

Abstract
Arteriolar endothelial cell–expressed (EC-expressed) α-globin binds endothelial NOS (eNOS) and degrades its enzymatic product, NO, via dioxygenation, thereby lessening the vasodilatory effects of NO on nearby vascular smooth muscle. Although this reaction potentially affects vascular physiology, the mechanisms that regulate α-globin expression and dioxygenase activity in ECs are unknown. Without β-globin, α-globin is unstable and cytotoxic, particularly in its oxidized form, which is generated by dioxygenation and recycled via endogenous reductases. We show that the molecular chaperone α-hemoglobin–stabilizing protein (AHSP) promotes arteriolar α-globin expression in vivo and facilitates its reduction by eNOS. In Ahsp−/− mice, EC α-globin was decreased by 70%. Ahsp−/− and Hba1−/− mice exhibited similar evidence of increased vascular NO signaling, including arteriolar dilation, blunted α1-adrenergic vasoconstriction, and reduced blood pressure. Purified α-globin bound eNOS or AHSP, but not both together. In ECs in culture, eNOS or AHSP enhanced α-globin expression posttranscriptionally. However, only AHSP prevented oxidized α-globin precipitation in solution. Finally, eNOS reduced AHSP-bound α-globin approximately 6-fold faster than did the major erythrocyte hemoglobin reductases (cytochrome B5 reductase plus cytochrome B5). Our data support a model whereby redox-sensitive shuttling of EC α-globin between AHSP and eNOS regulates EC NO degradation and vascular tone.
Keywords: Hematology, Vascular Biology
Keywords: Protein misfolding, endothelial cells
Introduction
Hemoglobin A (HbA), the RBC O2 carrier, is a heterotetramer of 2 α-globin and 2 β-globin proteins (α2β2), each bound to a planar heme prosthetic group containing a central Fe atom that binds O2 reversibly (1). Related globin proteins, including myoglobin, cytoglobin (Cygb), and neuroglobin, have unique patterns of tissue expression, subcellular localization, and affinity for external ligands, including O2, CO2, and NO (2). Although HbA is expressed abundantly in RBC, low-level α-globin protein expression occurs in numerous other cells, including neuronal, retinal, alveolar epithelial, endometrial, kidney mesangial, and liver cells (2). Most or all α-globin in these cells is probably heme bound, because the non–heme-bound (apo) form is unstable (3). The functions of α-globin, if any, in nonerythroid tissues are unknown, although the concentrations of α-globin are probably too low to support the canonical role of O2 transport. Primordial globin proteins are believed to have arisen in low-O2 environments as O2 sensors and/or enzymatic NO scavengers before evolving into RBC O2 transporters (4).
α-Globin expressed in arteriolar endothelial cells (ECs) regulates vasoconstriction (5, 6). Specifically, arteriolar α-globin is expressed predominantly in the myoendothelial junction (MEJ), also referred to as the myoendothelial projection, a specialized EC structure that penetrates the internal elastic lamina to contact vascular smooth muscle cells (VSMCs) and mediate cross-cellular signaling (7, 8). In response to VSMC signals, MEJ endothelial cell NOS (eNOS) generates NO. This diffuses into adjacent VSMCs and activates soluble guanylyl cyclase to generate cyclic GMP, which induces vasodilation via multiple mechanisms (9–11). Oxygenated, reduced (Fe2+) α-globin in the MEJ binds eNOS and degrades NO via dioxygenation [NO + O2 (Fe2+) α-globin → NO3– + (Fe3+) α-globin] (12–14). siRNA knockdown of α-globin expression in isolated arterioles increased local NO concentration and conferred resistance to phenylephrine-induced (PE-induced) vasoconstriction. Moreover, a peptide that disrupts the α-globin–eNOS interaction inhibited PE-induced vasoconstriction of isolated arterioles and reduced blood pressure after systemic administration (15). These vasomotor effects caused by α-globin RNA knockdown or disruption of the α-globin–eNOS complex depended on eNOS activity, suggesting that MEJ α-globin degrades eNOS-derived NO to enhance vascular tone. Mathematical modeling supports this mechanism, assuming that sufficient concentrations of (Fe2+) α-globin and eNOS are present at the MEJ (16). In this regard, it is necessary to stabilize and reduce (Fe3+) α-globin in ECs, because this redox form is particularly labile and unable to degrade NO. Cytochrome B5 reductase (CyB5R3) in ECs participates in α-globin reduction, although the mechanisms are undetermined (5, 6).
Arteriolar ECs express α-globin but not β-globin. However, α-globin monomers, particularly the oxidized (Fe3+) form, are unstable. In cells, α-globin monomers are either eliminated by protein quality-control mechanisms or form toxic precipitates (17, 18). In RBC precursors, α-globin is stabilized through interactions with β-globin or with the molecular chaperone α-hemoglobin–stabilizing protein (AHSP), which binds nascent apo–α-globin polypeptide to facilitate its folding for heme insertion and stabilizes the resultant holoprotein (α-globin) en route to HbA tetramer assembly (19, 20). AHSP is coexpressed with α-globin in arteriolar ECs (8). Therefore, EC α-globin could be stabilized through interactions with AHSP, eNOS, or both.
The current study addresses 3 major questions related to the roles of α-globin, eNOS, and AHSP in arterioles. First, what are the long-term effects, if any, of EC α-globin deficiency on blood vessel structure and function? Second, what are the roles of AHSP and eNOS in synthesizing and maintaining functional α-globin in ECs? Third, how is oxidized (Fe3+) α-globin reduced to regenerate (Fe2+) α-globin and restore dioxygenase activity? We addressed these questions by examining mice with disrupted Ahsp or α-globin (Hba1) genes and by studying the activities of α-globin, AHSP, and eNOS proteins in cells in culture and in solution. We show that ablation of the Hba1 gene leads to dilated arteries with reduced vasoreactivity and decreased systemic blood pressure. Moreover, we show that Ahsp gene disruption reduces the abundance of EC α-globin protein, causing changes in arterial anatomy and function that mimic those conferred by Hba1 loss. Finally, our study demonstrates that the reductase domain of eNOS rapidly reduces AHSP-bound (Fe3+) α-globin. Our findings support and extend previous models of α-globin function in ECs and demonstrate an essential role for AHSP in this process.
Results
α-Globin and AHSP are expressed in arterioles.
Immunofluorescence staining of thoracodorsal artery (TDA) and mesenteric arteriole sections detected α-globin, eNOS, and the MEJ marker PAI1 (21) (Figure 1A and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI99933DS1), consistent with our previous report (8). We were unable to costain sections for α-globin and AHSP, because both antibodies are derived from the same species (Supplemental Table 1). However, the distribution of α-globin and AHSP overlapped with that of PAI (Figure 1A and Supplemental Figure 1), suggesting that the 3 proteins colocalize to the MEJ. In human mesentery and heart sections from 7 nonthalassemic individuals, ECs showed staining for α-globin with 2 polyclonal antisera prepared against different regions of the protein, but not for β-globin (Figure 1, B and C; Supplemental Figure 2, A and B; and Supplemental Table 1). In contrast, intraluminal RBC showed staining for α-globin and β-globin (Supplemental Figure 2, C and D). We also detected eNOS, AHSP, and α-globin in human arteriolar VSMCs at a weaker staining intensity than was seen in ECs (Figure 1, B and C; Supplemental Figure 2, A and B).
