

Abstract
Our understanding of the role of the vascular endothelium has evolved over the past two decades, with the recognition that it is a dynamically regulated organ and that it plays a nodal role in a variety of physiological and pathological processes. Endothelial cells (ECs) are not only a barrier between the circulation and peripheral tissues, but also actively regulate vascular tone, blood flow and platelet function. Dysregulation of ECs contributes to pathological conditions such as vascular inflammation, atherosclerosis, hypertension, cardiomyopathy, retinopathy, neuropathy and cancer. The close anatomical relationship between vascular endothelium and highly vascularized metabolic organs/tissues suggests that the crosstalk between ECs and these organs is vital for both vascular and metabolic homeostasis. Numerous reports support that hyperlipidemia, hyperglycemia and other metabolic stresses result in endothelial dysfunction and vascular complications. However, how ECs may regulate metabolic homeostasis remains poorly understood. Emerging data suggest that the vascular endothelium plays an unexpected role in the regulation of metabolic homeostasis and that endothelial dysregulation directly contributes to the development of metabolic disorders. Here, we review recent studies about the pivotal role of ECs in glucose and lipid homeostasis. In particular, we introduce the concept that the endothelium adjusts its barrier function to control the trans-endothelial transport of fatty acids, lipoproteins, lipoprotein lipases, glucose and insulin. In addition, we summarize reports that ECs communicate with metabolic cells through EC-secreted factors and we discuss how endothelial dysregulation contributes directly to the development of obesity, insulin resistance, dyslipidemia, diabetes, cognitive defects and fatty liver disease.
Keywords: vascular endothelium, metabolic homeostasis, obesity, diabetes, insulin resistance
Subject Terms: Endothelium, Vascular Type, Nitric Oxide, Metabolism
Vascular endothelium is well-situated to regulate metabolic homeostasis
Blood vessels are broadly divided into two categories, macrovasculature and microvasculature1. The macrovasculature is composed of large vessels, such as arteries and veins, and is responsible for carrying blood towards or away from organs. The microvasculature--small arterioles, capillaries and venules--primarily participates in the regulation of local blood perfusion and metabolic exchange between circulation and peripheral tissues1, 2. In most organs, small arterioles (diameter 10–100 μm) are well innervated and highly responsive to sympathetic vasoconstriction. They play a major role in tuning the systemic vascular resistance and blood flow volume, ultimately influencing downstream capillary fluid exchange. Capillaries and post-capillary venules are main sites for fluid and macromolecular exchange. Due to the low shear stress force and selective expression of endothelial adhesion molecules, venules are also important sites for leukocyte adhesion3–5.
The vascular endothelium is a thin layer of simple squamous cells lining the interior surface of the entire cardiovascular system, from the largest arteries and veins to the smallest capillaries. It forms a critical boundary between the circulating blood and underlying tissues. In the larger vessels, such as veins and arteries, the vessel wall consists of endothelial cells (ECs), smooth muscle cells and elastic fibers. In the capillaries, however, ECs constitute the entire blood vessel wall. They create a large surface area with relatively high permeability for the exchange of gases, fluids, electrolytes and macromolecules. There are three types of capillaries: continuous, fenestrated and discontinuous capillaries. Continuous capillaries are low permeability conduits located in skeletal muscle, heart, adipose tissue, lung, skin and the central nervous system, where intercellular gaps are tight and the basement membrane is continuous for ECs. Fenestrated capillaries are located at the choroid plexus of brain and endocrine organs where intercellular clefts exist between neighboring ECs but the basement membrane surrounding ECs is continuous. In liver, bone marrow and spleen, discontinuous capillaries (sinusoids) display gaps in both endothelial layer and basement membrane, therefore providing the highest permeability.
ECs serve as “first responders” in the modulation of vascular tone, permeability and interaction with circulating macromolecules. Activation of ECs is an upstream event necessary to expand the vascular network through the processes of vasculogenesis, angiogenesis and arteriogenesis. The close anatomical association between metabolic tissues/cells and ECs, especially microvascular ECs that play a critical role in metabolic exchange, suggests that they may possess potential functional interdependence. Dysregulation of ECs contributes to many cardiovascular, circulatory and blood diseases in response to metabolic stress and vascular injury6–9. However, how ECs regulate metabolic homeostasis remains poorly understood. A growing body of evidence suggests that ECs are also actively involved in the regulation of metabolic processes. Dysregulation of ECs aggravates insulin resistance, adipose inflammation, diabetes, non-alcoholic fatty liver disease and other metabolic conditions. In this review, we will discuss about the emerging roles of ECs in the control of metabolic homeostasis and how endothelial dysregulation contributes to the development of metabolic disorders.
Metabolic processes involved in the regulation of energy homeostasis
Energy homeostasis is a balance of energy inflow (food intake) and energy expenditure, involving a variety of biological processes such as nutrient adsorption and transport, substrate metabolism, energy storage and clearance and appetite control. Energy provision (adenosine triphosphate, ATP) required for daily activities is mainly provided within the body by two types of macromolecules-carbohydrates and lipids. Major organs or tissues that are involved in the regulation of energy metabolism include the brain, gastrointestinal tract, liver, adipose tissue, pancreas and muscle. Tightly regulated cellular crosstalk in and between these organs or tissues plays a central role in maintaining energy homeostasis in vital organs and individual health. Excess calorie intake, stationary lifestyle, and genetic predisposition lead to disruption of energy balance, contributing to development of metabolic diseases such as obesity, metabolic syndrome, diabetes mellitus, insulin resistance and non-alcoholic fatty liver diseases.
Dietary carbohydrates and lipids are digested and processed into glucose and fatty acids (FAs). When glucose perfuses pancreatic endocrine tissue, it increases intracellular ATP/ADP in pancreatic beta cells and promotes insulin secretion10. Insulin in the circulation in turn activates insulin receptor-mediated signaling pathways in skeletal muscle, heart and adipose tissue, resulting in increased GLUT4-mediated glucose uptake11, 12. Additional glucose is stored in skeletal muscle and liver in the form of glycogen13–16. Glycogen breakdown in skeletal muscle meets the energy demands of muscle during exercise. In circumstances such as prolonged, low-intensity exercise, FA oxidation can also provide ATP to muscle cells17. In adipocytes, glucose is used to produce glycerol, along with free FAs (FFAs) delivered from the liver for TG synthesis. The liver acts as a hub to metabolically coordinate with various tissues, including skeletal and cardiac muscle and adipose tissue. Under feeding conditions when the liver is saturated with glycogen (~5% of liver mass), glucose is converted into FFAs, which are further esterified into TG, or amino acids in hepatocytes. Under fasted conditions or increased physical activity, glucagon secreted from pancreatic alpha cells activates glycogenolysis and gluconeogenesis signaling pathways in the liver to increase glucose production18. FFAs stored as TG in adipose tissue can also be mobilized through lipolysis and used by peripheral tissues, which is stimulated by glucagon and adrenaline and inhibited by insulin19. In addition, hormones released from peripheral tissues are integrated into the brain20, 21, providing signals for appetite control by neurotransmitters and modulators such as serotonin (reviewed by Yeo GS et al22). For the past several decades, it has been well recognized that the crosstalk between different metabolic organs/tissues plays an essential role in the regulation of metabolic homeostasis23, 24. The signaling network responsible for intercellular communication includes the hormonal and nervous communication. However, the underlying molecular pathogenesis of the communications between organs/tissues during the development of metabolic diseases remains to be explored. This review will discuss recent data supporting that the vascular endothelium plays a crucial role in these communications between metabolic organs/tissues.
Endothelial responses to metabolic dyshomeostasis
Although the microvascular endothelium is critical for metabolic exchange, the detailed regulatory mechanisms of trans-endothelial transport of FAs, lipoproteins and metabolic hormones and regulators remain incompletely understood. The following sections highlight recent advances that describe how ECs directly control the trans-endothelial transport of these macromolecules by regulating endothelial barrier function. We particularly focus on molecular pathways that regulate crucial transporters and receptors of these transcytosis machineries. Since ECs actively communicate with surrounding cells in metabolic tissues by secreting metabolic factors/regulators, we also summarize recent reports about these endothelial-derived metabolic regulators (nitric oxide, extracellular matrix proteins, hormones, growth factors and enzymes). Last, ECs also regulate metabolic tissues indirectly through its own activation, proliferation and differentiation. We will discuss these processes briefly here since they have been reviewed elsewhere.
1. Endothelial barrier function in metabolic regulation
Vascular endothelium is anatomically interposed between the vascular space and peripheral tissues and therefore acts as a barrier regulating the exchange of macromolecules. However, a significant role of the vascular endothelium in control of nutrients (FAs, glucose, amino acids) and metabolic regulators (i.e., hormones and enzymes) across endothelial barrier for their delivery to peripheral tissues has not been well appreciated. Endothelial permeability is regulated by two distinct pathways—the transcellular pathway through which solutes are actively transported across the endothelial layer, or the paracellular pathway by which solutes passively move through the intercellular space between neighboring ECs. Recent studies unveil specific transporters, receptors and signaling pathways required for the transcellular transport of a variety of macromolecules such as FAs, lipoproteins, glucose, hormones (i.e. insulin, leptin) and enzymes (i.e. lipoprotein lipase) in ECs (Fig. 1). In this section, we review these transcytosis machineries for maintaining metabolic homeostasis.
Figure 1.
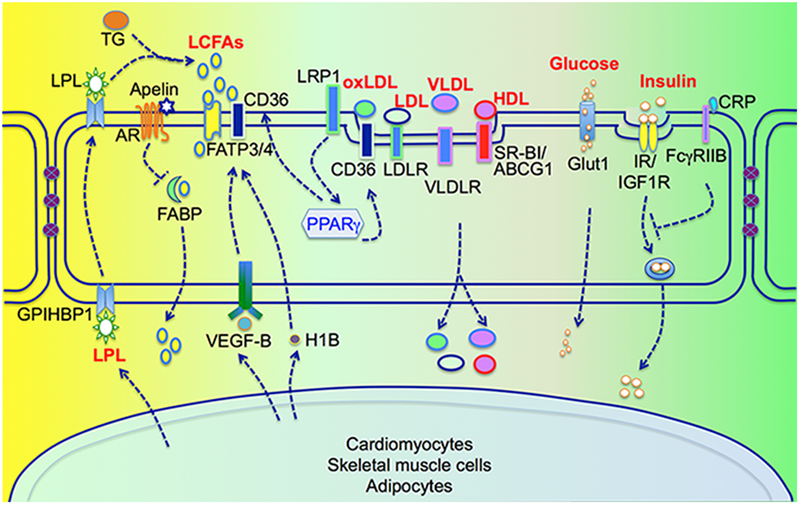
Trans-endothelial transport of FAs, lipoprotein, glucose and insulin.
1.1. Transport of FAs and lipoproteins
Most dietary lipids consist of long-chain FAs (LCFAs; ≥12 carbons), whereas FAs with short (<6 carbons) and medium-chains (with 6~12 carbons) are less frequently found in the diet and do not need active transport across cell membranes. It was once thought that FAs could enter cells by diffusion until the first putative FFA transporter (mAspAT/FABPPM) was discovered in rat aortic ECs in 199125. Since then, many different types of FA transport proteins including fatty acid translocase (FAT/CD36), FATPs (FATP1–6) and intracellular fatty acid binding proteins (FABPs) have been discovered.
In the liver, a fenestrated sinusoidal endothelial layer allows direct access of circulating FFAs to the space of Disse. However, ECs lining fat-utilizing peripheral tissues such as cardiac and red skeletal muscle form a continuous and nonfenestrated layer, which raises the need for an efficient trans-endothelial transport system for FAs. In ECs, FATP3, FATP4, FABP3–5, FABPPM and CD36 have been detected26–31. Hagberg et al32 demonstrated that co-expression of FATP3 and FATP4 synergistically increases lipid accumulation in ECs, suggesting that FATP3 and FATP4 are required for LCFA uptake by ECs. VEGF-B secreted by neighboring cardiomyocytes induces FATP3 and FATP4 expression in ECs. Mice lacking VEGF-B exhibit decreased LCFA uptake and lipid deposition in muscle, but gain more weight due to the increased lipid accumulation in white adipose tissue (WAT)32, 33. More interestingly, administration of anti-VEGF-B antibody 2H10 improves peripheral insulin sensitivity and decreases lipid accumulation in high-fat diet (HFD) fed rat or db/db diabetes mice. These studies for the first time demonstrate that targeting the barrier function of the vascular endothelium can be an effective strategy for treating insulin resistance and type 2 diabetes mellitus (T2DM). However, the translational path of inhibiting VEGF-B to treat dysglycemia requires further evaluation since VEGF-B has salutary effects on coronary arteriogenesis and is neuroprotective34–36. Another paracrine stimulus for FA trans-endothelial transport is 3-hydroxy-isobutyrate (3-HIB), a catabolic intermediate of the branch-chained amino acid (BCAA)-valine and generated by skeletal muscle cells. Increased circulating levels of BCAA are implicated in the progression of diabetes in animals and patients37. Elevated 3-HIB levels are also observed in skeletal muscle and serum of db/db mice or diabetic patients38. Knockdown of HIBC hydrolase, which blocks the generation of 3-HIB, in myotubes almost completely inhibits FA uptake in ECs38. In addition, knockdown of FATP3 and FATP4 reduces FA uptake in response to 3-HIB, suggesting that 3-HIB promotes FA transport across ECs through FATP3 and FATP438. However, how 3-HIB regulates the activity of FATP3 and FATP4 remains to be further explored.
