

Abstract
Mutations in the gene encoding dystrophin, a protein that maintains muscle integrity and function, cause Duchenne muscular dystrophy (DMD). The deltaE50-MD dog model of DMD harbors a mutation corresponding to a mutational “hotspot” in the human DMD gene. We used adeno-associated viruses to deliver CRISPR gene editing components to four dogs and examined dystrophin protein expression 6 weeks after intramuscular delivery (n = 2) or 8 weeks after systemic delivery (n = 2). After systemic delivery in skeletal muscle, dystrophin was restored to levels ranging from 3 to 90% of normal, depending on muscle type. In cardiac muscle, dystrophin levels in the dog receiving the highest dose reached 92% of normal. The treated dogs also showed improved muscle histology. These large-animal data support the concept that, with further development, gene editing approaches may prove clinically useful for the treatment of DMD.
The persistent contraction and relaxation of cardiac and skeletal muscles necessitates mechanisms that maintain the integrity of muscle membranes (1, 2). Dystrophin is a large scaffolding protein that supports muscle structure and function by linking the cytoskeleton with the sarcolemma of muscle tissue (1, 3, 4). Mutations in the dystrophin gene cause Duchenne muscular dystrophy (DMD), a disorder primarily affecting boys that is characterized by progressive muscle degeneration and atrophy, leading to premature death from cardiomyopathy and respiratory collapse (5). Thousands of mutations have been identified in the dystrophin gene, which spans ~2.5 megabases of DNA and contains 79 exons. Many of these mutations cluster into “hotspots,” most commonly in a region that spans exons 45 to 50, typically placing exon 51 out of frame with preceding exons and preventing expression of functional dystrophin protein (6, 7). Therapies that induce “skipping” of exon 51 restore the reading frame and in principle could benefit ~13% of DMD patients (8). An oligo-nucleotide that allows skipping of exon 51 can restore dystrophin expression to 0.22 to 0.32% of normal levels after 1 year of treatment and has been approved for DMD patients (9–13).
CRISPR-Cas9 gene editing can target DMD mutations and restore dystrophin expression in mice and muscle cells derived from human induced pluripotent stem cells (iPSCs) (14–22). An essential step toward clinical translation of gene editing as a therapeutic strategy for DMD is the demonstration of efficacy and safety of this approach in large mammals.
The mutation carried by the deltaE50-MD canine model of DMD leads to loss of exon 50 and moreover can be corrected by skipping of exon 51, making this a valuable model for translational studies. First identified as a naturally occurring, spontaneous mutation in Cavalier King Charles Spaniels (23) and now maintained on a beagle background, this model (in contrast to mice) exhibits many of the clinical and pathological features of the human disease, such as muscle weakness, atrophy, and fibrosis (24).
To correct the dystrophin reading frame in the deltaE50-MD canine model (henceforth referred to as ΔEx50) (Fig. 1A), we used Streptococcus pyogenes Cas9 coupled with a single guide RNA (sgRNA) to target a region adjacent to the exon 51 splice acceptor site (referred to as sgRNA-51) (Fig. 1B). The sgRNA-51 corresponded to a highly conserved sequence that differs by only one nucleotide between the human and dog genomes (fig. S1A). Cas9 coupled with each of these sgRNA-51 sequences introduced a genomic cut only in DNA of the respective species (fig. S1B).
Fig. 1. Single-cut CRISPR editing of canine exon 50 in vivo and in vitro.
