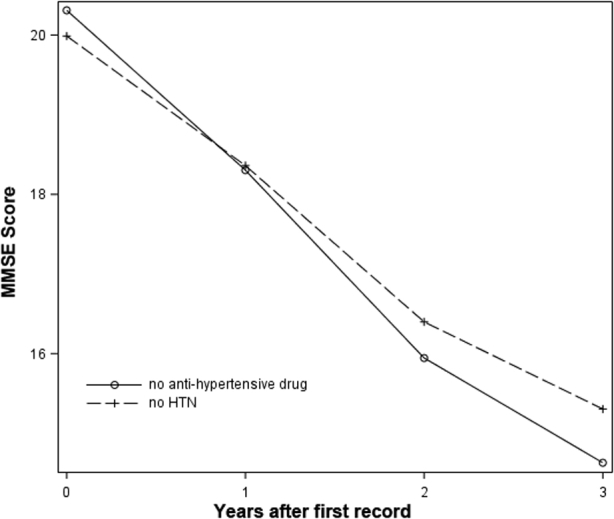
Cognitive decline of patients with no hypertension (HTN) versus patients with HTN that did not use any anti-hypertensive drugs. No significant difference was found between the two groups using the mixed-effect regression model.
References
- 1.Drugs for Alzheimer's disease: Best avoided. No therapeutic advantage. Prescrire Int. 2012;21:150. [PubMed] [Google Scholar]
- 2.Schmitt B., Bernhardt T., Moeller H.-J., Heuser I., Frölich L. Combination therapy in Alzheimer's disease. CNS Drugs. 2004;18:827–844. doi: 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]
- 3.Sakurai H., Hanyu H., Kanetaka H., Sato T., Shimizu S., Hirao K. Prevalence of coexisting diseases in patients with Alzheimer's disease. Geriatr Gerontol Int. 2010;10:216–217. doi: 10.1111/j.1447-0594.2010.00609.x. [DOI] [PubMed] [Google Scholar]
- 4.Zlokovic B.V. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu C., Winblad B., Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005;4:487–499. doi: 10.1016/S1474-4422(05)70141-1. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman R.F., Schneider A.L., Albert M., Alonso A., Bandeen-Roche K., Coker L. Midlife hypertension and 20-year cognitive change: The atherosclerosis risk in communities neurocognitive study. JAMA Neurol. 2014;71:1218–1227. doi: 10.1001/jamaneurol.2014.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruitenberg A., Skoog I., Ott A., Aevarsson O., Witteman J.C., Lernfelt B. Blood pressure and risk of dementia: Results from the Rotterdam study and the Gothenburg H-70 Study. Dement Geriatr Cogn Disord. 2001;12:33–39. doi: 10.1159/000051233. [DOI] [PubMed] [Google Scholar]
- 8.Verghese J., Lipton R.B., Hall C.B., Kuslansky G., Katz M.J. Low blood pressure and the risk of dementia in very old individuals. Neurology. 2003;61:1667–1672. doi: 10.1212/01.wnl.0000098934.18300.be. [DOI] [PubMed] [Google Scholar]
- 9.Kuller L.H., Lopez O.L., Newman A., Beauchamp N.J., Burke G., Dulberg C. Risk factors for dementia in the Cardiovascular Health Cognition Study. Neuroepidemiology. 2003;22:13–22. doi: 10.1159/000067109. [DOI] [PubMed] [Google Scholar]
- 10.Petitti D.B., Crooks V.C., Buckwalter J.G., Chiu V. Blood pressure levels before dementia. Arch Neurol. 2005;62:112. doi: 10.1001/archneur.62.1.112. [DOI] [PubMed] [Google Scholar]
- 11.Skoog I., Nilsson L., Persson G., Lernfelt B., Landahl S., Palmertz B. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 12.Qiu C., von Strauss E., Fastbom J., Winblad B., Fratiglioni L. Low blood pressure and risk of dementia in the Kungsholmen project: A 6-year follow-up study. Arch Neurol. 2003;60:223–228. doi: 10.1001/archneur.60.2.223. [DOI] [PubMed] [Google Scholar]
- 13.Gelber R.P., Ross G.W., Petrovitch H., Masaki K.H., Launer L.J., White L.R. Antihypertensive medication use and risk of cognitive impairment: The Honolulu-Asia Aging Study. Neurology. 2013;81:888–895. doi: 10.1212/WNL.0b013e3182a351d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soto M.E., Abellan van Kan G., Nourhashemi F., Gillette-Guyonnet S., Cesari M., Cantet C. Angiotensin-converting enzyme inhibitors and Alzheimer's disease progression in older adults: Results from the réseau sur la maladie d'Alzheimer français cohort. J Am Geriatr Soc. 2013;61:1482–1488. doi: 10.1111/jgs.12415. [DOI] [PubMed] [Google Scholar]