Figure 1. α-Globin and AHSP are coexpressed in small arteries.

(A) Immunofluorescence detection of the indicated proteins (white arrows) in TDA sections from 24-week-old WT mice. DAPI-stained nuclei are blue. Each row represents a single transverse section. The dashed line across the internal elastic lamina demarcates ECs and VSMCs. Scale bars: 5 μm. (B) Representative images of IHC detection of the indicated proteins (arrows) in human mesenteric arteries. Scale bars: 50 μm and 25 μm (insets). (C) Quantification of staining for the indicated proteins in studies of multiple human subjects (n = 4–6), performed as shown in B. Positivity scores represent the number of positive pixels (intensity of 1+, 2+, or 3+), divided by the total number of pixels (both positive and negative) in a selected area. *P < 0.05, ***P < 0.005, and ****P < 0.001, by unpaired t test.
Expression of α-globin in ECs is AHSP dependent.
We investigated whether AHSP regulates α-globin protein levels in small artery ECs, as in RBC (22). Immunofluorescence staining of α-globin was reduced substantially in TDAs from Ahsp−/− mice (Figure 2A). Consistent with this, Western blot analysis showed that α-globin was reduced by approximately 70% in Ahsp−/− mice as compared with expression in age-matched controls (Figure 2, B and C). Quantitative reverse transcription PCR (RT-qPCR) showed no difference between α-globin (Hba1 and Hba2) or eNOS (Nos3) mRNA levels in control and Ahsp−/− ECs (Figure 2D). Overall, these findings show that AHSP is required to maintain normal levels of α-globin in small artery ECs, probably by stabilizing the protein.
Figure 2. α-Globin protein expression is reduced in TDAs from Ahsp−/− mice.

(A) Immunofluorescence detection of the indicated proteins (white arrows) in TDA sections from 24-week-old Ahsp−/− mice. The dashed line across the internal elastic lamina demarcates ECs and VSMCs. Scale bars: 5 μm. (B) Western blot detection of the indicated proteins in TDAs. The relative intensity of the α-globin signals normalized to the β-actin signal is indicated in the top panel. The migration of molecular mass standards is shown on the right. RBC were used as a control for globin expression. h, human; m, mouse. (C) Quantification of α-globin protein expression by Western blotting as in B. The graph shows the average signal intensities from 3 separate experiments. (D) Relative mRNA levels in control and Ahsp−/− TDAs as measured by RT-qPCR and normalized to Actb mRNA. The graph shows data from 4 mice, with 2 TDAs analyzed from each one. *P < 0.05, by unpaired t test.
Disruption of Ahsp or Hba1 genes reduces small artery tone.
We hypothesized that EC α-globin degrades NO to promote vasoconstriction of arterioles in vivo (8). To investigate this possibility further, we evaluated baseline TDA diameters in Ahsp−/− and Hba1−/− mice. Mouse α-globin is encoded by 2 tandem genes, Hba1 and Hba2, which are expressed at a 40:60 ratio in adult RBC (23). The TDAs of Ahsp−/− and Hba1−/− mice appeared to be dilated compared with those of WT controls (Figure 3, A and B). The lumen diameters of randomly sampled cross sections were increased by 92% (P < 0.001) and 69% (P < 0.01), respectively, in Ahsp−/− and Hba1−/− mice (Figure 3, B and C). However, the mutant TDAs had normal thicknesses of the vascular smooth muscle and the internal elastic lamina layer (Supplemental Figure 3). Immunostaining for laminin and fibronectin showed similar subendothelial and adventitial deposition of these matrix proteins in all groups (data not shown).
Figure 3. Ahsp or Hba1 gene disruption causes steady-state TDA dilation in vivo.

(A) TDAs and veins in situ. Scale bars: 5 mm. (B) Electron micrographs of TDA cross sections from 24-week-old mice. Scale bars: 20 μm. (C) Histomorphometric analysis of randomly sampled TDA cross sections from WT control mice (n = 5, 15 random measurements), Ahsp−/− mice (n = 5, 13 random measurements), and Hba1−/− mice (n = 4, 5 random measurements). **P < 0.01 and ***P < 0.005, by unpaired t test.
Next, we examined TDA reactivity in response to PE. The Ahsp−/− and Hba1−/− genotypes cause mild hemolytic anemia (17, 22, 24), which could affect vascular physiology. To eliminate this potentially confounding effect, we transplanted normal mouse bone marrow into lethally irradiated Ahsp−/−, Hba1−/−, and WT control mice. In Ahsp−/− recipients 4 weeks after transplantation, more than 85% of the peripheral blood mononuclear cells contained WT alleles (Supplemental Figure 4). Six weeks after transplantation, the abnormal RBC indices (RBC distribution width index [RDW] and reticulocyte count) of both mutant genotypes resolved, suggesting that erythropoiesis was occurring largely through normal donor hematopoietic stem cells (Supplemental Table 2).
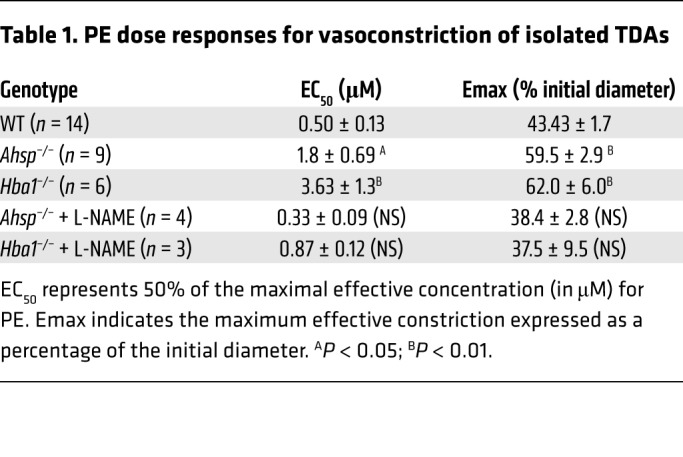
We used pressure myography to measure TDA lumen diameters after treatment with PE over a dose range of 10−9 to 10−3 M (Figure 4A). Compared with control arterioles, both Ahsp−/− and Hba1−/− TDAs had significantly blunted responses to PE (Figure 4A and Table 1). Mutant and WT TDAs showed similar responses to several drugs that bypass EC-derived NO signaling to VSMCs, including constriction after treatment with the eNOS inhibitor L-nitro–arginine methyl ester (L-NAME) (Figure 4A), constriction after treatment with KCl to depolarize VSMCs (Supplemental Figure 5A), dilation after treatment with the intermediate and small potassium (IK/SK) channel activator NS309 to enhance EC-VSMC coupling (Supplemental Figure 5B), and dilation after treatment with the NO donor sodium nitroprusside to activate VSMC guanylate cyclase directly (Supplemental Figure 5C). Together, these data indicate that VSMC communication with ECs and physiologic responses to NO are intact and support the hypothesis that EC α-globin deficiency or loss of AHSP (with resultant α-globin deficiency) inhibits TDA contractility by increasing NO diffusion to VSMCs. To assess these effects systemically, we implanted radiotelemetry blood pressure monitors into the left carotid arteries of Ahsp−/− and Hba1−/− mice that were transplanted previously with WT bone marrow. Compared with controls, both mutant strains had significantly lower mean arterial blood pressure (Figure 4B), with the loss of Hba1 having a stronger effect.