FABP4 and FABP5 are exclusively expressed in ECs of capillaries and venules of cardiac and skeletal muscles, but not in arterioles, arteries or aorta39. In addition, their expression is detectable in the capillary ECs of brown and white adipose tissue, but not in liver, brain and lung. In FABP4/5 double knockout mice, FA uptake in the heart and skeletal muscle is significantly decreased, whereas glucose uptake is dramatically elevated to compensate for the limited supply of FAs39. Consistent with these findings, reduced FA uptake is observed in heart or liver-specific FABP knockout mouse models40. However, about 60% of FA uptake remains in mouse hearts lacking both FABP4/5, indicating that additional trans-endothelial FA transport systems operate independently of FABP4/5. Nevertheless, the pivotal role of FABP4 in FA uptake and transfer through the endothelial layer is further supported by recent studies with apelin, EC-specific apelin receptor (APLNR), EC-specific FoxO1, and apelin; EC-specific FoxO1 double knockout mouse models41. In adult metabolic tissues, APLNR is expressed specifically in ECs. Apelin/APLNR triggers endothelial FoxO1 inactivation and blocks FABP4 expression. In vitro FA uptake and transfer studies indicate that the disruption of apelin signaling pathways increases FA transfer across microvascular ECs, which can be abrogated by the FABP4 inhibitor BMS309403. Consistent with these observations, increased FA accumulation occurs in the heart, skeletal muscle, liver and brown adipose tissue in apelin knockout mice41. EC-specific FoxO1 depletion could ‘rescue’ FA accumulation and increase glucose clearance in apelin; EC-specific FoxO1 double knockout mice. FAs are also accumulated in metabolic tissues of EC-specific APLNR knockout mice. Treatment with BMS309403 alleviates FA accumulation and thereby improves glucose utilization. Apelin also inhibits oleic acid-induced vascular hyperpermeability by stabilizing VE-cadherin-based adherens junctions of ECs42. In response to HFD feeding, apelin-deficient mice display an obese phenotype, whereas Apelin transgenic mice have reduced body mass and subcutaneous adipose tissue42. Collectively, these observations indicate that apelin signaling within ECs regulates metabolic homeostasis likely through multiple molecular mechanisms such as FA transcytosis and EC dysfunction.
CD36, also known as fatty acid translocase (FAT), was initially cloned as an adipocyte membrane protein that facilitates FA uptake43. Cell culture studies demonstrate that CD36 also promotes the transport of FA across endothelial layer44. CD36-null mice display a dramatic decrease of FA uptake and utilization in heart, skeletal muscle and adipose tissue45, 46. In contrast, mice with CD36 overexpression in muscle exhibit increased FA oxidation in response to contraction47. Cardiac-restricted overexpression of PPARα results in myocyte lipid accumulation and cardiac dysfunction, that can be rescued by the depletion of CD36 or heart-specific lipoprotein lipase (LPL)48, 49. These data suggest that CD36-mediated FA uptake is rate-limiting for FA utilization in these metabolic tissues. Paradoxically, mice with depletion of CD36 have increased FA uptake in the liver and developed hepatic steatosis50. The opposing effects of CD36 depletion on FA uptake by muscle, adipose tissue and liver still remain a mystery. It is likely that differential permeability of ECs in the liver (which are highly permeable) and muscle and adipose tissues (which are less permeable) contributes to this discrepancy. An EC-specific CD36 knockout mouse model will be required to dissect the role of CD36 in trans-endothelial transport of FAs and elucidate the role of this endothelial mechanism for the overall FA uptake in metabolic tissues.
PPARγ is a master transcriptional regulator of lipid metabolism, tuning gene expression involved in the release, transport and storage of FAs, such as LPL and CD36 (reviewed by Lehrke M et al51). The specific depletion of PPARγ in ECs leads to decreased expression of FA-handling proteins, including CD36 and FABP452, 53. Mice with PPARγ depletion in ECs exhibit increased serum levels of VLDL and FFAs. Uptake of FA analogues is significantly reduced in heart, red skeletal muscle and adipose tissues of these mice52, and lipid accumulation is reduced in adipose tissue and skeletal muscle, but not liver53. These data suggest that PPARγ also plays a crucial role in FA transport across the nonfenestrated endothelium in skeletal muscle and adipose tissue. PPARγ agonists (TZDs) are popular drugs for treating diabetes. However, substantial clinical and preclinical experiences have indicated that TZDs lead to increased cardiovascular risk54. PPARγ-induced FA accumulation and cardiac dysfunction may be responsible for this side effect, which could be tested in future studies by blocking FA transport with inhibitors of FABP4 or CD36.
The endothelial outer membrane surface is the location where most intravascular lipoprotein metabolism takes place. Chylomicrons are converted metabolically to chylomicron remnants and VLDL to LDL. Hydrolysis of TG-rich lipoproteins generates FAs, which are transported across the endothelium through multiple mechanisms as we discussed above. However, it is still not clear whether intact TG can be transported across the endothelium. It is known that lipoproteins are transported across the endothelium via coordinated receptor-mediated endocytosis and exocytosis. For example, LDL, acetylated LDL, oxidized LDL and VLDL can all be taken up by ECs through LDL receptors, the scavenge receptor and VLDL receptors55. LDL is cleared mainly by the liver where tissue parenchymal cells have direct contact with blood. However, tissues containing non-fenestrated ECs are responsible for ~15–35% of LDL metabolism56. LDLR is expressed on the apical side of microvascular ECs and promotes LDL transcytosis across the blood brain barrier57. CD36 is a key scavenger receptor that is required for internalization of oxidized LDL in ECs. It can be induced by LDLR-related protein 1 (LRP1) through increasing PPARγ transcriptional activity58. Depletion of LRP1 in ECs results in decreased oxidized LDL uptake. Mice with LRP1 depletion display improved lipid homeostasis. SR-BI, ATP binding cassette transporter G1 (ABCG1) and A1 are the main transporters for HDL. Recent studies have demonstrated that SR-BI and ABCG1 are involved in HDL/Apo-AI uptake and transcytosis in aortic ECs59. Knockdown of SR-BI blocks HDL internalization in human brain microvascular ECs60. However, the physiological role of SR-BI in endothelial transcytosis of HDL requires further characterization since SR-BI knockout does not alter the transfer of HDL-derived lipids into ECs of mouse liver61. Interestingly, SR-BI and ABCA1, but not ABCG1, are expressed in lymphatic ECs, a key player responsible for transporting cholesterol from subcutaneous tissues back to plasma and liver during reverse cholesterol transport. Lim et al62 performed studies suggesting that SR-BI but not ABCA1 is responsible for HDL uptake in lymphatic ECs. The transport of HDL to lymph nodes by lymphatic vessels is impaired in SR-BI knockout mice, resulting in cholesterol accumulation in the skin, strongly suggesting that lymphatic ECs play a critical role in HDL transport across endothelial layer and reverse cholesterol transport.
Currently the understanding of lipoprotein transport across the endothelium is still very limited, mainly because of the lack of good tools to visualize lipid transport directly. Fluorescently labeled lipids have been widely used to image lipids in live cells. However, the modification of lipids (i.e. BODIPY) might change the biochemical properties of the tagged lipids. Recently progress has been made to directly visualize HDL uptake in ECs in vitro and in vivo63. The further development of high-resolution imaging techniques for lipids in cells and tissues will surely expedite our understanding of the molecular machinery for lipid transport across the endothelial layer of blood vessels.
1.2. Transport of LPL
A group of lipases, including lipoprotein lipase (LPL), hepatic lipase (HL) and endothelial lipase (EL), are required for lipoprotein metabolism. They bind to heparin and are anchored to the cellular surface to hydrolyze TG and phospholipids within circulating lipoproteins. These lipases are expressed differentially in many tissues and have unique preferences for lipoprotein substrates64. EL mainly hydrolyzes phospholipids and is involved in HDL metabolism. HL hydrolyses both phospholipid and TG and is required for the metabolism of HDL, apolipoprotein (apo) B and LDL. LPL is the main lipase for TG and is responsible for the hydrolysis of VLDL and chylomicrons. LPL, secreted by adipocytes and myocytes, needs to be transported across the EC layer into capillary lumen for TG hydrolysis. A glycosylphosphatidyl-inositol-anchored protein called GPIHBP1 can bind LPL and is required for vesicle-mediated trans-endothelial transport of LPL and lipolysis of chylomicrons65, 66. PPARγ is the transcription factor required for GPIHBP1 induction in ECs. Mice with PPARγ knockout display decreased expression of GPIHBP1 in ECs, inhibited LPL activity and TG and FA metabolism53. A deficiency of GPIHBP1, due to missense mutations or blocked by its endogenous autoantibodies, prevents LPL from reaching the capillary lumen, resulting in low LPL plasma level and severe hypertriglyceridemia in patients (chylomicronemia, defined as a TG level >11.3 mM or 1000 mg/dL)67–74. Cell culture studies indicate that this GPIHBP1-dependent LPL movement across endothelial layer is a caveolin-1 independent process. However, additional studies are required for better understanding of the molecular mechanisms by which GPIHBP1 facilitates the transfer of LPL across endothelial layer. Interestingly, recent data suggest that GPIHBP1 can also stabilize LPL activation by blocking angiopoietin-like protein 4-catalyzed unfolding of LPL’s hydrolase domain75.
1.3. Transport of glucose
Glucose transport is regulated by adjusting local blood flow and trans-endothelial access, allowing the eventual delivery of glucose to metabolic cells. Vasodilation is induced by insulin through nitric oxide-dependent mechanisms in skeletal muscle, which contributes to both insulin sensitivity and responsiveness in non-obese humans76, 77. This endothelium-dependent vasodilation is impaired by elevated plasma FFAs78. Surprisingly, recent evidence indicates that trans-endothelial transport of glucose is a dynamically regulated process. Glucose uptake occurs primarily by facilitated diffusion, an energy-independent process that uses glucose transporters to move glucose across the cellular membrane79. In ECs, the main glucose transporter is GLUT1. It has been generally accepted that the endothelium does not play a rate-limiting role in glucose metabolism, due to the incapability of ECs to downregulate the rate of glucose transport when exposed to high glucose levels80. However, this concept has been challenged by recent studies. Prolonged exposure of ECs to high glucose decreases the rate of glucose transport in brain and heart ECs as a consequence of decreased GLUT1 expression81, 82. Patients with genetic GLUT1 deficiency syndrome display abnormalities attributed to decreased trans-endothelial glucose delivery to the brain83, 84. GLUT1 is a HIF1α-responsive gene in ECs85. HIF1α depletion in ECs decreases glucose uptake and overexpressed GLUT1 can inhibit this decrease. Importantly, Endothelial-specific HIF1α knockout mice display decreased CSF/blood glucose ratios85, which is a diagnostic criterion for clinical GLUT1 deficiency syndrome. These observations raise the intriguing possibility that the endothelium plays a critical role in regulating systemic glucose metabolism by controlling glucose transport across the endothelial barrier. Further investigation of the role of GLUT1 in ECs with emerging genetic tools will be vitally important for understanding the precise contributions of endothelial glucose transport in metabolic processes.
In insulin-sensitive tissues, glucose uptake is stimulated via insulin-dependent activation of its receptor (IR) and insulin receptor substrates (IRS), leading to translocation of glucose transporters86. In ECs, insulin signaling activates two major pathways, phosphoinositide 3 kinase (PI3K)-Akt and RAS-mitogen activated protein kinase, to regulate vasoreactivity (reviewed by Kubota T et al87). In vitro studies have shown divergent results about whether insulin signaling regulates glucose transport and metabolism in microvascular ECs. Insulin signaling does not regulate glucose transport in human micro- and macrovascular endothelial cells88 and bovine brain and retinal ECs89, 90, whereas glucose transport and glycogen synthesis are increased by insulin in bovine ECs isolated from adipose tissue or retinas or rabbit ECs91–93. Recent in vivo studies demonstrate that mice with endothelial depletion of IR display no changes in fed and fasting blood glucose levels94, which suggests that glucose uptake of ECs is unlikely regulated by insulin signaling.
1.4. Transport of hormones
Multiple pathways have been identified for the transport of hormones across the endothelial barrier. Compared to transcytosis, paracellular transport is a more passive mechanism for hormone transport. During inflammation, this passage is increased specifically in venules by opening junctions, which can be inhibited by hormones such as ghrelin95. Adiponectin can also block endothelial hyperpermeability induced by angiotensin II or TNFα96. In addition, studies indicate that transcytosis is required for trans-endothelial transport of hormones. For example, leptin is transported via transcytosis across capillary ECs of gastric intestinal tract or blood brain barrier97, 98. Insulin is one of the best-studied hormones that is transported across the endothelial barrier through transcytosis, which we will discuss here as an example of how ECs regulate the transcytosis of hormones.