- 15.Forette F., Seux M.-L., Staessen J.A., Thijs L., Babarskiene M.-R., Babeanu S. The prevention of dementia with antihypertensive treatment: New evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046–2052. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- 16.Peters R., Beckett N., Forette F., Tuomilehto J., Clarke R., Ritchie C. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): A double-blind, placebo controlled trial. Lancet Neurol. 2008;7:683–689. doi: 10.1016/S1474-4422(08)70143-1. [DOI] [PubMed] [Google Scholar]
- 17.Hajjar I., Catoe H., Sixta S., Boland R., Johnson D., Hirth V. Cross-sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J Gerontol A Biol Sci Med Sci. 2005;60:67–73. doi: 10.1093/gerona/60.1.67. [DOI] [PubMed] [Google Scholar]
- 18.Guo Z., Fratiglioni L., Zhu L., Fastbom J., Winblad B., Viitanen M. Occurrence and progression of dementia in a community population aged 75 years and older: Relationship of antihypertensive medication use. Arch Neurol. 1999;56:991–996. doi: 10.1001/archneur.56.8.991. [DOI] [PubMed] [Google Scholar]
- 19.Wang L., Xie X.-Q. Computational target fishing: What should chemogenomics researchers expect for the future of in silico drug design and discovery? Future Med Chem. 2014;6:247–249. doi: 10.4155/fmc.14.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie X.-Q., Wang L., Liu H., Ouyang Q., Fang C., Su W. Chemogenomics knowledgebased polypharmacology analyses of drug abuse related G-protein coupled receptors and their ligands. Front Pharmacol. 2014;5 doi: 10.3389/fphar.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L., Ma C., Wipf P., Liu H., Su W., Xie X.-Q. TargetHunter: An In Silico Target Identification Tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013;15:395–406. doi: 10.1208/s12248-012-9449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H., Wang L., Lv M., Pei R., Li P., Pei Z. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model. 2014;54:1050–1060. doi: 10.1021/ci500004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H., Ma S., Feng Z., Wang D., Li C., Cao Y. Cardiovascular Disease Chemogenomics Knowledgebase-guided Target Identification and Drug Synergy Mechanism Study of an herbal formula. Sci Rep. 2016;6:33963. doi: 10.1038/srep33963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez O.L., Becker J.T., Wahed A.S., Saxton J., Sweet R.A., Wolk D.A. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80:600–607. doi: 10.1136/jnnp.2008.158964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamboh M. The relationship of APOE polymorphism and cholesterol levels in normoglycemic and diabetic subjects in a biethnic population from the San Luis Valley, Colorado. Atherosclerosis. 1995;112:145–159. doi: 10.1016/0021-9150(94)05409-c. [DOI] [PubMed] [Google Scholar]
- 26.Chiou S.-Y., Lai G.-W., Tsai Y.-H., Lin L.-Y., Lin G. QSAR for acetylcholinesterase and butyrylcholinesterase inhibition by cardiovascular drugs and benzodiazepines. Med Chem Res. 2005;14:297–308. [Google Scholar]
- 27.Moore P.F. The effects of diazoxide and benzothiadiazine diuretics upon phosphodiesterase. Ann N Y Acad Sci. 1968;150:256–260. doi: 10.1111/j.1749-6632.1968.tb19050.x. [DOI] [PubMed] [Google Scholar]
- 28.Schupp M., Lee L.D., Frost N., Umbreen S., Schmidt B., Unger T. Regulation of peroxisome proliferator–activated receptor γ activity by losartan metabolites. Hypertension. 2006;47:586–589. doi: 10.1161/01.HYP.0000196946.79674.8b. [DOI] [PubMed] [Google Scholar]
- 29.Williamson JD, SPRINT Research Group. A Randomized Trial of Intensive Versus Standard Systolic Blood Pressure Control and the Risk of Mild Cognitive Impairment and Dementia: Results from SPRINT MIND. Presented at: Alzheimer's Association International Conference; July 14-18, 2018; Los Angeles, CA, USA.
