

Abstract
Previous findings showed that in mice, complete knockout of activity-dependent neuroprotective protein (ADNP) abolishes brain formation, while haploinsufficiency (Adnp+/–) causes cognitive impairments. We hypothesized that mutations in ADNP lead to a developmental/autistic syndrome in children. Indeed, recent phenotypic characterization of children harboring ADNP mutations (ADNP syndrome children) revealed global developmental delays and intellectual disabilities, including speech and motor dysfunctions. Mechanistically, ADNP includes a SIP motif embedded in the ADNP-derived snippet drug candidate NAP (NAPVSIPQ, also known as CP201), which binds to microtubule end–binding protein 3, essential for dendritic spine formation. Here, we established a unique neuronal membrane–tagged, GFP-expressing Adnp+/– mouse line allowing in vivo synaptic pathology quantification. We discovered that Adnp deficiency reduced dendritic spine density and altered synaptic gene expression, both of which were partly ameliorated by NAP treatment. Adnp+/–mice further exhibited global developmental delays, vocalization impediments, gait and motor dysfunctions, and social and object memory impairments, all of which were partially reversed by daily NAP administration (systemic/nasal). In conclusion, we have connected ADNP-related synaptic pathology to developmental and behavioral outcomes, establishing NAP in vivo target engagement and identifying potential biomarkers. Together, these studies pave a path toward the clinical development of NAP (CP201) for the treatment of ADNP syndrome.
Keywords: Therapeutics
Keywords: Drug therapy
Introduction
Essential for both brain formation (1, 2) and function (3), activity-dependent neuroprotective protein (ADNP) (4, 5) regulates hundreds of key genes (2, 6). ADNP is involved in chromatin function and transcription (2, 7, 8) by interacting with heterochromatin proteins (HP1) (2, 8, 9) and chromodomain helicase DNA-binding protein 4 (CHD4) (8) and with the SWItch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex (10), which is also linked to RNA alternative splicing (11). ADNP is further associated with protein translation (12), microtubule dynamics and axonal transport (6), and autophagy (6, 13). Although complete Adnp knockout is lethal, Adnp haploinsufficient (Adnp+/–) mice survive but exhibit cognitive and social deficits as well as aging-associated microtubule tubulin–associated unit (tau) pathology and neurodegeneration (3). This phenotype is preserved even if the deficient mice are outbred (6, 12), attesting to the strong impact of the Adnp gene on neuronal function.
By exploiting whole-exome sequencing to examine approximately 6,000 undiagnosed autistic children, ADNP was discovered as a major de novo mutated causal gene (14–16) for children suffering from intellectual disabilities (IDs) (15), among other afflictions. ADNP syndrome (Helsmoortel–Van der Aa syndrome) characterization is based on an existing large body of ADNP research and the availability of Adnp+/– mice (3, 17). Furthermore, premature primary tooth eruption was recently suggested as an early diagnostic biomarker for ADNP syndrome, with ADNP also regulating tooth eruption in mice (17, 18). In-depth case studies of ADNP syndrome provide a broad view of the phenotype and identified, among other impairments, global developmental delays, ID, speech impediments, and motor dysfunctions (17, 19, 20). With more than 160 children diagnosed (http://www.adnpkids-researchfoundation.org/research.html; http://www.coronisns.com/) and a projection of approximately 13,200 ADNP syndrome cases in the developed world, and given its association with other related autism-linked genes (6, 12), it is of interest to further understand ADNP functions in vivo and implement therapeutic regimens.
In this respect, an ADNP-derived neuroprotective 8-aa peptide, NAPVSIPQ (or NAP, which is also called davunetide or CP201), which enhances cognitive function in Adnp+/– mice (3), was discovered in the Gozes laboratory. NAP efficacy in cognitive enhancement is not limited to laboratory models but is also translated to humans, with favorable intranasal brain bioavailability and a broad safety profile (21) in addition to cognitive and functional protection shown in clinical trials involving amnestic mild cognitive impairment and in patients with schizophrenia (22–25).
Mechanistically, the SIP motif in NAP (NAPVSIPQ) is the signature motif that interacts with microtubule end–binding proteins, such as end-binding proteins 1 and 3 (EB1 and EB3), and enhances the capacity of microtubule plus-end (growing end) tracking proteins (+TIPs) (including ADNP) to bind to the dynamic microtubule (26). EB3 is essential for dendritic spine formation, and NAP enhancement of dendritic spine formation is EB3 dependent. The interaction of NAP with EB1 and EB3 dramatically enhances tau recruitment to the microtubule shaft, protecting against neurodegeneration, as excess free tau may be prone to hyperphosphorylation and aggregation leading to neurodegeneration (27). Furthermore, NAP enhances ADNP interaction with microtubule-associated protein 1 light chain 3 (13), which initiates autophagosome formation and protects the cells against the accumulation of misfolded proteins.
In the current study, we predicted that the Adnp+/– mouse model would mimic the developmental delays and synaptic dysfunctions exhibited by children with ADNP syndrome. This prediction is coupled with the fact that most children with ADNP syndrome suffer from heterozygous nonsense (STOP) or truncating frameshift mutations (15, 17, 28), with some showing almost complete deletions of 1 allele (19, 29), presenting a haploinsufficient genotype/phenotype. A comprehensive comparative analysis of 78 children with ADNP syndrome with mutations spanning the entire protein suggested a similar partial loss of function among the different phenotypic outcomes, with somewhat increased severity in children carrying the most abundant mutation p.Tyr719* (27). This, combined with previous results showing that both ADNP alleles are expressed in mutated human cells, with 1 allele missing important domains in the intact protein (15), strengthens the idea that the Adnp+/– mouse is a predictive model for ADNP heterozygous mutation deficiency in humans.
Here, by labeling neurons with GFP in the Adnp+/– mice, we show that haploinsufficiency negatively impacted hippocampal and cortical synaptic formations in vivo and that this was further rescued by NAP treatment. Previous RNA-sequencing (RNA-seq) experiments revealed that ADNP mutations in patient-derived lymphoblastoid cells and mouse Adnp haploinsufficiency affected the expression of shared multifunctional peripheral and hippocampal genes (6, 17). These findings were verified and extended in the current study, which showed regulation and partial reversal with NAP treatment. We further characterized, for the first time to our knowledge, developmental milestones in postnatal Adnp+/– mice. We discovered that Adnp+/– mice mimic the observed global developmental delays, including vocalization impediments, found in children with ADNP syndrome. Additionally, the mouse deficits were partially rescued by treatment with NAP. Considering that NAP showed broad safety in clinical trials in adult human cohorts with amnestic mild cognitive impairment, schizophrenia, and progressive supranuclear palsy (22–25), our current results now pave the path toward a clinical therapy for ADNP syndrome.
Results
Adnp+/– mice display a significantly decreased dendritic spine density, compared with Adnp+/+ mice, that is partly rescued by NAP treatment.
GFP-expressing mouse lines (30, 31) allow for the determination of dendritic spine morphology and synaptic structure (31). Here, Adnp+/– mice were bred with a membrane-bound GFP–expressing (mGFP-expressing) mouse line (L15) to produce an Adnp+/–-GFP mouse model.
Dendritic spines, which are small swellings of the dendritic tree, were evaluated in both hippocampal CA1 pyramidal cells and the L5 cortical layer controlling cognitive and motor functions, respectively (Figure 1, Figure 2, Supplemental Tables 1 and 2, and Supplemental Figures 1–4; supplemental material available online with this article; https://doi.org/10.1172/JCI98199DS1). Measurements were made for (a) density of dendritic spines (GFP puncta label), classified into subgroups on the basis of the following morphology types: stubby spines (<0.5 μm in length, lacking a clear head); mushroom spines (mushroom-shaped head, approximately 1 μm in length); and thin spines (with an elongated narrow neck with a distinctive head) (32, 33); (b) density of shaft synapses (postsynaptic density PSD95 puncta immunogold labeling, representing immature synapses); and (c) shaft synapse volume (PSD95, puncta volume, representing the degree of synapse maturity). Genotype, sex, and drug treatment effects were all evaluated.
Figure 1. Adnp+/– mice, compared with Adnp+/+ mice, display a significant decrease in hippocampal dendritic spine density that is ameliorated by NAP.

Average total spine density (males: Adnp+/+ n = 84, Adnp+/– n = 75, Adnp+/– NAP, n = 73; females: Adnp+/+ n = 48, Adnp+/– n = 45, Adnp+/– NAP, n = 45 dendrites/experimental group) and shaft synapse density (males: Adnp+/+ n = 11, Adnp+/– n = 15, Adnp+/– NAP, n = 10; females: Adnp+/+ n = 11, Adnp+/– n = 19, Adnp+/– NAP, n = 19 dendrites/experimental group). Total spine density was significantly decreased in both male and female Adnp+/– mice, with NAP significantly increasing spine density. Shaft synapse density was significantly increased in males and reduced by NAP in both sexes. A 2-way ANOVA with Tukey’s post hoc test was performed. Underlined numbers beneath the graphs represent the mean ± SEM. (A) In male mice, for total spine density, main genotype [F(1,290) = 62.278, P < 0.001] and interaction [F(1,290) = 31.385, P < 0.001] effects were found. For shaft synapses, main genotype [F(1,42) = 23.358, P < 0.001] and treatment [F(1,42) = 9.752, P = 0.003] effects were found. Tukey’s post hoc test revealed significant differences between Adnp+/+ and Adnp+/– mice (***P < 0.001) and between NAP- and vehicle-treated Adnp+/– mice (**P < 0.01 and ***P < 0.001). (B) In female mice, for total spine density, main genotype [F(1,183) = 9.327, P = 0.003], treatment [F(1,183) = 11.167, P = 0.001], and interaction [F(1,183) = 17.332, P < 0.001] effects were found. For shaft synapses, a main treatment effect was found [F(1,71) = 13.726, P < 0.001]. Tukey’s post hoc test revealed significant differences between Adnp+/+ and Adnp+/– mice (***P < 0.001) and between NAP- and vehicle-treated Adnp+/– mice (**P < 0.01 and ***P < 0.001). (A and B) Adnp+/+ data are reshown in Supplemental Figure 1, A and B. Scale bars: 2 μm.
