

Mesenchymal stem cells (MSCs) have been the subject of intense scientific interest in recent years. MSCs were discovered in 1970 as a resident population of cells in bone marrow (BM), where they play a pivotal role in hematopoiesis.1 MSCs are also found multiple tissues throughout the body including adipose, gut umbilical cord and the heart; however, BM-derived MSCs continue to be among the most widely studied, owing to their ease of isolation and expansion in culture. Multiple studies to date demonstrate the therapeutic potential of cardiac delivery of MSCs in the treatment of heart disease, including reduced myocyte apoptosis, stimulation of neoangeogenesis and improved cardiac function.2, 3
Several clinical trials of MSC based therapies have been conducted to date, and while results have been mixed, there is cause for cautious optimism, with numerous studies reporting improved cardiac function and reduced scar size in patients.4 However, the mechanisms underlying MSC-based therapy remain elusive. Reports of low retention rates of MSCs in the heart post-delivery may point to a paracrine mechanism of action.5 Indeed, conditioned media from MSCs conveys similar benefits as co-culture with MSCs themselves, including reduced cardiomyocyte apoptosis in vitro.6 In addition, others have shown that MSC-derived exosomes are cardioprotective in ischemia reperfusion injury.7
Autophagy is an evolutionarily conserved mechanism, wherein damaged proteins and organelles are degraded via delivery to the lysosome. In addition to removing potentially toxic components from the cellular milieu, autophagy is utilized to maintain energy homeostasis during times of nutrient deprivation. Stimulation of autophagy has been shown to improve cellular health, cardiac function and overall longevity in several models; however, other studies suggest that autophagy may directly contribute to myocyte death in certain cardiac injury models.8 In the current issue of Circulation Research, Xiao et al. impart fresh insight into the complex role of autophagy in cardiac injury, and provide compelling evidence for autophagy modulation being key to the therapeutic mechanism of action of MSCs based treatments.9
In agreement with previous studies, the authors found that administration of MSC directly into the heart thirty minutes post-myocardial infarction (MI), improved cardiac function and recovery in mice. Infarct size and apoptosis were both significantly decreased in treated animals compared to untreated. Interestingly, the authors noted changes in the levels of autophagy markers LC3-II and p62 following MI, and found significant differences between control and MSC treated animals post-MI. To assess if this was due to increased autophagic flux, which refers to autophagosome formation, maturation and degradation steps, the authors utilized Bafilomycin A1, an inhibitor of the vacuolar-type H+-ATPases, to block the fusion between the mature autophagosomes and lysosomes. If autophagic flux is increased and the fusion step is abrogated, then the autophagosomes will accumulate in the cytosol.
Indeed, the authors found that the presence of Bafilomycin A1 led to a significant increase in LC3II and p62 levels confirming increased autophagic flux in hearts following MI injury; however, flux was significantly reduced in MSC treated animals relative to controls. Furthermore, post-MI autophagosome number was significantly diminished in the hearts of animals that received MSC transplantation, as assessed via transmission electron microscopy. These results suggested that autophagic activity was elevated in the injured heart and that MSC treatment reduced this effect.
Next, the authors investigated the functional consequences of elevated autophagy in the heart. They found that treatment of mice with the autophagy inhibitor, 3-methyl adenine (3-MA), thirty minutes prior to MI also led to an attenuation of myocyte apoptosis, reduced remodeling and improved post-injury cardiac function overall. This suggested that autophagy inhibition might be, at least in part, responsible for the therapeutic benefits of MSC transplantation. These results were confirmed in vitro, using hypoxia in conjunction with serum deprivation (H/SD) to simulate the conditions of MI in isolated neonatal mouse cardiomyocytes (NMCs). Co-culture with MSCs reduced both autophagic flux and cell death in NMCs exposed to H/SD conditions.
Continuing with their cell culture model, the authors sought to better understand the mechanism by which MSCs can modulate autophagic activity in cardiomyocytes and found p53 and Bnip3, two known autophagy activators, to be upregulated following H/SD exposure in NMCs.10 Co-culture with MSCs significantly diminished their increase, pointing to this signaling pathway as a target of MSC therapy. Strengthening this conclusion, MSC co-culture elicited a partial blockade of increased autophagic flux in NMCs transduced with adenoviral p53 or Bnip3 constructs. Importantly, these results were corroborated in vivo; increases in p53 and Bnip3 protein levels observed in the border zone of MI injured mice were blocked in MSC treated animals.