Trans-endothelial transport of insulin is a rate-limiting step in insulin-induced glucose uptake by skeletal muscle87. Increased insulin action kinetics are observed in liver compared with skeletal muscle, suggesting that the fenestrated endothelium of liver capillaries allows free passage of insulin while continuous capillaries of skeletal muscle need an insulin transport mechanism. In ECs, insulin binds avidly to IR and also, at high concentrations, to insulin-like growth factor 1 receptor (IGF1R). Receptor-mediated transport of insulin is supported by in vitro studies indicating that transport of insulin across ECs is inhibited by blocking antibodies to IR or IGF1R99. Earlier studies with endothelial specific knockout mice of IR or IGF1R, generated by Tie2-Cre-mediated depletion, failed to show differences in plasma insulin levels and glucose or insulin tolerance tests94, 100, 101. However, recent studies using different genetic approaches have begun to unveil a pivotal role for receptor-mediated insulin transport in systemic glucose homeostasis. In an endothelial IR knockout mouse model generated by Cdh5-Cre-mediated depletion, the depletion of IR in ECs inhibits the delivery of insulin to target tissues where continuous endothelium barriers exist, such as skeletal muscle, brown fat, brain102. This inhibition results in impairment in glucose tolerance, brain-controlled appetite responses, and mild obesity. Intracellular insulin signaling mediators including PI3K and eNOS are required for insulin transport across ECs, indicated by their inhibitor studies103. EC-specific knockout mice of IRS-2 display similar metabolic defects including glucose intolerance and impaired insulin action on target tissues104.
Fcγ receptor IIB (FcγRIIB), the C-reactive protein (CRP) receptor, has recently been demonstrated to block endothelial insulin transport by antagonizing eNOS activity105. Mice with endothelial-specific FcγRIIB depletion display improved glucose homeostasis in response to CRP. Nitric oxide can promote insulin uptake and across through ECs via nitrosylating PTP1B, an inhibitory phosphatase of insulin signaling106. In addition, both caveolae- and clathrin-coated vesicles have been suggested to mediate the uptake and transcytosis of insulin across endothelial layer107, 108. However, it is still not clear how these two pathways are regulated and whether they are regulated differentially in the vessel beds of different tissues. Although insulin transport across endothelial layer is tightly regulated through receptor-dependent processes, at very high levels insulin transport may occur via non-saturable processes such as passive diffusion109, 110. However, it is unclear how this happens, and how much this independent diffusion contributes to the global regulation of insulin and glucose metabolism. Although the trans-endothelial transport of insulin remains to be further studied, available evidence has underscored the role of endothelial insulin signaling in the maintenance of whole body metabolic homeostasis. Targeting endothelial insulin transport could potentially become a therapeutic target for treating insulin resistance and T2DM.
Recent observations, especially with genetic approaches (Table 1), suggest that ECs play a critical role in controlling the transcytosis of FAs, lipoprotein, glucose and hormones such as insulin. However, our understanding is still very preliminary and a lot of questions remain to be answered. For example, is intact TG transcytosed through ECs? How do FABP, FATP and CD36 coordinate together for FA transport? Does the transport of these macromolecules take place bidirectionally? In addition, amino acids such as arginine and citrulline play an important role in metabolic homeostasis. The transport of amino acids across the endothelial monolayer also requires complicated transport systems, which has been reviewed elsewhere (i.e. Mann GE et al111) and will not be discussed in this review. Yet we also know that paracellular transport likely contributes to the increased transport of macromolecules in response to vascular inflammation associated diabetes112. In addition, the crosstalk of transcellular and paracellular routes exists and consequently alters endothelial barrier function113. The exact roles of paracellular transport of macromolecules in metabolic processes are still poorly understood and merit further investigation.
Table 1.
A summary of the genetic approaches used for evaluating the roles of trans-endothelial transport of macromolecules in metabolic homeostasis.
| Cargo | Model System | Signaling Effects and Metabolic Phenotype | Ref. |
|---|---|---|---|
| FA | |||
| HFD-fed PparγEC−/− mice | Decreased CD36 and FABP4 induction, increased VLDL and FFA levels, reduced lipid accumulation, improved glucose responses | Kanda et al.(2009)53, Goto et al. (2013)52 | |
| LDL | Ldlr−/− mice | LDLR-dependent transcytosis of LDL across BBB, CNS drug delivery | Molino et al. (2017)57 |
| oxLDL | HFD-fed Lrp1EC−/− mice | Decreased expression of CD36, oxLDL uptake by ECs, improved lipid and glucose responses | Mao et al. (2017)58 |
| HDL | SR-BI−/− mice | Compromised transport of HDL through lymphatic vessels, accumulation of cholesterol and HDL in plasma | Lim et al. (2013)62 |
| LPL | |||
| Human loss-of-function mutations of GPIHBP1, GPIHBP1 autoantibodies in patients with chylomicronemia | Familial chylomicronemia, low plasma level of LPL | Beigneux et al.(2009,2015,2017)67–69
Olivecrona et al.(2010)71 Plengpanich et al.(2014)72 Rios et al.(2012)73 Surendran et al.(2012)74 Buonuomo et al.(2015)70 |
|
| Glucose | HIF1αEC−/− mice | Decreased GLUT1 induction, glucose uptake and CSF/blood glucose ratios | Huang et al. (2012)85 |
| Leptin | Normal and STZ-diabetic mice | Glucose and insulin increase leptin across BBB of normal but not STZ-diabetic mice | Kastin AJ et al. (2001)98 |
| Insulin | |||
| Mice of FcγRIIBEC−/−, FcγRIIBEC−/−;TG-CRP, eNOS (S1176D);TG-CRP | The CRP/FcγRIIB axis inhibits eNOS activity. Depleted FcγRIIB or eNOS constitutive activation (S1176D knock-in) improves CRP-induced glucose intolerance and insulin resistance. | Tanigaki K et al. (2016)105 |
iEC, inducible EC; STZ, streptozotocin; CSF, cerebrospinal fluid; TG, transgenic.
2. Endothelial cell-secreted factors regulating metabolic signaling
ECs play a crucial role in survival, angiogenesis and inflammation by secreting regulatory cytokines. Recent studies, including numerous genetic analyses (Table 2), indicate that ECs can also secrete factors that act as paracrine or endocrine regulators for metabolic cells/tissues and are actively involved in the maintenance of metabolic homeostasis. We group these EC-secreted factors into four different categories based on their biochemical characteristics and cellular functionality: nitric oxide (NO), insulinotropic factors, growth factors and enzymes.
Table 2.
A summary of representative genetic approaches used for evaluating the roles of EC-secreted factors in metabolic homeostasis.
| Secreted Factor | Model System | Signaling Effect and Metabolic Phenotype | Ref. |
|---|---|---|---|
| NO | |||
| e/n/iNOS−/− mice | Visceral obesity, hypertriglyceridemia, glucose intolerance | Nakata et al.(2008)123 | |
| ECM proteins | |||
| NOD mice | Degradation of islet basement membrane at onset of insulitis and diabetes | Irving-Rodgers et al.(2008)125 | |
| TSP-1 | TSP-1−/− mice, islet transplant-ation from WT/TSP-1−/− mice | Decreased insulin secretion and glucose tolerance, TSP-1 derived from ECs is important for insulin secretion | Olerud et al.(2011)126 |
| HGF | CCl4-induced mouse liver injury model, injected with VEGF agonist (Flt/KDR selective) | VEGF/Flt1 induces HGF and reduces liver damage | LeCouter et al.(2003)127 |
| PDGF-CC | Vegfr2iEC−/− and Pdgf-c−/−mice, Adenoviral PDGF-C | Impaired WAT-beige transition, adenoviral PDGF-C improves glucose tolerance in HFD-obese mice. | Seki et al.(2016)128 |
| EL | |||
| EL gene variants | Strong association with HDL level | Ma et al.(2003)130, Edmondson et al. (2009)132, Singaraja et al. (2013)133 |
iEC, inducible EC; epi, epithelial; NOD, non-obese diabetic; FAO, fatty acid oxidation
2.1. Nitric oxide
NO is an essential determinant of cardiovascular homeostasis through its role in modulating vasomotor tone, maintaining integrity of the vasculature and protection from cellular injury. NO is predominantly synthesized by endothelial NO synthase (eNOS) in ECs. eNOS can be activated by fluid shear stress and numerous other vasoactive agonists through protein phosphorylation, protein-protein interactions, Ca2+ signaling and subcellular translocation of proteins. Interestingly, circulating lipoproteins such as HDL can also activate eNOS via a SR-BI-mediated caveolae signaling complex, whereas oxidized LDL inhibits eNOS by disrupting caveolae structure134, 135. In liver sinusoidal ECs, insulin significantly increases eNOS activity and NO production114. ECs with triple knockout of the three FoxO proteins have increased insulin-stimulated NO production, which in turn promotes tyrosine nitration of hepatic IR114, 136. Mice lacking FoxO proteins develop hepatic insulin resistance in response to high-fat diet, which can be inhibited by eNOS inhibitor L-NAME114. However, insulin sensitivity in skeletal muscle and adipose tissue is not affected in EC-specific FoxO knockout mice. Endothelial Shc homology 2–containing inositol phosphatase 2 (SHIP2) is another regulator of NO generation in response to insulin and shear stress115, 137. Interestingly, EC-specific SHIP2 knockout mice have impaired NO bioavailability and vasodilation, and develop insulin resistance in skeletal muscle and adipose tissue115. In addition, NO also increases food intake by regulating leptin and serotonergic signaling pathways in the brain138, 139. In the pancreas, NO affects both exocrine and endocrine functions140, although the physiologic role of NO in insulin secretion in beta islets remains incompletely understood (summarized by Broniowska et al141). eNOS has been suggested to be involved in obesity and metabolism through multiple processes, such as promoting fat oxidation, hepatic insulin sensitizing substance and adipocyte browning, decreasing gluconeogenesis etc. (Sansbury BE et al140). Genetic studies indicate a strong association of eNOS mutations with insulin resistance, diabetes, hypertriglyceridemia and HDL decrease116–118. However, studies with eNOS, eNOS/nNOS or eNOS/nNOS/iNOS triple knockout mice suggest a protective role of NO in obesity, insulin resistance, hyperlipidemia, hypertension and other metabolic diseases119–123. NO exerts multifunctional roles through cGMP-dependent protein kinase-mediated protein phosphorylation, S-nitrosylation of cysteine residues or nitration of tyrosine residues. Therefore, the identification of specific mediators for NO in hepatocytes, beta islets or neuronal cells may provide safer therapeutic tools for inhibiting hepatic insulin resistance, diabetes and other metabolic disorders.
2.2. Insulinotropic factors
Pancreatic islets in the adult are one of the most vascularized organs, containing fenestrated capillaries that constitute 8~10% of the islet volume and are organized into a glomerular-like network. While the high vascular density and endothelial function are maintained by VEGF secreted from beta cells142–144, recent studies indicate that EC-secreted factors and extracellular matrix proteins also play an important role in regulating insulin secretion from beta cells.
Extracellular matrix (ECM) proteins are the non-cellular component within all tissues and organs. Islet ECM contains classical basement membrane components such as collagens, laminins, heparin sulfate proteoglycans and hyaluronan125, 145. Intra-islet ECs synthesize ECM components to preserve the function and integrity of blood vessels. Interestingly, beta cells also depend on the ECM components from ECs124. These ECM proteins not only provide structural support for beta cells, they also promote beta cell growth, differentiation, survival, migration and potentiate insulin secretion. For example, collagen IV, fibronectin and heparin sulfate are crucial for beta cell migration, growth, survival and maturation124, 146. Collagen IV can also potentiate insulin secretion by interacting with α1β1 integrins on beta cells. Laminins and fibronectin was reported to promote insulin gene expression and beta cell proliferation124, 146.
Thrombospondin-1 (TSP-1) is an endogenous inhibitor of angiogenesis and tumor growth147. In pancreatic islets, TSP-1 is primarily expressed in islet ECs126. TSP-1-deficient mice demonstrate glucose intolerance and a defect in glucose-stimulated insulin secretion126. The islet transplantation experiments indicate that TSP-1 derived from ECs is important for beta cell function126. Although the underlying mechanisms are not well understood, recent studies suggest that TSP-1 promotes beta cell function by activating the TGFβ−1 pathway and protecting beta cells from lipotoxicity by activating PERK and NRF2148, 149. Endothelin-1 (ET-1), a potent vasoconstrictor, is expressed in both ECs and islet endocrine cells150. In vitro studies with inhibitors indicate that ET-1 can stimulate insulin secretion directly through its receptor ET(A) and the PKC signaling pathway151, 152.