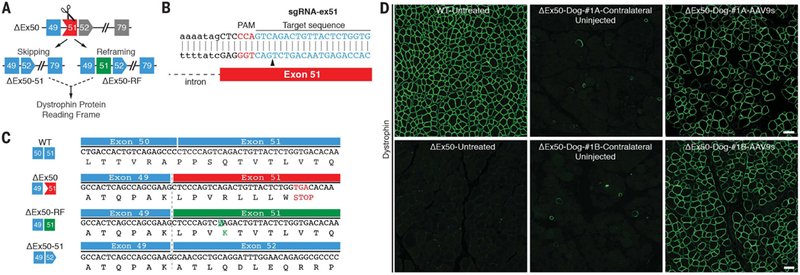
(A) Scheme showing the CRISPR-Cas9–mediated genome editing approach to correct the reading frame in ΔEx50 dogs by reframing and skipping of exon 51. Gray exons are out of frame. (B) Illustration of sgRNA binding position and sequence for sgRNA-ex51. PAM sequence for sgRNA is indicated in red. Black arrowhead indicates the cleavage site. (C) Sequence of the RT-PCR products of the ΔEx50–51 lower band confirmed that exon 49 spliced directly to exon 52, excluding exon 51. Sequence of RT-PCR products of ΔEx50 reframed (ΔEx50-RF). (D) Cranial tibialis muscles of ΔEx50 dogs were injected with AAV9s encoding sgRNA-51 and Cas9 and analyzed 6 weeks later. Dystrophin immunohistochemistry staining of cranial tibialis muscle of wild-type (WT) dog untreated, ΔEx50 dog untreated, ΔEx50 dogs contralateral (uninjected) muscle, and ΔEx50 dogs injected with AAV9-Cas9 and AAV9-sgRNA-51 (referred to as ΔEx50-#1A-AAV9s and ΔEx50-#1B-AAV9s). Scale bar: 50 μm.
For the in vivo delivery of Cas9 and sgRNA-51 to skeletal muscle and heart tissue in dogs, we used recombinant adeno-associated virus serotype 9 (referred to as AAV9), which displays preferential tropism for these tissues (25, 26). A muscle-specific creatine kinase (CK) regulatory cassette was used to drive expression of Cas9; three RNA polymerase III promoters (U6, H1, and 7SK) directed expression of the sgRNA, as described previously in mice (fig. S2) (18). AAV9-Cas9 and AAV9-sgRNA-51 were initially introduced into the cranial tibialis muscles of two 1-month-old dogs by intramuscular injection with 1.2 × 1013 AAV9 viral genomes (vg) of each virus. Muscles were analyzed 6 weeks after injection. In vivo targeting efficiency was estimated within muscle biopsy samples by reverse transcription–polymerase chain reaction (RT-PCR) with primers for sequences in exons 48 and 53, and genomic PCR amplification products spanning the target site were subjected to amplicon deep sequencing. The latter indicated that a mean of 9.96% of total reads contained changes at the targeted genomic site, including insertions, deletions, and substitutions (fig. S3). The most commonly identified mutations with a mean of 2.35% contained an adenosine (A) insertion immediately 3′ to the Cas9 genomic cutting site (Fig. 1C). The deletions identified with this method encompassed a highly predicted exonic splicing enhancer (ESE) site for exon 51 (18, 27, 28) (fig. S3A). However, this method does not identify larger deletions that might occur beyond the annealing sites of the primers used for PCR. Because these tissue samples contain a mixture of muscle and nonmuscle cells, the method likely underestimates the efficiency of gene editing within muscle cells.
Sequencing of RT-PCR products of RNA from muscle of ΔEx50 dogs injected intramuscularly with AAV9-Cas9 and AAV9-sgRNA-51 showed that deletion of exon 51 (ΔEx50–51) allowed splicing from exon 49 to 52, which restores the dystrophin open reading frame (fig. S3B). On gels, the PCR product with the A insertion was indistinguishable in size from non-edited cDNA products, so we performed deep-sequencing analysis to quantify its abundance compared to other small insertions. Deep sequencing of the upper band containing the non-edited cDNA product and reframed cDNA products indicated that a mean of 73.19% of total reads contained reframed cDNA products with an A insertion, a mean of 26.81% contained non-edited cDNA product, and the rest contained small deletions and insertions (fig. S3C). However, nonsense-mediated decay might affect the abundance of non-edited cDNA products. These data indicate that the two ΔEx50 dogs injected with AAV9-Cas9 and AAV9-sgRNA-51 had a high frequency of reframing events (with cDNA products containing an A insertion in the sequence of exon 51) and exon 51 skipping events resulting from deletion of the highly conserved ESE region.