Figure 4. Ahsp or Hba1 gene disruption inhibits vasoconstriction after α1-adrenergic stimulation and reduces systemic blood pressure.

Mice with the indicated genotypes were lethally irradiated, transplanted with WT hematopoietic stem and progenitor cells to restore normal RBC production, and examined 6 to 8 weeks later. (A) Vasoconstriction of TDAs from control (n = 7) and mutant (Ahsp−/−, n = 9; Hba1−/−, n = 6) mice after treatment with escalating doses of PE, with or without the eNOS inhibitor L-NAME (Ahsp−/− ± L-NAME, n = 4; Hba1−/− ± L-NAME, n = 3). (B) Mean arterial blood pressures of mice with the indicated genotypes measured by radiotelemetry (control, n = 11; Ahsp−/−, n = 7; Hba1−/−, n = 5). *P < 0.05 and ****P < 0.001, by 2-way ANOVA (A) and unpaired t test (B).
Table 1. PE dose responses for vasoconstriction of isolated TDAs.
AHSP and eNOS interactions with α-globin.
Free α-globin denatures rapidly in cells and is eliminated by protein quality control mechanisms (18) or forms toxic precipitates (22, 25). During RBC formation, AHSP acts as a molecular chaperone to stabilize α-globin before HbA assembly (19, 20). In ECs, α-globin could be stabilized through interactions with eNOS and/or AHSP (8). We expressed α-globin–GFP and AHSP-mCherry or eNOS-mCherry in cultures of human coronary artery ECs, which do not express endogenous α-globin or AHSP (data not shown). Consistent with our previous findings (17), AHSP enhanced α-globin protein expression, as evidenced by the increase in the GFP signal: 8-fold as measured by image analysis and 4-fold as measured by flow cytometry (Figure 5A and Supplemental Figure 6, A–C). Coexpressed eNOS also increased α-globin–GFP expression, although to a lesser extent than did AHSP (the increases were 5-fold and 2-fold, respectively). In cell images, α-globin protein expression largely overlapped with that of AHSP or eNOS (Figure 5A, far-right panels). Western blot analysis also showed enhancement of α-globin protein levels by coexpressed eNOS (a 2.6-fold increase) or AHSP (a 4.5-fold increase) (Figure 5, B and C). The steady-state levels of α-globin–GFP mRNA were not altered by coexpressed eNOS or AHSP (Figure 5D), consistent with our model in which these proteins bind α-globin and inhibit its degradation in cells. To study the protein interactions in solution, we combined purified human AHSP, CO–α-globin, and Myc-eNOS and performed anti-Myc IP followed by Western blotting (Figure 6). As reported previously (8), α-globin coimmunoprecipitated with eNOS. However, AHSP, which binds CO–α-globin in solution (association constant [KA]: 1 × 107/M) (17, 26), did not coimmunoprecipitate with eNOS or the α-globin–eNOS complex. Thus, AHSP and eNOS interactions with α-globin are probably mutually exclusive.
Figure 5. AHSP or eNOS stabilizes α-globin in cells.

(A) Human coronary ECs were transduced with lentiviral expression vectors encoding human α-globin–GFP, human AHSP-mCherry, or human eNOS-mCherry fusion proteins and visualized by fluorescence microscopy. Representative images of merged GFP and mCherry signals in single cells are shown (white rectangles and far-right panels). Scale bars: 25 μm (panels in first 3 columns) and 10 μm (panels at far right). (B) Western blot detection of the indicated proteins in cells shown in A. The indicated proteins are as follows: 1, eNOS-mCherry; 2, endogenous eNOS; 3, AHSP-mCherry; 4, recombinant human AHSP (2 ng); 5, α-globin–GFP; and 6, purified α-globin (2 ng). (C) Quantification of α-globin–GFP protein expression by Western blotting as in B (n = 3 independent experiments). (D) Quantitative RT-qPCR analysis of α-globin–GFP mRNA 4 days after lentiviral transduction with vectors encoding the indicated proteins (n = 4 independent experiments). *P < 0.05 and **P < 0.01, by unpaired t test.
Figure 6. Mutually exclusive binding of AHSP or eNOS to α-globin.

Myc-tagged eNOS (1 μg) was incubated with purified AHSP (25 ng) and/or α-globin (25 ng) for 90 minutes in TBS at 37°C and then immunoprecipitated with anti-Myc or IgG Dynabeads and analyzed by Western blotting using the indicated antibodies. The lanes were run on the same gel but were noncontiguous. This experiment was conducted 3 times.
To determine whether interactions with AHSP or eNOS stabilize oxidized (Fe3+) α-globin, we incubated oxygenated (Fe2+) α-globin with AHSP or eNOS and then added the oxidant potassium ferricyanide and measured the precipitation, as evidenced by light scattering at 700 nm. We observed that free α-globin precipitated rapidly, whereas eNOS remained in solution (Supplemental Figure 7A). AHSP, but not eNOS, inhibited the precipitation of oxidized α-globin (Figure 7A and ref. 17). Moreover, AHSP inhibited the precipitation of oxidized α-globin–eNOS. Thus, although AHSP and eNOS can each bind α-globin and enhance its accumulation in cells (Figure 5), only AHSP stabilizes oxidized α-globin.
Figure 7. AHSP stabilizes oxidized (Fe3+) α-globin and facilitates its reduction by eNOS.

(A) Purified oxygenated (Oxy) (Fe2+) α-globin (15 μM) was incubated with AHSP and/or full-length recombinant eNOS for 60 minutes at 37°C. Control refers to (Oxy) (Fe2+) α-globin alone. Potassium ferricyanide was added at t0 (to a final concentration of 50 μM) to induce heme oxidation. Protein precipitation was monitored by light scattering at 700 nm. D, density. (B) Reduction of the(Fe3+) α-globin–AHSP complex by the indicated proteins (either 0.15 μM CyB5R3, 0.15 μM CyB5R3 plus 1 μM CyB5, or 1 μM full-length eNOS) with 50 to 100 μM NADH. Reactions were performed in PBS with 250 U/ml catalase at 25°C at a physiologically low O2 concentration (10 Torr) with CO (100 μM) to prevent the eventual partial reoxidation of reduced α-globin. All reactions were performed 3 times. ΔA, change in light absorbance.
Reduction of (Fe3+) α-globin–AHSP by CyB5R3 plus CyB5 or eNOS.