- 30.Kherada N., Heimowitz T., Rosendorff C. Antihypertensive therapies and cognitive function: A review. Curr Hypertens Rep. 2015;17:79. doi: 10.1007/s11906-015-0592-7. [DOI] [PubMed] [Google Scholar]
- 31.Starr J.M., Whalley L.J., Deary I.J. The effects of antihypertensive treatment on cognitive function: Results from the HOPE study. J Am Geriatr Soc. 1996;44:411–415. doi: 10.1111/j.1532-5415.1996.tb06412.x. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative P., Neal B., MacMahon S. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069–1075. doi: 10.1001/archinte.163.9.1069. [DOI] [PubMed] [Google Scholar]
- 33.Yagi S., Akaike M., Aihara K-i, Iwase T., Yoshida S., Sumitomo-Ueda Y. High plasma aldosterone concentration is a novel risk factor of cognitive impairment in patients with hypertension. Hypertens Res. 2010;34:74–78. doi: 10.1038/hr.2010.179. [DOI] [PubMed] [Google Scholar]
- 34.Trompet S., Westendorp R.G., Kamper A.M., de Craen A.J. Use of calcium antagonists and cognitive decline in old age: The Leiden 85-Plus Study. Neurobiol Aging. 2008;29:306–308. doi: 10.1016/j.neurobiolaging.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 35.de Oliveira F.F., Chen E.S., Smith M.C., Bertolucci P.H.F. Associations of blood pressure with functional and cognitive changes in patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 2016;41:314–323. doi: 10.1159/000447585. [DOI] [PubMed] [Google Scholar]
- 36.Fogari R., Mugellini A., Zoppi A., Lazzari P., Destro M., Rinaldi A. Effect of telmisartan/hydrochlorothiazide vs lisinopril/hydrochlorothiazide combination on ambulatory blood pressure and cognitive function in elderly hypertensive patients. J Hum Hypertens. 2005;20:177–185. doi: 10.1038/sj.jhh.1001964. [DOI] [PubMed] [Google Scholar]
- 37.Douiri A., McKevitt C., Emmett E.S., Rudd A.G., Wolfe C.D.A. Long-term effects of secondary prevention on cognitive function in stroke patients. Circulation. 2013;128:1341–1348. doi: 10.1161/CIRCULATIONAHA.113.002236. [DOI] [PubMed] [Google Scholar]
- 38.Salloway S., Ferris S., Kluger A., Goldman R., Griesing T., Kumar D. Efficacy of donepezil in mild cognitive impairment: A randomized placebo-controlled trial. Neurology. 2004;63:651–657. doi: 10.1212/01.wnl.0000134664.80320.92. [DOI] [PubMed] [Google Scholar]
- 39.Koontz J., Baskys A. Effects of galantamine on working memory and global functioning in patients with mild cognitive impairment: A double-blind placebo-controlled study. Am J Alzheimers Dis Other Demen. 2005;20:295–302. doi: 10.1177/153331750502000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feldman H., Gauthier S., Hecker J., Vellas B., Subbiah P., Whalen E. A 24-week, randomized, double-blind study of donepezil in moderate to severe Alzheimer's disease. Neurology. 2001;57:613–620. doi: 10.1212/wnl.57.4.613. [DOI] [PubMed] [Google Scholar]
- 41.Courtney C., Farrell D., Gray R., Hills R., Lynch L., Sellwood E. Long-term donepezil treatment in 565 patients with Alzheimer's disease (AD2000): Randomised double-blind trial. Lancet. 2004;363:2105–2115. doi: 10.1016/S0140-6736(04)16499-4. [DOI] [PubMed] [Google Scholar]
- 42.Lopez O.L. Cholinesterase inhibitor treatment alters the natural history of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2002;72:310–314. doi: 10.1136/jnnp.72.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raschetti R., Maggini M., Sorrentino G.C., Martini N., Caffari B., Vanacore N. A cohort study of effectiveness of acetylcholinesterase inhibitors in Alzheimer’s disease. Eur J Clin Pharmacol. 2005;61:361–368. doi: 10.1007/s00228-005-0946-1. [DOI] [PubMed] [Google Scholar]
- 44.Miranda L.F., Gomes K.B., Silveira J.N., Pianetti G.A., Byrro R., Peles P.R. Predictive factors of clinical response to cholinesterase inhibitors in mild and moderate Alzheimer's disease and mixed dementia: A one-year naturalistic study. J Alzheimers Dis. 2015;45:609–620. doi: 10.3233/JAD-142148. [DOI] [PubMed] [Google Scholar]