2.3. Growth factors
A number of growth factors are secreted from ECs within metabolic organ/tissues. Among them include hepatocyte growth factor (HGF), which can be secreted from islet ECs153. HGF can stimulate beta cell proliferation and insulin production154. Beta cell-specific depletion of HGF receptor c-met results in impaired insulin secretion and glucose homeostasis155, 156. The secretion of HGF from islet ECs can be stimulated by VEGF-A and insulin. Islet mass is rather stable during adult life. However, it increases in response to obesity, insulin resistance or pregnancy. Interestingly, vascular growth is closely associated with beta cell proliferation during pregnancy, which is likely due to the induction of insulin and VEGF in beta cells in response to placental lactogens and prolactin153. Insulin and VEGF can stimulate HGF production in islet ECs through paracrine effects, which further enhances beta cell proliferation. The crosstalk between islet beta cells and ECs forms a feed forward signaling loop to coordinate the growth of beta cells and vasculature. Similarly, this crosstalk also exist for hepatocytes and liver sinusoidal ECs. In the liver sinusoidal ECs, HGF can be also induced by VEGF-A that is released from hepatocytes157. The secreted HGF further promotes hepatocyte proliferation127.
Connective tissue growth factor (CTGF/CCN2) is another growth factor secreted from islet ECs158. CTGF null embryos display defects in islet morphogenesis, beta cell proliferation and decreased insulin (+) cells159. Mice with EC-specific depletion of CTGF display impaired beta cell proliferation, while overexpressed CTGF enhances beta cell proliferation160–162. In addition, CTGF stimulates beta cell maturation, regeneration and modulates responses upon streptozotocin-induced diabetes. In adipose tissue, VEGF stimulates the production PDGF-CC via VEGFR2 activation in ECs128. Mice with depleted endothelial VEGFR2, knockout Pdgf-c gene or pharmacological blockade of PDGFR-β demonstrate impaired WAT-beige transition upon cold or adrenergic activation. These observations raise the provocative possibility that PDGF-CC secreted from ECs promotes WAT browning. This model proposes that the local crosstalk between metabolic cells and ECs, a critical process for metabolic homeostasis, is mediated by paracrine factors such as growth factors. As more EC-derived growth factors are identified to participate in the crosstalk of ECs and their neighboring metabolic cells, they provide new targets for the treatment of diabetes and other metabolic disorders.
2.4. The special case of endothelial lipase
Endothelial lipase (EL) shares substantial homology with LPL and HL and is the only known lipase expressed predominantly by ECs. EL is also secreted by smooth muscle cells, cardiomyocytes and macrophages but far less abundantly. In 1999, two laboratories independently cloned the EL gene from cultured ECs163, 164. In vitro studies demonstrate that EL can hydrolyze phospholipids in chylomicrons, very low density lipoprotein, intermediate density lipoprotein, LDL and HDL. However, opposite results were reported regarding the changes of LDL-C concentrations in EL knockout mice129, 130. The majority of studies in cultured cells and animals support that the lipase activity of EL is more specific to HDL than other lipoproteins165. However, genetic studies show that few SNPs of EL gene are related to changes in plasma HDL-C concentrations. Conflicting data have also been obtained from studies with human patients regarding the association of EL and HDL concentrations132, 133, 166, 167. In ECs, EL can be induced by inflammatory cytokines (i.e. IL-1β and TNFα) and biomechanical forces168, 169. Human plasma EL concentrations are strongly associated with inflammatory markers CRP and interleukin-6 (IL-6)170. Mice with EL depletion display severe heart failure due to decreased cardiac uptake of FAs131, suggesting the significant role of EL in FA metabolism. However, the specific roles of endothelial or cardiac EL remain to be further characterized.
Emerging data suggest that ECs communicate with neighboring cells within the microenvironment of metabolic organs through secreting regulatory factors (Fig. 2). Dysregulation of secretory pathways of ECs may play vital roles in the progression of metabolic disorders. Better understanding of these EC-specific secretory pathways may provide easy-to-access therapeutic tools for treating diabetes, pancreatic disorders and cardiomyopathy.
Figure 2.
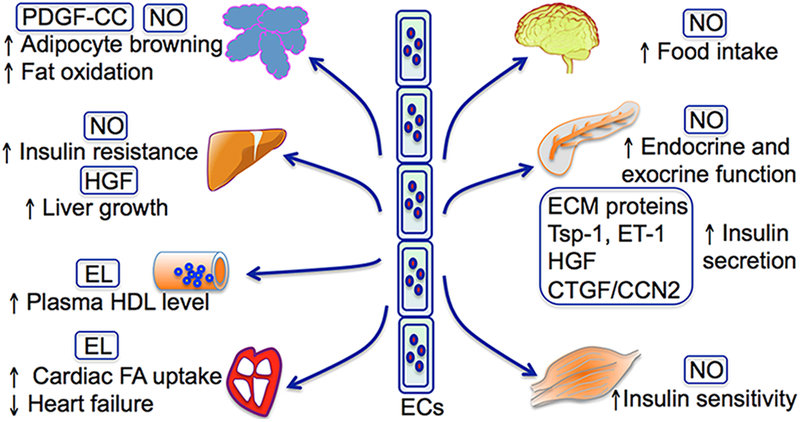
Metabolic regulation by endothelial cell-secreted factors.
3. Endothelial dedifferentiation in response to metabolic dyshomeostasis
Endothelial differentiation from pluripotent progenitor cells is a crucial process for embryonic development171. Liver sinusoidal ECs experience a differentiation process during the gestational stage that is tightly regulated by EC-derived VEGF and hepatoblast-derived humoral factors172. Following differentiation, liver sinusoidal endothelium becomes highly permeable due to the formation of fenestrae between liver sinusoidal ECs (LSECs) and the disappearance of cellular basement membrane. In pathologic and in vitro conditions, matured LSECs are dedifferentiated, demonstrating the loss of fenestrae and synthesis of basement membrane173. This process is called capillarization and has been recognized as an early event in chronic liver injury in animal models and patients174, 175. In cirrhotic liver, LSEC capillarization is induced, which in turn activates hepatic stellate cells and macrophages, leading to liver fibrosis176. Differentiated LSECs are considered gatekeepers of fibrosis and progression from a simple steatosis to the early stage of nonalcoholic steatohepatitis. In rat models, the restoration of LSEC differentiation results in regression of liver fibrosis, suggesting that differentiation of LSECs may play a role in fibrosis regression177. However, more human studies will be needed to support this hypothesis. Pseudocapillarization is similar to capillarization in morphology, but refers to phenotype changes of LSECs in aged animals and humans178, 179. Fenestrae structures of LSECs allow passage of lipoproteins and chylomicron remnants. Reduced fenestrae numbers during pseudocapillarization impairs lipoprotein metabolism, contributing to dyslipidemia and atherosclerosis associated with old age180, 181.
4. Endothelial dysfunction in response to metabolic events
Metabolic organs such as liver, islet, adipose tissue, cardiac and skeletal muscle contain extensive capillary networks. Endothelial dysfunction is observed during the development of metabolic syndrome, diabetes and insulin resistance182. NO is a major player in endothelial function. NO regulates organ blood flow and systemic blood pressure by acting as a potent vasodilator. NO bioavailability is diminished in obese and diabetic animals and in human patients183, 184. The expression and activity of eNOS are decreased and uncoupling of eNOS is induced in metabolic diseases (reviewed by Sansbury BE et al140), which may contribute to the disruption of metabolic homeostasis. Studies also suggest that endothelial dysfunction may contribute to the dysregulation of metabolic homeostasis by modulating vessel tone, NO bioavailability and inflammation.
In skeletal muscle, insulin stimulates vasodilatory effects through IR-dependent eNOS activation in ECs185. This vasodilatory effect of insulin is inhibited by inflammatory factors, FFAs, oxidative stress and adipokines186–190. Insulin, as well as other hormones such as GLP-1 and adiponectin, carbohydrate-containing meals, and exercise can all increase microvascular perfusion in skeletal muscle even in the absence of total blood flow increase, which precedes and correlates with the rise in muscle glucose uptake (reviewed by Sansbury BE et al140). The increase in microvascular blood volume of skeletal muscle and heart is blunted in obesity and insulin resistance. In the heart, the inhibition of endothelium-derived NO alters mitochondrial oxygen consumption and substrate utilization by the heart191, 192.
In islets, blood flow is impaired by the depletion of IRS-2 in ECs193. However, treatment with the angiotensin-converting enzyme inhibitor enalapril maleate, which increases islet blood flow, can restore insulin secretion by islet cells. In addition, the treatment of islets with conditioned media from dysfunctional islet ECs decreases glucose-stimulated insulin secretion and total insulin content194. These data suggest that ECs are critical for glucose uptake in muscle and insulin secretion from islets by regulating blood flow. Vasoactive drugs have been shown to enhance islet and muscle blood flow and promote glucose homeostasis195. Further translational studies with these vasoactive drugs could identify therapeutic applications in islet disorders.
In adipose tissue, endothelial dysfunction is found in obese mice and human subjects exhibiting inflammatory responses196. Sun et al recently show that miRNA-181b, a microRNA that inhibits endothelial inflammatory responses, is repressed in diabetic ECs197. Interestingly, the systemic delivery of miR-181b can reduce inflammation and EC dysfunction in white adipose tissue and improve glucose tolerance and insulin sensitivity in diet-induced obese mice. These observations suggest that reagents that inhibit endothelial dysfunction might be effective for treating obesity-induced insulin resistance.
LSECs are the main source of NO in the normal liver. Fluid shear stress-dependent eNOS activation promotes NO production198. Flow-responsive transcription factor Kruppel-like factor 2 (KLF2) is responsible for the induction of NO and downregulation of vasoconstrictive protein endothelin-1199. Endothelial dysfunction occurs early in chronic liver disease, precedes fibrosis and inflammation and persists in severe cirrhosis200, 201. In cirrhotic liver, capillarization of LSECs is associated with endothelial dysfunction, leading to increased intrahepatic vascular resistance. Statins improve LSEC function, leading to improvements in liver fibrosis and portal pressure decrease202. The roles of endothelial dysfunction in chronic liver diseases have been summarized in many reviews173, 203.
5. Crosstalk between metabolic events and endothelial-dependent vascular growth
Endothelial-dependent processes of vascular growth, including vasculogenesis and angiogenesis, play a key role in cardiovascular development during embryogenesis. Angiogenesis, a remodeling process of an established capillary network, is also required for pathological conditions such as wound healing. Interestingly, emerging data suggest that endothelial growth is also involved in regulating metabolic homeostasis. For example, a positive correlation exists between skeletal muscle capillarity and insulin sensitivity204, 205. Skeletal microvasculature recruitment is recognized as an important determinant of muscle glucose uptake and insulin sensitivity206. Endothelial-specific depletion of FoxO1/3/4 results in increased skeletal muscle microvascular density207. The expansion of the capillary network leads to increased insulin transport across the capillary endothelium, which may partially explains why EC-specific FoxO depletion protects mice from HFD-induced insulin resistance. In the endocrine pancreas, angiogenesis plays an important role in maintaining islet structure and function through regulation of local blood flow. The survival and optimal engraftment of transplanted islets in type 1 diabetes is dependent on the vascular density and blood flow of islet microvessels208. Pro-angiogenic factors (i.e. VEGF, GLP-1) play crucial roles in angiogenesis after islet transplantation209, 210.
In adipose tissue, angiogenesis is well recognized during adipose tissue expansion (reviewed by Cao Y211, 212). The crosstalk of adipocytes and ECs is a key event during the coordination of vessel growth and fat expansion. A group of angiogenic factors are secreted from adipocytes to stimulate new vessel formation, such as VEGF, FGF-2, HGF, TGF-β, leptin, resistin and visfatin211, 212. In addition, angiogenesis Inhibitors are shown to block fat mass expansion in mice213–215.
Hepatic angiogenesis is closely linked with liver fibrosis216. A vicious circle between fibrosis and pathological angiogenesis is likely to occur, where parenchymal hypoxia activates HIF-dependent liver revascularization and wound healing processes in the liver. However, pathologic angiogenesis provides immature and highly permeable vessels that fail to rescue liver hypoxia, which further promotes the development of liver fibrosis217. Consonant with this model, liver-specific knockout of HIF1α reduces liver fibrosis218. In addition, anti-angiogenic therapy is highly effective in reducing liver fibrogenic progression (summarized by Bocca C. et al216).
Taken together, these studies suggest that targeting angiogenesis can be a potential therapeutic intervention for obesity through blocking adipose tissue expansion or for chronic liver disease through reducing liver fibrosis. However, concerns exist since angiogenic blockers have detrimental effects on skeletal muscle insulin and glucose delivery, beta cell function and diabetes-related wound healing process. Dissecting specific angiogenic pathways for individual metabolic processes is required for safer drug targeting strategies.
Summary and perspectives
Our understanding of ECs, which were initially viewed as a thin layer of inactive cells lining in the inner surface of the circulatory system, has progressed significantly. It has been well recognized that ECs play active roles in a variety of pathological and physiological processes. However, most research has focused on the regulatory roles of endothelial dysfunction in atherosclerosis, coronary artery disease, stroke, hypertension, angiogenesis-related diseases and diabetes-associated vascular complications. Based on the emerging data we have discussed in this review, we can begin to appreciate that the crosstalk between ECs and metabolic tissues/cells is bidirectional. The vascular endothelium possesses underappreciated roles in maintaining systemic metabolic homeostasis through controlling the trans-endothelial transport and secretion of metabolic regulators, or adjusting its own functional status such as differentiation, dysfunction and growth, which indirectly affect the function of metabolic cells/tissues. Dysregulation of endothelial processes results in metabolic disturbance, which in turn exacerbates endothelial and vascular dysfunction and likely acts as a feedback loop for the development of metabolic disorders. Better understanding of these dynamically regulated processes may open up new avenues of the therapeutic approaches for metabolic disorders based on manipulation of these endothelial specific signaling events.