Dioxygenation of NO by (Fe2+) α-globin produces (Fe3+) α-globin, which must be recycled to the reduced form. We studied this process in vitro, focusing on potential roles for AHSP (Figure 7B). In solution with 1 μM electron donor and acceptor proteins, CyB5R3 plus cytochrome B5 (CyB5) reduced (Fe3+) α-globin–AHSP at 3 × 10−3/second (25°C) (Figure 7B and Table 2), as with the rates achieved by other redox systems studied previously (27). In contrast, CyB5R3 alone had no effect on (Fe3+) α-globin–AHSP. There is a similar requirement for both proteins in the reduction of oxidized HbA (met HbA) in RBC, in which CyB5 serves as the direct electron donor (Table 2) (28, 29). Remarkably, eNOS reduced (Fe3+) α-globin–AHSP 6-fold more rapidly than did CyB5R plus CyB5 (Figure 7B and Table 2). eNOS also reduced (Fe3+) Cygb at a rate similar to that of (Fe3+) α-globin–AHSP and reduced met HbA approximately 5-fold more slowly (Table 2 and Supplemental Figure 7, B and C). All reductase reactions included CO, which inhibits electron transfer from the eNOS oxygenase domain and the reduced globin reaction product. Thus, eNOS reduces globins via direct electron transfer from its flavin-associated reductase domain (30–32). Of note, we could not examine the effects of any reductase system on free (Fe3+) α-globin or (Fe3+) α-globin–eNOS, because these proteins precipitated rapidly in solution at 25°C (see Figure 7A).
Table 2. Reduction of globin proteins by CyB5R, CyB5, and eNOS.

Discussion
In arterioles, eNOS-derived NO diffuses across MEJs to inhibit VSMC contractility, thereby enhancing vasodilation and O2 delivery to tissues. This process is regulated physiologically at multiple levels, including that of NO destruction, which can be mediated by globin proteins (33, 34). Our findings provide new insights into this process by showing that α-globin and AHSP participate in NO degradation in vivo. The results of previous studies support a model in which eNOS-bound α-globin degrades NO, thereby reducing its delivery across the MEJ to cause VSMC contraction and vasoconstriction (7, 8, 35). Here, we confirm and extend those prior studies by showing that the molecular chaperone AHSP is required for α-globin protein expression in ECs and that Hba1−/− and Ahsp−/− mice exhibit similarly dilated arterioles (thoracodorsal and mesenteric), with blunted α1-adrenergic contractile responses and reduced systemic blood pressure.
Free α-globin is unstable, and numerous cellular mechanisms exist to prevent its precipitation and/or degradation (17, 19, 20, 36–39). In RBC precursors, AHSP binds nascent α-globin, stabilizes its folding, and facilitates heme insertion. Ultimately, α-globin is transferred from AHSP to β-globin. Once formed, α-globin–β-globin dimers dissociate at very low rates, and AHSP is eventually degraded as HbA synthesis declines during RBC maturation. Thus, most α-globin in mature RBC is stabilized by β-globin in heterodimers and HbA heterotetramers. Ablation of the Ahsp gene destabilizes α-globin in RBC but causes only modest impairment of HbA production (22). In contrast, Ahsp loss in ECs reduces α-globin protein by approximately 70%. Thus, AHSP stabilizes α-globin to a greater extent in ECs than in RBC, probably because ECs lack β-globin. In ECs in culture, eNOS stabilizes α-globin, but to a lesser extent than does AHSP (Figure 5 and Supplemental Figure 6). Moreover, AHSP, but not eNOS, inhibits the precipitation of oxidized (Fe3+) α-globin in solution (Figure 7A).
In the MEJ, oxygenated (Fe2+) α-globin bound to eNOS promotes vasoconstriction by degrading locally produced NO to generate NO3– and oxidized (Fe3+) α-globin, which is both unstable and unable to degrade NO. Previous studies showed that AHSP stabilizes bound (Fe3+) α-globin by converting it to a hexacoordinate structure, in which iron is bound on either side of the planar heme ring by histidine residues located at α-helical positions F8 and E7 (40, 41). Here, we verified this functional interaction and show that AHSP binding to (Fe3+) α-globin facilitates its reduction by eNOS (Figure 7B). Thus, we propose a model for α-globin regulation in ECs, whereby the oxidized (Fe3+) form generated by NO degradation is released from the oxygenase domain of eNOS (7, 8), captured by AHSP, reduced to the (Fe2+) form through a bimolecular reaction with the eNOS reductase domain, and then transferred back to eNOS for another cycle of NO degradation. This model predicts that AHSP deficiency will cause the accumulation of oxidized free α-globin and its subsequent degradation, NO stabilization, and vasodilation, as we observed in Ahsp–/– mice. The affinity of AHSP for (Fe3+) α-globin is approximately 100-fold greater than for oxygenated (Fe2+) α-globin (20, 42). Although the kinetics of α-globin binding to eNOS are unknown, our model predicts that equilibria favor the binding of (Fe2+) α-globin to eNOS and (Fe3+) α-globin to AHSP. This is supported by the finding that oxidant-induced precipitation of α-globin–eNOS is inhibited by the addition of AHSP (Figure 7A).
The discovery that eNOS can serve as a direct electron donor to (Fe3+) α-globin–AHSP highlights a previously unappreciated mechanism for globin protein reduction that probably contributes to EC physiology. Additionally, CyB5R3 binds the eNOS–α-globin complex and is essential for maintaining α-globin reduction in isolated TDAs (8). Direct reduction of free or AHSP-bound (Fe3+) α-globin by CyB5R3 in the MEJ would require the levels of CyB5 to be sufficient for it to serve as the final electron donor (Figure 7B). It is also possible that CyB5R3 can donate electrons directly to eNOS-bound (Fe3+) α-globin or promote its reduction indirectly by modulating the cellular redox status.
Overall, our model predicts that NO diffusion or scavenging in ECs is regulated by redox-dependent shuttling of α-globin between eNOS and AHSP. Thus, conditions that promote heme oxidation, such as a high rate of NO degradation by α-globin, low O2 tension, or acidosis (27, 43), would favor AHSP sequestration of (Fe3+) α-globin in an inert but stable state, so as to minimize NO degradation, promote vasodilation, and enhance tissue O2 delivery. Reducing conditions would favor the accumulation of (Fe2+) α-globin bound to eNOS, local NO degradation, and vasoconstriction. Additional factors may also influence α-globin dioxygenase activity, including other EC reductase systems, arteriolar shear forces that regulate eNOS activity, inflammation, and oxidative stress (44).
Active eNOS functions as a homodimer to generate NO. To maintain catalytic activity, the reductase domains transfer electrons from NADPH via FAD and FMN to heme iron in the oxidase domains of the opposing subunits (45, 46). The eNOS reductase domain can also transfer electrons to external substrates, with the best example being cytochrome c (32). Here, we extend this concept by showing that eNOS can reduce globin proteins, including α-globin (bound to AHSP), Cygb, and neuroglobin (data not shown). This finding may have broad implications, considering that the related proteins inducible NOS (iNOS) and neuronal NOS (nNOS) could also modulate the redox states of Cygb and neuroglobin to regulate their functions in various tissues (47).