- 45.Khachaturian A.S., Zandi P.P., Lyketsos C.G., Hayden K.M., Skoog I., Norton M.C. Antihypertensive medication use and incident Alzheimer disease: The Cache County Study. Arch Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- 46.Yasar S., Xia J., Yao W., Furberg C.D., Xue Q.L., Mercado C.I. Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology. 2013;81:896–903. doi: 10.1212/WNL.0b013e3182a35228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson L., Tabet N., Richardson C., Gard P. Antihypertensives, angiotensin, glucose and Alzheimer’s disease. Expert Rev Neurother. 2013;13:477–482. doi: 10.1586/ern.13.32. [DOI] [PubMed] [Google Scholar]
- 48.Rogerson F. Determinants of spironolactone binding specificity in the mineralocorticoid receptor. J Mol Endocrinol. 2003;31:573–582. doi: 10.1677/jme.0.0310573. [DOI] [PubMed] [Google Scholar]
- 49.Puscas I., Coltau M., Baican M., Domuta G., Hecht A. Vasodilatory effect of diuretics is dependent on inhibition of vascular smooth muscle carbonic anhydrase by a direct mechanism of action. Drugs Exp Clin Res. 1999;25:271–279. [PubMed] [Google Scholar]
- 50.Chen X. TTD: Therapeutic target database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fossati S., Giannoni P., Solesio M.E., Cocklin S.L., Cabrera E., Ghiso J. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis. 2016;86:29–40. doi: 10.1016/j.nbd.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fritze J., Walden J. Clinical findings with nimodipine in dementia: Test of the calcium hypothesis. J Neural Transm Suppl. 1995;46:439–453. [PubMed] [Google Scholar]
- 53.Anekonda T.S., Quinn J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: The case for isradipine. Biochim Biophys Acta. 2011;1812:1584–1590. doi: 10.1016/j.bbadis.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Birks J., López-Arrieta J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002:CD000147. doi: 10.1002/14651858.CD000147. John Wiley & Sons, Ltd. [DOI] [PubMed] [Google Scholar]
- 55.Nimmrich V., Eckert A. Calcium channel blockers and dementia. Br J Pharmacol. 2013;169:1203–1210. doi: 10.1111/bph.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willis M., Kaufmann W.A., Wietzorrek G., Hutter-Paier B., Moosmang S., Humpel C. L-type calcium channel CaV 1.2 in transgenic mice overexpressing human AβPP751 with the London (V717I) and Swedish (K670M/N671L) mutations. J Alzheimers Dis. 2010;20:1167–1180. doi: 10.3233/JAD-2010-091117. [DOI] [PubMed] [Google Scholar]
- 57.Samer C.F., Daali Y., Wagner M., Hopfgartner G., Eap C.B., Rebsamen M.C. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br J Pharmacol. 2010;160:907–918. doi: 10.1111/j.1476-5381.2010.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abadir P.M., Foster D.B., Crow M., Cooke C.A., Rucker J.J., Jain A. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011;108:14849–14854. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashby E.L., Kehoe P.G. Current status of renin–aldosterone angiotensin system-targeting anti-hypertensive drugs as therapeutic options for Alzheimer's disease. Expert Opin Investig Drugs. 2013;22:1229–1242. doi: 10.1517/13543784.2013.812631. [DOI] [PubMed] [Google Scholar]
- 60.Kehoe P.G., Miners S., Love S. Angiotensins in Alzheimer's disease–friend or foe? Trends Neurosci. 2009;32:619–628. doi: 10.1016/j.tins.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Kehoe P.G., Davies N.M., Martin R.M., Ben-Shlomo Y. Associations of angiotensin targeting antihypertensive drugs with mortality and hospitalization in primary care patients with dementia. J Alzheimers Dis. 2013;33:999–1008. doi: 10.3233/JAD-2012-121090. [DOI] [PubMed] [Google Scholar]