Figure 2. Adnp+/– mice, compared with Adnp+/+ mice, display significantly decreased cortical dendritic spine density that is ameliorated by NAP.

Average total spine density (males: Adnp+/+ n = 47, Adnp+/– n = 43, Adnp+/– NAP, n = 45; females: Adnp+/+ n = 47, Adnp+/– n = 47, Adnp+/– NAP, n = 49 dendrites/experimental group) and shaft synapse density (males: Adnp+/+ n = 14, Adnp+/– n = 14, Adnp+/– NAP, n = 17; females: Adnp+/+ n = 18, Adnp+/– n = 18, Adnp+/– NAP, n = 17 dendrites/experimental group). Total spine density was significantly decreased in both male and female Adnp+/– mice, with NAP significantly increasing spine density. Shaft synapse density was significantly increased in males and reduced by NAP in both sexes. A 2-way ANOVA with Tukey’s post hoc test was performed. Underlined numbers beneath the graphs represent the mean ± SEM. (A) In male mice, for total spine density, main genotype [F(1,178) = 26.892, P < 0.001], treatment [F(1,178) = 29.250, P = 0.001], and interaction [F(1,178) = 82.876, P < 0.001] effects were found. For shaft synapses, main genotype [F(1,55) = 5.967, P = 0.018)] and interaction [F(1,55) = 5.769, P = 0.020] effects were found. Tukey’s post hoc test revealed significant differences between Adnp+/+ and Adnp+/– mice (**P < 0.01 and ***P < 0.001) and between NAP- and vehicle-treated Adnp+/– mice (*P < 0.05 and ***P < 0.001). (B) In female mice, for total spine density, main genotype [F(1,188) = 53.088, P < 0.001], treatment [F(1,188) = 22.105, P < 0.001], and interaction [F(1,188) = 5.506, P = 0.020] effects were found. For shaft synapses, a main treatment effect was found [F(1,67) = 12.743, P < 0.001]. Tukey’s post hoc test revealed significant differences between Adnp+/+ and Adnp+/– mice (***P < 0.001) and between NAP- and vehicle-treated Adnp+/– mice (**P < 0.01). (A and B) Adnp+/+ data are reshown in Supplemental Figure 2, A and B. Scale bars: 2 μm.
The measurements showed similar patterns in both tested brain areas, with Adnp deficiency resulting in substantial decreases in spine density (male and female mice) and increases in PSD95-asymmetric shaft synapses (males only, as indicated by increased localization of PSD95 in dendritic shafts rather than spines), which were all rescued by NAP treatment. In female mice, NAP treatment significantly decreased shaft synapses. Closer inspection suggested a more severe Adnp+/– genotype effect on total spine density in the male cortex compared with the hippocampus (Figure 1A, –1.56-fold reduction compared with Figure 2A, –1.83-fold reduction compared with the Adnp+/+ genotype). In hippocampal CA1 pyramidal cells, all dendritic spine subtypes were reduced in the Adnp+/– mice, except for the thin spines observed in males. The spine loss was rescued by NAP treatment, except for the stubby spines seen in males (Supplemental Figure 1). Supplemental Figure 2 shows the cortical spine data indicating a significant genotype effect (P < 0.01) and NAP rescue for all subtypes in males (P < 0.05). In the female cortex (Supplemental Figure 2), we observed a significant genotype effect only on the mushroom spines, which was completely rescued by NAP treatment (P < 0.001).
Further sex comparisons revealed differences in excitatory synapse numbers, with the Adnp+/– male mice showing significantly reduced hippocampal spine density, coupled with increased immature pathologic excitatory shaft synapses compared with Adnp+/– female mice (P < 0.01, Supplemental Table 2). This genotype- and sex-dependent pathology also extended to the cortex, with increased PSD95 shaft synapse density in Adnp+/– males compared with Adnp+/– females (P < 0.01), and was rescued by NAP treatment.
Measurements of PSD95 for excitatory shaft synapse volumes (indicative of synaptic maturation) showed significant increases in Adnp+/– mice in both hippocampus and cortex (P < 0.05), but not in the male mouse cortical spines. We observed a further increase with NAP treatment in female mice only, suggesting a compensatory effect (Supplemental Figures 1 and 2, insets).
We also studied the Adnp+/+ genotype for NAP effects. Specifically, in Adnp+/+ male mice, we found that NAP treatment reduced hippocampal stubby, thin, and total spine densities (Supplemental Figure 3, P < 0.05) as well as cortical mushroom, stubby, and total densities (Supplemental Figure 4, P < 0.05). Importantly, in males, NAP treatment did not affect PSD95 shaft synapse density in either tested region (Supplemental Figures 3 and 4, insets). These results were extended and revealed no effect of NAP on shaft synapse volumes in the cortex. Furthermore, we found that NAP treatment led to significantly increased shaft synapse volumes (maturation, P < 0.01) in the male hippocampus.
In female mice, NAP treatment did not affect dendritic spines in the Adnp+/+ hippocampus, but significantly increased cortical mushroom- and stubby-shaped spines (Supplemental Figures 3 and 4, P < 0.05). Interestingly, we found that shaft synapse densities (immature synapses) were decreased in the female cortex following NAP treatment (Supplemental Figure 4, P < 0.05). Furthermore, NAP treatment increased PSD95 shaft synapse volume in both tested brain regions (Supplemental Figures 3 and 4, insets, P < 0.05).
Gene expression changes suggest mechanisms and peripheral biomarkers.
Expression levels of 93 genes (Supplemental Table 3) were simultaneously measured using high-throughput quantitative reverse transcription PCR (HT qRT-PCR). Measurements assessed genotype, sex, and treatment effects on the hippocampus, cerebral cortex, and spleen, the latter of which was examined for potential peripheral biomarkers. Genes were chosen on the basis of previous results using complete Affymetrix microarray analysis to address developmental gene expression linked to Adnp haploinsufficiency as well as expression of genes linked to NAP protection during stressful conditions (2, 3, 34). We chose additional RNA transcripts on the basis of Adnp genotype RNA-seq data, as previously described (6, 17). Additionally, given that ADNP and NAP are linked with autophagy (13), cell adhesion (35), immune response (36), autism (6, 13, 15, 17, 27), and synapse-related processes (6), the analysis included several representative genes pertaining to these processes. We thoroughly evaluated this method (Supplemental Table 4).
We assessed mice in 2 age groups: 19- to 27-day-old mice, representing young, developing mice, and 3-month-old mice, representing mature mice (Supplemental Tables 5–15). Hprt was used for both groups as a validated reference transcript. At the younger age, the results supported the Adnp+/– mouse model, with decreases in cortical Adnp levels in both sexes, and a female-specific reduction in the hippocampus and spleen (Supplemental Table 5). In the older mice, we found that the genotype-associated decrease was preserved in the female cortex but was increased in the female Adnp+/– spleen, and was reduced by NAP treatment in the male hippocampus (Supplemental Table 8).
We evaluated global changes in gene expression and found that 49 gene transcripts were affected by the Adnp genotype, NAP treatment, or sex in the young mice (Supplemental Table 5) and that 57 gene transcripts were affected in the older mice (Supplemental Table 8). A Venn diagram (http://bioinfogp.cnb.csic.es/tools/venny/) identified 31 shared gene transcripts across the 2 tested ages that were affected by 1 or more of the 3 measured variables. To further elucidate the impact of age, all transcripts were assessed for changes between the younger and the older ages, resulting in 64 affected transcripts (Supplemental Table 13). To understand the implications of global gene expression changes affected by ADNP, NAP treatment, sex, and age, we implemented STRING function enrichment for biological processes (Supplemental Tables 6 and 7 for the young age group, Supplemental Tables 9 and 10 for the older age group, Supplemental Tables 11 and 12 for the 31 shared genes, and Supplemental Tables 14 and 15 for the age effects, with the first table in each pair analyzed according to the mouse genome and the second one according to the human genome).
We evaluated the 31 transcripts regulated at both tested ages (see above) and found that the most enriched modified functions were related to nervous system development and activity including synapse assembly, positive regulation of synaptic transmission, glutamatergic, regulation of synapse organization, regulation of cell communication, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor clustering, learning or memory, social behavior, regulation of ion transport, vocalization behavior, and nervous system development (Figure 3, Supplemental Tables 11 and 12). Figure 3, Supplemental Figure 5 (younger age group), Supplemental Figure 6 (older age group), and Supplemental Tables 5 and 8 reveal an overall similar pattern of Adnp genotype– and NAP treatment–regulated human and mouse protein product interactions across ages with Akt1 (the mosaic mutations of which lead to the Proteus syndrome, characterized by the overgrowth of skin, connective tissue, brain, and other tissues; ref. 37) and discs large MAGUK scaffold protein 4 (Dlg4, also known as Psd95), a key regulator of synaptic plasticity (see above) that plays central roles associated with ADNP and NAP function.
Figure 3. Function enrichment and network analysis of genes regulated by genotype and drug treatment.

STRING protein-protein interaction network (77) (https://string-db.org) analysis was performed for common genes listed in Supplemental Table 5 and Supplemental Table 8 representing shared transcripts that changed as a consequence of genotype or NAP treatment in 19- to 27-day-old and 3-month-old mice, compared with either the mouse (A) or human (B) database. Enriched biological processes are marked on the network according to the color legend.
We analyzed specific transcripts that changed as a consequence of Adnp haploinsufficiency and that were rescued by NAP treatment and found 2 hippocampus- and 1 spleen-regulated transcript species at 19 to 27 days of age and 6 hippocampal, 1 cortical, and 9 splenic transcripts at 3 months of age. We noted sex-dependent regulation, with no overlap discovered between age groups or tissues (Figure 4). Quality control analysis showed a good separation between genotypes and treatment (Supplemental Figures 7 and 8).