Our studies elucidate one of several mechanisms by which globin proteins influence blood vessel tone by degrading NO. RBC HbA can either produce or degrade NO, with the latter process being particularly impactful during intravascular hemolysis (48). Additionally, VSMC-expressed Cygb degrades NO, resulting in vasoconstriction and reduced tissue blood flow (49). In isolated mesenteric arteries, the NO consumption by VSMC Cygb is greater than that by EC α-globin, although the relative activities of these 2 proteins with respect to NO metabolism may vary according to specific physiologic conditions, pathologies, and vascular beds. According to our findings, NOS proteins present in VSMCs may participate in the reduction of Cygb (Table 2 and ref. 50).
The detection of α-globin and AHSP in mouse and human ECs suggests that the roles of these proteins in blood vessels are conserved across species (Figure 1). However, Etyang et al. reported normal blood pressure in a cohort of Kenyan adolescents with α-thalassemia trait caused by the loss of 2 α-globin genes (51). This contrasts with our finding that the systemic blood pressure of Hba1−/− mice (which also lack 2 α-globin genes) was lower than that of control animals. Several possible explanations may account for this apparent inconsistency. First, human EC–expressed α-globin may have minimal effects on vascular tone compared with the effects we have observed in mice. Second, human EC α-globin may modulate blood pressure or regional blood flow under specific stresses. For example, α-globin is expressed in human and mouse pulmonary artery ECs during pulmonary hypertension and could contribute to its pathophysiology by degrading NO (52). Third, low-grade hemolysis in individuals with α-thalassemia trait could raise plasma free Hb levels to degrade NO and thereby oppose the effects of α-globin reduction in ECs. Here, we studied isolated EC α-globin deficiency by transplanting α-thalassemic mice with WT hematopoietic stem cells, which generate normal RBC. Fourth, although the humans studied by Etyang et al. and the mice described here both lack 2 of the 4 total α-globin genes, the mutations are distinct and could impair EC α-globin expression to different extents (Supplemental Figure 8).
Overall, our studies provide new insights into the regulation of blood vessel contractility by showing that AHSP and α-globin participate in the elimination of NO, a potent vasodilator. Our model predicts that dynamic, reversible binding of α-globin to AHSP or eNOS represents a redox-sensitive mechanism for regulating blood vessel NO concentrations according to physiologic requirements. Further studies are required to validate this model and to fully appreciate the physiologic implications of our findings in mice and humans.
Methods
Mice.
The breeding and analysis of Ahsp−/− and Hba1−/− mice have been described previously (17, 24). Experiments were conducted with 6- to 24-week-old mice, with WT littermates used as controls. Bone marrow transplantation was performed as described previously (53). Briefly, recipient mice were lethally irradiated, and after a 24-hour interval, they received a transplant of 2 × 106 bone marrow cells from donor mice via tail-vein injection. Engraftment was determined by regenotyping the mice 4 weeks later.
DNA constructs and lentiviral vectors.
EGFP or mCherry fluorescent reporter sequences were fused in-frame to human α-globin, AHSP, or eNOS and subcloned into pCl20c-MSCV (54, 55). Vector particles were prepared by transiently transfecting HEK293T cells with the vector genome plasmid and the packaging helper plasmids pCAGG-GP1.1R, pCAGG-VSVG, and pCAG4-RTR2 (56). The supernatant was concentrated by ultracentrifugation and titrated on HOS cells by serial dilution. Digital droplet PCR was then used to determine the vector genome copy number.
Cell culture and lentiviral transduction.
Human primary coronary ECs (Lonza) were grown in EGM-2 Bullet Kit Medium (Lonza). Lentiviruses were added to the ECs for 72 hours. Cells were analyzed by microscopy with a Nikon Eclipse Ti microscope and NIS-Elements software, by flow cytometry with an LSR/Fortessa cell analyzer from BD Biosciences, and by Western blotting.
Antibodies.
The source, application, concentration, and vendor of each antibody are listed in Supplemental Table 1.
Blood vessel isolation and vasoreactivity studies.
Mice were euthanized by CO2 asphyxiation. The TDAs and mesenteric arteries were isolated by dissection, cannulated, and washed with Krebs-HEPES buffer supplemented with 1% BSA to remove RBC. Pressure myography was performed as described previously (57), and the results were analyzed using SigmaPlot, version 10.0.
Blood pressure measurement.
Mice were anesthetized with isoflurane, and radiotelemetry units (TA11PA-C10; Data Sciences International [DSI]) were implanted into their left carotid artery. In each animal, the catheter was tunneled through to the radio transmitter, which was placed into a subcutaneous pouch along the flank. Mice were allowed to recover for 7 days after surgery in order to regain their normal circadian rhythms before arterial pressure measurements and experiments were initiated. Continuous blood pressure measurements were recorded using Dataquest ART 20 software (DSI).
Histology.
For standard histologic analysis, mice were anesthetized, the arteries were exposed by dissection, and the animals were perfused with 10% formalin. The arteries were removed, embedded in paraffin, sectioned at a thickness of 4 μm, and stained with H&E. For measurements of arterial lumen diameters, 3 sections 100 μm apart were prepared from each artery at a random site. Cross-sectional area measurements (in μm2) of arteries from control, Ahsp−/−, and Hba1−/− mice were made using a Nikon Eclipse Ni microscope and Nikon NIS-Elements software. Lumen diameters were determined in a blinded manner by a board-certified veterinary pathologist (HST). For electron microscopy, arteries from animals were perfused with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, and postfixed for 1.5 hours in 2% osmium tetroxide in 0.1 M cacodylate buffer with 0.3% potassium ferrocyanide. The arteries were dehydrated through a graded ethanol series, infiltrated with propylene oxide, and embedded in epoxy resin, which was polymerized at 70°C overnight. Semithin sections (0.5-μm thick) were stained with toluidine blue.
Histochemical staining and immunostaining.
Arteries were isolated from anesthetized, formalin-perfused mice, and 4-μm paraffin sections were prepared. For flat mounts, the arteries were sectioned longitudinally and processed whole. Whenever possible, we analyzed multiple antibodies prepared against different regions of the same protein. All antibodies used (Supplemental Table 1) were tested for specificity by Western blot analysis and immunostaining against a combination of positive control (spleen and bone marrow) and negative control (adult liver) tissues. For immunofluorescence, sections were permeabilized with 0.1% Triton X-100 in PBS for 15 minutes at room temperature; treated with 3% BSA and 3% goat serum, 0.1% Triton, and 0.05% Tween-20 in PBS for 1 hour; and then incubated with a primary antibody overnight at 4°C. Sections were then washed extensively, incubated with the secondary antibody for 2 hours at room temperature, mounted on glass slides, and visualized with a confocal laser scanning microscope (Nikon Eclipse TE2000-E). Bright-field images were acquired with a Nikon Eclipse Ni microscope. All slides were initially analyzed in a blinded manner by a board-certified veterinary pathologist (HST).
