

Abstract
Although treatment with immune checkpoint inhibitors provides promising benefit for cancer patients, optimal use is encumbered by high resistance rates and requires a thorough understanding of resistance mechanisms. We observed that tumors treated with PD-1/PD-L1 blocking antibodies develop resistance through the up-regulation of CD38, which is induced by all-trans retinoic acid (ATRA) and IFN-β in the tumor microenvironment. In vitro and in vivo studies demonstrate that CD38 inhibits CD8+ T cell function via adenosine receptor signaling, and that CD38 or adenosine receptor blockade are effective strategies to overcome the resistance. Large datasets of human tumors reveal expression of CD38 in a subset of tumors with high levels of basal or treatment-induced T cell infiltration, where immune checkpoint therapies are thought to be most effective. These findings provide a novel mechanism of acquired resistance to immune checkpoint therapy and an opportunity to expand their efficacy in cancer treatment.
INTRODUCTION
Although strategies incorporating immune checkpoint inhibition, e.g. PD-1/PD-L1 blockade, are achieving unprecedented success, high rates of resistance still limit their efficacy (1–3). Using K-ras/p53-mutant genetically-engineered murine (KP tumor) and Lewis lung cancer (LLC tumor) models of NSCLC we have previously shown that the response to anti-PD-L1 treatment is dependent upon epigenetic regulation of tumor cell PD-L1 (4,5). We used these well-established NSCLC tumor models and melanoma tumor models to further study immunotherapy resistance.
CD38 is a member of the ribosyl cyclase family that is widely expressed on the surface of non-hematopoietic cells and diverse immune cells. As an ectozyme, CD38 converts NAD+ to ADP-ribose (ADPR) and cADPR, which are essential for the regulation of extracellular metabolites, intracellular Ca2+, cell adhesion, and signal transduction pathways (6). The receptor/ligand activity of CD38 has been documented in multiple immune cell types and the function varies during lymphocyte development, activation, and differentiation (7). However, its potential function on tumor cells has not been fully elaborated.
Here we report that tumors gain resistance to PD-L1/PD-1 blockade over time, and that CD38 up-regulation on tumor cells is induced by ATRA and IFN-β. CD38 expression mediates suppression via adenosine receptor signaling on cytotoxic T cells. CD38 manipulation was sufficient to regulate CD8+ T cell proliferation, antitumor cytokine secretion, and killing capability. Pathologic analysis of lung cancer specimens revealed positive immunohistochemical staining for CD38 on tumor cells in 15–23% of cases and bioinformatic analyses of patient datasets of NSCLC and melanoma revealed a strong correlation between CD38 expression and an inflamed microenvironment. To test whether CD38 blockade might be efficacious to counter the resistance, we used combination therapy with anti-CD38, or alternatively with an adenosine receptor antagonist, and anti-PD-L1 in lung cancer animal models and demonstrated therapeutic benefit.
RESULTS
Resistance to PD-1/PD-L1 Blockade Results From CD38 Up-regulation Mediated by ATRA and IFN-β in Tumors
Our previous data demonstrated suppression of tumor growth and metastases with anti-PD-L1 antibody treatment, but the lack of complete durable responses (4,5), suggesting the existence of antecedent or acquired resistance mechanisms that subvert enhanced infiltration of effector T cells. We adopted a parallel approach to discover these additional mechanisms, first by long-term pharmacologic treatment of animals with anti-PD-L1 or -PD1 antibody, and second by testing of syngeneic tumors with PD-L1 knockout (KO). Despite initial suppression of tumors by anti-PD-L1, progressive resistance developed in tumor models over 5–7 weeks of treatment (Fig. 1A and Supplemental Fig. 1A). By week 7 for the KP and week 5 for the LLC tumor models there were no significant differences between the isotype control and the treatment groups (Fig. 1A). Week 5 and 7 samples from the 344SQ tumors were used for mRNA profiling (Supplemental Figs. 2A-E), representing the respective time points of greatest observed difference in tumor growth and the point at which the anti-PD-L1-treated samples displayed complete resistance. Using a fold-change cutoff of 2.0, a total of 412 genes with FDR ≤ 0.05 (p < 0.035) were found to be differentially expressed between anti-PD-L1 and isotype treated tumors at week 5 (Supplemental Table 1). Comparing the top 200 differentially-expressed genes (100 up- and 100 down-regulated; as depicted in the volcano plot in Supplemental Fig. 2C) versus the results of gene-set enrichment analysis (GSEA) and Ingenuity pathway analysis (IPA) on the entire set of differentially expressed genes, and finally versus results of proteomic analysis by reverse-phase protein array (RPPA), we identified CD38 as the only prominently up-regulated gene/protein identified in all analyses (Fig. 1B, Supplemental Figs. 2C-M, and Supplemental Tables 2–6). Its expression also temporally occurred by week 5 of anti-PD-L1 antibody treatment, along with consistent mRNA changes of CD38-related genes (6,8–14) and therefore preceded the observed acquisition of tumor resistance (Fig. 1B). Both qPCR and FACS analyses confirmed that CD38 mRNA and protein levels were significantly increased on anti-PD-L1 resistant tumor cells (Figs. 1B, 1C, and Supplemental Fig. 1B). We found similar resistance and upregulation of CD38 with several KP lung tumor models and the B16 melanoma model treated with either anti-PD-1 or anti-PD-L1 (Supplemental Figs. 3A-E).
Figure 1.
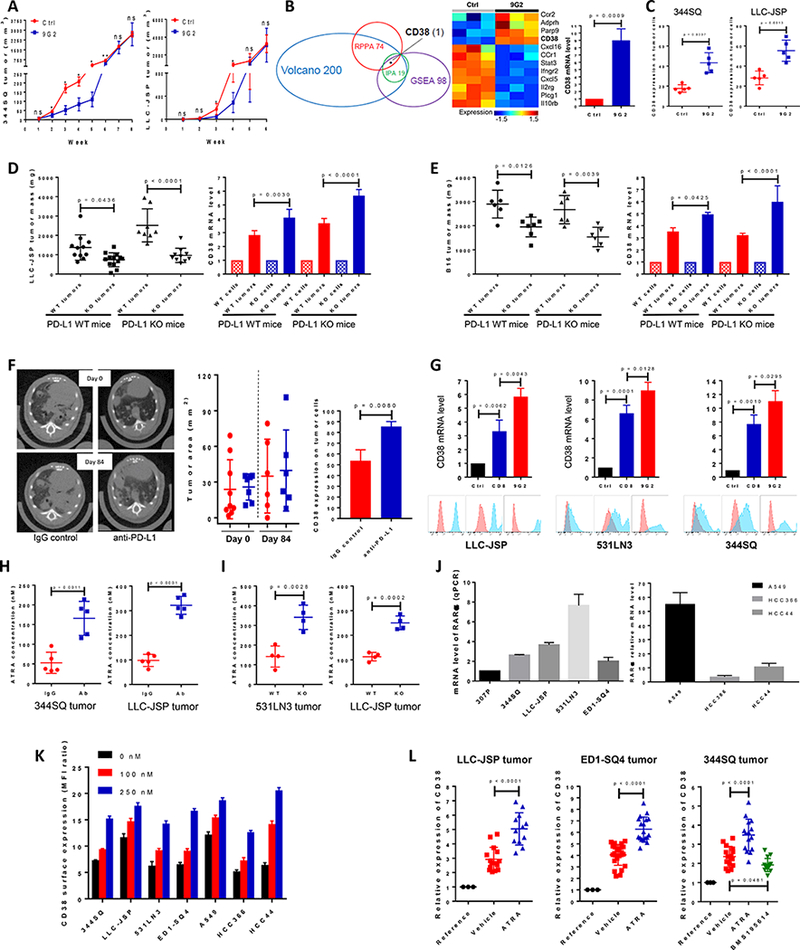
PD-1/PD-L1 blockade resistance results from CD38 up-regulation due to the enrichment of ATRA in tumors.
(A) (Left panel) anti-PD-L1 antibody or an IgG control was injected into 129/Sv mice (200 μg; intraperitoneally) once a week for 7 weeks beginning on day 7 after 344SQ tumor cells were subcutaneously implanted (1 × 106 cells per mouse). Tumors were measured once a week for 8 weeks. The tumor growth curve is shown, with tumor sizes (n = 6 or 7) presented as mean ± SEM. ns, no significant difference, *p < 0.05, **p < 0.01. (Right panel) anti-PD-L1 antibody or an IgG control was injected into C57BL/6 mice (200 μg; intraperitoneally) once a week for 5 weeks beginning on day 7 after the subcutaneous implantation of LLC-JSP tumor cells (0.5 × 106 cells per mouse). Tumors were measured once a week for 6 weeks. The tumor growth curve is shown, with tumor sizes (n = 10 or 11) presented as mean ± SEM. ns, no significant difference, *p < 0.05.
(B) (Left panel) Venn diagram of genes changed upon anti-PD-L1 antibody treatment in 344SQ tumors (n = 3) at week 5. The top 100 up-regulated genes and top 100 down-regulated genes were included from the Volcano plot analysis, 98 genes involved in T cell activity from Gene Set Enrichment Analysis (GSEA), and the top 19 networks identified with Ingenuity Pathway Analysis (IPA) software. 74 protein markers involved in immune signaling pathways, cell cycle signaling, tumor metabolism signaling were included for RPPA. CD38 is the only molecule overlapping in all 4 analyses. (Middle panel) Heat map showing differentially expressed mRNAs related to CD38 from the two profiled groups. (Right panel) Relative CD38 mRNA levels in sorted 344SQ tumor cells (CD31-CD45-EpCAM+ for sorting) were quantified by qPCR assays using the tumor samples at week 5 (n = 3) from the control and anti-PD-L1 groups. mRNA levels are normalized to L32.
(C) 344SQ tumors in Figure 1A were harvested and CD38 expression on sorted tumor cells was analyzed by FACS at week 5, and represented in the left panel. LLC-JSP tumors in Figure 1A were harvested and CD38 expression on tumor cells was analyzed by FACS at week 4, and represented in the right panel.
(D) In immune competent C57BL/6 PD-L1 WT mice (n = 11 or 13), Lewis lung LLC-JSP cells with wildtype PD-L1 or PD-L1 KO (0.5 × 106 cells per mouse) were subcutaneously injected. Mice were sacrificed 4 weeks post-injection. The primary tumor mass is shown in the left panel, presented as mean ± SEM. CD38 mRNA levels quantified with qPCR assay in sorted tumor cells are shown in the right panel. In C57BL/6 PD-L1 KO mice (n = 8 or 10), Lewis lung LLC-JSP cells with wildtype PD-L1 or PD-L1 KO (1 × 106 cells per mouse) were subcutaneously injected. Mice were sacrificed 4 weeks post-injection. The primary tumor mass is shown in the left panel, with tumor sizes presented as mean ± SEM. CD38 mRNA levels quantified with qPCR assay in sorted tumor cells are shown in the right panel. mRNA levels are normalized to L32. ANOVA test was used to analyze.