To evaluate the specificity of our gene editing approach, we analyzed predicted off-target genomic sites for possible promiscuous editing. A total of three potential genome-wide off-target sites (OT1 to OT3) (fig. S4) were predicted in coding exons and four in noncoding regions (fig. S4) by the CRISPR design tool (http://crispr.mit.edu/). We performed deep sequencing at the top predicted off-target sites within protein-coding exons. None of these sites revealed notably more sequence alterations than the background analysis performed with other regions of the amplicons (fig. S5).
To evaluate dystrophin correction at the protein level, we performed histological analysis of AAV9-injected cranial tibialis muscles 6 weeks after AAV9 injection. Dystrophin immunohistochemistry of muscle from ΔEx50 dogs injected with AAV9-Cas9 and AAV9-sgRNA-51 revealed widespread expression: The majority of fibers within the injected muscles expressed sarcolemmal dystrophin, albeit to varying levels (Fig. 1D). Western blot analysis confirmed the restoration of dystrophin expression in skeletal muscle (Fig. 2, A and B) to ~60% of wild-type (WT) levels. On average, 2% of WT levels of dystrophin were detected in the uninjected contralateral muscles, far more than could be attributed to rare revertant events, which typically represent fewer than 0.001% of fibers and are undetectable by Western blot in ΔEx50 muscle. We attribute expression in uninjected contralateral muscles to leakage of AAV9 into circulation. As assessed by hematoxylin and eosin (H&E) staining, the injected muscles appeared to be normalized relative to muscles of untreated animals, with fewer hypercontracted or necrotic fibers, reduced edema and fibrosis, and fewer regions of inflammatory cellular infiltration (Fig. 2C). Immunohistochemistry for developmental myosin heavy chain (dMHC), a marker of regenerating fibers, revealed a marked reduction in dMHC-positive fibers within injected muscles (fig. S6).
Fig. 2. Dystrophin correction after intramuscular delivery of AAV9-encoded gene editing components.
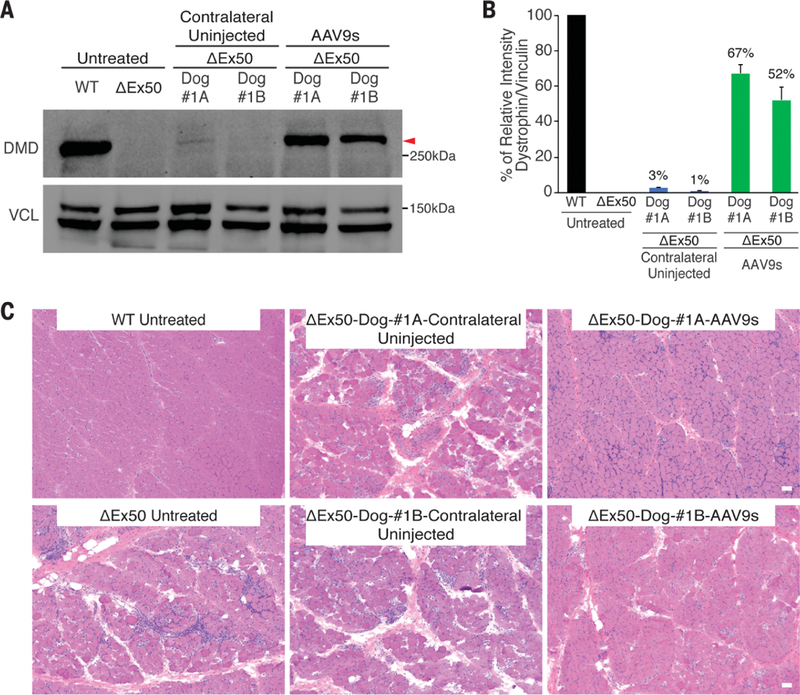
(A) Western blot analysis of dystrophin (DMD) and vinculin (VCL) expression in cranial tibialis muscles 6 weeks after intramuscular injection in 2 dogs (#1A and #1B). (B) Quantification of dystrophin expression from blots after normalization to vinculin. (C) Histochemistry by H&E staining of cranial tibialis muscle from a WT dog, ΔEx50 dog untreated, ΔEx50 contralateral uninjected, and ΔEx50 dogs injected intramuscularly with AAV9-Cas9 and AAV9-sgRNA-51 (referred to as ΔEx50-Dog-#1A-AAV9s and ΔEx50-Dog-#1B-AAV9s). Scale bar: 50 µm.