Figure 4. The Adnp genotype affects gene expression in the brain and spleen in 19- and 27-day-old and 3-month-old mice, with significant amelioration following NAP treatment.

HT qRT-PCR was performed on mRNA extracted from hippocampus, cortex, and spleens of 19- to 27-day-old mice (males: Adnp+/+ n = 5, Adnp+/– n = 5, Adnp+/– NAP, n = 5; females: Adnp+/+ n = 6, Adnp+/– n = 4, Adnp+/– NAP, n = 5) and of 3-month-old male (M) and female (F) mice (males: Adnp+/+ n = 3, Adnp+/– n = 4, Adnp+/– NAP, n = 4; females: Adnp+/+ n = 6, Adnp+/– n = 7, Adnp+/– NAP, n = 8). Results were normalized to Hprt. Significantly affected genes in 19- to 27-day-old mice (A) and 3-month-old mice (B) are presented. An unpaired Student’s t test revealed significant differences between vehicle-treated Adnp+/+ and Adnp+/– mice and between NAP- and vehicle-treated Adnp+/–mice (*P < 0.05 and **P < 0.01). Additional Student’s t tests were performed, comparing data between male and female mice to determine sex differences. All reported P values were also significant after multiple comparisons correction at a FDR of 10%.
In the young, developing mouse, specific Adnp genotype– and NAP-regulated hippocampal transcripts included a reduction and rescue of formyl peptide receptor 3 (Fpr-rs3) in males only, in agreement with the previous genotype-associated reduction we observed in the developing embryo (2). Tubulin β 1 class VI (Tubb1) increased in the Adnp+/– female mouse and was rescued by NAP treatment, thus correlating with our genotype-related RNA-seq data (6) (Figure 4A). Surprisingly, we found that the model-related Adnp transcript haploinsufficiency was corrected in the young female spleen with NAP treatment (Figure 4A).
In the 3-month-old hippocampi (Figure 4B), we found significant sex-dependent changes for Adnp+/– gene regulation and NAP rescue in the following genes in male mice: (a) apolipoprotein E (Apoe), the lead gene for Alzheimer’s disease risk, which was shown before to be a major gene regulated by ADNP (10, 13); (b) Gm21949, which is suggested to play a role in calcium-mediated responses, action potential conduction in myelinated cells, and axonal outgrowth and guidance (6); (c) lipase A (Lipa), which is related to lipid metabolism and was previously shown to be regulated by the Adnp genotype in mice (3); (d) autism-associated neuroligin 2 (Nlgn2), a postsynaptic membrane cell adhesion protein that mediates the formation and maintenance of synapses between neurons (12); (e) paired box protein 6 (Pax6), a key regulator in glutamatergic neuronal differentiation (38) and cortical development (39), which was shown before by us to be regulated by ADNP (complete knockout of Adnp rendered Pax6 expression undetectable in the brain primordium, contrasting with increased expression in Adnp+/– embryos [ref. 1] and in subcortical brain domains of 2-month-old male Adnp+/– mice [ref. 3]); and (f) Wolframin endoplasmic reticulum transmembrane glycoprotein (Wfs1), which is associated with neurodegeneration and cellular calcium homeostasis regulation and was previously shown to be regulated by NAP (34).
In the mature cerebral cortex, only histone cluster 1 H3 family member B (Hist1h3b), which was one of the major transcripts downregulated in the hippocampi of 5-month-old Adnp+/–mice compared with Adnp+/+ mice (6, 17), was found here to be downregulated in the female Adnp+/– mouse. This effect was now shown to be reversed by NAP treatment (Figure 4B).
ADNP expression in lymphocytes correlates with inflammation levels (36), disease state, and autophagy (13), as well as intelligence (40). We therefore searched for splenic genotype–specific gene expression changes that were also normalized by NAP treatment in the mature mouse (Figure 4B). Notably, splenic maturation occurs in the postnatal mouse, which is suggestive of age-dependent gene regulation (41).
In male mice, the ATP-binding cassette subfamily F member 3 (Abcf3), bone morphogenetic protein 4 (Bmp4), cadherin 17 (Cdh17), lysine demethylase 5d (Kdm5d), Kruppel-like factor 1 (Klf1), and period circadian regulator 1 (Per1) were upregulated as a consequence of Adnp haploinsufficiency and rescued by NAP. In female mice, Akt1 (above) and ionized calcium–binding adapter molecule 1 (Iba1), a marker of microglial activation that crosslinks actin (42), were markedly increased in the Adnp+/– mouse spleen and normalized by NAP treatment, suggesting a potential peripheral inflammation–linked biomarker. Likewise, mechanistic target of rapamycin (Mtor), which has been linked to cellular regulation, protein translation, autophagy, and the actin cytoskeleton (43–45), was also found to be regulated by ADNP and NAP. Importantly, transcripts affected by Adnp deficiency and reversed by NAP treatment in the mouse spleen (Figure 4) were also found to be affected by various human ADNP mutations in lymphoblastoid cells at the RNA-seq level (18) (Supplemental Figure 9).
Several of the genes affected in 3-month-old mice (Figure 4B) were also found to be regulated by either the Adnp genotype or NAP treatment in the 19- to 27-day-old mice. Akt1, Gm21949, Mtor, Nlgn2, and Per1 were all affected by NAP treatment in the younger mouse cortex, specifically in males. Apoe was affected by NAP treatment in the male cortex and female spleen. Female cortical Hist1h3b was found to be regulated only by the Adnp+/– genotype. Splenic Bmp4 was found to be affected by NAP treatment, specifically in females (Supplemental Table 5).
Adnp+/– genotype exhibits significant decreases in ultrasonic vocalizations and developmental milestone delays, and daily NAP treatment partially reverses the phenotype.
Ultrasonic vocalization (USV) calls of distress (40–70 kHz) were induced on P8 following separation from the dams (46, 47). Results showed a 2-fold reduction in the number of all USVs in the Adnp+/– mice, which was dramatically increased by NAP treatment (merged male and female data, Figure 5A). Separating males and females revealed a substantial inhibition in female pups that was completely reversed by daily NAP treatment. In male pups, the NAP-induced increase in vocalizations was even more significant (Figure 5A, inset). We observed no effect of NAP on vocalizations in Adnp+/+ mice (Supplemental Figure 10A).
Figure 5. Adnp haploinsufficiency causes significant decreases in USVs and delays in developmental milestones, partially reversed with daily NAP treatment.

(A) For USV measurement, a 2-way ANOVA with Tukey’s post hoc test was performed. Main treatment [F(1,460) = 26.095, P < 0.001] and interaction [F(1,460) = 17.463, P < 0.001] effects were found. Adnp+/– pups produced significantly fewer USVs per minute compared with Adnp+/+ pups (*P < 0.05), with NAP increasing vocalization production by 3-fold compared with Adnp+/– mice (***P < 0.001) and by 2-fold compared with Adnp+/+ mice (***P < 0.001, Mann-Whitney U test). When comparing sexes (inset graph), NAP treatment had the most profound effect on Adnp+/– males (4-fold increase) compared with females (2-fold increase) (#P < 0.05, Mann-Whitney U test). Results are presented as the mean ± SEM USVs per minute (males: Adnp+/+ n = 11, Adnp+/– n = 9, Adnp+/– NAP, n = 6; females: Adnp+/+ n = 11, Adnp+/– n = 8, Adnp+/– NAP, n = 5, 6 USV calls per mouse). (B–D) For developmental milestone measurements, a 2-way ANOVA with Tukey’s post hoc test was performed, with data expressed as the mean ± SEM of the first neonatal day of success in the test (Adnp+/+ n = 35–51, Adnp+/– n = 19–29, Adnp+/– NAP, n = 28–43; exact numbers are detailed in Supplemental Table 16). For the ear twitch reflex, a significant main genotype effect was found [F(1,109) = 11.851, P < 0.001], with Adnp+/– pups displaying a significantly earlier response compared with Adnp+/+ pups (*P < 0.05). For the air righting reflex, a significant main treatment effect was found [F(1,164) = 24.838, P < 0.001], implying that NAP-treated Adnp+/– pups acquired an air righting reflex significantly earlier than did Adnp+/– pups (**P < 0.01). For negative geotaxis, significant genotype [F(1,163) = 36.780, P < 0.001] and treatment [F(1,163) = 7.684, P = 0.006] effects were found. Adnp+/– mice presented a significant delay in negative geotaxis compared with Adnp+/+ littermates (***P < 0.001), with NAP treatment significantly improving the phenotype (**P < 0.01). (A–D) Adnp+/+ data are reshown in Supplemental Figure 10, A–D.
Developmental tests characterize early markers of behavior and assess the impact of insults and inborn errors (4, 48, 49). Here, we analyzed the impact of Adnp haploinsufficiency and daily NAP applications as a potential therapy. Male and female mice were grouped together, as no sex differences were observed. Figure 5B shows that the ear twitch reflex appeared a day earlier in the Adnp+/– mice when compared with Adnp+/– littermates, which may be related to genotype-linked increased irritation. No ear twitch reflex effect was observed in Adnp+/+ mice (Supplemental Figure 10B). While the air righting reflex (acquired innate ability of the mouse pup to orient itself as it falls to land on its feet) was not significantly delayed as a consequence of Adnp deficiency, NAP treatment resulted in an earlier (1 day) acquisition of this behavior (Figure 5C), with a similar improvement observed in Adnp+/+ mice (Supplemental Figure 10C). Importantly, negative geotaxis, a test used to investigate motor coordination and vestibular sensitivity, showed delayed development in Adnp+/– mice and normalization with NAP treatment (Figure 5D). We observed no effect for negative geotaxis in Adnp+/+ mice (Supplemental Figure 10D).
Adnp+/– mice exhibit sex-related growth delays as well as impaired gait, which are partially ameliorated by NAP treatment.