In murine cardiac injury models, MSC-derived exosomes confer many of the therapeutic benefits mediated by MSC transplant.7 Here too, Xiao et al. found that NCMs treated with MSC-derived exosomes recapitulated the results obtained using their MSC/NMC co-culture model, including decreased autophagic flux and cytoprotection in cells exposed to H/SD, as well as downregulation of the p53/Bnip3-signaling pathway. These findings demonstrate that the anti-autophagic effects of MSCs are in large part mediated by exosome release.
Although exosome cargo can include various cellular macromolecules, previous studies have shown that pretreatment of MSCs with RNase can nullify their efficacy, pointing to an important role for microRNAs carried by exosomes.11 Xiao and colleagues identified miR-125b-5p to be one of the most abundant microRNAs in the MSC-derived exosomes. MiR-125b-5p is known to target p53 and was increased in NMCs that were cultured with MSC derived exosomes over a 24-hour period, suggesting exosomal transfer of this miRNA. Making use of an anti-miR-125b-5p oligonucleotide, the authors elegantly demonstrate that the effects mediated by MSC-derived exosomes, are entirely dependent on this miRNA. Exosomes derived from MSCs pretreated with anti-miR125b did not inhibit autophagy nor offer cardioprotection in either H/SD exposed NCMs, or MI-injured mice.
This study provides persuasive evidence for a mechanism of action dependent upon exosome-mediated delivery of miR125b from MSCs to cardiomyocytes (Figure). However, it also raises several critical questions that need to be investigated in future studies. Autophagy can be either beneficial or detrimental to the heart following injury, with outcomes depending on the type and duration of insult. Here, Xiao et al. found a moderate decrease in autophagic flux mediated by miR125b or the autophagy inhibitor 3-MA to be beneficial. However, it is important to note that the authors also found that disrupting autophagy more robustly, by knocking down the critical autophagy protein Atg7 produced the opposite effect and was damaging to NMCs. These results highlight the narrow therapeutic window in targeting autophagic activity in myocytes and provide a cautionary note to others pursuing autophagy modulation as a therapeutic strategy. Moreover, given the notoriously promiscuous nature of miRNAs, future studies should also examine other miR125b targets as potential mechanisms of action. For example, among its many targets, miR125b is known to regulate myeloid cell leukemia-1 (MCL-1).12 MCL-1 is an anti-apoptotic BCL-2 protein and a critical regulator of both cell survival and autophagy.13 Other targets of miR125b include several regulators of apoptosis and the immune response, which could also explain the therapeutic effect elicited by this miRNA. Finally, while the positive in vivo results presented by the authors are encouraging, enthusiasm should be tempered by the timing of treatments. MSC and exosome treatments were delivered to mice thirty minutes post-MI, while 3-MA was administered thirty minutes prior to injury. It would be of interest to administer treatments at later time points post-injury in future studies, more congruent with incidence and treatment of cardiac events in the clinical setting. Again, this may be of great importance given the temporal and injury-type dependent variability of autophagy inhibition on outcome.
Figure. Mesenchymal stem cell-mediated cardioprotection.