Dystrophin nucleates a series of proteins into the dystrophin-associated glycoprotein complex (DGC) to link the cytoskeleton and extracellular matrix (3, 4). In ΔEx50 mice, dogs, and DMD patients, these proteins are destabilized and do not localize to the subsarcolemmal region (4). Muscles injected with AAV9-Cas9 and AAV9-sgRNA-51 showed recovery of the DGC protein β-dystroglycan compared to contralateral uninjected muscles (fig. S7). We conclude that—at least in a short time frame of 6 weeks—single-cut genomic editing using AAV9-Cas9 and AAV9-sgRNA-51 can efficiently restore dystrophin expression and assembly of the DGC in dystrophic muscles. Immunohistochemistry using canine-specific CD4 and CD8 T cell markers (fig. S8) showed no evidence of enhanced mono-nuclear cellular infiltration or relevant hematological abnormalities in the treated animals compared to untreated controls or reference ranges (fig. S9).
Based on the high dystrophin-correction efficiency observed after intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51, we tested for rescue of dystrophin expression in two ΔEx50 dogs after systemic delivery of gene editing components. The dogs at 1 month of age were injected intravenously with the viruses and analyzed 8 weeks later. We tested two doses (2 × 1013 vg/kg and 1 × 1014 vg/kg) of each of the two viruses (AAV9-Cas9 and AAV9-sgRNA-51). To avoid a possible immune reaction, we included a transient regimen of immune suppression with the high dose. Systemic delivery of 2 × 1013 vg/kg of each virus (total virus 4 × 1013 vg/kg) in ΔEx50-Dog-#2A resulted in expression of virus in peripheral skeletal muscle samples and, to a lower extent, in heart samples, as shown by quantitative PCR (qPCR) analysis (fig. S10A). The delivery of 1 × 1014 vg/kg of each virus (total virus 2 × 1014 vg/kg) in ΔEx50-Dog-#2B (via infusion) allowed more widespread expression of viral constructs in the peripheral skeletal muscle samples and in heart samples (fig. S10B). Systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 led to dystrophin expression in a broad range of muscles, including the heart, in gene-edited ΔEx50 dogs at 8 weeks after injection— and to a markedly greater extent than that achieved with the lower dose (Fig. 3).
Fig. 3. Immunostaining of dystrophin after intravenous delivery of AAV9-encoded gene editing components.