(E) In immune competent C57BL/6 PD-L1 WT mice (n = 6 or 7), melanoma B16 cells with wildtype PD-L1 or PD-L1 KO (2 × 106 cells per mouse) were subcutaneously injected. Mice were sacrificed 4 weeks post-injection. The primary tumor mass is shown in the left panel, presented as mean ± SEM. CD38 mRNA levels quantified with qPCR assay in sorted tumor cells are shown in the right panel. In C57BL/6 PD-L1 KO mice (n = 6), melanoma B16 cells with wildtype PD-L1 or PD-L1 KO (2 × 106 cells per mouse) were subcutaneously injected. Mice were sacrificed 4 weeks post-injection. The primary tumor mass is shown in the left panel, presented as mean ± SEM. CD38 mRNA levels quantified with qPCR assay in sorted tumor cells are shown in the right panel. mRNA levels are normalized to L32. ANOVA test was used to analyze.
(F) K-rasLA1/+p53R172HΔg/+ mice were intraperitoneally injected with anti-PD-L1 antibody (200 μg per mouse) or an isotype-matched IgG control once a week for 12 weeks. The initial lung tumor area measured by micro-CT was ~28 mm2 in each group, with representative sections shown in the left panel. Tumors were monitored by micro-CT scanning and plots of tumor size at indicated times are shown in the middle panel (red dots, IgG control group; blue squares, anti-PD-L1 group). Representative histograms of CD38 expression on tumor cells (CD31-CD45-EpCAM+) at the endpoint are shown in the right panel.
(G) Indicated cancer cells (0.5 × 106 cells per mouse) were subcutaneously injected into mice to challenge and activate T cells. Tumors, blood, spleen were harvested to isolated CD8 T cells 2 weeks post-injection. 2000 cancer cells were co-cultured with 0.1 million CD8 T cells in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) or in the presence of anti-CD3 (5 μg/ml), anti-CD28 (5 μg/ml), and anti-PD-L1 (20 μg/ml) for 3 days. The non-adherent CD8 T cells were washed away and cancer cells were harvested for qRT-PCR and FACS analysis. The experiments were repeated three times. Data was analyzed using ANOVA test.
(H) The indicated tumor-bearing mice were treated with anti-PD-L1 antibody (Ab) or an IgG control (IgG) (200 μg; intraperitoneally) once a week for 4 weeks beginning on day 7 after tumor cells were subcutaneously implanted (1 × 106 cells per mouse). The tumors were harvested to measure the concentration of ATRA on week 5 after tumor cell inoculation. Tumor lysates were used to measure the concentration of ATRA using the Thermo Orbitrap Fusion Tribrid Mass Spectrometer. The concentrations of ATRA in tumors are presented as mean ± SD with p values.
(I) Tumors were harvested on week 5 after PD-L1WT531LN3, PD-L1KO531LN3, PD-L1WTLLC-JSP, PD-L1KOLLC-JSP cells (1 × 106 cells per mouse) were subcutaneously implanted into syngeneic mice. Tumor lysates were used to measure the concentration of ATRA using the Thermo Orbitrap Fusion Tribrid Mass Spectrometer. The concentrations of ATRA in tumors are presented as mean ± SD with p values.
(J) The retinoic acid receptor alpha (RARα) mRNA levels in a panel of lung cancer cell lines (Left panel: murine cancer lines; right panel: human cancer lines) was measured by qPCR assays. mRNA levels were normalized to L32. The summarized data from three independent experiments are shown.
(K) Cells were incubated with ATRA at different concentrations (0 nM, 100 nM, and 250 nM) for 3 days and stained with anti-CD38 antibody for FACS analysis. CD38 surface expression was quantified by the ratio of mean fluorescence intensity (MFI). The experiments were repeated three times.
(L) The indicated tumor-bearing mice (LLC-JSP bearing C57BL/6 mice; ED1-SQ4 bearing FVB mice; 344SQ bearing 129/Sv mice) were treated with vehicle, ATRA (45 μg in 100 μl 1% methylcellulose; oral administration) or RARα antagonist BMS195614 (67 μg in 100 μl 1% methylcellulose; oral administration) once a day for 2 weeks beginning on day 4 after tumor cells were subcutaneously implanted (1 × 106 cells per mouse). At the endpoint, CD38 mRNA levels in sorted tumor cells were measured by qPCR assays. The respective parental cell lines were included as the reference. mRNA levels were normalized to L32. The summarized data from three independent experiments are shown with p values calculated by ANOVA test. Reference, cell line; Vehicle, sorted tumor cells from control vehicle treated tumors; ATRA, sorted tumor cells from ATRA treated tumors; BMS195614, sorted tumor cells from BMS195614 treated tumors.
Because our previous reports and work from other labs emphasize the dominant role of PD-L1 expression on tumor cells in mediating tumor immune escape (4,15,16) (Supplemental Figs. 4A and 4B), we also used a genetic approach to block PD-L1-mediated signaling. We generated lung cancer cell lines (LLC-JSP and the KP model 531LN3) and the melanoma cell line B16 with PD-L1 knockout by CRISPR/Cas9 editing and tested them in syngeneic PD-L1 wildtype or PD-L1 knockout mice. Both partial PD-L1 signaling blockade (PD-L1 knockout cancer cells implanted in PD-L1 wildtype mice) and complete blockade (PD-L1 knockout cancer cells implanted in PD-L1 knockout mice) partially suppressed tumor growth in a CD8+ T cell-dependent manner (Supplemental Figs. 4C–4F, and 5), but resulted in ~4–6 fold CD38 up-regulation versus the same cells grown in vitro (Figs. 1D-E, Supplemental Fig. 3F). Consistent with these findings, anti-PD-L1 antibody treatment in the autochthonous KP model over 12 weeks showed no durable effect on tumor growth or animal survival, but we observed a significant increase in CD38 on tumor cells in the PD-L1 treatment group (Fig. 1F and Supplemental Figs. 1C-D). The consistency of the results between pharmacologic and genetic blockade of PD-1/PD-L1 in syngeneic and autochthonous models of lung cancer and melanoma indicated that CD38 could represent an important pathway in the development of resistance.
To investigate how CD38 is upregulated on tumor cells, we tested co-cultures of tumor cells with activated CD8+ T cells and found a significant increase of CD38 mRNA and protein (Fig. 1G), which was further enhanced by addition of anti-PD-L1 and similar to the upregulation observed in tumors (Figs. 1D-E and Supplemental Fig. 3). Altogether the data suggest that the activated T cells in the inflammatory tumor microenvironment stimulate CD38 expression. This finding prompted us to explore the potential mechanism(s) of CD38 up-regulation. Prior literature suggests that CD38 is regulated by several soluble factors that may be present in tumor microenvironment, including ATRA and IFN-β (17–20). Analysis of the metabolites in anti-PD-L1 treated or PD-L1 KO tumors demonstrated an enrichment of ATRA and an increase in the mRNA for Rbp4 and Stra6 that regulate cellular retinol uptake (21) (Figs. 1H-I and Supplemental Figs. 6A-B). When human or murine lung cancer lines expressing retinoic acid receptor alpha (RARα) were treated with ATRA for 3 days, CD38 was up-regulated in a dose-dependent manner (Figs. 1J-K and Supplemental Fig. 6C). In syngeneic animal tumor models, CD38 on tumor cells was significantly up-regulated after 2 weeks of ATRA treatment versus vehicle control, while treatment with the RARα antagonist, BMS195614, inhibited the CD38 upregulation (Fig. 1L). In addition, we used the tumor lysates to perform ELISA-based assays and found a significant increase of IFN-β in anti-PD-L1 treated tumors (Supplemental Fig. 7A). Upon culturing with IFN-β for 3 days, surface CD38 was significantly increased on multiple cancer lines (Supplemental Fig. 7B). When KP-derived 344SQ tumor bearing mice were treated with anti-PD-L1 and mRNA profiling was performed by Nanostring, we observed an increase of IRF1, which was confirmed by qPCR for IRF1, IRF2 and IRF3 (Supplemental Fig. 8). IRF-1 is a transcription factor and tumor suppressor involved in cell growth regulation and immune responses that is induced by ATRA and is essential for the induced expression of IFN-β (22–25). This provides a connection between the independent observations that ATRA and IFN-β are upregulated and produce upregulation of CD38 expression on tumor cells. IFN-γ, TNF-α, IL-2, and IL-1β are potent antitumor cytokines also documented to induce CD38 in other cell types. We tested if these cytokines modulate CD38 expression in our models, but did not observe effects on CD38 expression in two lung cancer models (Supplemental Fig. 9). Taken together, PD-1/PD-L1 blockade results in an infiltration of activated T cells and inflammatory changes that lead to ATRA and IFN-β-mediated CD38 upregulation.
CD38 Suppresses CD8+ T Cell Function via Adenosine Receptor Signaling
Despite the CD8+ T cell–dependent effect of anti-PD-L1 antibody during the first 2–5 week treatment period (Fig. 2A), which we previously published (4), over time the treatment group showed reduced CD8+ T cell infiltration into tumors, accompanied by a decrease in CD44highCD62Llow memory (48.6% vs 28.4%) and Ki67+ proliferative CD8+ T cells (18.8% vs 6.46%), and an increase in exhausted CD8+ T cells (PD-1+LAG3+ and PD-1+TIM3+) (Fig. 2B, and Supplemental Fig. 10). The initial antitumor efficacy of short-term anti-PD-L1 antibody treatment and the temporal CD38 upregulation on resistant tumor cells caused by the infiltration of activated T cells suggests that simultaneous blockade of CD38 and PD-L1 might be required for tumor rejection. We tested this by sorting the PD-L1KO531LN3 cells or PD-L1KOLLC-JSP cells for high and negative CD38 surface staining and found that the PD-L1KOCD38negative cells did not form tumors upon implantation into immune competent mice, while PD-L1KOCD38high cancer cells resulted in large primary tumors, numerous metastases and rapidly aggressive disease (Fig. 2C, Supplemental Figs. 11A-F). To test the effect of CD38 on cancer cell growth, we measured in vitro growth rate and cell cycle, but found no difference between PD-L1KOCD38negative and PD-L1KOCD38high cancer cells (Supplemental Figs. 11G-H). By contrast, when CD8+ T cells were depleted, PD-L1KOCD38negative cancer cells developed tumors and metastasized while PD-L1KOCD38high cancer cells developed tumors similarly in both groups (Fig. 2D and Supplemental Fig. 12A). When PD-L1KOCD38negative and PD-L1KOCD38high cancer cells were injected into Rag2−/− mice, both tumor cell types formed tumors without significant differences (Supplemental Fig. 12B). Additionally, the development of resistance to the anti-PD-L1 treatment was inhibited by adoptive transfer of CD8+ T cells (Fig. 2E). Collectively, these data indicate that CD38 promotes tumor progression via the suppression of CD8+ T cell function.
Figure 2.