- 62.Carson J.A., Turner A.J. β-Amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- 63.Fyhrquist F., Saijonmaa O. Renin-angiotensin system revisited. J Intern Med. 2008;264:224–236. doi: 10.1111/j.1365-2796.2008.01981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wright J.W., Harding J.W. Brain renin-angiotensin—a new look at an old system. Prog Neurobiol. 2011;95:49–67. doi: 10.1016/j.pneurobio.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 65.Stragier B., De Bundel D., Sarre S., Smolders I., Vauquelin G., Dupont A. Involvement of insulin-regulated aminopeptidase in the effects of the renin–angiotensin fragment angiotensin IV: A review. Heart Fail Rev. 2007;13:321–337. doi: 10.1007/s10741-007-9062-x. [DOI] [PubMed] [Google Scholar]
- 66.Vanderheyden P.M.L. From angiotensin IV binding site to AT4 receptor. Mol Cell Endocrinol. 2009;302:159–166. doi: 10.1016/j.mce.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 67.Hua F., Zhou J., Liu J., Zhu C., Cui B., Lin H. Glycogen synthase kinase-3β negatively regulates TGF-β1 and angiotensin II-mediated cellular activity through interaction with Smad3. Eur J Pharmacol. 2010;644:17–23. doi: 10.1016/j.ejphar.2010.06.042. [DOI] [PubMed] [Google Scholar]
- 68.Pohanka M. Acetylcholinesterase inhibitors: A patent review (2008—present) Expert Opin Ther Pat. 2012;22:871–886. doi: 10.1517/13543776.2012.701620. [DOI] [PubMed] [Google Scholar]
- 69.Greig N.H., Utsuki T., Yu Q.-S., Zhu X., Holloway H.W., Perry T. A new therapeutic target in Alzheimer's disease treatment: Attention to butyrylcholinesterase. Curr Med Res Opin. 2001;17:159–165. doi: 10.1185/0300799039117057. [DOI] [PubMed] [Google Scholar]
- 70.Riederer P., Danielczyk W., Grünblatt E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology. 2004;25:271–277. doi: 10.1016/S0161-813X(03)00106-2. [DOI] [PubMed] [Google Scholar]
- 71.Andreyev A.Y., Kushnareva Y.E., Starkov A. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 72.Huang W.J., Zhang X., Chen W.W. Role of oxidative stress in Alzheimer's disease. Biomed Rep. 2016;4:519–522. doi: 10.3892/br.2016.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weyler W., Hsu Y.-P.P., Breakafield X.O. Biochemistry and genetics of monoamine oxidase. Pharmacol Ther. 1990;47:391–417. doi: 10.1016/0163-7258(90)90064-9. [DOI] [PubMed] [Google Scholar]
- 74.Ciccone C.D. Free-radical toxicity and antioxidant medications in Parkinson's disease. Phys Ther. 1998;78:313–319. doi: 10.1093/ptj/78.3.313. [DOI] [PubMed] [Google Scholar]
- 75.Law A., Gauthier S., Quirion R. Say NO to Alzheimer’s disease: The putative links between nitric oxide and dementia of the Alzheimer’s type. Brain Res Rev. 2001;35:73–96. doi: 10.1016/s0165-0173(00)00051-5. [DOI] [PubMed] [Google Scholar]
- 76.Eliasson M.J., Huang Z., Ferrante R.J., Sasamata M., Molliver M.E., Snyder S.H. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Segarra V., Crespo M.I., Pujol F., Beleta J., Doménech T., Miralpeix M. Phosphodiesterase inhibitory properties of losartan. Design and synthesis of new lead compounds. Bioorg Med Chem Lett. 1998;8:505–510. doi: 10.1016/s0960-894x(98)00058-4. [DOI] [PubMed] [Google Scholar]
- 78.Impey S., Mark M., Villacres E.C., Poser S., Chavkin C., Storm D.R. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 79.García-Osta A., Cuadrado-Tejedor M., García-Barroso C., Oyarźbal J., Franco R. Phosphodiesterases as therapeutic targets for Alzheimer's disease. ACS Chem Neurosci. 2012;3:832–844. doi: 10.1021/cn3000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tully T. Regulation of gene expression and its role in long-term memory and synaptic plasticity. Proc Natl Acad Sci U S A. 1997;94:4239–4241. doi: 10.1073/pnas.94.9.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munoz L., Ammit A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 82.Schieven G.L. The biology of p38 kinase: A central role in inflammation. Curr Top Med Chem. 2005;5:921–928. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- 83.Bodles A.M., Barger S.W. Secreted β-amyloid precursor protein activates microglia via JNK and p38-MAPK. Neurobiol Aging. 2005;26:9–16. doi: 10.1016/j.neurobiolaging.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 84.Bhat N.R., Feinstein D.L., Shen Q., Bhat A.N. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells. Roles of nuclear factors, nuclear factor kappa B, cAMP response element-binding protein, CCAAT/enhancer-binding protein-beta, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- 85.Sheng J.G., Jones R.A., Zhou X.Q., McGinness J.M., Van Eldik L.J., Mrak R.E. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: Potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- 87.Aisen P.S. Evaluation of selective COX-2 inhibitors for the treatment of Alzheimer's disease. J Pain Symptom Manage. 2002;23:S35–S40. doi: 10.1016/s0885-3924(02)00374-3. [DOI] [PubMed] [Google Scholar]
- 88.Aisen P.S., Schafer K.A., Grundman M., Pfeiffer E., Sano M., Davis K.L. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: A randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 89.Mash D.C., Flynn D.D., Potter L.T. Loss of M2 muscarine receptors in the cerebral cortex in Alzheimer's disease and experimental cholinergic denervation. Science. 1985;228:1115–1118. doi: 10.1126/science.3992249. [DOI] [PubMed] [Google Scholar]
- 90.Ni Y., Zhao X., Bao G., Zou L., Teng L., Wang Z. Activation of β2-adrenergic receptor stimulates γ-secretase activity and accelerates amyloid plaque formation. Nat Med. 2006;12:1390–1396. doi: 10.1038/nm1485. [DOI] [PubMed] [Google Scholar]
- 91.Gong C.-X., Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lau L., Schachter J.B., Seymour P.A., Sanner M.A. Tau protein phosphorylation as a therapeutic target in Alzheimer's disease. Curr Top Med Chem. 2002;2:395–415. doi: 10.2174/1568026024607526. [DOI] [PubMed] [Google Scholar]
- 93.Weingarten M.D., Lockwood A.H., Hwo S.-Y., Kirschner M.W. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alonso A.D.C., Zaidi T., Grundke-Iqbal I., Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kersten S., Desvergne B., Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 96.Spiegelman B. PPAR-gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 97.Landreth G., Jiang Q., Mandrekar S., Heneka M. PPARγ agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics. 2008;5:481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Waldstein S.R., Giggey P.P., Thayer J.F., Zonderman A.B. Nonlinear relations of blood pressure to cognitive function: The Baltimore Longitudinal Study of Aging. Hypertension. 2005;45:374–379. doi: 10.1161/01.HYP.0000156744.44218.74. [DOI] [PubMed] [Google Scholar]
- 99.Reinprecht F., Elmståhl S., Janzon L., André-Petersson L. Hypertension and changes of cognitive function in 81-year-old men: A 13-year follow-up of the population study “Men born in 1914”, Sweden. J Hypertens. 2003;21:57–66. doi: 10.1097/00004872-200301000-00014. [DOI] [PubMed] [Google Scholar]
- 100.Guo Z., Fratiglioni L., Winblad B., Viitanen M. Blood pressure and performance on the Mini-Mental State Examination in the very old: Cross-sectional and longitudinal data from the Kungsholmen Project. Am J Epidemiol. 1997;145:1106–1113. doi: 10.1093/oxfordjournals.aje.a009073. [DOI] [PubMed] [Google Scholar]
- 101.Bohannon A.D., Fillenbaum G.G., Pieper C.F., Hanlon J.T., Blazer D.G. Relationship of race/ethnicity and blood pressure to change in cognitive function. J Am Geriatr Soc. 2002;50:424–429. doi: 10.1046/j.1532-5415.2002.50104.x. [DOI] [PubMed] [Google Scholar]
- 102.de Oliveira F.F., Bertolucci P.H.F., Chen E.S., Smith M.C. Brain-penetrating angiotensin-converting enzyme inhibitors and cognitive change in patients with dementia due to Alzheimer's disease. J Alzheimers Dis. 2014;42:S321–S324. doi: 10.3233/JAD-132189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.