This review focuses on recent progress regarding the endothelial regulation of metabolic processes at least partially through local interplay of microvascular ECs and metabolic cells within their residing organs. Accumulated in vivo evidence gathered from EC-specific knockout mouse models reveals a causal relationship of malfunctional endothelial actions in the disruption of systemic metabolic homeostasis. We speculate that microvascular ECs are the main endothelial cells responsible for crosstalk with metabolic cells/tissues, which is essential for both vascular and metabolic homeostasis. However, our knowledge of these functional roles of microvascular endothelium remains limited. Technical hurdles to the study of microvascular endothelial cells in vivo still exist, such as the difficulty to perform quantitative analysis and direct in vivo imaging on microvasular endothelium. In addition, the specific roles of microvascular vs macrovascular dysregulation in the development of metabolic disorders needs to be further characterized.
During the study of the crosstalk between microvascular ECs and metabolic cells/tissues, one important issue we need be aware of is the phenotypic heterogeneity of ECs. Difference exists between arteries, veins and capillaries, as well as within a variety of vessel beds of specific organs219, 220. In addition, endothelial heterogeneity might even exist between distinct regions of the same organ. For example, ~84% of vascular receptors of the islet ECs are unique to the islet and are not detected in ECs in the surrounding exocrine pancreatic tissue221. It makes islet ECs unique in response to glucose-dependent regulation of nitric oxide and other angiogenic or angiostatic factors such as VEGF, PDGF, endostatin etc (reviewed by Olsson R et al208). It raises needs to identify endothelial factors specific for microvascular ECs (arteriole ECs, capillary ECs or venule ECs) in different metabolic organs such as skeletal muscle, adipose tissue, pancreas and liver. In addition, cell culture studies needs to be performed with microvascular ECs isolated specifically from the designated vessels beds. Given that the in vivo microenvironment is an important determinant for metabolic phenotype, cell culture studies can be only used as supportive evidence and needs to be confirmed with in vivo studies. These studies will help us better understand the unique roles of microvascular ECs within individual metabolic organs. More importantly, it will provide new insights into designing safer therapeutic strategies and specific drug delivery. In addition, macrovascular function might also impact metabolic processes through regulating vascular tone, endocrine pathways or other unknown mechanisms. It suggests the possible involvement of the interaction of ECs with other cells such as smooth muscle cells, which might become another interesting research area for understanding the etiology of metabolic disorders.
It is generally recognized that ECs rely mainly on glycolysis as an energy source during their quiescent and active status such as sprouting angiogenesis222, 223. Metabolic stress such as hyperglycemia can inhibit endothelial glycolytic flux, and glycolytic intermediates are diverted into polyol pathway, hexosamine biosynthesis pathway and methyglyoxal production224. It remains unclear how endothelial metabolism is coordinated with whole body metabolic homeostasis. Further studies can be directed to test whether specific connections exist between these two processes. If specific dysregulation of endothelial metabolic signaling pathways contributes to certain aspects of disrupted whole body glucose or lipid homeostasis, targeting endothelial metabolic process could become another potential therapeutic targeting strategy for treating metabolic disorders.
Acknowledgments
SOURCES OF FUNDING
This work was supported by NIH R01s HL112890 and HL061656 (to X.P.) and HL122736 (to X.L.).
Non-Standard Abbreviations and Acronyms
- 3-HIB
3-hydroxy-isobutyrate
- ABCA1
adenosine triphosphate binding cassette transporter A1
- ABCG1
adenosine triphosphate binding cassette transporter G1
- APLNR
apelin receptor
- Apo
apolipoprotein
- ATP
adenosine triphosphate
- BCAA
branched-chain amino acid
- BODIPY
boron dipyrromethene
- CRP
C-reactive protein
- CTGF
connective tissue growth factor
- EC
endothelial cell
- ECM
extracellular matrix
- EL
endothelial lipase
- eNOS
endothelial nitric oxide synthase
- ET-1
endothelin-1
- FA
fatty acid
- FABP
fatty acid binding protein
- FATP
fatty acid transport protein
- FcγRIIB
Fcγ receptor IIB
- FFA
free fatty acid
- FoxO
Forkhead box O
- GLP-1
glucagon-like peptide 1
- GLUT
glucose transporter
- GPIHBP1
glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1
- HDL
high density lipoprotein
- HFD
high-fat diet
- HGF
hepatocyte growth factor
- HIF1
hypoxia-inducible factor 1
- IGF1R
insulin-like growth factor 1 receptor
- IL-6
interleukin-6
- IR
insulin receptor
- IRS
insulin receptor substrate
- LCFA
long-chain fatty acid
- LDL
low density lipoprotein
- LDLR
low density lipoprotein receptor
- HL
hepatic lipase
- LPL
lipoprotein lipase
- LRP1
low density lipoprotein receptor-related protein 1
- LSEC
liver sinusoidal endothelial cell
- NO
nitric oxide
- NRF2
nuclear factor erythroid 2–related factor 2
- PDGF
platelet-derived growth factor
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- PI3K
phosphoinositide 3 kinase
- PPAR
peroxisome proliferator-activated receptor
- PTP1B
protein tyrosine phosphatase 1B
- SHIP2
Shc homology 2–containing inositol phosphatase 2
- SR-BI
scavenger receptor class B type I
- T2DM
type 2 diabetes mellitus
- TNF
tumor necrosis factor
- TSP-1
thrombospondin-1
- TZD
thiazolidinedione
- VEGF
vascular endothelial growth factor
- VLDL
very low density lipoprotein
- WAT
white adipose tissue
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Levick JR. An introduction to cardiovascular physiology. 5th ed. London: Hodder Arnold; 2010. [Google Scholar]
- 2.Sherwood L and Cengage Learning (Firm). Human physiology: from cells to systems. 7th ed. Australia; United States: Brooks/Cole, Cengage Learning; 2010. [Google Scholar]
- 3.Tuma RF, Durán WN and Ley K. Microcirculation. 2nd ed. Amsterdam; Boston: Elsevier/Academic Press; 2008. [Google Scholar]
- 4.Granger DN and Senchenkova E. Inflammation and the microcirculation San Rafael (CA); 2010. [PubMed] [Google Scholar]
- 5.Simon SI, Sarantos MR, Green CE and Schaff UY. Leucocyte recruitment under fluid shear: mechanical and molecular regulation within the inflammatory synapse. Clin Exp Pharmacol Physiol. 2009;36:217–24. [DOI] [PubMed] [Google Scholar]
- 6.Virchow R Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr Rev. 1989;47:23–5. [DOI] [PubMed] [Google Scholar]
- 7.Ceriello A and Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004;24:816–23. [DOI] [PubMed] [Google Scholar]
- 8.Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, Wenzel P, Munzel T and Keaney JF Jr. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation. 2008;118:1347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higashi Y, Noma K, Yoshizumi M and Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411–8. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu M, Takei M, Ishii H and Sato Y. Glucose-stimulated insulin secretion: A newer perspective. J Diabetes Investig. 2013;4:511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohn AD, Summers SA, Birnbaum MJ and Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–8. [DOI] [PubMed] [Google Scholar]
- 12.Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR and Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–8. [DOI] [PubMed] [Google Scholar]
- 13.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414:1–18. [DOI] [PubMed] [Google Scholar]
- 14.Syed NA and Khandelwal RL. Reciprocal regulation of glycogen phosphorylase and glycogen synthase by insulin involving phosphatidylinositol-3 kinase and protein phosphatase-1 in HepG2 cells. Mol Cell Biochem. 2000;211:123–36. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Wang S, Lin Y, Xu W, Ye D, Xiong Y, Zhao S and Guan KL. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 2012;15:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen J, Rustad PI, Kolnes AJ and Lai YC. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol. 2011;2:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonen A, Han XX, Habets DD, Febbraio M, Glatz JF and Luiken JJ. A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin- and AICAR-stimulated fatty acid metabolism. Am J Physiol Endocrinol Metab. 2007;292:E1740–9. [DOI] [PubMed] [Google Scholar]
- 18.Freychet L, Rizkalla SW, Desplanque N, Basdevant A, Zirinis P, Tchobroutsky G and Slama G. Effect of intranasal glucagon on blood glucose levels in healthy subjects and hypoglycaemic patients with insulin-dependent diabetes. Lancet. 1988;1:1364–6. [DOI] [PubMed] [Google Scholar]
- 19.Frayn KN, Arner P and Yki-Jarvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem. 2006;42:89–103. [DOI] [PubMed] [Google Scholar]
- 20.Davis JF, Choi DL and Benoit SC. Insulin, leptin and reward. Trends Endocrinol Metab. 2010;21:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterson MJ and Horvath TL. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015;22:962–70. [DOI] [PubMed] [Google Scholar]
- 22.Yeo GS and Heisler LK. Unraveling the brain regulation of appetite: lessons from genetics. Nat Neurosci. 2012;15:1343–9. [DOI] [PubMed] [Google Scholar]
- 23.Hardie DG. Organismal carbohydrate and lipid homeostasis. Cold Spring Harb Perspect Biol. 2012;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Alexander RK and Lee CH. Lipid metabolites as metabolic messengers in inter-organ communication. Trends Endocrinol Metab. 2014;25:356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vyska K, Meyer W, Stremmel W, Notohamiprodjo G, Minami K, Machulla HJ, Gleichmann U, Meyer H and Korfer R. Fatty acid uptake in normal human myocardium. Circ Res. 1991;69:857–70. [DOI] [PubMed] [Google Scholar]
- 26.Antohe F, Popov D, Radulescu L, Simionescu N, Borchers T, Spener F and Simionescu M. Heart microvessels and aortic endothelial cells express the 15 kDa heart-type fatty acid-binding proteins. Eur J Cell Biol. 1998;76:102–9. [DOI] [PubMed] [Google Scholar]
- 27.Cechetto JD, Sadacharan SK, Berk PD and Gupta RS. Immunogold localization of mitochondrial aspartate aminotransferase in mitochondria and on the cell surface in normal rat tissues. Histol Histopathol. 2002;17:353–64. [DOI] [PubMed] [Google Scholar]
- 28.Elmasri H, Karaaslan C, Teper Y, Ghelfi E, Weng M, Ince TA, Kozakewich H, Bischoff J and Cataltepe S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009;23:3865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenwalt DE, Watt KW, Hasler T, Howard RJ and Patel S. Structural, functional, and antigenic differences between bovine heart endothelial CD36 and human platelet CD36. J Biol Chem. 1990;265:16296–9. [PubMed] [Google Scholar]
- 30.Masouye I, Hagens G, Van Kuppevelt TH, Madsen P, Saurat JH, Veerkamp JH, Pepper MS and Siegenthaler G. Endothelial cells of the human microvasculature express epidermal fatty acid-binding protein. Circ Res. 1997;81:297–303. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe K, Wakabayashi H, Veerkamp JH, Ono T and Suzuki T. Immunohistochemical distribution of heart-type fatty acid-binding protein immunoreactivity in normal human tissues and in acute myocardial infarct. J Pathol. 1993;170:59–65. [DOI] [PubMed] [Google Scholar]
- 32.Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone-Elander S, Claesson-Welsh L, Yla-Herttuala S, Lindahl P and Eriksson U. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–21. [DOI] [PubMed] [Google Scholar]
- 33.Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsater H, Scotney P, Nyqvist D, Samen E, Lu L, Stone-Elander S, Proietto J, Andrikopoulos S, Sjoholm A, Nash A and Eriksson U. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426–30. [DOI] [PubMed] [Google Scholar]
- 34.Bry M, Kivela R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, Carmeliet P, Lemstrom KB, Shibuya M, Yla-Herttuala S, Alhonen L, Mervaala E, Andersson LC, Knuuti J and Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–33. [DOI] [PubMed] [Google Scholar]
- 35.Poesen K, Lambrechts D, Van Damme P, Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen A, Raes K, Tjwa M, Eriksson U, Shibuya M, Nuydens R, Van Den Bosch L, Meert T, D’Hooge R, Sendtner M, Robberecht W and Carmeliet P. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J Neurosci. 2008;28:10451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, Hua J, Matsuo O, Fong GH, Ding H, Cao Y, Becker KG, Nash A, Heldin CH and Li X. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. 2008;118:913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS Jr., Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD and Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu Q, Forman DE, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL and Arany Z. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iso T, Maeda K, Hanaoka H, Suga T, Goto K, Syamsunarno MR, Hishiki T, Nagahata Y, Matsui H, Arai M, Yamaguchi A, Abumrad NA, Sano M, Suematsu M, Endo K, Hotamisligil GS and Kurabayashi M. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arterioscler Thromb Vasc Biol. 2013;33:2549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Binas B and Erol E. FABPs as determinants of myocellular and hepatic fuel metabolism. Mol Cell Biochem. 2007;299:75–84. [DOI] [PubMed] [Google Scholar]