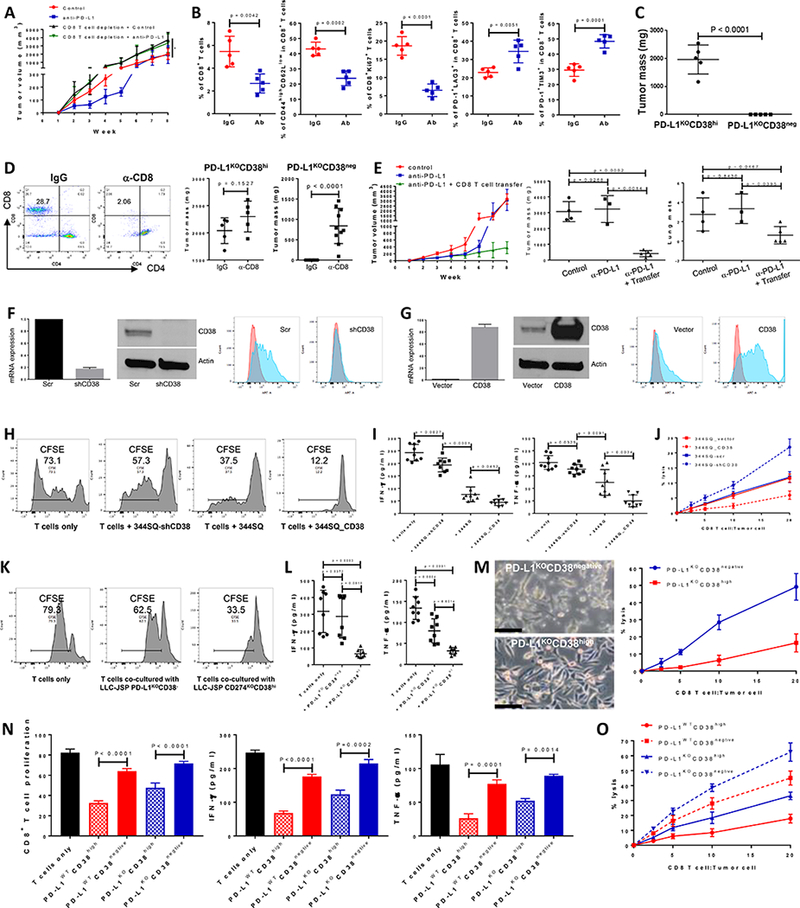
CD38 on tumor cells suppresses CD8+ T cell function.
(A) Growth of subcutaneous 344SQ tumors in immune competent 129/Sv mice treated with IgG control, anti-PD-L1 (clone 9G2; 200 μg per mouse), anti-PD-L1 plus anti-CD8 (clone 2.43; 200 μg per mouse), respectively. Mice (n = 5 or 8) were intraperitoneally treated with the antibody once a week for 7 weeks beginning on day 7 after the tumor cell injection (1 × 106 cells per mouse). In the group of anti-PD-L1 plus anti-CD8 treatment, mice were pretreated with anti-CD8 antibody (400 μg per mouse) one week before tumor cell injection. Tumor sizes are presented as mean ± SEM and statistical significance (ns, no significant difference; *p < 0.05; **p < 0.01) determined on indicated weeks. The statistically significant differences between the groups of control and anti-PD-L1 are as follows: week 1, ns; week 2, *; week 3, **; week 4, *; week 5, **; week 6, *; week 7, ns; week 8, ns. ANOVA test was used to calculate the significant difference between two groups. For the analysis among multiple groups at the end point (week 8), ANOVA was used to analyze, *p < 0.05.
(B) FACS analysis of % CD8+TIL, the proliferation marker Ki67, surface CD44, CD62L, PD1, LAG3, and TIM3 marker expression levels on CD8+ T cells from primary tumors in 129/Sv mice (n = 5) treated with anti-PD-L1 antibody at week 5. Data are shown as mean ± sd. t-test was used to analyze.
(C) PD-L1KOCD38high531LN3 cells or PD-L1KOCD38negative531LN3 cells (5 × 106 cells per mouse) were subcutaneously injected into immune competent 129/Sv mice (n = 5). The primary tumor sizes at week 4 are shown with mean ± SEM. t-test was used to analyze.
(D) (Left panel) CD8 T cells in spleen were determined 2 weeks post initial anti-CD8 antibody injection to test CD8 T cell depletion efficiency. (Middle panel) 5 × 106 of PD-L1KOCD38high531LN3 cells were subcutaneously injected into 129/Sv mice (n = 5) after CD8 T cells were depleted (α-CD8). The T cell un-depleted group was included as the control (IgG). The total tumors were measured 4 weeks post tumor cell transplantation and are shown with mean ± SEM. t-test was used to analyze. (Right panel) 5 × 106 of PD-L1KOCD38negative 531LN3 cells were subcutaneously injected into 129/Sv mice (n = 10) after CD8 T cells were depleted (α-CD8). The T cell un-depleted group was included as the control (IgG). The total tumors from primary site and peritoneal cavity were measured 5 weeks post tumor cell transplantation and are shown with mean ± SEM. t-test was used to analyze.
(E) To prepare CD8+ T cells, 129/Sv mice were challenged with 0.5 × 106 344SQ for 2 weeks. CD8+ T cells were isolated from these tumors, blood, and spleens. A separate cohort of 344SQ tumor-bearing mice were treated with anti-PD-L1 antibody or control (as described in Figure 1A), then used as recipients for the CD8 T cell adoptive transfer assay. At week 4, mice received cyclophosphamide at 100 mg/kg intravenously 6 hours before CD8 T cell transfer (6 × 106 per mouse, intravenously), followed by IL-2 (20,000 units, intraperitoneally) at 8 hours after T cell transfer then every 12 hours for 3 days. The tumor growth curves are shown in the left panel. At the endpoint, mice were necropsied to harvest primary tumors and lungs, which were weighed, and to quantify distant metastases. The primary tumor weights and lung metastatic nodules are shown in the middle and right panels. ANOVA test was used to analyze.
(F) Cell lines with stable expression of a scramble control (344SQ-scr) or shRNA against CD38 (344SQ-shCD38) were generated and subjected to qPCR assays. Relative CD38 mRNA levels are normalized to L32 and shown in the left panel, the Western blot assay for protein in the middle panel, versus β-actin as a loading control. The surface expression of CD38 on the cell lines was determined using FACS and are shown in the right panel.
(G) The stable cell lines 344SQ_vector (empty vector control) and 344SQ_CD38 (CD38 overexpression) were generated and subjected to qPCR assays. mRNA levels of CD38 are normalized to L32 and shown in the left panel, Western blot assay for protein in the middle panel, versus β-actin as a loading control. The surface expression of CD38 was determined using FACS and is in the right panel.
(H-J) To prepare tumor specific CD8+ T cells, 129/Sv mice were challenged with 0.5 × 106 344SQ for 2 weeks. CD8+ T cells were isolated from these tumors, blood, and spleens. CFSE-labeled CD8+ T cells were co-cultured with indicated cancer cells in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. CD8+ T cells only was included as the control. T cell proliferation was quantified using FACS analysis (H). The supernatants from each co-culture were subjected to ELISA assay to measure IFN-γ and TNF-α (I). Tumor cells and CD8+ T cells were co-cultured at the indicated ratios in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. Tumor cells only were used as the control for calculation. At day 4, CD8+ T cells and dead tumor cells were washed away and viable tumor cells were counted with 0.4% Trypan Blue staining. The CD8+ T cell killing efficiency is shown in (J). The experiments were repeated at least three times. p values were calculated with ANOVA test.
(K-M) To prepare tumor specific CD8+ T cells, C57BL/6 mice were challenged with 0.2 × 106 LLC-JSP for 2 weeks. CD8+ T cells were isolated from these tumors, blood, and spleens. CFSE-labeled CD8+ T cells were co-cultured with indicated cancer cells in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. CD8+ T cells only was included as the control. T cell proliferation was quantified using FACS analysis (K). The supernatants from each co-culture were subjected to ELISA assay to measure IFN-γ and TNF-α (L). Tumor cells and CD8+ T cells were co-cultured at the indicated ratios in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. Tumor cells only were used as the control for calculation. At day 4, after taking photos, CD8+ T cells and some dead tumor cells were washed away and viable tumor cells were counted with 0.4% Trypan Blue staining. The CD8+ T cell killing efficiency is shown in (M). The experiments were repeated at least three times. p values were calculated with ANOVA test. Scale bars represent 100 μm.
(N and O) To prepare tumor specific CD8+ T cells, 129/Sv mice were challenged with 0.5 × 106 531LN3 for 2 weeks. CD8+ T cells were isolated from these tumors, blood, and spleens. CFSE-labeled CD8+ T cells were co-cultured with indicated cancer cells (sorted from PD-L1WT531LN3 or PD-L1KO531LN3) in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. CD8+ T cells only was included as the control. T cell proliferation was quantified using FACS analysis (N). The supernatants from each co-culture were subjected to ELISA assay to measure IFN-γ and TNF-α (N). Tumor cells and CD8+ T cells were co-cultured at the indicated ratios in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. Tumor cells only were used as the control for calculation. At day 4, CD8+ T cells and some dead tumor cells were washed away and viable tumor cells were counted with 0.4% Trypan Blue staining. The CD8+ T cell killing efficiency is shown in (O). The experiments were repeated at least three times. p values were calculated with ANOVA test.
To further determine whether manipulating CD38 is sufficient to suppress CD8+ T cell function and control tumor, we used KP and LLC lung cancer lines to generate models with CD38 knockdown or constitutive expression and confirmed the CD38 expression levels by qRT-PCR, western blotting and FACS analysis (Figs. 2F-G, Supplemental Figs. 13A, B, and G). The effect of these cancer cells on T cell activity was first tested in the co-culture assay where isogenic tumor cells with different levels of CD38 expression were cultured with specific CD8+ T cells. T cell proliferation was measured by CFSE dilution, antitumor cytokine secretion by CD8+ T cells was assayed for IFN-γ and TNF-α, and tumor cell killing capacity was determined. Consistent with the in vivo suppressive effects of CD38 on CD8+ T cell function, the in vitro results provide direct evidence of CD38 inhibition on CD8+ T cell function (Figs. 2H-M, Supplemental Figs. 13C-E, and 13H-J). In KP-derived lung cancer line 531LN3 we observed similar results with co-culture assays using the sorted populations for PD-L1WTCD38high, PD-L1WTCD38negative, PD-L1KOCD38high, and PD-L1KOCD38negative (Figs. 2N-O).