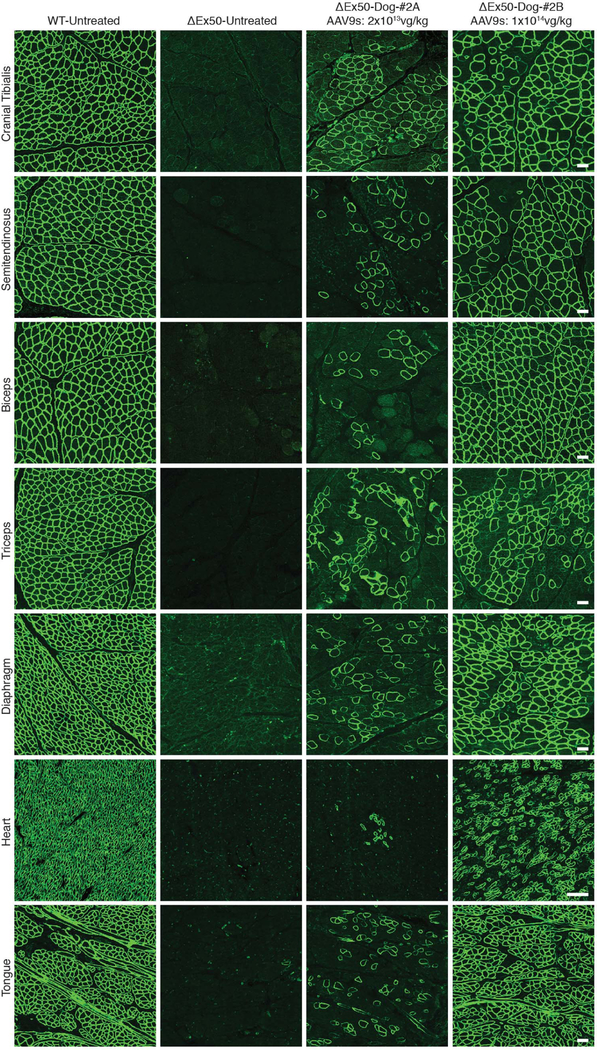
Dystrophin immunohistochemistry staining of cranial tibialis, semitendinosus, biceps, triceps, diaphragm, heart, and tongue muscles of WT dog, untreated ΔEx50 dog, and ΔEx50 dogs injected systemically with AAV9-Cas9 and AAV9-sgRNA-51 at 2 × 1013vg/kg (total virus 4 × 1013 vg/kg, referred to as ΔEx50-Dog #2A-AAV9s) and 1 × 1014 vg/kg (total virus 2 × 1014 vg/kg, referred to as ΔEx50-Dog #2B-AAV9s) for each virus.
To investigate the proportions of various insertions and deletions (indels) generated by systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51, we performed amplicon deep-sequencing analysis of the genomic DNA from heart, triceps, and biceps muscles. The genomic deep-sequencing analysis revealed an increased percentage of reads containing changes at the targeted genomic site, especially of the 1A insertion mutation in the samples from Dog-#2B compared to Dog-#2A (fig. S11).
Additionally, we performed tracking indels by decomposition (TIDE) (30) analysis at the genomic and cDNA levels, which showed an increase in numbers of indels in the samples from Dog-#2B compared to the samples from Dog-#2A (fig. S12). Testes analysis and Western blot analysis showed no activity of Cas9 and confirmed muscle-specific expression of gene editing machinery (fig. S13). Western blot analysis confirmed the restoration of dystrophin expression in skeletal muscle (Fig. 4, A and B) to ~50, 20, and 3% of WT levels for the cranial tibialis, triceps, and biceps, respectively, after systemic delivery of 2 × 1013 vg/kg of each virus (total virus 4 × 1013 vg/kg). For Dog-#2B, which received 1 × 1014 vg/kg of each virus (total virus 2 × 1014 vg/kg), Western blot analysis showed restoration of dystrophin expression (Fig. 4, C and D, and fig. S14) to ~70, 25, 64, 58, 92, and 5% of WT levels for the cranial tibialis, triceps, biceps, diaphragm, heart, and tongue muscles, respectively. Similar to what was seen after intramuscular injection, muscles appeared normalized via H&E staining (Fig. 4E). Immunostaining of muscle sections from treated ΔEx50 dogs also showed recovery of β-dystroglycan expression (fig. S15) and wide-spread reduction in dMHC (fig. S16).
Fig. 4. Western blot of dystrophin and muscle histology after intravenous delivery of AAV9-encoded gene editing components.