- 41.Hwangbo C, Wu J, Papangeli I, Adachi T, Sharma B, Park S, Zhao L, Ju H, Go GW, Cui G, Inayathullah M, Job JK, Rajadas J, Kwei SL, Li MO, Morrison AR, Quertermous T, Mani A, Red-Horse K and Chun HJ. Endothelial APLNR regulates tissue fatty acid uptake and is essential for apelin’s glucose-lowering effects. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawane M, Kajiya K, Kidoya H, Takagi M, Muramatsu F and Takakura N. Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes. 2013;62:1970–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E and Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–8. [PubMed] [Google Scholar]
- 44.Harmon CM and Abumrad NA. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J Membr Biol. 1993;133:43–9. [DOI] [PubMed] [Google Scholar]
- 45.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF and Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–62. [DOI] [PubMed] [Google Scholar]
- 46.Coburn CT, Knapp FF Jr., Febbraio M, Beets AL, Silverstein RL and Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–9. [DOI] [PubMed] [Google Scholar]
- 47.Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R and Abumrad NA. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem. 1999;274:26761–6. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN and Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–17. [DOI] [PubMed] [Google Scholar]
- 49.Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ and Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goudriaan JR, Dahlmans VE, Teusink B, Ouwens DM, Febbraio M, Maassen JA, Romijn JA, Havekes LM and Voshol PJ. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res. 2003;44:2270–7. [DOI] [PubMed] [Google Scholar]
- 51.Lehrke M and Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–9. [DOI] [PubMed] [Google Scholar]
- 52.Goto K, Iso T, Hanaoka H, Yamaguchi A, Suga T, Hattori A, Irie Y, Shinagawa Y, Matsui H, Syamsunarno MR, Matsui M, Haque A, Arai M, Kunimoto F, Yokoyama T, Endo K, Gonzalez FJ and Kurabayashi M. Peroxisome proliferator-activated receptor-gamma in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J Am Heart Assoc. 2013;2:e004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, Michel T and Plutzky J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest. 2009;119:110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nissen SE and Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. [DOI] [PubMed] [Google Scholar]
- 55.Baker DP, Van Lenten BJ, Fogelman AM, Edwards PA, Kean C and Berliner JA. LDL, scavenger, and beta-VLDL receptors on aortic endothelial cells. Arteriosclerosis. 1984;4:248–55. [DOI] [PubMed] [Google Scholar]
- 56.van Hinsbergh VW, Havekes L, Emeis JJ, van Corven E and Scheffer M. Low density lipoprotein metabolism by endothelial cells from human umbilical cord arteries and veins. Arteriosclerosis. 1983;3:547–59. [DOI] [PubMed] [Google Scholar]
- 57.Molino Y, David M, Varini K, Jabes F, Gaudin N, Fortoul A, Bakloul K, Masse M, Bernard A, Drobecq L, Lecorche P, Temsamani J, Jacquot G and Khrestchatisky M. Use of LDL receptor-targeting peptide vectors for in vitro and in vivo cargo transport across the blood-brain barrier. FASEB J. 2017;31:1807–1827. [DOI] [PubMed] [Google Scholar]
- 58.Mao H, Lockyer P, Li L, Ballantyne CM, Patterson C, Xie L and Pi X. Endothelial LRP1 regulates metabolic responses by acting as a co-activator of PPARgamma. Nat Commun. 2017;8:14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rohrer L, Ohnsorg PM, Lehner M, Landolt F, Rinninger F and von Eckardstein A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ Res. 2009;104:1142–50. [DOI] [PubMed] [Google Scholar]
- 60.Fung KY, Wang C, Nyegaard S, Heit B, Fairn GD and Lee WL. SR-BI Mediated Transcytosis of HDL in Brain Microvascular Endothelial Cells Is Independent of Caveolin, Clathrin, and PDZK1. Front Physiol. 2017;8:841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Out R, Hoekstra M, Spijkers JA, Kruijt JK, van Eck M, Bos IS, Twisk J and Van Berkel TJ. Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res. 2004;45:2088–95. [DOI] [PubMed] [Google Scholar]
- 62.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, Tan KW, Heather A, Alexander JS and Angeli V. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013;17:671–84. [DOI] [PubMed] [Google Scholar]
- 63.Fruhwurth S, Pavelka M, Bittman R, Kovacs WJ, Walter KM, Rohrl C and Stangl H. High-density lipoprotein endocytosis in endothelial cells. World J Biol Chem. 2013;4:131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang H and Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–88. [DOI] [PubMed] [Google Scholar]
- 65.Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A and Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davies BS, Goulbourne CN, Barnes RH 2nd, Turlo KA, Gin P, Vaughan S, Vaux DJ, Bensadoun A, Beigneux AP, Fong LG and Young SG. Assessing mechanisms of GPIHBP1 and lipoprotein lipase movement across endothelial cells. J Lipid Res. 2012;53:2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beigneux AP, Fong LG, Bensadoun A, Davies BS, Oberer M, Gardsvoll H, Ploug M and Young SG. GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding. Circ Res. 2015;116:624–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beigneux AP, Franssen R, Bensadoun A, Gin P, Melford K, Peter J, Walzem RL, Weinstein MM, Davies BS, Kuivenhoven JA, Kastelein JJ, Fong LG, Dallinga-Thie GM and Young SG. Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler Thromb Vasc Biol. 2009;29:956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beigneux AP, Miyashita K, Ploug M, Blom DJ, Ai M, Linton MF, Khovidhunkit W, Dufour R, Garg A, McMahon MA, Pullinger CR, Sandoval NP, Hu X, Allan CM, Larsson M, Machida T, Murakami M, Reue K, Tontonoz P, Goldberg IJ, Moulin P, Charriere S, Fong LG, Nakajima K and Young SG. Autoantibodies against GPIHBP1 as a Cause of Hypertriglyceridemia. N Engl J Med. 2017;376:1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buonuomo PS, Bartuli A, Rabacchi C, Bertolini S and Calandra S. A 3-day-old neonate with severe hypertriglyceridemia from novel mutations of the GPIHBP1 gene. J Clin Lipidol. 2015;9:265–70. [DOI] [PubMed] [Google Scholar]
- 71.Olivecrona G, Ehrenborg E, Semb H, Makoveichuk E, Lindberg A, Hayden MR, Gin P, Davies BS, Weinstein MM, Fong LG, Beigneux AP, Young SG, Olivecrona T and Hernell O. Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J Lipid Res. 2010;51:1535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plengpanich W, Young SG, Khovidhunkit W, Bensadoun A, Karnman H, Ploug M, Gardsvoll H, Leung CS, Adeyo O, Larsson M, Muanpetch S, Charoen S, Fong LG, Niramitmahapanya S and Beigneux AP. Multimerization of glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and familial chylomicronemia from a serine-to-cysteine substitution in GPIHBP1 Ly6 domain. J Biol Chem. 2014;289:19491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rios JJ, Shastry S, Jasso J, Hauser N, Garg A, Bensadoun A, Cohen JC and Hobbs HH. Deletion of GPIHBP1 causing severe chylomicronemia. J Inherit Metab Dis. 2012;35:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Surendran RP, Visser ME, Heemelaar S, Wang J, Peter J, Defesche JC, Kuivenhoven JA, Hosseini M, Peterfy M, Kastelein JJ, Johansen CT, Hegele RA, Stroes ES and Dallinga-Thie GM. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mysling S, Kristensen KK, Larsson M, Kovrov O, Bensadouen A, Jorgensen TJ, Olivecrona G, Young SG and Ploug M. The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane protein GPIHBP1 counteracts this unfolding. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinberg HO, Brechtel G, Johnson A, Fineberg N and Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baron AD, Steinberg HO, Chaker H, Leaming R, Johnson A and Brechtel G. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J Clin Invest. 1995;96:786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, Bayazeed B and Baron AD. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest. 1997;100:1230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorens B and Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, Davidheiser S, Przybylski RJ and King GL. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes. 1993;42:80–9. [DOI] [PubMed] [Google Scholar]
- 81.Alpert E, Gruzman A, Riahi Y, Blejter R, Aharoni P, Weisinger G, Eckel J, Kaiser N and Sasson S. Delayed autoregulation of glucose transport in vascular endothelial cells. Diabetologia. 2005;48:752–5. [DOI] [PubMed] [Google Scholar]
- 82.Rajah TT, Olson AL and Grammas P. Differential glucose uptake in retina- and brain-derived endothelial cells. Microvasc Res. 2001;62:236–42. [DOI] [PubMed] [Google Scholar]
- 83.Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ and De Vivo DC. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18:188–91. [DOI] [PubMed] [Google Scholar]
- 84.Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP and De Vivo DC. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005;57:111–8. [DOI] [PubMed] [Google Scholar]
- 85.Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A and Giordano FJ. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc Natl Acad Sci U S A. 2012;109:17478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd and Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–9. [DOI] [PubMed] [Google Scholar]
- 87.Kubota T, Kubota N and Kadowaki T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev Endocr Metab Disord. 2013;14:207–16. [DOI] [PubMed] [Google Scholar]
- 88.Artwohl M, Brunmair B, Furnsinn C, Holzenbein T, Rainer G, Freudenthaler A, Porod EM, Huttary N and Baumgartner-Parzer SM. Insulin does not regulate glucose transport and metabolism in human endothelium. Eur J Clin Invest. 2007;37:643–50. [DOI] [PubMed] [Google Scholar]
- 89.Takakura Y, Kuentzel SL, Raub TJ, Davies A, Baldwin SA and Borchardt RT. Hexose uptake in primary cultures of bovine brain microvessel endothelial cells. I. Basic characteristics and effects of D-glucose and insulin. Biochim Biophys Acta. 1991;1070:1–10. [DOI] [PubMed] [Google Scholar]
- 90.Betz AL, Bowman PD and Goldstein GW. Hexose transport in microvascular endothelial cells cultured from bovine retina. Exp Eye Res. 1983;36:269–77. [DOI] [PubMed] [Google Scholar]
- 91.Bar RS, Siddle K, Dolash S, Boes M and Dake B. Actions of insulin and insulinlike growth factors I and II in cultured microvessel endothelial cells from bovine adipose tissue. Metabolism. 1988;37:714–20. [DOI] [PubMed] [Google Scholar]
- 92.Gerritsen ME, Burke TM and Allen LA. Glucose starvation is required for insulin stimulation of glucose uptake and metabolism in cultured microvascular endothelial cells. Microvasc Res. 1988;35:153–66. [DOI] [PubMed] [Google Scholar]
- 93.King GL, Buzney SM, Kahn CR, Hetu N, Buchwald S, Macdonald SG and Rand LI. Differential responsiveness to insulin of endothelial and support cells from micro- and macrovessels. J Clin Invest. 1983;71:974–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL and Kahn CR. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest. 2003;111:1373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwan RO, Cureton E, Dozier K, Curran B, Sadjadi J and Victorino GP. Ghrelin decreases microvascular leak during inflammation. J Trauma. 2010;68:1186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mahadev K, Wu X, Donnelly S, Ouedraogo R, Eckhart AD and Goldstein BJ. Adiponectin inhibits vascular endothelial growth factor-induced migration of human coronary artery endothelial cells. Cardiovasc Res. 2008;78:376–84. [DOI] [PubMed] [Google Scholar]
- 97.Cammisotto PG, Gingras D and Bendayan M. Transcytosis of gastric leptin through the rat duodenal mucosa. Am J Physiol Gastrointest Liver Physiol. 2007;293:G773–9. [DOI] [PubMed] [Google Scholar]
- 98.Kastin AJ and Akerstrom V. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology. 2001;73:237–42. [DOI] [PubMed] [Google Scholar]
- 99.Wang H, Liu Z, Li G and Barrett EJ. The vascular endothelial cell mediates insulin transport into skeletal muscle. Am J Physiol Endocrinol Metab. 2006;291:E323–32. [DOI] [PubMed] [Google Scholar]
- 100.Kondo T, Vicent D, Suzuma K, Yanagisawa M, King GL, Holzenberger M and Kahn CR. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111:1835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, Clermont A, Geraldes P, Dall’Osso C, Wagers AJ, Huang PL, Rekhter M, Scalia R, Kahn CR and King GL. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Konishi M, Sakaguchi M, Lockhart SM, Cai W, Li ME, Homan EP, Rask-Madsen C and Kahn CR. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A. 2017;114:E8478–E8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang H, Wang AX, Liu Z and Barrett EJ. Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes. 2008;57:540–7. [DOI] [PubMed] [Google Scholar]