Given that patients with ADNP syndrome have short stature (19) and motor delays (17, 19, 20), we set out to test these traits in Adnp+/– mice during early development. Initially, we checked for mouse length (potentially corresponding to stature in an ADNP syndrome child) and discovered shorter lengths as the Adnp+/– mice matured, starting earlier in males (Figure 6, A and B). We observed no significant NAP treatment effect on Adnp+/– (Figure 6, A and B) or Adnp+/+ (Supplemental Figure 11) mice. Supplemental Figure 12 shows marked delays in weight gain that were also sex dependent (apparent earlier in females).
Figure 6. Adnp+/– pups exhibit significantly delayed growth as well as significantly impaired gait at 18 to 40 days of age, affected by NAP treatment, in a sex-dependent manner.

(A and B) A 2-way, repeated-measures ANOVA with Bonferroni’s means separation test revealed significant differences in length between Adnp+/+ and Adnp+/– littermate mice (males: Adnp+/+ n = 18, Adnp+/– n = 12, Adnp+/– NAP, n = 12; females: Adnp+/+ n = 20, Adnp+/– n = 9, Adnp+/– NAP, n = 23). For males, main effects for group [F(1,28) = 24.025, P < 0.001], day [F(21,573) = 2122.663, P < 0.001], and interaction [F(21,573) = 3.703, P < 0.001] were found, with significant differences between Adnp+/+ and Adnp+/– mice (*P < 0.05 and **P < 0.01). For females, main effects for group [F(1,27) = 16.178, P < 0.001], day [F(21,554) = 1487.989, P < 0.001], and interaction [F(21,554) = 5.267, P < 0.001] were found, with significant differences between Adnp+/+ and Adnp+/– mice (*P < 0.05 and ***P < 0.001). NAP treatment did not affect length acquisition in male or female Adnp+/– pups. (C–E) For gait analysis, a 2-way ANOVA with Tukey’s post hoc test was performed. An unpaired Student’s t test was also used to determine sex differences (males: Adnp+/+ n = 104, Adnp+/– n = 72, Adnp+/– NAP, n = 64; females: Adnp+/+ n = 96, Adnp+/– n = 44, Adnp+/– NAP, n = 116 paw replicates per experimental group). For standing (seconds), main genotype [F(1,316) = 23.683, P < 0.001] and interaction [F(1,316) = 18.030, P <0.001] effects were found in males and main interaction effect in females (F(1,352) = 4.894, P < 0.05). For step cycles (seconds), main genotype [F(1,316) = 32.116, P < 0.001] and interaction [F(1,316) = 18.086, P < 0.001] effects were found in males. In females, a main interaction effect was found [F(1,352) = 4.974, P = 0.026]. For stride length (cm), main genotype [F(1,316) = 38.359, P <0.001] and treatment [F(1,316) = 6.152, P = 0.014] effects were found in males. In females, main genotype [F(1,352) = 21.286, P < 0.001], treatment [F(1,352) = 5.371, P = 0.021], and interaction [F(1,352) = 7.117, P = 0.008] effects were found. For standing, step cycle, and stride length parameters, significant differences between Adnp+/+ and Adnp+/– mice (***P < 0.001) and NAP- versus vehicle-treated Adnp+/– mice (***P < 0.001) were found. Sex differences were observed for all 3 gait parameters (**P < 0.01 and ***P < 0.001, Student’s t test). (A and B) Adnp+/+ data are reshown in Supplemental Figure 11, A and B; (C–E) Adnp+/+ data are also shown in Supplemental Figure 13, A–C.
We performed gait analysis of 1-month-old mice using the CatWalk XT (Noldus Information Technology). In general, the results showed that the Adnp+/– genotype–inflicted impairments were partly ameliorated by NAP treatment. We also observed sex differences. Specifically, the standing time and step cycle parameters indicated better performance in males, with significant impairments seen in Adnp+/– mice and amelioration with NAP treatment (Figure 6, C and D). Stride length (cm) was significantly reduced in the Adnp+/– mice, with NAP treatment causing a slight reduction in stride length in Adnp+/– males and an apparent increase in females (Figure 6E). Supplemental Figure 13 shows a sexual dichotomy among Adnp+/+ mice (male mice had better standing times and step cycles, with NAP treatment slightly reducing the standing times for males, while increasing both the standing times and step cycles for females). We observed no effect of sex on stride lengths. Furthermore, we observed reduced performance of Adnp+/– mice in single stance (males) and swing (both sexes) tests, with NAP treatment causing a reduction in single stance performance in females and improving swing in males. Taken together, the results showed that gait was affected by the Adnp genotype (more severely in males), with an overall amelioration with NAP treatment.
NAP protects against motor, cognitive, and social deficits in Adnp+/– mice.
On P21, Adnp+/– mice were divided into groups according to sex, genotype, and treatment. We then measured the latency to fall off an inverted cage lid (hanging wire) and found a highly significant impairment (decreased latency) in male Adnp+/– mice and a complete reversal with NAP treatment (Figure 7A). The females were not affected in this behavior, indicating sex differences in motor behavior and development in the haploinsufficient mice. Likewise, Adnp+/– males, but not females, showed significantly reduced grip strength that was completely reversed by NAP treatment (Figure 7B). In the uncompromised Adnp+/+ mice, NAP showed no effects on these behaviors (Supplemental Figure 14).
Figure 7. Influence of the Adnp genotype on motor, memory, and social aspects, all of which improve with NAP treatment.

A 2-way ANOVA or 2-way, repeated-measures ANOVA with Tukey’s post hoc test was performed (males: Adnp+/+ n = 20, Adnp+/– n = 12, Adnp+/– NAP, n = 9; females: Adnp+/+ n = 14–15, Adnp+/– n = 7, Adnp+/– NAP, n = 20). (A) To assess neuromuscular ability, the latency (seconds) to fall off an inverted cage lid was determined by a hanging wire test. For males, a main group effect was found [F(3,49) = 8.186, P < 0.001], with significant differences between Adnp+/+ and Adnp+/– mice and NAP- versus vehicle-treated Adnp+/– mice (**P < 0.01, 2-way ANOVA). Sex differences were found among Adnp+/– mice (**P < 0.01, Mann-Whitney U test). (B) To assess forelimb grip strength, peak grip force was evaluated. For males, a main group effect was found [F(3,49) = 6.154, P = 0.001], with significant differences between Adnp+/+ and Adnp+/– mice and NAP- versus vehicle-treated Adnp+/– mice (**P < 0.01, 2-way ANOVA). Sex differences were found in Adnp+/– mice (*P < 0.05, Mann-Whitney U test). FG, grip force. (C) For object recognition assessment, a main genotype effect in males [F(1,49) = 7.037, P = 0.011] and a main interaction effect in females [F(1,56) = 5.386, P = 0.024] were found. In both sexes, significant differences between Adnp+/+ and Adnp+/– mice and NAP- versus vehicle-treated Adnp+/– mice were observed (*P < 0.05 and ***P < 0.001). The discrimination 2 score (D2) was determined using the following equation: D2 = (b – a)/(b + a), where a = time spent exploring the familiar object, and b = time spent exploring the novel object. (D) For social recognition among female mice, a main effect for the sniffed item was found [F(1,56) = 30.447, P < 0.001], with significant differences between the time spent sniffing the cup and the mouse for Adnp+/+ mice (**P < 0.01) and NAP-treated Adnp+/– mice (***P < 0.001 vs. cup). (E) Female Adnp+/– mice exhibited impaired olfactory function, which was restored by NAP treatment. *P < 0.05, **P < 0.01, and ***P < 0.001 versus previous sniffing (novel vs. familiar odor), by paired Student’s t test (Adnp+/+ n = 11, Adnp+/– n = 5, Adnp+/– NAP n = 15). (F) Social memory was assessed. For males, main treatment [F(1,49) = 4.573, P = 0.037] and interaction [F(1,49) = 4.473, P = 0.040] effects were found. For females, a significant main interaction effect [F(1,56) = 4.463, P = 0.039] was found. Among both sexes, significant differences between Adnp+/+ and Adnp+/– mice and NAP- versus vehicle-treated Adnp+/– mice were observed (*P < 0.05 and **P < 0.01). (A–C and F) Adnp+/+ data are reshown in Supplemental Figure 14, A–D. (D and E) Adnp+/+ data are reshown in Supplemental Figure 14, E and G.
Our previous data primarily indicated object and social memory deficits in the Adnp+/– mice (6, 13). Furthermore, while Adnp+/+ mice prefer novel objects and novel mice, Adnp+/– mice are either indifferent or prefer familiarity (6, 13). Here, we found that object recognition memory was normalized following NAP treatment in Adnp+/– mice (Figure 7C) and that NAP treatment did not change the behavior of normal Adnp+/+ mice (Supplemental Figure 14).
Interestingly, we detected sex-specific differences in object/mouse preference in the female mice, which did not prefer mice over objects (potential autistic behavior). The indifference phenotype was ameliorated by NAP treatment (Figure 7D). This was coupled with deficits in olfactory function in the Adnp+/– females, but not males, with the female mice exhibiting impaired odor discrimination that was also restored by NAP treatment (Figure 7E; for more detail, see Supplemental Figure 14).
As with object recognition memory, the deficient social memory of Adnp+/– mice (males and females) was normalized by NAP treatment (Figure 7F).
The Adnp+/– mouse mimics the human ADNP syndrome patient.