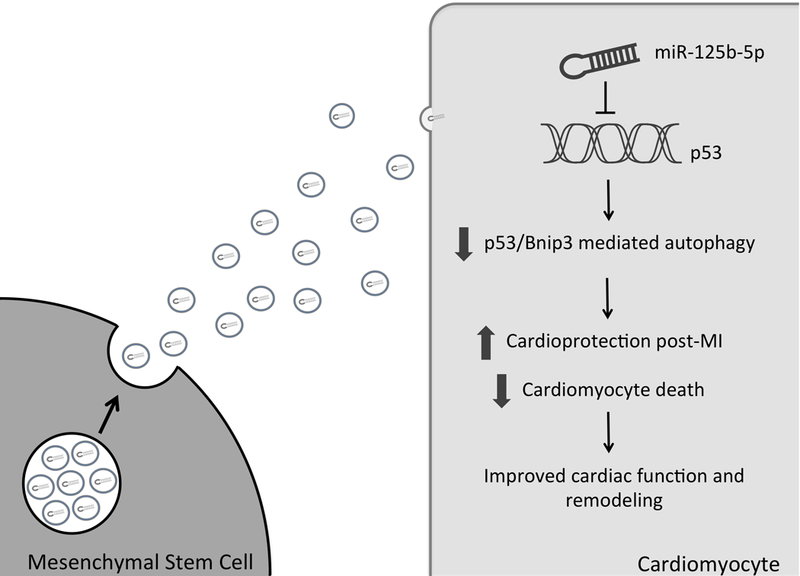
Exosome release from transplanted mesenchymal stem cells mediates transfer of miR-125–5p into cardiomyocytes. Downregulation of p53 expression suppresses p53/Bnip3-mediated autophagy, promoting a cardioprotective phenotype in the post-MI heart.
Overall, the findings by Xiao et al. represent a significant contribution to an exciting field. The authors shed new light on the therapeutic potential of MSCs and describe a mechanism of action dependent upon exosome-mediated transfer of miRNA and suppression of autophagy.
Acknowledgments
Sources of Funding
ÅB Gustafsson is supported by an AHA Established Investigator Award and by NIH R01HL132300, R01HL138560 and P01HL085577. B Woodall is supported by NIH T32HL007444.
Footnotes
Disclosures
None
References
- 1.Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet . 1970; 3:393–403. [DOI] [PubMed] [Google Scholar]
- 2.Schuleri KH, Feigenbaum GS, Centola M, Weiss ES, Zimmet JM, Turney J, Kellner J, Zviman MM, Hatzistergos KE, Detrick B, Conte JV, McNiece I, Steenbergen C, Lardo AC, Hare JM. Autologous mesenchymal stem cells produce reverse remodelling in chronic ischaemic cardiomyopathy. Eur Heart J . 2009; 30:2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, Coulter SC, Lin J, Ober J, Vaughn WK, Branco RV, Oliveira EM, He R, Geng YJ, Willerson JT, Perin EC. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation . 2005; 111:150–156. [DOI] [PubMed] [Google Scholar]
- 4.Sanina C, Hare JM. Mesenchymal Stem Cells as a Biological Drug for Heart Disease: Where Are We With Cardiac Cell-Based Therapy? Circ Res . 2015; 117:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation . 2002; 105:93–98. [DOI] [PubMed] [Google Scholar]
- 6.Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, Dzau VJ. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med . 2005; 11:367–368. [DOI] [PubMed] [Google Scholar]
- 7.Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res . 2013; 10:301–312. [DOI] [PubMed] [Google Scholar]
- 8.Nah J, Fernandez AF, Kitsis RN, Levine B, Sadoshima J. Does Autophagy Mediate Cardiac Myocyte Death During Stress? Circ Res . 2016; 119:893–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, Zhong Z, Zhao J, Li Q, Zhu D, Ke C, Zhong S, Wu X, Yu H, Zhu W, Chen J, Zhang J, Wang J, Hu X. Transplanted Mesenchymal Stem Cells Reduce Autophagic Flux in Infarcted Hearts via the Exosomal Transfer of mir-125b. Circ Res . 2018. [DOI] [PubMed] [Google Scholar]
- 10.Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension (Dallas, Tex. : 1979) . 2013; 62:70–77. [DOI] [PubMed] [Google Scholar]
- 11.Reis LA, Borges FT, Simoes MJ, Borges AA, Sinigaglia-Coimbra R, Schor N. Bone marrow-derived mesenchymal stem cells repaired but did not prevent gentamicin-induced acute kidney injury through paracrine effects in rats. PloS one . 2012; 7:e44092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S, Liu F, Xie L, Peng Y, Lv X, Zhu Y, Zhang Z, He X. miR-125b Suppresses Proliferation and Invasion by Targeting MCL1 in Gastric Cancer. Biomed Res Int . 2015; 2015:365273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson AB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev . 2013; 27:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]