We further observed that the growth of CD38 knockdown tumors (344SQ-shCD38) was slowed, with significant reduction in primary tumor size and lung metastatic lesions compared with control 344SQ_scr tumors (Fig. 3A). In contrast, 344SQ tumors with constitutive CD38 overexpression (344SQ_CD38) grew faster, produced larger primary tumors and more lung metastases than the vector control (344SQ_vector; Fig. 3B). In addition, tumor microenvironment of each model displayed distinct and consistent immune repertoire changes upon genetic manipulation of CD38 expression on tumor cells. We observed significantly lower levels of total CD8+ T and IFN-γ+CD8+ T cell infiltrates, and higher percentages of exhausted PD-1+TIM3+CD8+ T cells in CD38-expressing tumors (Figs. 3C-H). Similar results of tumor growth and immune cell profiling were obtained in C57BL/6 animals with LLC-JSP tumors and in 129/Sv animal model with KP-derived 531LN3 tumors (Figs. 3I, Supplemental Figs. 13F, 13K, and Table 1). To exclude that these observations resulted from an impact of tumor size on immune phenotype, we tested adjusted cancer cell numbers of CD38 KD (knockdown), CD38 WT (wildtype), and CD38 OE (overexpression) and chose tumors of similar size to analyze the CD8 T cell infiltration and their function. The data indicates that CD38 has a significant impact on CD8+ T cell function regardless of tumor size or growth rate (Supplemental Fig. 14).
Figure 3.
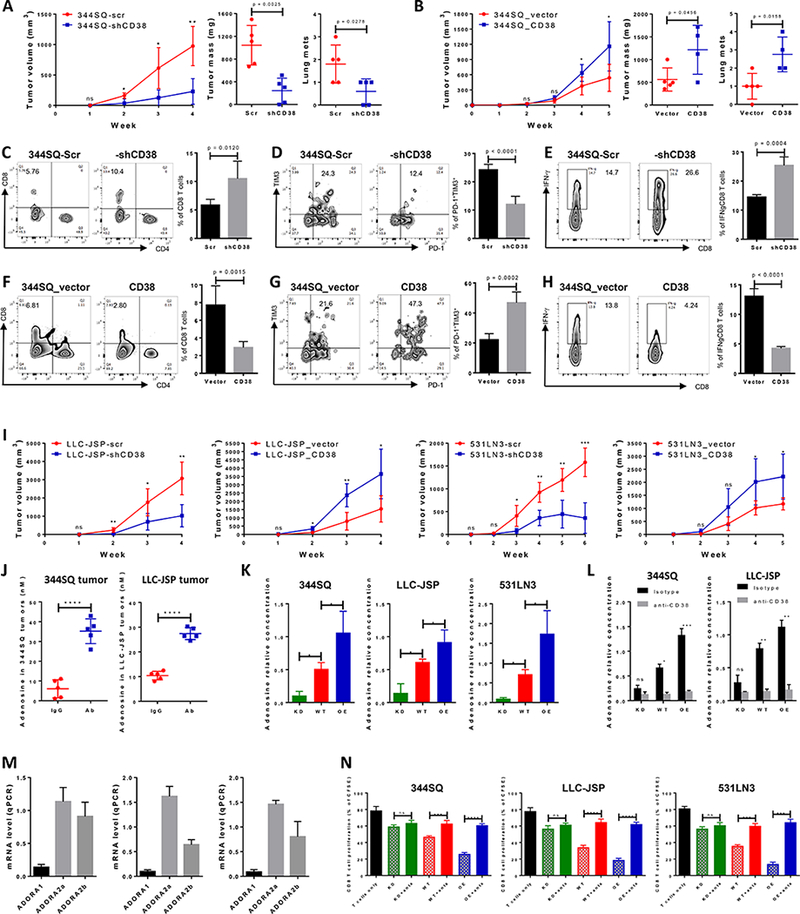
CD38 regulates tumor growth and metastasis by adenosine-mediated CD8+ T cell suppression.
(A) 344SQ-scr or 344SQ-shCD38 cells (2 × 106 cells per mouse) were subcutaneously injected into immune competent 129/Sv mice (n = 5). Tumor size was measured weekly and tumor growth curves are shown in the left panel, with tumor sizes presented as mean ± SEM. ns, no significant difference, *p < 0.05, **p < 0.01. The primary tumor mass and lung metastatic nodules are shown in the middle and right panels 4 weeks post-injection.
(B) 344SQ_vector or 344SQ_CD38 cells (1 × 106 cells per mouse) were subcutaneously injected into immune competent 129/Sv mice (n = 5). Tumor size was measured weekly and tumor growth curves are shown in the left panel, with tumor sizes presented as mean ± SEM. ns, no significant difference, *p < 0.05. The primary tumor mass and lung metastatic nodules are shown in the middle and right panels 5 weeks post-injection.
(C-E) At the endpoint, CD8+TILs in primary tumors (344SQ-scr or 344SQ-shCD38) were analyzed by FACS and are shown in (C). The percent of exhausted CD8+ T cells measured by PD-1+TIM3+ is shown in (D). The percent of antitumor IFN-γ+CD8+ T population is shown in (E). Representative plots of individual tumor are shown on the left and bar graphs of the summary data for all tumors on the right (n=5/group).
(F-H) At the endpoint, CD8+TILs in primary tumors (344SQ_vector or 344SQ_CD38) were analyzed by FACS and are shown in (F). The percent of exhausted CD8+ T cells measured by PD-1+TIM3+ is shown in (G). The percent of antitumor IFN-γ+CD8+ T population is shown in (H). Representative plots of individual tumor are shown on the left and bar graphs of the summary data for all tumors on the right (n=5/group).</p/.(I) Indicated isogenic cancer cells (1 × 106 cells per mouse for LLC-JSP-scr or LLC-JSP-shCD38 injection; 0.5 × 106 cells per mouse for LLC-JSP_vector or LLC-JSP_CD38 injection; 2 × 106 cells per mouse for 531LN3-scr, 531LN3-shCD38, 531LN3_vector, or 531LN3_CD38 injection) were subcutaneously injected into the syngeneic mice. Tumor sizes were measured weekly and tumor growth curves are shown. Tumor sizes are presented as mean ± SEM. t-test is used to analyze the difference. ns, no significant difference, *p < 0.05, **p < 0.01, ***p < 0.001.
(J) The indicated tumor-bearing mice were treated with anti-PD-L1 antibody (Ab) or an IgG control (IgG) (200 μg; intraperitoneally) once a week for 4 weeks beginning on day 7 after tumor cells were subcutaneously implanted (1 × 106 cells per mouse). Tumor lysates were used to measure the concentration of adenosine using the Agilent Triple Quad (QQQ) 6460 Mass Spectrometer. The concentrations of adenosine in tumors are presented as mean ± SD. ****p < 0.0001.
(K) 1 × 106 of indicated cells were cultured in 100 mm tissue culture dishes for 3 days. Cells were then treated for 30 min with 100 μM adenosine deaminase inhibitor EHNA before being cultured in the presence of 50 μM NAD+. Supernatants were collected after 1 hour incubation with NAD+ for determining adenosine concentration by Mass Spectrometry. The data from triplicates are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ANOVA test was used to analyze. KD, CD38 knockdown; WT, CD38 wildtype; OE, CD38 overexpression.
(L) 1 × 106 of indicated cells were cultured in the presence of anti-CD38 (30 μg/ml) or isotype control for 3 days. Cells were then treated for 30 min with 100 μM adenosine deaminase inhibitor EHNA before being cultured in the presence of 50 μM NAD+. Supernatants were collected after 1 hour incubation with NAD+ for determining adenosine concentration by Mass Spectrometry. The data from triplicates are shown as mean ± SEM. ANOVA test was used to analyze. ns, no significance, *p < 0.05, **p < 0.01, ***p < 0.001. KD, CD38 knockdown; WT, CD38 wildtype; OE, CD38 overexpression.
(M) 1 × 106 of tumor cells (left panel for 344SQ, middle panel for LLC-JSP, and right panel for 531LN3 respectively) were subcutaneously injected into syngeneic mice. 2 weeks later, CD8+ T cells were sorted from tumors for determining mRNA level of adenosine receptors ADORA1, ADORA2a, and ADORA2b by qPCR assays. mRNA levels were normalized to L32. The experiments were repeated three times.
(N) To prepare tumor specific CD8+ T cells, syngeneic immune competent mice were challenged with 0.5 × 106 tumor cells for 2 weeks. CD8+ T cells were isolated from these tumors, blood, and spleens. CFSE-labeled CD8+ T cells were co-cultured with indicated cancer cells in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. As indicated, the cocktail of specific adenosine receptor antagonists (500 nM ADORA1 antagonist PSB36, 1 μM ADORA2a antagonist SCH58261, and 1 μM ADORA2b antagonist PSB1115) was used to block adenosine receptor signaling. CD8+ T cells only was included as the control. T cell proliferation was quantified using FACS analysis. The pooled data from three independent experiments are shown as mean ± SEM. ns, no significant difference, **p < 0.01, ***p < 0.001, ****p < 0.0001. ANOVA test was used to analyze. KD, CD38 knockdown; WT, CD38 wildtype; OE, CD38 overexpression; anta, indicates the addition of the antagonist cocktail against ADORA1, ADORA2a, and ADORA2b.
Table 1.
Genetic manipulation CD38 on tumor cells dramatically changes the tumor immune microenvironment.
| Immune profile | LLC-JSP tumors |
531LN3 tumors |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD38 knockdown |
CD38 overexpression |
CD38 knockdown |
CD38 overexpression |
|||||||||
| Scr | shCD38 | p value | Vector | CD38 | p value | Scr | shCD38 | p value | Vector | CD38 | p value | |
| % of CD8+ T cells | 9.23 ± 1.39 | 16.36 ± 0.71 | (0.0018) | 10.41 ± 0.95 | 5.90 ± 0.92 | (0.0093) | 12.02 ± 1.57 | 18.08 ± 0.89 | (0.0100) | 14.60 ± 1.31 | 7.91 ± 1.02 | (0.0037) |
| % of PD-1+TIM3+CD8+ T cells | 23.44 ± 1.19 | 15.00 ± 1.56 | (0.0026) | 20.90 ± 1.63 | 30.78 ± 2.77 | (0.0153) | 19.26 ± 1.59 | 11.87 ± 1.41 | (0.0084) | 9.88 ± 1.98 | 52.36 ± 2.18 | (0.0001) |
| % of IFN-γ+CD8+ T cells | 7.62 ± 0.76 | 18.55 ± 0.84 | (< 0.0001) | 9.55 ± 0.65 | 5.09 ± 0.13 | (0.0005) | 12.80 ± 0.44 | 21.55 ± 0.82 | (< 0.0001) | 13.58 ± 0.49 | 4.14 ± 0.28 | (0.0001) |
Our data demonstrate that CD38 expressing tumor cells impair CD8+ T cell function. Based on the previously reported enzymatic functions of CD38 as part of an ectoenzyme complex that plays an important role in adenosine production (6), we used mass spectrometry analysis of tumor samples and found a ~2.5–6 fold increase in adenosine concentration in anti-PD-L1 vs control treated tumors (Fig. 3J). Since adenosine can suppresses T cell function in tumor microenvironment (6,26–29), we focused on the effects of adenosine for further study. We first compared adenosine concentrations in the supernatants of three different tumor cell cultures with models that had CD38 knockdown (KD), wildtype expression (WT), or constitutive CD38 expression (OE). In all three tumor models, CD38 expression level strongly correlated with increased adenosine (Fig. 3K), while adenosine production was blocked with anti-CD38 antibody (Fig. 3L), establishing a causal association between CD38 and adenosine. Previous reports suggested that inhibition of CD8+ T cell function by adenosine occurs through interaction with adenosine receptors ADORA2a and ADORA2b (26). We next challenged mice with different cancer lines and sorted the CD8+ TIL cells to perform qRT-PCR for ADORA1, ADORA2a, and ADORA2b expression. ADORA1 was expressed at low levels, but ADORA2a and ADORA2b were highly expressed on the tumor infiltrating CD8+ T cells (Fig. 3M). We functionally tested whether receptor antagonists could block CD38-mediated T cell suppression. T cell co-culture assays with the three tumor models demonstrated that the combined adenosine receptor antagonists effectively reversed the suppressive effect of tumor cell CD38 on T cell proliferation (Fig. 3N), indicating that CD38-mediated production of adenosine inhibits CD8+ T cell proliferation through adenosine receptor signaling on CD8+ T cells.