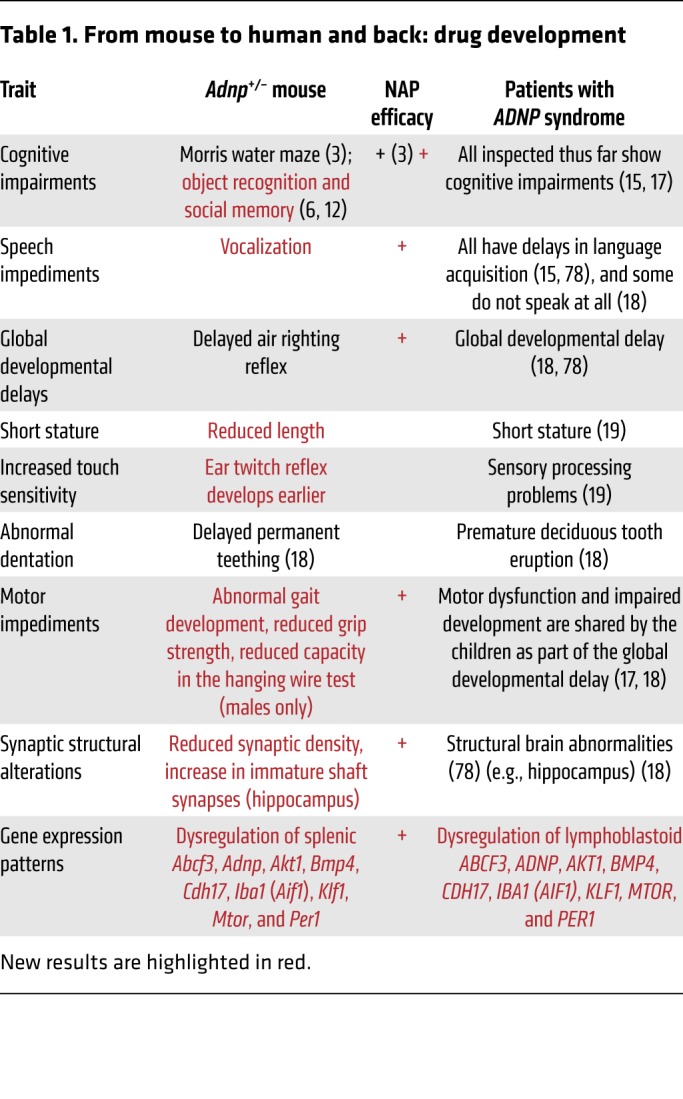
In the current study, we discovered parallels between the Adnp-deficient rodent model and ADNP syndrome patients, at developmental, motor, and cognitive levels (Table 1). Furthermore, the rodent model allowed precise quantitation of excitatory synapse density in the hippocampus and motor cortex and evaluation of gene expression patterns, thus correlating molecular, structural, and functional outcomes (see above and Table 1). Interestingly, we discovered peripheral biomarkers and sex-specific differences that may serve to guide in the clinical development of therapeutics. Most important, we showed a protective effect of NAP at all levels, indicating efficacy for this drug candidate.
Table 1. From mouse to human and back: drug development.
Discussion
We discovered ADNP 19 years ago (4, 5) and predicted that ADNP deficiency or mutations could lead to an autism-related intellectual disability syndrome in humans on the basis of findings in our mouse models (1, 3, 10, 15, 50, 51). However, in children with ADNP mutations, important phenotypic outcomes have been revealed that required further in-depth study of the Adnp haploinsufficient mouse model (6, 13) as a potential predictor of effects in humans (17). As indicated above, humans with ADNP mutations suffer from global developmental delays that affect motor and vocal (language) development, whereas these developmental delays have not been measured previously in the Adnp haploinsufficient mouse (15, 20). Target engagement (i.e., synaptic modulation and regulation of gene expression) in a relevant model for ADNP syndrome is imperative for the successful understanding and clinical development of the ADNP-enhancing drug candidate NAP (davunetide, CP201) (3, 13, 26, 27).
Here, in a unique mouse model of GFP labeled neurons, we demonstrated for the first time to our knowledge that ADNP was tightly linked to dendritic spine formation in vivo and that NAP corrected Adnp deficiency at the dendritic spine level. These in vivo results corroborated our in vitro data showing that NAP increases dendritic spine formation and maturation (PSD95 expression in dendritic spines) through interaction with the microtubule end–binding protein EB3 (26). This takes into consideration that microtubule insertion into dendritic spines drives spine maturation during long-term potentiation, a cellular model of learning and memory, and may therefore play a role in synaptic plasticity and memory formation (52). Furthermore, mushroom spines in particular, affected here by both Adnp deficiency as well as NAP treatment, are the stronger, more stable excitatory synapses and have been suggested to be associated with spatial and working memory (53), which connects with our current and previous data (3, 6, 13). Our results thus prove NAP target engagement in vivo. With regard to human clinical outcomes, NAP neuroprotective activity was tested in 2 human cerebral cortical cultures showing neuronal degeneration that were: (a) treated with 50 μM H2O2 (oxidative stress, 60% viability loss) and (b) of Down syndrome (DS) origin. Addition of NAP at femtomolar concentrations resulted in substantial increases in the survival of normal neurons treated with H2O2. Femtomolar concentrations of NAP exhibited a more potent neuroprotective efficacy that was comparable to that of the antioxidant N-tert-butyl-2-sulpho-phenylnitrone (s-PBN) at 100 μM. Treatment of DS cortical neurons with NAP resulted in a marked increase in neuronal survival as well as a reduction in degenerative morphological changes. The results suggest that NAP possesses potent neuroprotective properties against oxidative damage in human neurons that may be useful for preserving neuronal function and preventing neuronal death associated with chronic autistic and neurodegenerative disorders (54). Importantly, DS postmortem samples present a decrease in dendritic spines (55), attesting to the relevance to humans of NAP treatment effects on dendritic spines. Additionally, in clinical trials involving patients suffering from schizophrenia, magnetic resonance spectroscopy (MRS) revealed that NAP (davunetide) provided neuroprotection (23), a finding that may be further associated with dendritic spine measurements (56).
In general, and in association with previous in vitro (26) and in vivo (3) results, we found that NAP treatment rescued the Adnp+/– phenotype. Surprisingly, we observed a significant 10%–20% reduction (P < 0.001) in dendritic spine density in the Adnp+/+ male mouse (hippocampus and cortex) following NAP treatment. This reduction was neither coupled to an increase in pathological shaft synapses, nor to a decrease in shaft synapse volumes, suggesting a possible physiological function. Interestingly, NAP treatment specifically increased the expression of Nlgn2 and Nlgn3 (neuroligins) in the young Adnp+/– male mouse cortex (Supplemental Table 5), while it decreased these transcripts in the older Adnp+/– male mouse hippocampus but did not affect expression levels in the females (Supplemental Table 8). As neuroligins antagonize neurite outgrowth in the male Caenorhabditis elegans sexually dimorphic neurons to regulate sexual behavior, it is now suggested that ADNP and NAP play a part in sexually dichotomous brain plasticity, which is reflected in the above-mentioned changes in dendritic spine densities (57). Notably, sex differences were observed in ADNP expression in the human hippocampus (12), and ADNP promoter and transcript levels were found to be autoregulated by ADNP (2, 58) as well as by NAP treatment (Figure 4). Here, NAP treatment slightly affected standing and step cycle gait parameters (reduced standing times for males and increased step cycles for females, Supplemental Figure 13), without affecting other tested behavioral phenotypic outcomes in Adnp+/+ mice (Supplemental Figures 13 and 14), and NAP was previously shown to increase cognitive performance in normal rodents (4, 59). It should also be noted that NAP provides neuroprotection over a broad range of concentrations (4) and has a proven safety profile based on past clinical trials involving more than 500 patients (22–24).
Our previous data also indicated several Adnp+/–-associated impairments, including slower axonal transport in the male olfactory bulb compared with females, that were partially recovered by the NAP-derivative, EB3-interacting molecule SKIP (6). These results may partly explain the sex-dependent differences found in odor discrimination (indicated above). Furthermore, NAP was also found to repair axonal transport deficits in models of neurodegeneration (60).
Interestingly, ADNP is found in both the neuronal nucleus and cytoplasm (50). In the nucleus, ADNP interacts with HP1α (2), which in turn recruits it to histone-marked H3 lysine 9 trimethylation (H3K9me3) sites of pericentromeric heterochromatin silencing of major satellite repeats (9). Furthermore, H3K9me3 downregulation, which mediates gene transcription in the hippocampus, promotes spine formation and reverses age-dependent deficits in hippocampal memory (61). This is also in line with the most recent work identifying ADNP recruitment of HP1 and CHD4 to control lineage-specifying genes (8). Importantly, an ADNP-interacting DNA sequence was identified, GCGCCCTCCAG, which is similar to the neuronal CCCTC binding factor CTCF that was previously identified as a regulator of remote memory and cortical synaptic plasticity (62).
Using HT qRT-PCR to quantify 93 gene transcripts (Supplemental Table 3), we identified multiple changes centered on synaptic activity and organ shaping (Figure 3). We observed significant Adnp genotype-, age-, and treatment-related changes in expression patterns, including expression of hippocampal Pax6 (Figure 4B). Recent work (8) corroborated our original finding of ADNP regulation of PAX6 (1). Pax6 has been associated with the specification of the subcortical domains of the limbic system (63) and with neurogenesis in general. Increases in Pax6 expression may suggest aberrant developmental processes and potential compensatory mechanisms associated with Adnp deficiencies. Partial loss of function of PAX6 in humans has been associated with verbal working memory impairments, ID, and autism (39). In animal models, PAX6 affects social behavior (38), and Pax6-mutated rats exhibit abnormal USVs (64) and significant USV decreases in females. This effect mimics our current data and is further corroborated by evidence in humans suggesting greater communication deficits in autistic girls than in autistic boys (64). Here, the abnormal increase in Pax6 expression in the hippocampus of 3-month-old male mice due to Adnp deficiency was reversed by NAP treatment.
Specifically, we discovered a substantial reduction in the number of USVs produced by Adnp+/– pups compared with those produced by their Adnp+/+ littermates. The effects of NAP treatment suggested a significant sex-dependent difference in the number of USVs, with a dramatic increase seen in males. The mechanisms underlying the vocal effect observed in Adnp+/– mice, as well as the sex-specific differences in response to NAP treatment, require further investigation. One possible explanation may reside in the sexually dichotomous ADNP expression patterns in the brain (also noted above), as shown in a variety of adult vertebrates: humans (hippocampus, with transcript levels higher in males) (12), mice (hypothalamus/arcuate nucleus changes with the estrous cycle) (65), and the songbird (cerebrum, with higher levels in males) (66).