- 104.Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, Kumagai K, Kawai T, Hashimoto S, Kobayashi T, Sato M, Tokuyama K, Nishimura S, Tsunoda M, Ide T, Murakami K, Yamazaki T, Ezaki O, Kawamura K, Masuda H, Moroi M, Sugi K, Oike Y, Shimokawa H, Yanagihara N, Tsutsui M, Terauchi Y, Tobe K, Nagai R, Kamata K, Inoue K, Kodama T, Ueki K and Kadowaki T. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. [DOI] [PubMed] [Google Scholar]
- 105.Tanigaki K, Chambliss KL, Yuhanna IS, Sacharidou A, Ahmed M, Atochin DN, Huang PL, Shaul PW and Mineo C. Endothelial Fcgamma receptor IIB activation blunts insulin delivery to skeletal muscle to cause insulin sesistance in mice. Diabetes. 2016;65:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang H, Wang AX, Aylor K and Barrett EJ. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes. 2013;62:4030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Azizi PM, Zyla RE, Guan S, Wang C, Liu J, Bolz SS, Heit B, Klip A and Lee WL. Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol Biol Cell. 2015;26:740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang H, Wang AX and Barrett EJ. Caveolin-1 is required for vascular endothelial insulin uptake. Am J Physiol Endocrinol Metab. 2011;300:E134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hamilton-Wessler M, Ader M, Dea MK, Moore D, Loftager M, Markussen J and Bergman RN. Mode of transcapillary transport of insulin and insulin analog NN304 in dog hindlimb: evidence for passive diffusion. Diabetes. 2002;51:574–82. [DOI] [PubMed] [Google Scholar]
- 110.Steil GM, Ader M, Moore DM, Rebrin K and Bergman RN. Transendothelial insulin transport is not saturable in vivo. No evidence for a receptor-mediated process. J Clin Invest. 1996;97:1497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mann GE, Yudilevich DL and Sobrevia L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol Rev. 2003;83:183–252. [DOI] [PubMed] [Google Scholar]
- 112.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH and Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;11:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yoon N, Dang TQ, Chasiotis H, Kelly SP and Sweeney G. Altered transendothelial transport of hormones as a contributor to diabetes. Diabetes Metab J. 2014;38:92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tsuchiya K and Accili D. Liver sinusoidal endothelial cells link hyperinsulinemia to hepatic insulin resistance. Diabetes. 2013;62:1478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Watt NT, Gage MC, Patel PA, Viswambharan H, Sukumar P, Galloway S, Yuldasheva NY, Imrie H, Walker AMN, Griffin KJ, Makava N, Skromna A, Bridge K, Beech DJ, Schurmans S, Wheatcroft SB, Kearney MT and Cubbon RM. Endothelial SHIP2 Suppresses Nox2 NADPH Oxidase-Dependent Vascular Oxidative Stress, Endothelial Dysfunction, and Systemic Insulin Resistance. Diabetes. 2017;66:2808–2821. [DOI] [PubMed] [Google Scholar]
- 116.Gonzalez-Sanchez JL, Martinez-Larrad MT, Saez ME, Zabena C, Martinez-Calatrava MJ and Serrano-Rios M. Endothelial nitric oxide synthase haplotypes are associated with features of metabolic syndrome. Clin Chem. 2007;53:91–7. [DOI] [PubMed] [Google Scholar]
- 117.Monti LD, Barlassina C, Citterio L, Galluccio E, Berzuini C, Setola E, Valsecchi G, Lucotti P, Pozza G, Bernardinelli L, Casari G and Piatti P. Endothelial nitric oxide synthase polymorphisms are associated with type 2 diabetes and the insulin resistance syndrome. Diabetes. 2003;52:1270–5. [DOI] [PubMed] [Google Scholar]
- 118.Rittig K, Holder K, Stock J, Tschritter O, Peter A, Stefan N, Fritsche A, Machicao F, Haring HU and Balletshofer B. Endothelial NO-synthase intron 4 polymorphism is associated with disturbed in vivo nitric oxide production in individuals prone to type 2 diabetes. Horm Metab Res. 2008;40:13–7. [DOI] [PubMed] [Google Scholar]
- 119.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B and Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–5. [DOI] [PubMed] [Google Scholar]
- 120.Cook S, Hugli O, Egli M, Menard B, Thalmann S, Sartori C, Perrin C, Nicod P, Thorens B, Vollenweider P, Scherrer U and Burcelin R. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes. 2004;53:2067–72. [DOI] [PubMed] [Google Scholar]
- 121.Cook S, Hugli O, Egli M, Vollenweider P, Burcelin R, Nicod P, Thorens B and Scherrer U. Clustering of cardiovascular risk factors mimicking the human metabolic syndrome × in eNOS null mice. Swiss Med Wkly. 2003;133:360–3. [DOI] [PubMed] [Google Scholar]
- 122.Shankar RR, Wu Y, Shen HQ, Zhu JS and Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49:684–7. [DOI] [PubMed] [Google Scholar]
- 123.Nakata S, Tsutsui M, Shimokawa H, Suda O, Morishita T, Shibata K, Yatera Y, Sabanai K, Tanimoto A, Nagasaki M, Tasaki H, Sasaguri Y, Nakashima Y, Otsuji Y and Yanagihara N. Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms. Circulation. 2008;117:2211–23. [DOI] [PubMed] [Google Scholar]
- 124.Nikolova G, Jabs N, Konstantinova I, Domogatskaya A, Tryggvason K, Sorokin L, Fassler R, Gu G, Gerber HP, Ferrara N, Melton DA and Lammert E. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell. 2006;10:397–405. [DOI] [PubMed] [Google Scholar]
- 125.Irving-Rodgers HF, Ziolkowski AF, Parish CR, Sado Y, Ninomiya Y, Simeonovic CJ and Rodgers RJ. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia. 2008;51:1680–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Olerud J, Mokhtari D, Johansson M, Christoffersson G, Lawler J, Welsh N and Carlsson PO. Thrombospondin-1: an islet endothelial cell signal of importance for beta-cell function. Diabetes. 2011;60:1946–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ and Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–3. [DOI] [PubMed] [Google Scholar]
- 128.Seki T, Hosaka K, Lim S, Fischer C, Honek J, Yang Y, Andersson P, Nakamura M, Naslund E, Yla-Herttuala S, Sun M, Iwamoto H, Li X, Liu Y, Samani NJ and Cao Y. Endothelial PDGF-CC regulates angiogenesis-dependent thermogenesis in beige fat. Nat Commun. 2016;7:12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD and Quertermous T. Endothelial lipase is a major determinant of HDL level. J Clin Invest. 2003;111:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ and Chan L. Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc Natl Acad Sci U S A. 2003;100:2748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nakajima H, Ishida T, Satomi-Kobayashi S, Mori K, Hara T, Sasaki N, Yasuda T, Toh R, Tanaka H, Kawai H and Hirata K. Endothelial lipase modulates pressure overload-induced heart failure through alternative pathway for fatty acid uptake. Hypertension. 2013;61:1002–7. [DOI] [PubMed] [Google Scholar]
- 132.Edmondson AC, Brown RJ, Kathiresan S, Cupples LA, Demissie S, Manning AK, Jensen MK, Rimm EB, Wang J, Rodrigues A, Bamba V, Khetarpal SA, Wolfe ML, Derohannessian S, Li M, Reilly MP, Aberle J, Evans D, Hegele RA and Rader DJ. Loss-of-function variants in endothelial lipase are a cause of elevated HDL cholesterol in humans. J Clin Invest. 2009;119:1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Singaraja RR, Sivapalaratnam S, Hovingh K, Dube MP, Castro-Perez J, Collins HL, Adelman SJ, Riwanto M, Manz J, Hubbard B, Tietjen I, Wong K, Mitnaul LJ, van Heek M, Lin L, Roddy TA, McEwen J, Dallinge-Thie G, van Vark-van der Zee L, Verwoert G, Winther M, van Duijn C, Hofman A, Trip MD, Marais AD, Asztalos B, Landmesser U, Sijbrands E, Kastelein JJ and Hayden MR. The impact of partial and complete loss-of-function mutations in endothelial lipase on high-density lipoprotein levels and functionality in humans. Circ Cardiovasc Genet. 2013;6:54–62. [DOI] [PubMed] [Google Scholar]
- 134.Blair A, Shaul PW, Yuhanna IS, Conrad PA and Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. 1999;274:32512–9. [DOI] [PubMed] [Google Scholar]
- 135.Mineo C and Shaul PW. HDL stimulation of endothelial nitric oxide synthase: a novel mechanism of HDL action. Trends Cardiovasc Med. 2003;13:226–31. [DOI] [PubMed] [Google Scholar]
- 136.Charbonneau A and Marette A. Inducible nitric oxide synthase induction underlies lipid-induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes. 2010;59:861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tai LK, Zheng Q, Pan S, Jin ZG and Berk BC. Flow activates ERK1/2 and endothelial nitric oxide synthase via a pathway involving PECAM1, SHP2, and Tie2. J Biol Chem. 2005;280:29620–4. [DOI] [PubMed] [Google Scholar]
- 138.Calapai G, Corica F, Allegra A, Corsonello A, Sautebin L, De Gregorio T, Di Rosa M, Costantino G, Buemi M and Caputi AP. Effects of intracerebroventricular leptin administration on food intake, body weight gain and diencephalic nitric oxide synthase activity in the mouse. Br J Pharmacol. 1998;125:798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Iuras A, Telles MM, Bertoncini CR, Ko GM, de Andrade IS, Silveira VL and Ribeiro EB. Central administration of a nitric oxide precursor abolishes both the hypothalamic serotonin release and the hypophagia induced by interleukin-1beta in obese Zucker rats. Regul Pept. 2005;124:145–50. [DOI] [PubMed] [Google Scholar]
- 140.Sansbury BE and Hill BG. Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med. 2014;73:383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Broniowska KA, Oleson BJ and Corbett JA. beta-Cell responses to nitric oxide. Vitam Horm. 2014;95:299–322. [DOI] [PubMed] [Google Scholar]
- 142.Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ and McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–76. [DOI] [PubMed] [Google Scholar]
- 143.Lammert E, Gu G, McLaughlin M, Brown D, Brekken R, Murtaugh LC, Gerber HP, Ferrara N and Melton DA. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2003;13:1070–4. [DOI] [PubMed] [Google Scholar]
- 144.Brissova M, Shostak A, Shiota M, Wiebe PO, Poffenberger G, Kantz J, Chen Z, Carr C, Jerome WG, Chen J, Baldwin HS, Nicholson W, Bader DM, Jetton T, Gannon M and Powers AC. Pancreatic islet production of vascular endothelial growth factor--a is essential for islet vascularization, revascularization, and function. Diabetes. 2006;55:2974–85. [DOI] [PubMed] [Google Scholar]
- 145.Myllyharju J Extracellular matrix and developing growth plate. Curr Osteoporos Rep. 2014;12:439–45. [DOI] [PubMed] [Google Scholar]
- 146.Parnaud G, Hammar E, Rouiller DG, Armanet M, Halban PA and Bosco D. Blockade of beta1 integrin-laminin-5 interaction affects spreading and insulin secretion of rat beta-cells attached on extracellular matrix. Diabetes. 2006;55:1413–20. [DOI] [PubMed] [Google Scholar]
- 147.Lawler J Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP and Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–70. [DOI] [PubMed] [Google Scholar]
- 149.Cunha DA, Cito M, Carlsson PO, Vanderwinden JM, Molkentin JD, Bugliani M, Marchetti P, Eizirik DL and Cnop M. Thrombospondin 1 protects pancreatic beta-cells from lipotoxicity via the PERK-NRF2 pathway. Cell Death Differ. 2016;23:1995–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kugelmeier P, Nett PC, Zullig R, Lehmann R, Weber M and Moritz W. Expression and hypoxic regulation of the endothelin system in endocrine cells of human and rat pancreatic islets. JOP. 2008;9:133–49. [PubMed] [Google Scholar]
- 151.Gregersen S, Thomsen JL, Brock B and Hermansen K. Endothelin-1 stimulates insulin secretion by direct action on the islets of Langerhans in mice. Diabetologia. 1996;39:1030–5. [DOI] [PubMed] [Google Scholar]
- 152.Gregersen S, Thomsen JL and Hermansen K. Endothelin-1 (ET-1)-potentiated insulin secretion: involvement of protein kinase C and the ET(A) receptor subtype. Metabolism. 2000;49:264–9. [DOI] [PubMed] [Google Scholar]
- 153.Johansson M, Mattsson G, Andersson A, Jansson L and Carlsson PO. Islet endothelial cells and pancreatic beta-cell proliferation: studies in vitro and during pregnancy in adult rats. Endocrinology. 2006;147:2315–24. [DOI] [PubMed] [Google Scholar]
- 154.Garcia-Ocana A, Vasavada RC, Cebrian A, Reddy V, Takane KK, Lopez-Talavera JC and Stewart AF. Transgenic overexpression of hepatocyte growth factor in the beta-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes. 2001;50:2752–62. [DOI] [PubMed] [Google Scholar]
- 155.Roccisana J, Reddy V, Vasavada RC, Gonzalez-Pertusa JA, Magnuson MA and Garcia-Ocana A. Targeted inactivation of hepatocyte growth factor receptor c-met in beta-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of beta-cell mass. Diabetes. 2005;54:2090–102. [DOI] [PubMed] [Google Scholar]
- 156.Dai C, Huh CG, Thorgeirsson SS and Liu Y. Beta-cell-specific ablation of the hepatocyte growth factor receptor results in reduced islet size, impaired insulin secretion, and glucose intolerance. Am J Pathol. 2005;167:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Moniaux N and Faivre J. Key role of sinusoidal endothelial cells in the triggering of liver regeneration. J Hepatol. 2011;55:488–90. [DOI] [PubMed] [Google Scholar]