Tumor Cell Lines and Patient Tumors Express CD38, Associated With Expression of Multiple Immune Checkpoints and an Active Intratumoral Immune Cell Infiltrate
The data from the murine models suggested that increased CD38 expression on tumor cells may represent an escape mechanism from the infiltrating cytotoxic T cells induced by anti-PD-L1/PD-1 therapy. To understand if this is a generalizable phenomenon, we stained for CD38 and performed FACS analysis on a panel of cancer cell lines representing lung, melanoma, breast, and sarcoma. Notably, 12 of 13 murine lines highly express CD38 (Fig. 4A). Additional FACS and western blotting analysis of NSCLC lines derived from a variety of lung cancer patients showed surface CD38 expression on most of the lines (Fig. 4A and Supplemental Fig. 15A). We next used two independent tissue microarrays of early-stage lung cancer specimens to analyze CD38 expression by immunohistochemical staining (259 from the MD Anderson PROSPECT dataset (TMA3) and 534 specimens (TMA4)). We validated and used a monoclonal antibody specifically recognizing CD38 to determine the membranous protein expression only on cancer cells in the TMA specimens (Supplemental Fig. 15B). Of the 259 TMA3 specimens, 209 had qualified staining, and 23% exhibited positive staining for CD38 on tumor cells; while of the 534 TMA4 specimens, 471 had qualified staining, and 15% exhibited positive staining for CD38 on tumor cells (Figs. 4B, 4F, Supplemental Figs. 15F, Supplemental Tables 7 and 8). We have corresponding total tumor mRNA expression data for 165 samples in TMA3 cohort and found a broad distribution of CD38 mRNA expression in the samples. Importantly there was a strong correlation between IHC score of protein levels and mRNA expression (p = 6.13e-7; Fig. 4C). The fact that some tumors with low tumor cell membrane staining displayed high mRNA levels is probably due to the presence of CD38 on infiltrating cell populations in the tumors.
Figure 4.
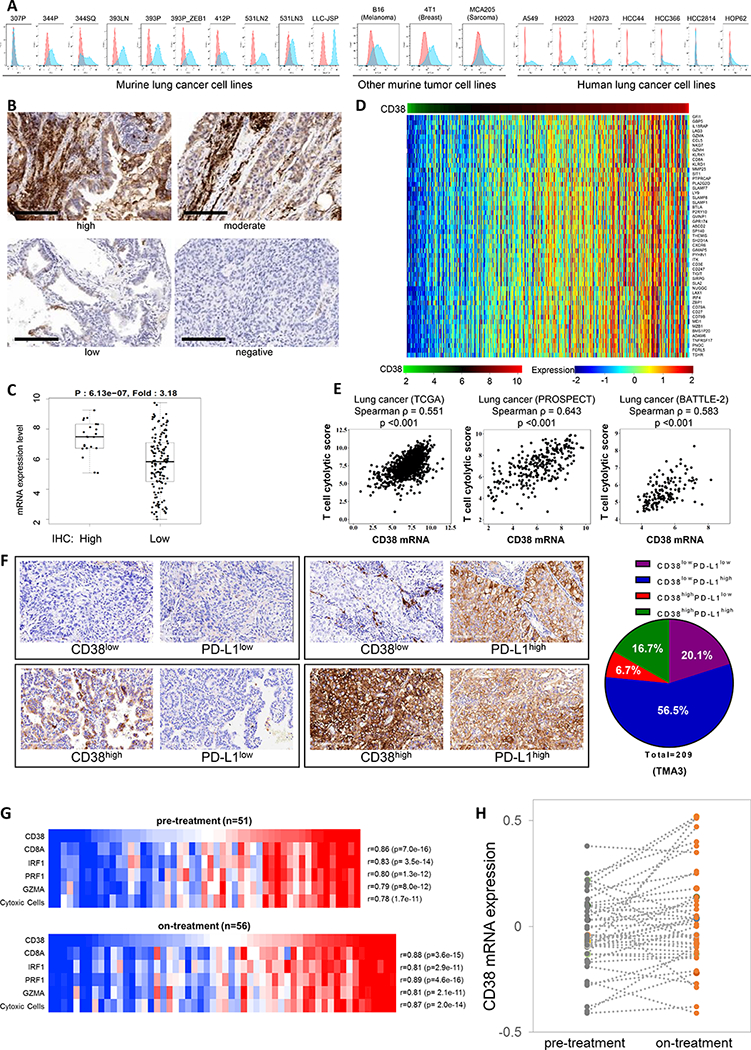
Tumor cell lines and patient tumors express CD38, which is associated with an active intratumoral immune cell infiltrate.
(A) Surface expression of CD38 on multiple murine and human tumor cell lines by FACS analysis.
(B) IHC staining of CD38 was performed in tumors from the lung cancer patient tissue microarray bank TMA3 (n = 259). Representative images of cell membrane staining intensity are shown. The cell membrane staining intensity and percentage of positive cells were analyzed and used to generate an H-score for each sample that passed quality control (n = 211). Scale bars represent 200 μm.
(C) Dot plot showing the correlation of CD38 mRNA expression level and IHC H-score for tumors in which both were available (n = 165). Samples were stratified as H-score of 0 (negative staining), or >0 (positive staining). p value by t-test is shown.
(D) Heat map of association between CD38 mRNA levels and a list of immune-related genes in the TCGA lung adenocarcinoma dataset. Spearman correlation test is applied on each gene to check the association with mRNA levels of CD38. Adjusted p value < 0.05 and spearman rho ≥ 0.5 were used as the criteria to select the most significant immune markers for generating the heat map.
(E) Spearman’s rank correlation (ρ) was used to assess the association between T cell cytolytic score and CD38 expression in lung cancer patients’ samples from TCGA (n = 1008), MD Anderson PROSPECT (n = 275), and MD Anderson BATTLE-2 (n =144) datasets. T cell cytolytic score was computed as described in (49).
(F) IHC staining and scoring for CD38 and PD-L1 were performed in tumors from the lung cancer patient tissue microarray bank TMA3 (n = 259). Representative images of cell membrane staining intensity are shown in the left panel. The cell membrane staining intensity and percentage of positive cells were analyzed and used to generate an H-score for each sample that passed quality control. Samples were stratified as CD38low (H-score < 2.5), CD38high (H-score > 2.5), PD-L1low (H-score < 15), or PD-L1high (H-score > 15). Percent distribution (pie chart) of each co-expression pattern of CD38 and PD-L1 IHC staining was summarized for the lung cancer patient tissue microarray bank TMA3 (209 of 259 IHC staining qualified samples).
(G) Co-expression of CD38 with key T cell and cytolytic markers in melanoma tumors pre- and on-anti-PD1 treatment. Heat map for pre-treatment (n=51, upper panel) and on-treatment (n=56, lower panel) melanoma tumors (48), highlighting mRNA expression levels of CD38 and cytolytic T-cell markers (CD8A, PRF1 and GZMA), interferon regulatory factor 1 (IRF1) and a composite cytolytic marker cell profile. Samples were sorted by CD38 expression level from lowest (left) to highest (right). CD38 levels showed a correlation coefficient of ≥ 0.78 with each factor (p-values all < 10e-10)
(H) CD38 levels increase in melanoma tumors treated with anti-PD-1. Pre- and on-anti-PD-1 treatment CD38 mRNA expression levels from patient matched melanoma tumors (n=43). CD38 expression is shown in dark gray dots for pre-treatment samples and orange dots for on-treatment samples. Each of the matched sample are connected by a dashed gray line. CD38 levels are significantly higher in the on-treatment group compared with pre-treatment group (p = 0.019, paired, two-tailed t test). Median/mean pre-treatment (−0.05/−0.03); on-treatment (0.01/0.04).
Given the strong correlation between CD38 mRNA and CD38 protein levels, we turned to other available patient datasets for which only mRNA expression data is available to perform additional analyses, including the lung cancer and melanoma datasets from TCGA (lung adenocarcinoma, n = 512; lung squamous carcinoma, n = 496; melanoma dataset, n = 469), and the MD Anderson BATTLE-2 trial (metastatic lung cancer, n = 144). Stratification of the samples by CD38 mRNA levels revealed relatively high levels in about ~25–30% of cases, with a strong correlation between CD38 expression and a previously described immune inflammatory signature (30) that includes multiple markers of immune suppressive cell types or known immune checkpoint molecules and cytokines, e.g., Foxp3, CTLA-4, PD-1, LAG3, TIM3, PD-L2, HVEM, BTLA, IDO, and CCL2 (Fig. 4D, Supplemental Figs. 15C-D, Supplemental Tables 9 and 10). The data from lung cancer and melanoma datasets demonstrates a strong correlation between CD38 expression and a cytolytic T cell tumor infiltrate (Fig. 4E, Supplemental Fig. 15E), consistent with the animal and in vitro co-culture studies (Figs. 1D-H, Supplemental Fig. 3). To further define the immune subsets of untreated tumors based on CD38 and PD-L1 expression we performed IHC staining of PD-L1 in the same 793 early-stage tumors from the tissue microarrays (TMA3 (259 cases) and TMA4 (534 cases)) and found that 10.2–16.7% expressed high levels of both, while 5.1–6.7% expressed high CD38 but low PD-L1. Roughly 50% of these untreated tumors had high PD-L1 staining and low CD38 (Fig. 4F, Supplemental Fig. 15F, Supplemental Tables 7 and 8). Interestingly, although CD38 was reported as a prognostic biomarker in multiple myeloma (7), and that PD-1/PD-L1 treatment is an effective therapy for certain metastatic NSCLC cases, we did not find an association between CD38/PD-L1 expression and overall survival in the early-stage lung cancer cohorts (Supplemental Fig. 16). Similarly, in a small group of metastatic NSCLC patients (n=50) who received anti-PD-1 therapy after prior chemotherapy, CD38 and PD-L1 levels on the tumor cells or in the tumor stroma were assessed by IHC in the pre-treatment tumor samples. The level of CD38 on tumor or infiltrating cells in the stroma showed a non-significant trend toward predicting clinical radiographic response (PR versus PD+SD) to PD-1 checkpoint blockade, with higher CD38 levels predicting lack of response (Supplemental Figs. 17A-C). Subcategorization by both PD-L1 and CD38 levels was not predictive of response (Supplemental Fig. 17D, Supplemental Table 11), but interpretation from these analyses is significantly limited by the small numbers in each group and will require additional investigation.