We observed male-specific Adnp+/– impairments in the expression of genes associated with synaptic function (e.g., Dbn1) in the mature hippocampus. This was coupled with significantly increased cortical synaptic impairments compared with Adnp+/– females (Figure 2) and reduced motor functions (Figure 7, A and B), all of which were ameliorated by NAP treatment. Interestingly, while most children with ADNP syndrome eventually walk independently (17, 19, 20), future studies are required to evaluate the possibility of sex differences and the severity of motor impairments in this patient population (17).
Previous studies in other mouse models (e.g., ALS mouse model) indicated an earlier onset with a more severe motor phenotype in males (67), which was linked to microtubule-dependent axonal transport and ameliorated in part by NAP treatment (60). In contrast, while sex-dependent cognitive differences were previously described in approximately 8-month-old Adnp+/– mice (6, 13), these differences were not observed here in 3-month-old mice, suggesting a sex-dependent difference in the development of cognitive abilities in mice similar to that of muscle skills. These results are in agreement with the sex-dependent development of behavioral, physiological, and autistic phenotypes observed in other mouse strains (68).
In conclusion, our findings directly associate ADNP and NAP with neurodevelopment, dendritic spine plasticity, cytoskeleton- and spine-linked gene regulation and binding proteins, language and communication, learning and memory, and muscle strength and gait development. Our studies provide further evidence for a drug-target association in vivo, which was also inferred by neuroprotection using MRS in patients with schizophrenia (23, 24). Additionally, a reduction in ADNP content in peripheral blood cells has been previously linked to inflammation coupled with NAP protection against it (36). Here, we identified potential surrogate peripheral blood biomarkers that have been partly associated with inflammation (e.g., splenic Iba1 and Mtor) and that were regulated by both ADNP syndrome–causing mutations (Supplemental Figure 9) and Adnp haploinsufficiency (in female mice). Furthermore, changes in the above-mentioned potential peripheral biomarkers were ameliorated with NAP treatment.
Last, our analysis of all tested genes in both age groups that exhibit expression changes associated with the Adnp genotype and NAP treatment revealed an important central role for Akt1 and Dlg4 (Psd95) (Figure 3, Supplemental Figure 5, and Supplemental Figure 6). As indicated above, Akt1 is linked to morphogenesis and to ADNP syndrome children who show specific facial characteristics (19), which translates the mouse findings to the human condition. Furthermore, PSD95 is an ADNP target at both the gene expression level and, most important, the synaptic level, with autism being a disease of the synapse (69). Thus, the current study sets the stage for the clinical development of NAP to treat children with ADNP syndrome and beyond.
Methods
Experimental design.
Animal group sizes were determined in a pilot study. Adnp pups were randomly allocated to experimental groups on P1. Half of the mice from each tested litter were assigned to the NAP treatment group, while the other half were assigned to the vehicle treatment group. The study was performed in a semi-blinded manner. Before genotyping, the investigators were blinded to the sex and genotype of the mice when measuring USVs and developmental milestones. Technical replicates were used for dendritic spine and gait analyses, whereas biological replicates were used for gene expression, USV, developmental, gait, motor, and cognitive analyses. All the experiments were replicated successfully and the results substantiated. Raw data for all experiments described here are presented in Supplemental Tables 17–29. Outlier values were determined and excluded by Grubbs’ test (as described below in the Statistics section). The accession numbers for all analyzed genes analyzed by HT qRT-PCR are provided in Supplemental Table 3. The exact experimental group allocations are detailed in Supplemental Table 16 and are included in each figure panel presenting multiple samples.
Animals.
The Adnp+/– mice on a mixed C57BL and 129/Sv background have been previously described (1, 3, 12). For continuous breeding, an ICR outbred mouse line was used (6, 12). Animals were housed in an animal facility under a 12-hour light/12-hour dark cycle, with free access to rodent chow and water. Mouse pups were subcutaneously administrated NAP (25 μg NAP/1 ml saline) for 21 consecutive days. NAP was injected in increasing volumes at 20- and 40-μl doses on days 1–4 and 5–7, respectively (3, 48). USV analysis was performed on P8 (46). Otherwise, pups tested for developmental milestones were further injected with NAP at the following doses: 40 μl, 80 μl, 120 μl, and 160 μl on P9–P10, P11–P14, P15–P18, and P19–P21, respectively (3, 48), after which the mice were weaned and genotyped. Mice at 21 days of age were treated daily with intranasal NAP in vehicle solution (termed DD) (0.5 μg NAP/5 μl DD) (3, 6, 70). These mice were further analyzed for motor and behavioral functions as detailed below. As the behavioral experiments required chronic daily administration, we chose the noninvasive intranasal route, which has been and is further planned to be used in clinical trials (see refs. 22 and 24 for examples). Importantly, our pharmacokinetic and bioavailability studies showed that after either intranasal or systemic administration, NAP (davunetide) rapidly appeared in the cerebrospinal fluid (CSF). These results, combined with timed tissue distribution data, indicate that intranasal NAP enters the CNS via the systemic circulation (21, 22, 71), and thus the different routes of administration do not affect NAP distribution or effects.
For developmental gene expression analysis, 2 subsets of mice were used. The first subset included 19- to 27-day-old mice (following NAP or vehicle administration as detailed above). The second subset of mice were sacrificed after 1 month of daily intranasal administrations (starting at 2 months of age) and processed as detailed below.
The Adnp+/–-GFP mouse model was used for the determination of dendritic spine morphology. For this purpose, Adnp+/– female mice were crossed with L15 male mice (on a C57BL/6 background) to produce an Adnp mouse model with neuronal GFP expression (72). In short, variegated mice were generated using standard techniques. A construct was generated, in which the cDNA for EGFP was fused to the membrane-anchoring domain (first 41 aa) of a palmitoylated mutant of MARCKS29 under the Thy1 promoter. Twenty-five distinct mouse lines were produced, each with subtly different patterns of expression (30). Of these, L15 mice were chosen, because they had a low, but consistent, number of mGFP-labeled cells within the CA1 area of the hippocampus (73–75). Three-month-old Adnp-GFP mice were treated for nine consecutive days with either intraperitoneal NAP injection (0.4 μg) diluted in 0.1 ml saline or with 0.1 ml saline as a vehicle. On day 9, mice were perfused, and brains were subjected to immunohistochemical analysis as described below.
Epstein-Barr virus–transformed human lymphoblastoid cells.
A representative lymphoblastoid cell line (LCL) from healthy adult donors was obtained from the National Laboratory for the Genetics of Israeli Populations (NLGIP; http://nlgip.tau.ac.il/) at Tel Aviv University. Two ADNP-mutated LCLs were purchased from the Simon Simplex Collection ( SSC04121 = ADNP [protein] p.Lys408Valfs*31 and SSC08311 = p.Tyr719*) (15), and one LCL was generated from peripheral blood lymphocytes donated by consenting patients, guardians, or physicians (18). RNA-seq results were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE81268) (6, 18).
Immunohistochemistry.
Histological staining and immunohistochemistry were performed on coronal 100-μm serial hippocampal free-floating brain sections as described in the Supplemental Methods.
Confocal imaging.
See the Supplemental Methods for details.
3D reconstructions and spine quantification.
See the Supplemental Methods for details.
HT qRT-PCR.
Ninety-three genes of interest were chosen, several of which were based on previous results using complete Affymetrix microarray analysis during embryogenesis and stress (GEO GSE4068 and GSE10923, respectively) (2, 3, 34), and from RNA-seq data comparing Adnp+/+ and Adnp+/– male and female mouse hippocampi at one month and five months of age, addressing developmental changes (GEO GSE72664) (6, 18). RNA from 19- to 27-day-old and 3-month-old mouse hippocampal, cortical, and splenic tissue samples was extracted using TRI Reagent (T9424, Sigma-Aldrich). RNA (1 μg/sample) was then subjected to reverse transcription (RT) using a qScript cDNA Synthesis Kit (Quanta Biosciences). Each cDNA sample was preamplified and further subjected to qPCR performed with the HT platform BioMark HD (Fluidigm) using 96.96 Dynamic Array IFC for Gene Expression, as detailed in the Supplemental Methods. Hprt was selected as a stable reference gene for both age groups. All data were normalized. Missing data were replaced by the highest Cq value within the assay +2. Data were further transformed into relative quantities and log scale transformation was used to ensure normal data distribution. Statistical analysis was performed by GenEx qPCR data analysis software (MultiD). Analysis was conducted separately for each tissue. P-values were corrected for multiple comparisons testing at FDR of 10%. Results of the two groups were also compared for potential age effect (Supplemental Table 13).
USVs.
Adnp mouse pups from 9 different litters were included in this study and treated daily with subcutaneous injections of NAP, as described in the Animals section (25 μg/1 ml saline) (3, 4, 48). The USV test is detailed in the Supplemental Methods. Results are presented as the mean number of USVs recorded per minute of the trial compared with saline-treated Adnp pup littermates (controls).
Developmental milestones.
Adnp mouse pups from 16 litters were included in the study and tested for neonatal development, as previously described (4, 48, 49). As animal randomization ended up with unequal male/female ratios for certain male/female groups, the sexes could not be separated, and therefore male and female data were merged. Pup paws were tattooed using a 31-gauge needle and tattoo ink (Ketchum Manufacturing Co., catalog 329AA) for identification on the day of birth and, until P21, underwent daily measurements for length and weight and observation for eye opening. Adnp pups were injected daily with NAP, 1 hour prior to the tests (3, 4, 48). The developmental milestone behavioral tests are detailed in the Supplemental Methods.
Gait analysis.