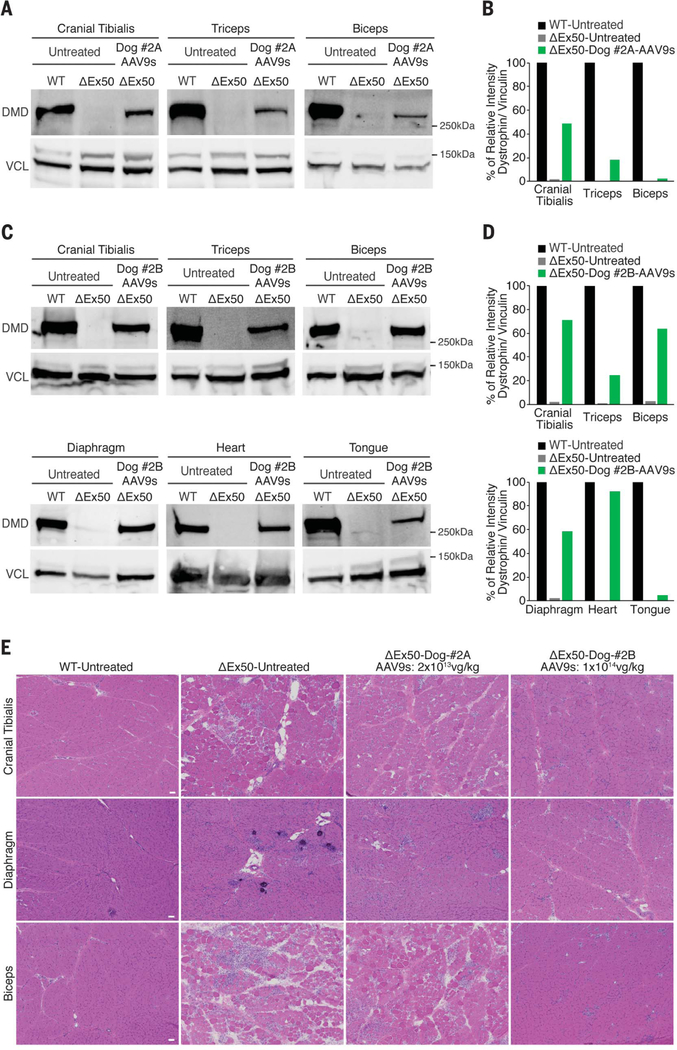
(A) Western blot analysis of dystrophin (DMD) and vinculin (VCL) of cranial tibialis, triceps, and biceps muscles of WT, untreated ΔEx50, and ΔEx50 injected with AAV9-Cas9 and AAV9-sgRNA-51 at 2 × 1013 vg/kg for each virus (total virus 4 × 1013 vg/kg, referred to as ΔEx50-Dog #2A-AAV9s). (B) Quantification of dystrophin expression from blots after normalization to vinculin. (C) Western blot analysis of DMD and VCL of cranial tibialis, triceps, biceps, diaphragm, heart, and tongue muscles of WT, untreated DEx50, and DEx50 injected with AAV9-Cas9 and AAV9-sgRNA-51 at 1 × 1014 vg/kg (total virus 2 × 1014 vg/kg, referred to as ΔEx50-Dog #2B-AAV9s). (D) Quantification of dystrophin expression from blots after normalization to vinculin. (E) H&E staining of cranial tibialis, diaphragm, and biceps muscles of WT, untreated ΔEx50, and ΔEx50 injected with AAV9-Cas9 and AAV9-sgRNA-51 at 2 × 1013 vg/kg for each virus (total virus 4 × 1013 vg/kg) and 1 × 1014vg/kg for each virus (total virus 2 × 1014 vg/kg). Scale bar: 50 μm.