We further evaluated a melanoma dataset where RNAseq data are available for tumor biopsy specimens (n=43 pairs) pre- and on-treatment with nivolumab (anti-PD-1), and observed that CD38 expression correlates with the activated CD8 T cell status of the tumors in both pre- (n=51) and on-treatment samples (n=56; Fig. 4G). Additionally, CD38 levels are increased in response to treatment (mean: pre-treatment −0.03, post-treatment 0.04, p=0.019 by paired t-test) (Fig. 4H). Overall, these findings from more than 2500 primary or metastatic lung and melanoma tumors demonstrate that CD38 is found at moderate to high levels in a large percentage of tumors, is expressed in tumors with an active immune cell infiltrate and in which multiple immune modulators (e.g., PD-L1) are expressed, and are consistent with CD38 being upregulated as a consequence of the natural or treatment-mediated immune/inflammatory reaction in tumor microenvironment.
Combination Blockade of PD-L1 and CD38 Improves Antitumor Immune Responses
Due to the upregulation of CD38 expression after PD-L1/PD-1 blockade and the subsequent suppressive effect on CD8+ T cells, we assessed the therapeutic efficacy of CD38 inhibitors (anti-CD38 antibody or the biological flavonoid Rhein, which is an enzymatic inhibitor (31)) in combination with anti-PD-L1 antibody in tumor models. Concurrent CD38 inhibition with anti-CD38 antibody (clone NIMR-5) and anti-PD-L1 antibody suppressed primary tumor growth and metastases more than either monotherapy alone or isotype treatment (Figs. 5A-C and Supplemental Table 12). When tumor-bearing mice were treated with anti-CD38 antibody, tumor growth was significantly inhibited. However, this inhibition was reversed after CD8 T cell depletion, demonstrating that CD38 works in a CD8 T cell-dependent manner (Supplemental Fig. 18). Although neither CD38 nor PD-L1 inhibition alone significantly reduced the number of metastatic lung nodules, they did reduce the metastatic tumor size, while the combination treatment reduced both metastatic tumor number and metastatic tumor size. Similar results were obtained with combination therapy of anti-PD-L1 antibody and the flavonoid Rhein, which inhibits CD38 enzymatic activity (Fig. 5D, Supplemental Fig. 19A, and Supplemental Table 12). Strikingly, combination genetic and pharmacologic blockade using the PD-L1KO531LN3 cells sorted for high CD38 expression and treated with Rhein completely and rapidly eradicated the tumors (Fig. 5E), similar to the results with the PD-L1KO531LN3 cells sorted for negative CD38 expression (Fig. 2D). Immune profiling of tumors from each single and combination treatment revealed that more total and activated CD8+ T cells, more memory CD8+ T cells (CD44highCD62Llow), increased CD4+ICOS+ T cells, and less infiltration of CD4+ Tregs and MDSCs (myeloid-derived suppressor cells), were observed in the combination therapies with anti-CD38 than in single or control treatments (Fig. 5F, Supplemental Figs. 19B and 20, and Table 2). Additionally, in a KP tumor model naturally CD38-deficient and unable to upregulate CD38 expression (307P), we observed that anti-PD-1/PD-L1 monotherapy dramatically inhibited tumor growth or eliminated tumors (Figs. 5G-H). Although therapeutic treatment with anti-CD38 antibody or the enzymatic inhibitor does not distinguish between the effects of tumor cell and host cell CD38, the data demonstrate that systemic treatment has similar efficacy in reprogramming the immune microenvironment of the tumor as genetic manipulation of CD38 on tumor cells, and does not engender systemic toxicity or antagonism that compromises the therapeutic effects.
Figure 5.
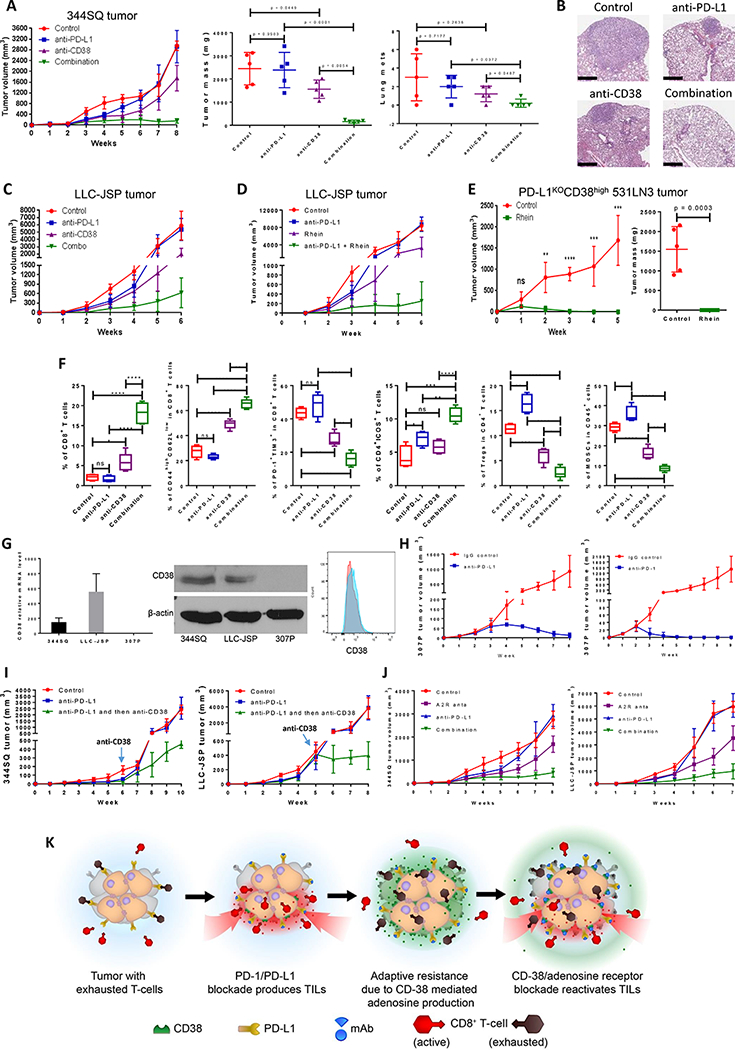
Co-inhibition of PD-L1 and CD38 or adenosine signaling improves antitumor immune responses.
(A) The indicated antibody or the isotype-matched IgG control was injected into 129/Sv mice (intraperitoneally) once a week for 7 weeks beginning on day 7 after subcutaneous 344SQ tumor cell injection (1 × 106 cells per mouse; n = 5/group). Dosing per injection was 200 μg of anti-PD-L1, 250 μg of anti-CD38. Tumors were measured once a week for 8 weeks. The tumor growth curves are shown in the left panel. The final tumor weights and metastatic lung nodules are shown in the middle and right panel. p values were calculated with ANOVA test.
(B) Representative H & E-stained lung tissues from each group of Figure 5A are shown, indicating metastatic nodules. Scale bars represent 600 μm.
(C) The indicated antibody was injected into C57BL/6 mice (intraperitoneally) once a week for 5 weeks beginning on day 7 after the subcutaneous LLC-JSP tumor cell injection (1 × 106 cells per mouse). Dosing per injection was 200 μg of anti-PD-L1, 250 μg of anti-CD38. Combo represents the combination of 200 μg anti-PD-L1 and 250 μg of anti-CD38. Tumors were measured once a week for 6 weeks. The tumor growth curves are shown.
(D) C57BL/6 mice were treated with anti-PD-L1 and Rhein (CD38 inhibitor) once a week for 5 weeks beginning on day 7 after the subcutaneous LLC-JSP tumor cell injection (1 × 106 cells per mouse). Dosing per intraperitoneal injection was 200 μg of anti-PD-L1, 50 mg/kg of Rhein. Tumors were measured once a week for 6 weeks. The tumor growth curves are shown (n = 5/group).
(E) PD-L1KOCD38high531LN3 cells (2 × 106 cells per mouse) were subcutaneously injected into immune competent 129/Sv mice (n = 5/group). Mice were treated with Rhein (CD38 inhibitor) once a week for 4 weeks beginning on day 1 after tumor cell injection. Dosing per intraperitoneal injection was 50 mg/kg. Tumors were measured once a week for 5 weeks. The tumor growth curves are shown in the left panel and the final tumor weights are shown in the right panel. Tumor sizes are presented as mean ± SEM. ns, no significant difference, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(F) FACS analysis of CD4+ICOS+TIL and CD8+TIL frequency, percent of memory CD8 T cells and exhausted CD8 T cells, and tumor-infiltrating Tregs and MDSCs from the endpoint primary tumors of Figure 5A. The statistical summary is shown. ns, no significant difference, **p < 0.01, ***p < 0.001, ****p < 0.0001. p values were calculated with ANOVA test. The gating strategies are included in the legend of Supplemental Figure S20.
(G) (Left) CD38 mRNA levels in murine lung cancer cell lines 344SQ, LLC-JSP, and 307P were determined by qPCR assays. mRNA levels are normalized to L32. The experiments were repeated at least three times. (Middle) CD38 protein levels in murine lung cancer cell lines 344SQ, LLC-JSP, and 307P were determined by Western blotting. β-actin was used as the loading control. (Right) 4 × 106 of 307P cells were subcutaneously injected into 129/Sv mice (n = 3). CD38 expression on sorted 307P tumor cells (CD31-CD45-EpCAM+ for sorting) was determined by FACS analysis 2 weeks post-cancer cell injection. The representative histogram is shown. Red, isotype staining; Light blue, CD38 staining.
(H) 129/Sv mice were treated weekly with anti-PD-L1 (200 μg per mouse), anti-PD-1 (200 μg per mouse), or their IgG control beginning on day 7 after a subcutaneous 307P cancer cell injection (4 × 106 cells per mouse; n = 5 or 7) for indicated weeks. The tumor growth was monitored once a week. The tumor growth curves are shown.
(I) (Left) 129/Sv mice were treated weekly with anti-PD-L1 (200 μg per mouse) or IgG control (Control) beginning on day 7 after a subcutaneous 344SQ cancer cell injection (0. 05 × 106 cells per mouse; n = 5) for 6 weeks. Mice in one of anti-PD-L1 treatment groups were sequentially treated with anti-CD38 (250 μg per mouse) once a week for 4 weeks. The tumor growth was monitored once a week. The tumor growth curves are shown. (Right) C57BL/6 mice were treated weekly with anti-PD-L1 (200 μg per mouse) or IgG control (Control) beginning on day 7 after a subcutaneous LLC-JSP cancer cell injection (0. 05 × 106 cells per mouse; n = 5) for 5 weeks. Mice in one of anti-PD-L1 treatment groups were sequentially treated with anti-CD38 (250 μg per mouse) once a week for 3 weeks. The tumor growth was monitored once a week. The tumor growth curves are shown.