The CatWalk XT (Noldus Information Technology) was used to analyze the gait of unforced, moving mice (76). The mice had to cross the runway of the CatWalk XT apparatus in a consistent manner, and a successful run was defined when an animal ran the track without any interruption or hesitation. Every mouse was tested until 3 to 5 complete successful runs were achieved. The graphs represent pooled raw data for 4 paws of each mouse, as obtained from the successful runs on the CatWalk walkway plate. The definitions of the presented gait parameters are provided in the Supplemental Methods.
Hanging wire test.
Three trials were performed for each mouse. An assessment of the mouse paws’ strength was performed by measuring their latency to fall off an inverted cage lid (placed 50 cm above the surface) onto soft bedding (maximum time of 90 s).
Forelimb grip strength test.
Five trials were performed for each mouse. A Grip Strength Meter (UgoBasile, catalog 47200) was used to measure forelimb grip strength. Mice were allowed to grasp a bar mounted on the force gauge, and then the peak pull force in grams was recorded on a digital force transducer. Five consecutive trials were conducted for each mouse. At the beginning of each trial, the gauge was reset to 0 g after stabilization, and the mouse’s tail was slowly pulled back. Tension was recorded by the gauge at the time the mouse released its forepaws from the bar.
Object recognition.
See Supplemental Methods and ref. 12 for details.
Social approach task.
Mice used as novel (target) mice to be explored by the subject mice were from the C57BL/6 strain (in our colony), known for their docile nature (3-month-old). The social approach task was previously reported (6, 12) (see also Supplemental Methods).
Odor discrimination test.
See Supplemental Methods and refs. 6 and 12.
Statistics.
Results are presented as the mean ± SEM. Data were checked for normal distribution by normality test. For 2 different categorically independent variables, a 2-way ANOVA or a 2-way, repeated-measures ANOVA followed by Tukey’s post hoc or Bonferroni’s means separation methods was performed. An unpaired Student’s t test or Mann-Whitney U test was performed when applicable. P values of less than 0.05 were considered statistically significant, and all tests were 2 tailed. For developmental milestone measurements, in vivo behavioral tests, and dendritic spine quantification, outlier values were excluded using the GraphPad QuickCalcs outlier calculator (https://graphpad.com/quickcalcs/Grubbs1.cfm). The specific excluded outlier values are highlighted in red in the relevant raw data presented in Supplemental Tables 17–29. For gene expression analysis, no data were excluded. All statistical analyses were conducted using either SigmaPlot software (version 11) for Windows or GraphPad Prism (versions 5 and 6) for Windows.
Study approval.
All procedures involving animals were conducted under the supervision and approval of the IACUC of Tel Aviv University and the Israeli Ministry of Health (01-17-029; M-15-059).
Author contributions
GHK and SS provided input on project design and performed experiments (GHK implemented the GFP mouse model, and SS was critical to all facets of the experimental protocols). AYLG performed immunohistochemistry and imaging. I. Grigg and GK performed gene expression and behavioral analyses, respectively. MPC performed bioinformatics analysis. AL performed immunohistochemistry. VK provided funding, performed gene expression experiments, and analyzed data. RAM provided the GFP mouse line, designed the morphology experiments, provided funding, and contributed to the writing of the manuscript. I. Gozes supervised the entire project, provided funding, designed experiments, analyzed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Segev Barak and Yarden Ziv (Tel Aviv University) for performing the NAP intraperitoneal administrations and to Eliezer Giladi (Tel Aviv University) for help with the mouse colony. We thank Lior Bikovski at the Myers Neuro-Behavioral Core Facility for support with in vivo mouse behavioral studies. We also thank Ran Elkon (Tel Aviv University) and Vendula Novosadova (BIOCEV) for their help with the statistical analysis, as well as Lucie Langerová (BIOCEV) for the developmental gene expression analysis. GHK and SS are supported by Eshkol fellowships from the Israeli Ministry of Science and Technology. SS is also supported by a Tel Aviv University Global Research and Training Fellowship and The Naomi Foundation, as well as by The Eldee Foundation/Bloomfield Family of Montreal Awards for Student Exchange (Tel Aviv University/McGill University). VK is supported by the CAS (RVO 86652036) and LQ1604 NPU II, project BIOCEV (CZ.1.05/1.1.00/02.0109) from the European Regional Development Fund (ERDF), and the Ministry of Education, Youth and Sports of the Czech Republic (MEYS CR). I. Gozes is supported by grants from the Israel Science Foundation (ISF) (grant 1424/14), the European Research Area Network (ERA-NET) Neuron AUTISYN, the Alberto Moscona Nisim (AMN) Foundation for the Advancement of Science, Art and Culture in Israel, as well as by Ronith and Armand Stemmer and Arthur Gerbi (French Friends of Tel Aviv University) and Spanish Friends of Tel Aviv University. RAM is supported by the Canadian Institutes of Health Research (CIHR). Funding was also provided by the US–Israel Binational Science Foundation — US National Science Foundation (BSF-NSF 2016746).
Version 1. 08/14/2018
In-Press Preview
Version 2. 09/24/2018
Electronic publication
Version 3. 11/01/2018
Print issue publication
Funding Statement
G.H.K. and S.S. are supported by Eshkol fellowships, the Israel Ministry of Science and Technology. S.S. is further supported by the Tel Aviv University Global Research & Training Fellowship and The Naomi Foundation, as well as The Eldee Foundation/Bloomfield Family of Montreal awards for student exchange (Tel Aviv University/McGill University). V.K. is supported by the CAS (RVO: 86652036) and LQ1604 NPU II, project BIOCEV (CZ.1.05/1.1.00/02.0109) from the ERDF, and MEYS CR. I. Gozes is supported by the following grants, ISF 1424/14, ERA-NET neuron AUTYSM, AMN Foundation as well as Drs. Ronith and Armand Stemmer, Mr Arthur Gerbi (French Friends of Tel Aviv University), and Spanish Friends of Tel Aviv University. R.A. McKinney is supported by CIHR.
Footnotes
Conflict of interest: IG is the chief scientific officer of Coronis Neurosciences, which is developing NAP (CP201) for ADNP syndrome. NAP (CP201) use is under patent protection (US patent nos. US7960334, US8618043, and US7452867; patent publication no. WO2017130190A1.)
Reference information: J Clin Invest. 2018;128(11):4956–4969.https://doi.org/10.1172/JCI98199.
Contributor Information
Gal Hacohen-Kleiman, Email: gal-kl@hotmail.com.
Shlomo Sragovich, Email: srshlomo@gmail.com.
Gidon Karmon, Email: gidikarmon@gmail.com.
Andy Y. L. Gao, Email: andy.gao@mail.mcgill.ca.
Iris Grigg, Email: iris.grigg5@gmail.com.
Metsada Pasmanik-Chor, Email: metsada@post.tau.ac.il.
Albert Le, Email: albert.le@mail.mcgill.ca.
Vlasta Korenková, Email: vlasta.korenkova@ibt.cas.cz.
R. Anne McKinney, Email: anne.mckinney@mcgill.ca.
Illana Gozes, Email: igozes@post.tau.ac.il.