To determine hematological and biochemical parameters of the treated dogs compared to the controls, we collected blood samples the day before injection and then at 1, 2, 4, 6, and 8 weeks after injection. The blood samples from all four dogs [healthy untreated, ΔEx50 untreated, and ΔEx50 dogs receiving 2 × 1013 vg/kg of each virus (total virus 4 × 1013 vg/kg) and 1 × 1014 vg/kg of each virus (total virus 2 × 1014 vg/kg)] before and after injection were unremarkable (fig. S17). Hematology counts, serum electrolytes, and kidney and liver function parameters remained within the normal ranges in all dogs. Additionally, blood samples were collected weekly for CK assessment: We observed a modest decline in serum CK activity in Dog-#2B treated with 1 × 1014 vg/kg of each virus (total virus 2 × 1014 vg/kg) (fig. S18) compared to the ΔEx50 untreated dog.
To evaluate the targeting efficiency of a human DMD mutation, we used a DMD iPSC line carrying a deletion from exon 48 to 50. Deletion of exons 48 to 50 leads to a frameshift mutation and appearance of a premature stop codon in exon 51. To correct the dystrophin reading frame, we introduced two concentrations of Cas9 and sgRNA-51 (26 ng/μl, referred to as high, and 13 ng/μl, referred to as low). Indel analysis showed 55.8 and 31.9% of indels for the high and low concentrations, respectively (fig. S19). Genomic deep-sequencing analysis revealed that 27.94% of mutations contained a single A insertion 3′ to the protospacer adjacent motif (PAM) sequence for the high concentration condition and 19.03% for the low concentration condition (fig. S20A), as observed in mouse and dog cells with a similar sgRNA directed against the same genomic locus. DMD iPSCs treated with Cas9 and sgRNA-51 and induced to form cardiomyocytes (iCMs) showed restoration of dystrophin immunostaining (fig. S20B) and expression of dystrophin protein to 67 to 100% of the levels of WT cardiomyocytes, as measured by Western blot (fig. S20, C and D).
It has been estimated that even 15% of normal levels of dystrophin would provide substantial therapeutic benefits for DMD patients (30–32). Our results demonstrate the efficacy of single-cut genome editing for restoration of dystrophin expression in a large-animal model of DMD, reaching up to ~80% of WT levels in some muscles after 8 weeks. Longer-term studies are required to establish whether the expression of dystrophin and the maintenance of muscle integrity that we observed are sustained.
This study, while encouraging, is preliminary and has several limitations, including the relatively small number of animals analyzed and the short duration of the analysis (6 to 8 weeks). The possibility of off-target effects of in vivo gene editing is a further potential safety concern. Although our initial deep-sequencing analysis of the top predicted off-target sites revealed no specific off-target gene editing above background levels in treated animals, it will be important to further assess possible off-target mutagenesis in longer-term studies with greater numbers of animals. Recent studies reported large deletions and complex genomic rearrangements at target sites of CRISPR-Cas9 in mouse embryonic cells, hematopoietic progenitors, and human immortalized epithelial cells (33). However, these cells are highly proliferative, are more genomically unstable in culture, and use different DNA repair pathways than somatic postmitotic cells (such as muscle and heart cells) (34). Future studies will be required to investigate the long-term genomic stability of gene-edited muscle tissues in vivo. Another potential concern with CRISPR-Cas9– mediated gene editing in vivo is immunogenicity of Cas9, particularly as expression persists after initial treatment. In this short-term study, we did not observe CD4- or CD8-positive cell infiltration of treated muscle, but longer-term studies with more sensitive assays must be performed. Additionally, although production of large quantities of AAV9 poses a challenge, doses of 2 × 1014vg/kg have been successfully used in human gene therapy trials (35).
Finally, although gene editing and exon-skipping oligonucleotides can both restore production of internally deleted dystrophin proteins, similar to those expressed in Becker muscular dystrophy, there are key distinctions between these approaches. Most notably, CRISPR gene editing may be permanent and not require redelivery, whereas oligonucleotides require continuous treatment. A corollary of this is that CRISPR treatment may be difficult to terminate if safety concerns arise.