(J) 200 μg of anti-PD-L1 antibody or the isotype-matched IgG control was intraperitoneally injected into mice (n = 5 or 7) once a week, while A2R anta (2 mg/kg of SCH 58261 and 1 mg/kg of PSB 1115) in 100 μl of carrier solution were intraperitoneally injected every other day, for indicated weeks beginning on day 7 after subcutaneous tumor cell injection (1 × 106 cells per mouse). The mice in control group received both IgG control and carrier solution. Tumors were measured once a week and the tumor growth curves are shown. A2R anta: adenosine receptor 2 antagonists; SCH 58261: A2a adenosine receptor antagonist; PSB 1115: A2b adenosine receptor antagonist.
(K) The working model of CD38 as a major mechanism of the resistance to PD-1/PD-L1 blockade.
Table 2.
Co-inhibition of PD-L1 and CD38 produces a favorable anti-tumor microenvironment.
| Infiltrating immune cells in LLC-JSP-bearing tumors | Combination therapy of anti-PD-L1 and anti-CD38 | Combination therapy of anti-PD-L1 and Rhein | ||||||
|---|---|---|---|---|---|---|---|---|
| Control | anti-PD-L1 | anti-CD38 | Combination | Control | anti-PD-L1 | Rhein | Combination | |
| % of CD8+ T cells | 5.05 ± 0.49 | 4.70 ± 0.32 | 9.15 ± 1.19 | 20.80 ± 1.21 | 4.99 ± 0.55 | 4.78 ± 0.39 | 7.13 ± 0.79 | 23.15 ± 0.82 |
| % of CD44highCD62Llow in CD8+ T cells | 50.90 ± 1.90 | 46.10 ± 1.18 | 64.48 ± 3.29 | 75.10 ± 1.04 | 50.68 ± 1.59 | 41.55 ± 3.97 | 55.84 ± 2.98 | 70.36 ± 1.07 |
| % of PD-1+TIM3+ in CD8+ T cells | 30.43 ± 2.17 | 38.53 ± 2.98 | 23.84 ± 1.67 | 10.12 ± 0.86 | 30.40 ± 1.44 | 40.03 ± 4.39 | 22.66 ± 1.96 | 13.68 ± 1.47 |
| % of CD4+ICOS+ T cells | 4.27 ± 0.46 | 6.26 ± 0.53 | 6.70 ± 0.65 | 9.20 ± 0.58 | 4.44 ± 0.47 | 6.72 ± 0.31 | 5.45 ± 0.50 | 8.78 ± 0.70 |
| % of Tregs in CD4+ T cells | 10.63 ± 0.45 | 13.90 ± 0.54 | 5.13 ± 0.30 | 2.83 ± 0.47 | 10.75 ± 0.56 | 15.25 ± 0.97 | 8.47 ± 0.72 | 4.77 ± 0.63 |
| % of MDCSs in CD45+ cells | 24.38 ± 1.98 | 30.53 ± 1.01 | 11.50 ± 1.40 | 5.17 ± 1.14 | 25.13 ± 2.10 | 30.78 ± 1.18 | 14.95 ± 1.94 | 8.66 ± 1.52 |
To model how CD38-blocking strategies might be translated into the clinic for patients with disease refractory to anti-PD-1/PD-L1, we tested sequential treatment after the development of resistance to anti-PD-L1 by treating animals with anti-CD38 antibody alone. We observed a substantial inhibition of tumor growth with an associated enhancement of the effector CD8+ and CD4+ T cell responses and blunting of the suppressor CD4+ T regulatory and MDSC populations, highlighting that CD38 is an independent factor in treatment-induced resistance (Fig. 5I, Supplemental Fig. 21). Because the anti-human CD38 antibody daratumumab, which is approved for the treatment of patients with multiple myeloma (32), was reported to kill cancer cells through antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) (33), we tested if the anti-mouse CD38 (NIMR-5) antibody used in our studies works similarly. However, we did not observe ADCC or CDC in the lung cancer models (Supplemental Figs. 22A-B). Interestingly, we found that CD38 was internalized after treating lung cancer cells with NIMR-5 (Supplemental Fig. 22C), consistent with the abrogation of adenosine levels upon treatment noted in Figure 3L. Finally, given our data that CD38 suppresses CD8+ T cell function via adenosine receptor signaling, and the on-going development of A2R inhibitors for cancer treatment (34), we tested the potential benefits from combination A2R antagonists and anti-PD-L1. We observed significant improvements of antitumor effect with A2R antagonists alone or combined with anti-PD-L1 (Fig. 5J), which confirms the strong role of adenosine signaling on CD8+ T cell function. Overall the therapeutic studies demonstrate that combination CD38 and PD-L1 blockade substantially reduces primary tumor burden and metastases, that CD38 can be used in combination or in a sequential manner upon the acquisition of resistance, and that targeting CD38 or the downstream adenosine signaling is highly effective.
DISCUSSION
Immune checkpoint therapies have gained significant attention for their clinical potential to improve durable outcomes for cancer patients (35,36). However, only a fraction of patients derive long-term benefit and extensive efforts are ongoing to understand the underlying mechanisms of response/resistance. Analyses of experimental models and patient tumors has demonstrated that both de novo and acquired immune checkpoint inhibitor resistance arise from a number of tumor-cell intrinsic and extrinsic mechanisms (3). Many of these mechanisms occur as a dynamic response of the tumor or microenvironment to an effective T cell infiltration. Herein we found that when animal models were treated for an extended period with anti-PD-1 or -PD-L1 therapy, acquired resistance developed after an initial suppression of tumor growth and metastases, independent of the tumor type and strain background. This resistance was observed in response to both pharmacologic and genetic PD-1/PD-L1 blockade in lung cancer and melanoma models, and in each case we found that CD38 was upregulated on tumor cells. We also found associated functional impairment of CD8+ T cells after their initial activation and expansion, demonstrating a novel parallel role for CD38 within the microenvironment of T-cell-inflamed tumors to drive adaptive immune escape. The data demonstrates that CD38 up-regulation after PD-1/PD-L1 blockade is neither genotype- nor histology-dependent, suggesting that CD38-mediated resistance could occur in a broad variety of cancer types.
Since CD38 expression was induced upon PD-1/PD-L1 blockade, we hypothesized that CD38 upregulation results from intratumoral T-cell infiltration and associated changes in the cytokine/metabolite milieu. Indeed, we found a strong correlation between CD8+ T cells and CD38 expression in NSCLC and melanoma. Additionally, when tumor cells were cultured in vitro with activated CD8+ T cells, CD38 was upregulated. In anti-PD-L1 antibody treated tumors, we observed increased ATRA levels, which regulates IRF1 and downstream IFN-β production (24,25), and could recapitulate the effect of ATRA on CD38 up-regulation by pharmacologic dosing or antagonism, consistent with previous reports that ATRA and IFN-β are enriched in inflammatory tumors and serve as potent inducers of CD38 (17,18,24,25). This study uncovered a new insight into how ATRA regulates T cell immunity via CD38 and the production of adenosine. Taken together, our current study emphasizes that the tumor immune microenvironment undergoes an adaptive reprogramming under the continued pressure of PD-L1/PD-1 axis blockade. Either pre-existent or upregulated CD38 expression in tumor cells is responsible for this adaptive immune shift in response to anti-PD-1/anti-PD-L1 antibody treatment, and over time the immunosuppressive effect of CD38high tumor cells becomes dominant over PD-L1.
CD38 has long been considered an immune molecule since it is expressed on activated B, T, and NK cells (7). However, several recent studies have suggested a broader distribution and more complex role, based upon its multifunctional activities (6). For example, a report showed that CD38high CD8+ T lymphocytes have strong immunosuppressive capabilities. This subset possesses a regulatory potential that could work together with the innate immune response and control immune homeostasis (37). Another group reported that CD38high MDSCs possess the capacity to suppress activated T cells and promote tumor growth to a greater degree than CD38low MDSCs (11). CD38 has also been associated with functions exerted by regulatory T cells (Tregs), in which high CD38 expression in Foxp3+/CD4+ T cell populations correlates with extremely powerful modulatory properties of CD4+ regulatory T lymphocytes (38). Consistent with these findings, when Anderson and colleagues evaluated percentages of CD38-expressing Treg subsets from normal donors and myeloma patients, they found a high expression of CD38 on CD4+CD25highFoxp3+ regulatory T cells, and that targeting CD38 could block this immunosuppressive population (39). In addition, the non-canonical adenosinergic pathway led by CD38/CD203a provides substrates to CD73 and consequently feeds the production of the potent immunosuppressor adenosine, which is normally essential in maintaining tissue homeostasis and preventing an overzealous immune response (7,40,41). Malavasi’s group has shown that primary human melanoma cell lines can take advantage of this ectoenzyme complex to suppress T cell proliferation through adenosine production (26). Another recent study demonstrated a role for NAD+ levels, which are regulated by CD38 on T cells, as an important factor in the reprogramming of intratumoral CD4+ cells into hybrid effector Th1/17 cells for enhanced adoptive T cell therapy (42). In this study, we have identified CD38 as an immune suppressive molecule expressed on cancer cells allowing their adaptive escape from checkpoint inhibitor-mediated immune attack through the adenosine production pathway, thereby promoting resistance to anti-PD-L1/PD-1 therapy, as shown schematically in the model (Fig. 5K). The emerging evidence suggests a multi-faceted immunosuppressive role for CD38 in regulating tumor immune microenvironment.
The identification of biomarkers that predict response to immune therapy or identify individuals most likely to develop resistance is a key goal in the clinical use of single-agent and combination therapies. Our observations from the clinical datasets of a strong correlation between CD38 expression and an immune inflamed tumor suggest that CD38 might serve as an appropriate marker of adaptive immune resistance to tumor specific T-cell infiltration rather than as a static constitutive biomarker. Although our animal model data suggests multiple potential translational strategies to overcome immune checkpoint inhibitor resistance mediated by CD38, many questions about how best to incorporate CD38 into immunotherapy strategies will need to be studied in prospective clinical trials of CD38 blockade. While daratumumab (anti-CD38 monoclonal antibody, DARZALEX®) is approved by the U.S. FDA for the treatment of multiple myeloma and shows pronounced efficacy as a single agent or combination therapy with an acceptable adverse event profile (32), there is currently no data regarding its efficacy in solid tumors. The mechanism of immune resistance to anti-PD-L1/PD-1 therapy caused by CD38 provides an evident rationale for recruitment of cancer patients for clinical trials of anti-CD38 in combination with anti-PD-L1/PD-1 to prevent therapy resistance and further enhance anti-tumor efficacy.
METHODS
Tumor Models and Tumor micro-CT Scanning
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at MD Anderson Cancer Center. To study primary tumor growth and lung metastases, cancer cells (if not indicated, 0.5 × 106 for LLC-JSP; 1 × 106 for 344SQ; 2 × 106 for 531LN3) in 100 μl of phosphate-buffered saline (PBS) were injected subcutaneously into the mouse flank. Tumor sizes were calculated using the formula: ½(length × width × width) at indicated time points. For lung metastasis measurement, the lungs were removed and immersed in cold PBS, nodules on the lung surface were counted as described previously (4,5).