Tissue microarray pathology data.
A tissue microarray (TMA) containing nonthalassemic human vessels (n = 7) from 4 anatomic locations (the mesentery, heart, lung, and kidney) was constructed at St. Jude Children’s Research Hospital. The IHC labeling conditions for anti–α-globin, anti–β-globin, and anti-AHSP are described in Supplemental Table 1. IHC-labeled TMAs were digitized using an Aperio ScanScope scanner. Both vascular endothelium and vascular smooth muscle cores containing either mesenteric or coronary vessels were separately annotated with Aperio ImageScope software. Chromogenic labeling was analyzed using an optimized Aperio Positive Pixel Count Algorithm, version 9 (Leica Biosystems), for the batch analysis of annotated areas of interest across all TMAs. An algorithm output provided 1+, 2+, and 3+ intensity values for pixels that were positive, along with the total number of pixels (excluding white areas) in each annotated layer, to generate a separate positivity score for the vascular endothelium and smooth muscle.
RNA extraction and RT-qPCR.
Total RNA was extracted from arteries as described previously (58). Isolated arteries were cannulated and washed extensively with Krebs-HEPES buffer to remove RBC. The RNA purity and concentration were quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad), followed by RT-qPCR analysis using SYBR Green (Applied Biosystems). Gene-specific primers were designed using Primer3 (Supplemental Table 3). RT-qPCRs were performed in duplicate and normalized against Actb mRNA; the comparative ΔΔCt method was used to determine the relative mRNA levels for each gene studied.
Western blot analysis.
Washed, isolated arteries were homogenized in RIPA buffer with protease inhibitor cocktail (Sigma-Aldrich) at 4°C, using a 200-μl hand-driven glass-glass Potter-Elvehjem Micro Tissue Grinder. Large cellular debris was concentrated by centrifugation (1,000 g for 5 minutes at 4°C) and discarded, followed by quantification of the protein in the supernatants using a Pierce BCA Kit (Thermo Fisher Scientific). Next, 30 μg of each arterial sample or 10 μg of transduced ECs was resolved by 12% SDS-PAGE, transferred onto a PVDF membrane, and incubated with the indicated antibodies (Supplemental Figure 1). Immunoreactive bands were labeled with the appropriate secondary antibodies coupled to HRP (Supplemental Table 1) and then detected with Pierce ECL Plus Western Blotting Substrate (Thermo Fisher Scientific). The apparent molecular mass of each protein was estimated by comparison with the PageRuler Precision Plus Protein Ladder (Bio-Rad). Signals were quantified using AlphaVIEW SA, version 3.4.0.0 software (ProteinSimple). Membranes were treated with Restore Plus Western Blot Stripping Solution (Thermo Fisher Scientific) to enable the sequential analysis of different proteins on the same membrane.
Co-IP.
Purified Myc-tagged eNOS was purchased from Origene. Purified α-globin and AHSP proteins were generated as previously described (20). Protein fractions were each incubated at equimolar concentrations for 90 minutes at 37ºC on a shaker, and then anti-Myc Dynabeads were added and the mixture incubated for 2 hours at 4°C. The beads were subjected to extensive washing with TBST buffer complemented with 1% BSA, and then the bound proteins were removed by incubation with Laemmli buffer for 10 minutes at 95°C and subjected to Western blot analysis.
Precipitation of oxidized α-globin.
α-Globin was diluted with 50 mM Tris, pH 7.4, 150 mM NaCl, and 1 mM EDTA to a final concentration of 15 μM and preincubated at 4°C with recombinant AHSP (20 μM or 40 μM) and/or eNOS (15 μM or 30 μM) (a gift from Huiying Li and Thomas L. Poulos, University of California, Irvine, Irvine, California, USA) in a volume of 90 μl for 60 minutes. The samples were warmed to 37°C, followed by addition of 10 μl of 0.5 mM potassium ferricyanide. Protein precipitation was quantified by light scattering at 700 nm using a Synergy H1 Plate Reader (BioTek) (17).
Reduction of oxidized (Fe3+) α-Hb bound to AHSP.
Oxygenated (Fe2+) α-globin (50 μM) was incubated with recombinant human AHSP (50 μM) at 25°C for 10 minutes. Under these conditions, AHSP-bound α-globin undergoes auto-oxidation to hexacoordinate (Fe3+) α-globin, as monitored spectrophotometrically. Globin proteins were oxidized by adding potassium ferricyanide, which was then immediately removed by Sephadex G-25 (GE Healthcare Life Sciences) chromatography. Experiments performed under anaerobic conditions included 100 μM CO. Potential reducing proteins were added at the following concentrations: 0.2 μM soluble recombinant CyB5R3 (LSBio, LS-G1599); 1 μM CyB5 (a gift from Gilles Truan, Institut National des Sciences Appliquées, Toulouse, France) (27); and 1 μM recombinant soluble eNOS (a gift from Huiying Li and Thomas L. Poulos, University of California, Irvine) premixed with 50 μM reduced NADPH or 100 μM reduced NADH. All reactions were performed in PBS with 250 U/ml bovine liver catalase (Sigma-Aldrich, catalog C40) and 10% glycerol at pH 7.4 and 25°C. For reactions under aerobic conditions, 50 U/ml superoxide dismutase from Aspergillus niger (Sigma-Aldrich, catalog S2515) was added, whereas under anaerobic conditions, 3 U/ml glucose oxidase (Sigma-Aldrich, catalog G2133) and 1 mM β-D(+)-glucose were added. The reactions were performed under hypoxic conditions with CO to prevent reoxidation of the reduced product by O2. The reaction kinetics were recorded as described previously (27).
Statistics.
All data represent the mean ± SEM. Statistical analyses were performed using GraphPad Prism 6.0 software. For multiple comparisons, statistical analyses were performed using a 1-way ANOVA with pairwise comparisons. A 2-tailed Student’s t test was used for individual comparisons if they were normally distributed. For vascular reactivity cumulative concentration-response curves, a 2-way ANOVA was performed, followed by a Bonferroni post hoc test. P values of less than 0.05 were considered statistically significant.
Study approval.
Mouse experimental protocols were approved by the IACUCs of St. Jude Children’s Research Hospital and the University of Virginia. Human specimens collected from patients identified as not having thalassemia were provided in a deidentified format. The study was reviewed and approved by the IRB of St. Jude Children’s Research Hospital.