Supplementary Material
ACKNOWLEDGMENTS
We thank V. Malladi (UT Southwestern Department of Clinical Sciences) for bioinformatics analysis; D. Church, S. Niessen, and D. Wells (RVC) for helpful research and clinical advice; staff and veterinarians within the RVC Biological Sciences Unit for technical assistance and animal care; C. Rodriguez for assistance; and J. Cabrera for graphics. We are grateful to S. Hauschka (University of Washington) for the CK8 promoter.
Funding: This work was supported by grants from the NIH (HL130253 and AR-067294), a Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (U54 HD 087351), the Robert A. Welch Foundation (grant 1-0025 to E.N.O.), Cure Duchenne, and Exonics Therapeutics. The RVC dog colony program was established and/or is supported by grants from the Wellcome Trust (101550/Z/13/Z), Muscular Dystrophy UK (RA3/3077), Joining Jack, and Duchenne Ireland.
Footnotes
Competing interests: L.A., R.B.-D., R.J.P., and E.N.O. are consultants for Exonics Therapeutics. L.A. and E.N.O. are co-inventors on a patent application (provisional filing number 62/442,606) related to the strategy presented in this study. The other authors declare no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and supplementary materials.
REFERENCES AND NOTES
- 1.O’Brien KF, Kunkel LM, Mol. Genet. Metab 74, 75–88 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Guiraud S et al. , Annu. Rev. Genomics Hum. Genet 16, 281–308 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Campbell KP, Kahl SD, Nature 338, 259–262 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG,Campbell KP, Nature 345, 315–319 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Muntoni F, Torelli S, Ferlini A, Lancet Neurol 2, 731–740 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Bladen CL et al. , Hum. Mutat 36, 395–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y et al. , J. Int. Med. Res 44, 99–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A et al. , Hum. Mutat 30, 293–299 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S,Aoki Y, Am. J. Transl. Res 8, 2471–2489 (2016). [PMC free article] [PubMed] [Google Scholar]
- 10.Stein CA, Mol. Ther 24, 1884–1885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aartsma-Rus A, Krieg AM, Nucleic Acid Ther 27, 1–3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowling JJ, Nat. Rev. Neurol 12, 675–676 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Mendell JR et al. , Ann. Neurol 79, 257–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long C et al. , Science 351, 400–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabebordbar M et al. , Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson CE et al. , Science 351, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long C et al. , Sci. Adv 4, p9004 (2018). [Google Scholar]
- 18.Amoasii L et al. , Sci. Transl. Med 9, eaan8081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bengtsson NE et al. , Nat. Commun 8, 14454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tremblay JP, Iyombe-Engembe J-P, Duchêne B,Ouellet DL, Mol. Ther 24, 1888–1889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young CS et al. , Cell Stem Cell 18, 533–540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ousterout DG et al. , Nat. Commun 6, 6244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walmsley GL et al. , PLOS ONE 5, e8647 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hildyard J et al. , Neuromuscul. Disord 28, S18 (2018). [Google Scholar]
- 25.Yue Y et al. , Hum. Mol. Genet 24, 5880–5890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE, Mol. Ther 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Smith PJ et al. , Hum. Mol. Genet 15, 2490–2508 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR, Nucleic Acids Res 31, 3568–3571 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brinkman EK, Chen T, Amendola M, van Steensel B, Nucleic Acids Res 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godfrey C et al. , Hum. Mol. Genet 24, 4225–4237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Putten M et al. , J. Mol. Cell. Cardiol 69, 17–23 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Beggs AH et al. , Am. J. Hum. Genet 49, 54–67 (1991). [PMC free article] [PubMed] [Google Scholar]
- 33.Kosicki M, Tomberg K, Bradley A, Nat. Biotechnol 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tichy ED et al. , Stem Cells Dev 19, 1699–1711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendell JR et al. , N. Engl. J. Med 377, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.