Spontaneous KrasLA1/+ and K-ras LA1/+/p53 R172HΔg/+ mice were bred in our laboratory and tumor growth was measured by micro-CT scanning. Multiple transverse cross-sectional CT images were provided for analysis by ImageJ. The largest cross-sectional tumor areas were selected for quantification. Tumor diameters were measured with the ImageJ line tool and areas were calculated using the formula for the area of an ellipse divided by 4.
In vivo Treatments
Mice were treated with antibodies (200 μg of anti-PD-L1 per mouse; 250 μg of anti-CD38 per mouse; or combination) and their IgG control via IP injection once a week for indicated weeks. Rhein (CD38 inhibitor) was used at 50 mg/kg intraperitoneally per dose once a week for indicated weeks. 200 μg/mouse of anti-PD-1 antibody or an IgG control was intraperitoneally injected into mice twice a week (or 300 μg/mouse once a week) for indicated weeks beginning on day 7 after tumor cells were subcutaneously implanted.
For ATRA and its receptor antagonist treatment, tumor-bearing mice were treated with ATRA (45 μg in 100 μL 1% methylcellulose; oral administration) or retinoic acid receptor alpha (RARα) antagonist BMS195614 (67 μg in 100 μL 1% methylcellulose; oral administration) once a day for 1–2 weeks beginning on day 4 after tumor cells were subcutaneously implanted (1 × 106 cells per mouse).
For the combination treatment of A2R antagonists and anti-PD-L1, 200 μg of anti-PD-L1 antibody or the IgG control was intraperitoneally injected into mice once a week, while 2 mg/kg of SCH 58261 (A2a adenosine receptor antagonist) and 1 mg/kg of PSB 1115 (A2b adenosine receptor antagonist) in 100 μL of carrier solution (15% DMSO, 15% Cremophore EL, 70% H2O) were intraperitoneally injected every other day, for indicated weeks beginning on day 7 after subcutaneous cell injection. The mice in control group received both IgG control and carrier solution.
Co-culture Assay
To prepare tumor-specific CD8+ T cells, 129/Sv mice were challenged by subcutaneous tumor cell injection with 0.5 × 106 344SQ or C57BL/6 mice with 0.2 × 106 LLC-JSP for 2 weeks. CD8+ T cells were isolated from the tumors, blood, and spleens of these animals, and labeled with CFSE following the kit’s instruction (CellTrace™ CFSE Cell Proliferation Kit, Catalog# C34554, Life Technologies). CFSE-labeled CD8+ T cells were co-cultured with the indicated cells in the presence of anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) for 4 days. T cell proliferation was quantified using FACS analysis. In some experiments, the cocktail of specific adenosine receptor antagonists (500 nM ADORA1 antagonist PSB36, 1 μM ADORA2a antagonist SCH58261, and 1 μM ADORA2b antagonist PSB1115) was used to block adenosine receptor signaling.
mRNA Expression Profiling of Cancer Patient Samples
Experimental details regarding TCGA datasets including RNA extraction, mRNA library preparation, sequencing (Illumina HiSeq platform), quality control, data processing and quantification of gene expression are previously published (43,44). For the PROSPECT and BATTLE-2 samples, the mRNA was extracted from frozen tumor tissue corresponding to the same specimen from which the formalin fixed, paraffin embedded blocks were made. Array based expression profiling of PROSPECT tumors was performed using the Illumina Human WG-6 v3 BeadChip, according to the manufacturer’s protocol. Gene expression data for the PROSPECT dataset have been previously deposited in the GEO repository (GSE42127) (30,45). The raw data files of transcriptomes were analyzed using Bioconductor R packages. Scatter plots were generated through the cBioPortal (46,47). Gene expression values (mRNA z-Scores; RNA Seq V2 RSEM) for the skin/cutaneous melanoma (SKCM) TCGA cohort (n=469) were derived from the cbioportal platform (44,46,47). The pre- and on-anti-PD1 treatment melanoma cohort and RNAseq analysis with gene-level FPKM values have been published (48), and gene expression levels were derived from the interface provided by the study: http://ioexplorer.org/shinyipop/bms038/, allowing for the generation of a Gene Set Variation Analysis (GSVA) enrichment score.
Supplementary Material
SIGNIFICANCE:
CD38 is a major mechanism of acquired resistance to PD-1/PD-L1 blockade, causing CD8+ T cell suppression. Co-inhibition of CD38 and PD-L1 improves antitumor immune response. Biomarker assessment in patient cohorts suggests that a combination strategy is applicable to a large percentage of patients in whom PD-1/PD-L1 blockade is currently indicated.
Acknowledgments
This work was supported by P50CA070907–16A1, P50CA070907–18, and Elsa U. Pardee Foundation to L.Chen; CPRIT RP150405 and CPRIT-MIRA RP160652-P3 to D.L. Gibbons; CPRIT RP130397 to P.L. Lorenzi; CA087546 and CA190722 to E. Dmitrovsky; CAMS Initiative for Innovative Medicine (2016-I2M-1–005), National Natural Science Foundation (81590765) to F.X. Qin. The work was also supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program. We thank the members of our labs for critical discussions.
Footnotes
No potential conflicts of interest are disclosed by the authors.
REFERENCES
- 1.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501 doi 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014;5:5241 doi 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Yi X, Goswami S, Ahn YH, Roybal JD, Yang Y, et al. Growth and metastasis of lung adenocarcinoma is potentiated by BMP4-mediated immunosuppression. Oncoimmunology 2016;5(11):e1234570 doi 10.1080/2162402X.2016.1234570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 2008;88(3):841–86 doi 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 7.Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom 2013;84(4):207–17 doi 10.1002/cyto.b.21092. [DOI] [PubMed] [Google Scholar]
- 8.Zucchetto A, Benedetti D, Tripodo C, Bomben R, Dal Bo M, Marconi D, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res 2009;69(9):4001–9 doi 10.1158/0008-5472.CAN-08-4173. [DOI] [PubMed] [Google Scholar]
- 9.Oh IH, Eaves CJ. Overexpression of a dominant negative form of STAT3 selectively impairs hematopoietic stem cell activity. Oncogene 2002;21(31):4778–87 doi 10.1038/sj.onc.1205592. [DOI] [PubMed] [Google Scholar]
- 10.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 2005;18(1):61–9 doi 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 11.Karakasheva TA, Waldron TJ, Eruslanov E, Kim SB, Lee JS, O’Brien S, et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res 2015;75(19):4074–85 doi 10.1158/0008-5472.CAN-14-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandele A, Sewatanon J, Gunisetty S, Singla M, Onlamoon N, Akondy RS, et al. Characterization of Human CD8 T Cell Responses in Dengue Virus-Infected Patients from India. J Virol 2016;90(24):11259–78 doi 10.1128/JVI.01424-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canto C, Sauve AA, Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med 2013;34(6):1168–201 doi 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF, et al. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. FASEB J 2006;20(7):1000–2 doi 10.1096/fj.05-4585fje. [DOI] [PubMed] [Google Scholar]
- 15.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520(7547):373–7 doi 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8(8):793–800 doi 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 17.Drach J, McQueen T, Engel H, Andreeff M, Robertson KA, Collins SJ, et al. Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res 1994;54(7):1746–52. [PubMed] [Google Scholar]
- 18.Mehta K, McQueen T, Manshouri T, Andreeff M, Collins S, Albitar M. Involvement of retinoic acid receptor-alpha-mediated signaling pathway in induction of CD38 cell-surface antigen. Blood 1997;89(10):3607–14. [PubMed] [Google Scholar]
- 19.Henig N, Avidan N, Mandel I, Staun-Ram E, Ginzburg E, Paperna T, et al. Interferon-beta induces distinct gene expression response patterns in human monocytes versus T cells. PLoS One 2013;8(4):e62366 doi 10.1371/journal.pone.0062366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016;16(3):131–44 doi 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- 21.Duester G Retinoic acid synthesis and signaling during early organogenesis. Cell 2008;134(6):921–31 doi 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis LF, Harada H, Wolchok JD, Taniguchi T, Vilcek J. Critical role of a common transcription factor, IRF-1, in the regulation of IFN-beta and IFN-inducible genes. EMBO J 1992;11(1):185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo XM, Ross AC. Retinoic acid exerts dual regulatory actions on the expression and nuclear localization of interferon regulatory factor-1. Exp Biol Med (Maywood) 2006;231(5):619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauvois B, Durant L, Laboureau J, Barthelemy E, Rouillard D, Boulla G, et al. Upregulation of CD38 gene expression in leukemic B cells by interferon types I and II. J Interferon Cytokine Res 1999;19(9):1059–66 doi 10.1089/107999099313299. [DOI] [PubMed] [Google Scholar]
- 25.Matikainen S, Ronni T, Hurme M, Pine R, Julkunen I. Retinoic acid activates interferon regulatory factor-1 gene expression in myeloid cells. Blood 1996;88(1):114–23. [PubMed] [Google Scholar]
- 26.Morandi F, Morandi B, Horenstein AL, Chillemi A, Quarona V, Zaccarello G, et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015;6(28):25602–18 doi 10.18632/oncotarget.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16-NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J Immunol 2015;195(3):965–72 doi 10.4049/jimmunol.1500591. [DOI] [PubMed] [Google Scholar]
- 28.Horenstein AL, Quarona V, Toscani D, Costa F, Chillemi A, Pistoia V, et al. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol Med 2016;22 doi 10.2119/molmed.2016.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013;2(9):e26246 doi 10.4161/onci.26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res 2016;22(14):3630–42 doi 10.1158/1078-0432.CCR-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol Sci 2018;39(4):424–36 doi 10.1016/j.tips.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med 2015;373(13):1207–19 doi 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 33.de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol 2011;186(3):1840–8 doi 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 34.Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restores Tumor-Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia 2017;19(7):530–6 doi 10.1016/j.neo.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348(6230):56–61 doi 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125(9):3384–91 doi 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bahri R, Bollinger A, Bollinger T, Orinska Z, Bulfone-Paus S. Ectonucleotidase CD38 demarcates regulatory, memory-like CD8+ T cells with IFN-gamma-mediated suppressor activities. PLoS One 2012;7(9):e45234 doi 10.1371/journal.pone.0045234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patton DT, Wilson MD, Rowan WC, Soond DR, Okkenhaug K. The PI3K p110delta regulates expression of CD38 on regulatory T cells. PLoS One 2011;6(3):e17359 doi 10.1371/journal.pone.0017359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin Cancer Res 2017;23(15):4290–300 doi 10.1158/1078-0432.CCR-16-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 2016;30(3):391–403 doi 10.1016/j.ccell.2016.06.025. [DOI] [PubMed] [Google Scholar]
- 41.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001;414(6866):916–20 doi 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 42.Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab 2018;27(1):85–100 e8 doi 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96 doi 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res 2013;19(6):1577–86 doi 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4 doi 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171(4):934–49 e15 doi 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.