Author contributions
CL, BEI, and MJW conceived and designed the experiments. CL, JTB, AF, LAB, JMK, MEG, HST, HA, and LK performed the experiments and collected data. CL, BEI, and MJW interpreted data and wrote the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Stephanie Fowler and Kalin Mayberry (Department of Hematology, St. Jude Children’s Research Hospital) for mouse studies; Victoria Frohlich, Sharon Frase, Linda Horner, and Randall Wakefield (St. Jude Cellular Imaging Shared Research Core); Sean Savage, Michael Anderson, and Joseph Emmons (St. Jude Veterinary and Pathology Core); Shao Youming and Heath Richard (St. Jude Protein Production Core); Cecilia Western (St. Jude Vector Core); and Keith A. Laycock (St. Jude Department of Scientific Editing). We thank Huiying Li and Thomas L. Poulos (University of California, Irvine) for providing full-length recombinant eNOS protein (supported by NIH grant GM57353), for helpful discussions on eNOS biochemistry, and for reviewing this manuscript. We thank Gilles Truan (Institut National des Sciences Appliquées de Toulouse, Toulouse, France) for providing the CyB5 expression plasmid. We thank John Olson (Rice University, Houston, Texas, USA) for helpful discussions. This work was supported by NIH grants R01 DK61692 (to MJW) and R01 HL088554 (to BEI and MJW); an American Heart Association Postdoctoral Fellowship (to CL); and the American Lebanese Syrian Associated Charities (ALSAC).
Version 1. 10/08/2018
Electronic publication
Version 2. 11/01/2018
Print issue publication
Funding Statement
Mitchell J. Weiss
Brant E. Isakson and Mitchell J. Weiss
Christophe Lechauve
Mitchell J. Weiss
Huiying Li and Thomas L. Poulos
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(11):5073–5082.https://doi.org/10.1172/JCI99933.
See the related Commentary at Escorting α-globin to eNOS: α-globin–stabilizing protein paves the way.
Contributor Information
Christophe Lechauve, Email: christophe.lechauve@stjude.org.
Joshua T. Butcher, Email: jbutcher@augusta.edu.
Abdullah Freiwan, Email: Abdullah.Freiwan@STJUDE.ORG.
Lauren A. Biwer, Email: biwer@virginia.edu.
Julia M. Keith, Email: Julia.Keith@STJUDE.ORG.
Miranda E. Good, Email: mgood@virginia.edu.
Hans Ackerman, Email: hans.ackerman@nih.gov.
Laurent Kiger, Email: laurent.kiger@inserm.fr.
Brant E. Isakson, Email: brant@virginia.edu.
Mitchell J. Weiss, Email: mitch.weiss@stjude.org.
References
- 1.Mettananda S, Gibbons RJ, Higgs DR. Understanding α-globin gene regulation and implications for the treatment of β-thalassemia. Ann N Y Acad Sci. 2016;1368(1):16–24. doi: 10.1111/nyas.12988. [DOI] [PubMed] [Google Scholar]
- 2.Saha D, Patgaonkar M, Shroff A, Ayyar K, Bashir T, Reddy KV. Hemoglobin expression in nonerythroid cells: novel or ubiquitous? Int J Inflam. 2014;2014:803237. doi: 10.1155/2014/803237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weatherall DJ. Thalassaemia: the long road from bedside to genome. Nat Rev Genet. 2004;5(8):625–631. doi: 10.1038/nrg1406. [DOI] [PubMed] [Google Scholar]
- 4.Vinogradov SN, Moens L. Diversity of globin function: enzymatic, transport, storage, and sensing. J Biol Chem. 2008;283(14):8773–8777. doi: 10.1074/jbc.R700029200. [DOI] [PubMed] [Google Scholar]
- 5.Heberlein KR, Straub AC, Isakson BE. The myoendothelial junction: breaking through the matrix? Microcirculation. 2009;16(4):307–322. doi: 10.1080/10739680902744404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandow SL, et al. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin Exp Pharmacol Physiol. 2009;36(1):67–76. doi: 10.1111/j.1440-1681.2008.05076.x. [DOI] [PubMed] [Google Scholar]
- 7.Straub AC, et al. Hemoglobin α/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol. 2014;34(12):2594–2600. doi: 10.1161/ATVBAHA.114.303974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straub AC, et al. Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature. 2012;491(7424):473–477. doi: 10.1038/nature11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgado M, Cairrão E, Santos-Silva AJ, Verde I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci. 2012;69(2):247–266. doi: 10.1007/s00018-011-0815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang PL, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377(6546):239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 11.Scotland RS, et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111(6):796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- 12.Doyle MP, Hoekstra JW. Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Inorg Biochem. 1981;14(4):351–358. doi: 10.1016/S0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 13.Angelo M, Hausladen A, Singel DJ, Stamler JS. Interactions of NO with hemoglobin: from microbes to man. Meth Enzymol. 2008;436:131–168. doi: 10.1016/S0076-6879(08)36008-X. [DOI] [PubMed] [Google Scholar]
- 14.Gladwin MT, Lancaster JR, Freeman BA, Schechter AN. Nitric oxide’s reactions with hemoglobin: a view through the SNO-storm. Nat Med. 2003;9(5):496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- 15.Keller TC, et al. Modulating vascular hemodynamics with an alpha globin mimetic peptide (HbαX) Hypertension. 2016;68(6):1494–1503. doi: 10.1161/HYPERTENSIONAHA.116.08171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parikh J, Kapela A, Tsoukias NM. Can endothelial hemoglobin-α regulate nitric oxide vasodilatory signaling? Am J Physiol Heart Circ Physiol. 2017;312(4):H854–H866. doi: 10.1152/ajpheart.00315.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kihm AJ, et al. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature. 2002;417(6890):758–763. doi: 10.1038/nature00803. [DOI] [PubMed] [Google Scholar]
- 18.Khandros E, Weiss MJ. Protein quality control during erythropoiesis and hemoglobin synthesis. Hematol Oncol Clin North Am. 2010;24(6):1071–1088. doi: 10.1016/j.hoc.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khandros E, et al. Insights into hemoglobin assembly through in vivo mutagenesis of α-hemoglobin stabilizing protein. J Biol Chem. 2012;287(14):11325–11337. doi: 10.1074/jbc.M111.313205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mollan TL, Khandros E, Weiss MJ, Olson JS. Kinetics of α-globin binding to α-hemoglobin stabilizing protein (AHSP) indicate preferential stabilization of hemichrome folding intermediate. J Biol Chem. 2012;287(14):11338–11350. doi: 10.1074/jbc.M111.313247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heberlein KR, et al. Plasminogen activator inhibitor-1 regulates myoendothelial junction formation. Circ Res. 2010;106(6):1092–1102. doi: 10.1161/CIRCRESAHA.109.215723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong Y, et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114(10):1457–1466. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Surney SJ, Popp RA. Changes in alpha-globin gene expression in mice of two alpha-globin haplotypes during development. Biochem Genet. 1990;28(9-10):445–457. doi: 10.1007/BF00554373. [DOI] [PubMed] [Google Scholar]
- 24.Chang J, et al. Inactivation of mouse alpha-globin gene by homologous recombination: mouse model of hemoglobin H disease. Blood. 1996;88(5):1846–1851. [PubMed] [Google Scholar]