- 158.Hall-Glenn F and Lyons KM. Roles for CCN2 in normal physiological processes. Cell Mol Life Sci. 2011;68:3209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crawford LA, Guney MA, Oh YA, Deyoung RA, Valenzuela DM, Murphy AJ, Yancopoulos GD, Lyons KM, Brigstock DR, Economides A and Gannon M. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol Endocrinol. 2009;23:324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guney MA, Petersen CP, Boustani A, Duncan MR, Gunasekaran U, Menon R, Warfield C, Grotendorst GR, Means AL, Economides AN and Gannon M. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc Natl Acad Sci U S A. 2011;108:15242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.James LR, Le C, Doherty H, Kim HS and Maeda N. Connective tissue growth factor (CTGF) expression modulates response to high glucose. PLoS One. 2013;8:e70441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Riley KG, Pasek RC, Maulis MF, Peek J, Thorel F, Brigstock DR, Herrera PL and Gannon M. Connective tissue growth factor modulates adult beta-cell maturity and proliferation to promote beta-cell regeneration in mice. Diabetes. 2015;64:1284–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hirata K, Dichek HL, Cioffi JA, Choi SY, Leeper NJ, Quintana L, Kronmal GS, Cooper AD and Quertermous T. Cloning of a unique lipase from endothelial cells extends the lipase gene family. J Biol Chem. 1999;274:14170–5. [DOI] [PubMed] [Google Scholar]
- 164.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M and Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet. 1999;21:424–8. [DOI] [PubMed] [Google Scholar]
- 165.McCoy MG, Sun GS, Marchadier D, Maugeais C, Glick JM and Rader DJ. Characterization of the lipolytic activity of endothelial lipase. J Lipid Res. 2002;43:921–9. [PubMed] [Google Scholar]
- 166.Badellino KO, Wolfe ML, Reilly MP and Rader DJ. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med. 2006;3:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Potocnjak I, Trbusic M, Teresak SD, Radulovic B, Pregartner G, Berghold A, Tiran B, Marsche G, Degoricija V and Frank S. Metabolic Syndrome Modulates Association between Endothelial Lipase and Lipid/Lipoprotein Plasma Levels in Acute Heart Failure Patients. Sci Rep. 2017;7:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jin W, Sun GS, Marchadier D, Octtaviani E, Glick JM and Rader DJ. Endothelial cells secrete triglyceride lipase and phospholipase activities in response to cytokines as a result of endothelial lipase. Circ Res. 2003;92:644–50. [DOI] [PubMed] [Google Scholar]
- 169.Hirata K, Ishida T, Matsushita H, Tsao PS and Quertermous T. Regulated expression of endothelial cell-derived lipase. Biochem Biophys Res Commun. 2000;272:90–3. [DOI] [PubMed] [Google Scholar]
- 170.Paradis ME, Badellino KO, Rader DJ, Deshaies Y, Couture P, Archer WR, Bergeron N and Lamarche B. Endothelial lipase is associated with inflammation in humans. J Lipid Res. 2006;47:2808–13. [DOI] [PubMed] [Google Scholar]
- 171.Ferguson JE 3rd, Kelley RW and Patterson C. Mechanisms of endothelial differentiation in embryonic vasculogenesis. Arterioscler Thromb Vasc Biol. 2005;25:2246–54. [DOI] [PubMed] [Google Scholar]
- 172.Walter TJ, Cast AE, Huppert KA and Huppert SS. Epithelial VEGF signaling is required in the mouse liver for proper sinusoid endothelial cell identity and hepatocyte zonation in vivo. Am J Physiol Gastrointest Liver Physiol. 2014;306:G849–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D and Rautou PE. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66:212–227. [DOI] [PubMed] [Google Scholar]
- 174.Mori T, Okanoue T, Sawa Y, Hori N, Ohta M and Kagawa K. Defenestration of the sinusoidal endothelial cell in a rat model of cirrhosis. Hepatology. 1993;17:891–7. [PubMed] [Google Scholar]
- 175.Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, Hultenby K, Christensson B, Ericzon BG, Holgersson J and Sumitran-Holgersson S. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163:1275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H and Tamaki K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 2015;95:1130–44. [DOI] [PubMed] [Google Scholar]
- 177.Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA and Deleve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918–927e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Le Couteur DG, Cogger VC, Markus AM, Harvey PJ, Yin ZL, Ansselin AD and McLean AJ. Pseudocapillarization and associated energy limitation in the aged rat liver. Hepatology. 2001;33:537–43. [DOI] [PubMed] [Google Scholar]
- 179.McLean AJ, Cogger VC, Chong GC, Warren A, Markus AM, Dahlstrom JE and Le Couteur DG. Age-related pseudocapillarization of the human liver. J Pathol. 2003;200:112–7. [DOI] [PubMed] [Google Scholar]
- 180.DG LEC, Cogger VC, McCuskey RS, R DEC, Smedsrod B, Sorensen KK, Warren A and Fraser R. Age-related changes in the liver sinusoidal endothelium: a mechanism for dyslipidemia. Ann N Y Acad Sci. 2007;1114:79–87. [DOI] [PubMed] [Google Scholar]
- 181.Le Couteur DG, Fraser R, Cogger VC and McLean AJ. Hepatic pseudocapillarisation and atherosclerosis in ageing. Lancet. 2002;359:1612–5. [DOI] [PubMed] [Google Scholar]
- 182.van den Oever IA, Raterman HG, Nurmohamed MT and Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflamm. 2010;2010:792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bender SB, Herrick EK, Lott ND and Klabunde RE. Diet-induced obesity and diabetes reduce coronary responses to nitric oxide due to reduced bioavailability in isolated mouse hearts. Diabetes Obes Metab. 2007;9:688–96. [DOI] [PubMed] [Google Scholar]
- 184.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A and Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zeng G and Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jonk AM, Houben AJ, de Jongh RT, Serne EH, Schaper NC and Stehouwer CD. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology (Bethesda). 2007;22:252–60. [DOI] [PubMed] [Google Scholar]
- 187.Ko SH, Cao W and Liu Z. Hypertension management and microvascular insulin resistance in diabetes. Curr Hypertens Rep. 2010;12:243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lteif A, Vaishnava P, Baron AD and Mather KJ. Endothelin limits insulin action in obese/insulin-resistant humans. Diabetes. 2007;56:728–34. [DOI] [PubMed] [Google Scholar]
- 189.Meijer RI, Bakker W, Alta CL, Sipkema P, Yudkin JS, Viollet B, Richter EA, Smulders YM, van Hinsbergh VW, Serne EH and Eringa EC. Perivascular adipose tissue control of insulin-induced vasoreactivity in muscle is impaired in db/db mice. Diabetes. 2013;62:590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yudkin JS, Eringa E and Stehouwer CD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. 2005;365:1817–20. [DOI] [PubMed] [Google Scholar]
- 191.Zhao G, Shen W, Zhang X, Smith CJ and Hintze TH. Loss of nitric oxide production in the coronary circulation after the development of dilated cardiomyopathy: a specific defect in the neural regulation of coronary blood flow. Clin Exp Pharmacol Physiol. 1996;23:715–21. [DOI] [PubMed] [Google Scholar]
- 192.Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X and Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83:969–79. [DOI] [PubMed] [Google Scholar]
- 193.Hashimoto S, Kubota N, Sato H, Sasaki M, Takamoto I, Kubota T, Nakaya K, Noda M, Ueki K and Kadowaki T. Insulin receptor substrate-2 (Irs2) in endothelial cells plays a crucial role in insulin secretion. Diabetes. 2015;64:876–86. [DOI] [PubMed] [Google Scholar]
- 194.Hogan MF, Liu AW, Peters MJ, Willard JR, Rabbani Z, Bartholomew EC, Ottley A and Hull RL. Markers of Islet Endothelial Dysfunction Occur in Male B6.BKS(D)-Leprdb/J Mice and May Contribute to Reduced Insulin Release. Endocrinology. 2017;158:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Huang Z, Jansson L and Sjoholm A. Vasoactive drugs enhance pancreatic islet blood flow, augment insulin secretion and improve glucose tolerance in female rats. Clin Sci (Lond). 2007;112:69–76. [DOI] [PubMed] [Google Scholar]
- 196.Villaret A, Galitzky J, Decaunes P, Esteve D, Marques MA, Sengenes C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL and Bouloumie A. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59:2755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sun X, Lin J, Zhang Y, Kang S, Belkin N, Wara AK, Icli B, Hamburg NM, Li D and Feinberg MW. MicroRNA-181b Improves Glucose Homeostasis and Insulin Sensitivity by Regulating Endothelial Function in White Adipose Tissue. Circ Res. 2016;118:810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ and Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100:2923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr. and Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Francque S, Laleman W, Verbeke L, Van Steenkiste C, Casteleyn C, Kwanten W, Van Dyck C, D’Hondt M, Ramon A, Vermeulen W, De Winter B, Van Marck E, Van Marck V, Pelckmans P and Michielsen P. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92:1428–39. [DOI] [PubMed] [Google Scholar]
- 201.Pasarin M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, Bosch J and Abraldes JG. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One. 2012;7:e32785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Abraldes JG, Albillos A, Banares R, Turnes J, Gonzalez R, Garcia-Pagan JC and Bosch J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology. 2009;136:1651–8. [DOI] [PubMed] [Google Scholar]
- 203.Marrone G, Shah VH and Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol. 2016;65:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lillioja S, Young AA, Culter CL, Ivy JL, Abbott WG, Zawadzki JK, Yki-Jarvinen H, Christin L, Secomb TW and Bogardus C. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest. 1987;80:415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hedman A, Berglund L, Essen-Gustavsson B, Reneland R and Lithell H. Relationships between muscle morphology and insulin sensitivity are improved after adjustment for intra-individual variability in 70-year-old men. Acta Physiol Scand. 2000;169:125–32. [DOI] [PubMed] [Google Scholar]
- 206.Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S and Barrett EJ. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes. 2004;53:1418–23. [DOI] [PubMed] [Google Scholar]
- 207.Nwadozi E, Roudier E, Rullman E, Tharmalingam S, Liu HY, Gustafsson T and Haas TL. Endothelial FoxO proteins impair insulin sensitivity and restrain muscle angiogenesis in response to a high-fat diet. FASEB J. 2016;30:3039–52. [DOI] [PubMed] [Google Scholar]
- 208.Olsson R and Carlsson PO. The pancreatic islet endothelial cell: emerging roles in islet function and disease. Int J Biochem Cell Biol. 2006;38:710–4. [DOI] [PubMed] [Google Scholar]
- 209.Sigrist S, Mechine-Neuville A, Mandes K, Calenda V, Braun S, Legeay G, Bellocq JP, Pinget M and Kessler L. Influence of VEGF on the viability of encapsulated pancreatic rat islets after transplantation in diabetic mice. Cell Transplant. 2003;12:627–35. [DOI] [PubMed] [Google Scholar]
- 210.Langlois A, Mura C, Bietiger W, Seyfritz E, Dollinger C, Peronet C, Maillard E, Pinget M, Jeandidier N and Sigrist S. In Vitro and In Vivo Investigation of the Angiogenic Effects of Liraglutide during Islet Transplantation. PLoS One. 2016;11:e0147068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Cao Y Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov. 2010;9:107–15. [DOI] [PubMed] [Google Scholar]
- 212.Cao Y Angiogenesis modulates adipogenesis and obesity. J Clin Invest. 2007;117:2362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Brakenhielm E, Cao R, Gao B, Angelin B, Cannon B, Parini P and Cao Y. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res. 2004;94:1579–88. [DOI] [PubMed] [Google Scholar]
- 214.Neels JG, Thinnes T and Loskutoff DJ. Angiogenesis in an in vivo model of adipose tissue development. FASEB J. 2004;18:983–5. [DOI] [PubMed] [Google Scholar]
- 215.Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R and Folkman MJ. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99:10730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bocca C, Novo E, Miglietta A and Parola M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol Gastroenterol Hepatol. 2015;1:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhang Z, Zhang F, Lu Y and Zheng S. Update on implications and mechanisms of angiogenesis in liver fibrosis. Hepatol Res. 2015;45:162–78. [DOI] [PubMed] [Google Scholar]
- 218.Moon JO, Welch TP, Gonzalez FJ and Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100:174–90. [DOI] [PubMed] [Google Scholar]
- 220.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–73. [DOI] [PubMed] [Google Scholar]
- 221.Yao VJ, Ozawa MG, Trepel M, Arap W, McDonald DM and Pasqualini R. Targeting pancreatic islets with phage display assisted by laser pressure catapult microdissection. Am J Pathol. 2005;166:625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M and Carmeliet P. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–63. [DOI] [PubMed] [Google Scholar]
- 223.Draoui N, de Zeeuw P and Carmeliet P. Angiogenesis revisited from a metabolic perspective: role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Eelen G, de Zeeuw P, Simons M and Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]