- 25.Khandros E, Thom CS, D’Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood. 2012;119(22):5265–5275. doi: 10.1182/blood-2011-12-397729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gell D, Kong Y, Eaton SA, Weiss MJ, Mackay JP. Biophysical characterization of the alpha-globin binding protein alpha-hemoglobin stabilizing protein. J Biol Chem. 2002;277(43):40602–40609. doi: 10.1074/jbc.M206084200. [DOI] [PubMed] [Google Scholar]
- 27.Kiger L, Vasseur C, Domingues-Hamdi E, Truan G, Marden MC, Baudin-Creuza V. Dynamics of α-Hb chain binding to its chaperone AHSP depends on heme coordination and redox state. Biochim Biophys Acta. 2014;1840(1):277–287. doi: 10.1016/j.bbagen.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Hultquist DE, Passon PG. Catalysis of methaemoglobin reduction by erythrocyte cytochrome B5 and cytochrome B5 reductase. Nature New Biol. 1971;229(8):252–254. doi: 10.1038/newbio229252a0. [DOI] [PubMed] [Google Scholar]
- 29.Kuma F. Properties of methemoglobin reductase and kinetic study of methemoglobin reduction. J Biol Chem. 1981;256(11):5518–5523. [PubMed] [Google Scholar]
- 30.Iyanagi T. Structure and function of NADPH-cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem Biophys Res Commun. 2005;338(1):520–528. doi: 10.1016/j.bbrc.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 31.Spencer AL, Bagai I, Becker DF, Zuiderweg ER, Ragsdale SW. Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J Biol Chem. 2014;289(43):29836–29858. doi: 10.1074/jbc.M114.582783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klatt P, Heinzel B, John M, Kastner M, Böhme E, Mayer B. Ca2+/calmodulin-dependent cytochrome c reductase activity of brain nitric oxide synthase. J Biol Chem. 1992;267(16):11374–11378. [PubMed] [Google Scholar]
- 33.Gödecke A. On the impact of NO-globin interactions in the cardiovascular system. Cardiovasc Res. 2006;69(2):309–317. doi: 10.1016/j.cardiores.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Gardner PR, Martin LA, Hall D, Gardner AM. Dioxygen-dependent metabolism of nitric oxide in mammalian cells. Free Radic Biol Med. 2001;31(2):191–204. doi: 10.1016/S0891-5849(01)00569-X. [DOI] [PubMed] [Google Scholar]
- 35.Butcher JT, Johnson T, Beers J, Columbus L, Isakson BE. Hemoglobin α in the blood vessel wall. Free Radic Biol Med. 2014;73:136–142. doi: 10.1016/j.freeradbiomed.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou S, Olson JS, Fabian M, Weiss MJ, Gow AJ. Biochemical fates of alpha hemoglobin bound to alpha hemoglobin-stabilizing protein AHSP. J Biol Chem. 2006;281(43):32611–32618. doi: 10.1074/jbc.M607311200. [DOI] [PubMed] [Google Scholar]
- 37.Mollan TL, Yu X, Weiss MJ, Olson JS. The role of alpha-hemoglobin stabilizing protein in redox chemistry, denaturation, and hemoglobin assembly. Antioxid Redox Signal. 2010;12(2):219–231. doi: 10.1089/ars.2009.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mollan TL, et al. α-Hemoglobin stabilizing protein (AHSP) markedly decreases the redox potential and reactivity of α-subunits of human HbA with hydrogen peroxide. J Biol Chem. 2013;288(6):4288–4298. doi: 10.1074/jbc.M112.412064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dickson CF, et al. α-Hemoglobin-stabilizing protein (AHSP) perturbs the proximal heme pocket of oxy-α-hemoglobin and weakens the iron-oxygen bond. J Biol Chem. 2013;288(27):19986–20001. doi: 10.1074/jbc.M112.437509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng L, et al. Structure of oxidized alpha-haemoglobin bound to AHSP reveals a protective mechanism for haem. Nature. 2005;435(7042):697–701. doi: 10.1038/nature03609. [DOI] [PubMed] [Google Scholar]
- 41.Feng L, et al. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119(5):629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 42.Brillet T, et al. Alpha-hemoglobin stabilizing protein (AHSP), a kinetic scheme of the action of a human mutant, AHSPV56G. J Biol Chem. 2010;285(23):17986–17992. doi: 10.1074/jbc.M109.098491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brantley RE, Smerdon SJ, Wilkinson AJ, Singleton EW, Olson JS. The mechanism of autooxidation of myoglobin. J Biol Chem. 1993;268(10):6995–7010. [PubMed] [Google Scholar]
- 44.Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol (Oxf) 2017;219(2):382–408. doi: 10.1111/apha.12725. [DOI] [PubMed] [Google Scholar]
- 45.Daff S. NO synthase: structures and mechanisms. Nitric Oxide. 2010;23(1):1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 46.Stuehr DJ, Tejero J, Haque MM. Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276(15):3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reeder BJ. Redox and peroxidase activities of the hemoglobin superfamily: relevance to health and disease. Antioxid Redox Signal. 2017;26(14):763–776. doi: 10.1089/ars.2016.6803. [DOI] [PubMed] [Google Scholar]
- 48.Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free Radic Biol Med. 2013;61:464–472. doi: 10.1016/j.freeradbiomed.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, et al. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat Commun. 2017;8:14807. doi: 10.1038/ncomms14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brophy CM, Knoepp L, Xin J, Pollock JS. Functional expression of NOS 1 in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2000;278(3):H991–H997. doi: 10.1152/ajpheart.2000.278.3.H991. [DOI] [PubMed] [Google Scholar]
- 51.Etyang AO, et al. Blood pressure and arterial stiffness in Kenyan adolescents with α+thalassemia. J Am Heart Assoc. 2017;6(4):e005613. doi: 10.1161/JAHA.117.005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alvarez RA, et al. Targeting pulmonary endothelial hemoglobin α improves nitric oxide signaling and reverses pulmonary artery endothelial dysfunction. Am J Respir Cell Mol Biol. 2017;57(6):733–744. doi: 10.1165/rcmb.2016-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA. Correction of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther. 2009;17(2):245–252. doi: 10.1038/mt.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanawa H, et al. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transduce primitive hematopoietic cells from human blood. Mol Ther. 2002;5(3):242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- 55.Hanawa H, et al. Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood. 2004;103(11):4062–4069. doi: 10.1182/blood-2004-01-0045. [DOI] [PubMed] [Google Scholar]
- 56.Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW, Persons DA. Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood. 2009;113(23):5747–5756. doi: 10.1182/blood-2008-10-186684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Billaud M, Lohman AW, Straub AC, Parpaite T, Johnstone SR, Isakson BE. Characterization of the thoracodorsal artery: morphology and reactivity. Microcirculation. 2012;19(4):360–372. doi: 10.1111/j.1549-8719.2012.00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bridenbaugh EA. Isolation and preparation of RNA from rat blood and lymphatic microvessels for use in microarray analysis. Methods Mol Biol. 2012;843:265–289. doi: 10.1007/978-1-61779-523-7_24. [DOI] [PubMed] [Google Scholar]
- 59.Amdahl MB, Sparacino-Watkins CE, Corti P, Gladwin MT, Tejero J. Efficient reduction of vertebrate cytoglobins by the cytochrome b5/cytochrome b5 reductase/NADH system. Biochemistry. 2017;56(30):3993–4004. doi: 10.1021/acs.biochem.7b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.