References
- 1.Pinhasov A, et al. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res. 2003;144(1):83–90. doi: 10.1016/S0165-3806(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 2.Mandel S, Rechavi G, Gozes I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol. 2007;303(2):814–824. doi: 10.1016/j.ydbio.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 3.Vulih-Shultzman I, et al. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323(2):438–449. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- 4.Bassan M, et al. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72(3):1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- 5.Zamostiano R, et al. Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem. 2001;276(1):708–714. doi: 10.1074/jbc.M007416200. [DOI] [PubMed] [Google Scholar]
- 6.Amram N, et al. Sexual divergence in microtubule function: the novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol Psychiatry. 2016;21(10):1467–1476. doi: 10.1038/mp.2015.208. [DOI] [PubMed] [Google Scholar]
- 7.Dresner E, et al. Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J Biol Chem. 2012;287(48):40173–40185. doi: 10.1074/jbc.M112.387027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostapcuk V, et al. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature. 2018;557(7707):739–743. doi: 10.1038/s41586-018-0153-8. [DOI] [PubMed] [Google Scholar]
- 9.Mosch K, Franz H, Soeroes S, Singh PB, Fischle W. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS ONE. 2011;6(1):e15894. doi: 10.1371/journal.pone.0015894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem. 2007;282(47):34448–34456. doi: 10.1074/jbc.M704756200. [DOI] [PubMed] [Google Scholar]
- 11.Schirer Y, Malishkevich A, Ophir Y, Lewis J, Giladi E, Gozes I. Novel marker for the onset of frontotemporal dementia: early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS ONE. 2014;9(1):e87383. doi: 10.1371/journal.pone.0087383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malishkevich A, Amram N, Hacohen-Kleiman G, Magen I, Giladi E, Gozes I. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry. 2015;5:e501. doi: 10.1038/tp.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merenlender-Wagner A, et al. Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiatry. 2015;20(1):126–132. doi: 10.1038/mp.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helsmoortel C, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014;46(4):380–384. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen E, Menashe I, Ziats MN, Pereanu W, Packer A, Banerjee-Basu S. A systematic variant annotation approach for ranking genes associated with autism spectrum disorders. Mol Autism. 2016;7:44. doi: 10.1186/s13229-016-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gozes I. Sexual divergence in activity-dependent neuroprotective protein impacting autism, schizophrenia, and Alzheimer’s disease. J Neurosci Res. 2017;95(1-2):652–660. doi: 10.1002/jnr.23808. [DOI] [PubMed] [Google Scholar]
- 18.Gozes I, et al. Premature primary tooth eruption in cognitive/motor-delayed ADNP-mutated children. Transl Psychiatry. 2017;7(2):e1043. doi: 10.1038/tp.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Dijck A, et al. Clinical Presentation of a Complex Neurodevelopmental Disorder Caused by Mutations in ADNP. Biol Psychiatry. doi: 10.1016/j.biopsych.2018.02.1173. doi: 10.1016/j.biopsych.2018.02.1173. [published online ahead of print March 15, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gozes I, et al. The Eight and a Half Year Journey of Undiagnosed AD: Gene Sequencing and Funding of Advanced Genetic Testing Has Led to Hope and New Beginnings. Front Endocrinol (Lausanne) 2017;8:107. doi: 10.3389/fendo.2017.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gozes I, et al. NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP) CNS Drug Rev. 2005;11(4):353–368. doi: 10.1111/j.1527-3458.2005.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morimoto BH, et al. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord. 2013;35(5-6):325–336. doi: 10.1159/000348347. [DOI] [PubMed] [Google Scholar]
- 23.Jarskog LF, et al. Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology. 2013;38(7):1245–1252. doi: 10.1038/npp.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Javitt DC, et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res. 2012;136(1-3):25–31. doi: 10.1016/j.schres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto BH, Fox AW, Stewart AJ, Gold M. Davunetide: a review of safety and efficacy data with a focus on neurodegenerative diseases. Expert Rev Clin Pharmacol. 2013;6(5):483–502. doi: 10.1586/17512433.2013.827403. [DOI] [PubMed] [Google Scholar]
- 26.Oz S, et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol Psychiatry. 2014;19(10):1115–1124. doi: 10.1038/mp.2014.97. [DOI] [PubMed] [Google Scholar]
- 27.Ivashko-Pachima Y, Sayas CL, Malishkevich A, Gozes I. ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: a novel avenue for protection against tauopathy. Mol Psychiatry. 2017;22(9):1335–1344. doi: 10.1038/mp.2016.255. [DOI] [PubMed] [Google Scholar]
- 28. doi: 10.1038/s41431-018-0165-8. Huynh MT, et al. A heterozygous microdeletion of 20q13.13 encompassing ADNP gene in a child with Helsmoortel-van der Aa syndrome [June 13, 2018]. Eur J Hum Genet . [DOI] [PMC free article] [PubMed]
- 29.Vandeweyer G, et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet. 2014;166C(3):315–326. doi: 10.1002/ajmg.c.31413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Paola V, Arber S, Caroni P. AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat Neurosci. 2003;6(5):491–500. doi: 10.1038/nn1046. [DOI] [PubMed] [Google Scholar]
- 31.Chang PK, Khatchadourian A, McKinney RA, Maysinger D. Docosahexaenoic acid (DHA): a modulator of microglia activity and dendritic spine morphology. J Neuroinflammation. 2015;12:34. doi: 10.1186/s12974-015-0244-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2(1):44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- 33.McKinney RA. Excitatory amino acid involvement in dendritic spine formation, maintenance and remodelling. J Physiol (Lond) 2010;588(Pt 1):107–116. doi: 10.1113/jphysiol.2009.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zemlyak I, et al. The microtubule interacting drug candidate NAP protects against kainic acid toxicity in a rat model of epilepsy. J Neurochem. 2009;111(5):1252–1263. doi: 10.1111/j.1471-4159.2009.06415.x. [DOI] [PubMed] [Google Scholar]
- 35.Fitzgerald DM, Charness ME, Leite-Morris KA, Chen S. Effects of ethanol and NAP on cerebellar expression of the neural cell adhesion molecule L1. PLoS ONE. 2011;6(9):e24364. doi: 10.1371/journal.pone.0024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braitch M, et al. Expression of activity-dependent neuroprotective protein in the immune system: possible functions and relevance to multiple sclerosis. Neuroimmunomodulation. 2010;17(2):120–125. doi: 10.1159/000258695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KC, et al. Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol Neurobiol. 2014;49(1):512–528. doi: 10.1007/s12035-013-8535-2. [DOI] [PubMed] [Google Scholar]
- 39.Manuel MN, Mi D, Mason JO, Price DJ. Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front Cell Neurosci. 2015;9:70. doi: 10.3389/fncel.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malishkevich A, Marshall GA, Schultz AP, Sperling RA, Aharon-Peretz J, Gozes I. Blood-Borne Activity-Dependent Neuroprotective Protein (ADNP) is Correlated with Premorbid Intelligence, Clinical Stage, and Alzheimer’s Disease Biomarkers. J Alzheimers Dis. 2016;50(1):249–260. doi: 10.3233/JAD-150799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuper CF, van Bilsen J, Cnossen H, Houben G, Garthoff J, Wolterbeek A. Development of immune organs and functioning in humans and test animals: Implications for immune intervention studies. Reprod Toxicol. 2016;64:180–190. doi: 10.1016/j.reprotox.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S, Imai Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem Biophys Res Commun. 2001;286(2):292–297. doi: 10.1006/bbrc.2001.5388. [DOI] [PubMed] [Google Scholar]
- 43.Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–291. doi: 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 45.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 46.Shu W, et al. Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci U S A. 2005;102(27):9643–9648. doi: 10.1073/pnas.0503739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hodges SL, Nolan SO, Reynolds CD, Lugo JN. Spectral and temporal properties of calls reveal deficits in ultrasonic vocalizations of adult Fmr1 knockout mice. Behav Brain Res. 2017;332:50–58. doi: 10.1016/j.bbr.2017.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rotstein M, Bassan H, Kariv N, Speiser Z, Harel S, Gozes I. NAP enhances neurodevelopment of newborn apolipoprotein E-deficient mice subjected to hypoxia. J Pharmacol Exp Ther. 2006;319(1):332–339. doi: 10.1124/jpet.106.106898. [DOI] [PubMed] [Google Scholar]
- 49. Gozes I. Neuropeptide Techniques. New York, NY; Humana Press: 2008. [Google Scholar]
- 50.Mandel S, Spivak-Pohis I, Gozes I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J Mol Neurosci. 2008;35(2):127–141. doi: 10.1007/s12031-007-9013-y. [DOI] [PubMed] [Google Scholar]
- 51.Borozdin W, et al. Multigene deletions on chromosome 20q13.13-q13.2 including SALL4 result in an expanded phenotype of Okihiro syndrome plus developmental delay. Hum Mutat. 2007;28(8):830. doi: 10.1002/humu.9502. [DOI] [PubMed] [Google Scholar]
- 52.Gordon-Weeks PR. The role of the drebrin/EB3/Cdk5 pathway in dendritic spine plasticity, implications for Alzheimer’s disease. Brain Res Bull. 2016;126(Pt 3):293–299. doi: 10.1016/j.brainresbull.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Mahmmoud RR, et al. Spatial and Working Memory Is Linked to Spine Density and Mushroom Spines. PLoS One. 2015;10(10):e0139739. doi: 10.1371/journal.pone.0139739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Busciglio J, et al. NAP and ADNF-9 protect normal and Down’s syndrome cortical neurons from oxidative damage and apoptosis. Curr Pharm Des. 2007;13(11):1091–1098. doi: 10.2174/138161207780618957. [DOI] [PubMed] [Google Scholar]
- 55.Ferrer I, Gullotta F. Down’s syndrome and Alzheimer’s disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 1990;79(6):680–685. doi: 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- 56.Hayashi-Takagi A, Barker PB, Sawa A. Readdressing synaptic pruning theory for schizophrenia: Combination of brain imaging and cell biology. Commun Integr Biol. 2011;4(2):211–212. doi: 10.4161/cib.4.2.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hart MP, Hobert O. Neurexin controls plasticity of a mature, sexually dimorphic neuron. Nature. 2018;553(7687):165–170. doi: 10.1038/nature25192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aboonq MS, Vasiliou SA, Haddley K, Quinn JP, Bubb VJ. Activity-dependent neuroprotective protein modulates its own gene expression. J Mol Neurosci. 2012;46(1):33–39. doi: 10.1007/s12031-011-9562-y. [DOI] [PubMed] [Google Scholar]
- 59.Gozes I, Alcalay R, Giladi E, Pinhasov A, Furman S, Brenneman DE. NAP accelerates the performance of normal rats in the water maze. J Mol Neurosci. 2002;19(1-2):167–170. doi: 10.1007/s12031-002-0028-0. [DOI] [PubMed] [Google Scholar]
- 60.Jouroukhin Y, Ostritsky R, Assaf Y, Pelled G, Giladi E, Gozes I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: protection against impairments in axonal transport. Neurobiol Dis. 2013;56:79–94. doi: 10.1016/j.nbd.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 61.Snigdha S, et al. H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J Neurosci. 2016;36(12):3611–3622. doi: 10.1523/JNEUROSCI.2693-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim S, et al. Remote Memory and Cortical Synaptic Plasticity Require Neuronal CCCTC-Binding Factor (CTCF) J Neurosci. 2018;38(22):5042–5052. doi: 10.1523/JNEUROSCI.2738-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoykova A, Gruss P. Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J Neurosci. 1994;14(3 Pt 2):1395–1412. doi: 10.1523/JNEUROSCI.14-03-01395.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Umeda T, et al. Evaluation of Pax6 mutant rat as a model for autism. PLoS ONE. 2010;5(12):e15500. doi: 10.1371/journal.pone.0015500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furman S, et al. Sexual dimorphism of activity-dependent neuroprotective protein in the mouse arcuate nucleus. Neurosci Lett. 2005;373(1):73–78. doi: 10.1016/j.neulet.2004.09.077. [DOI] [PubMed] [Google Scholar]
- 66.Hacohen Kleiman G, Barnea A, Gozes I. ADNP: A major autism mutated gene is differentially distributed (age and gender) in the songbird brain. Peptides. 2015;72:75–79. doi: 10.1016/j.peptides.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 67.Alves CJ, et al. Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 2011;1394:90–104. doi: 10.1016/j.brainres.2011.02.060. [DOI] [PubMed] [Google Scholar]
- 68.Dere E, et al. Heterozygous ambra1 deficiency in mice: a genetic trait with autism-like behavior restricted to the female gender. Front Behav Neurosci. 2014;8:181. doi: 10.3389/fnbeh.2014.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4(3):a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alcalay RN, Giladi E, Pick CG, Gozes I. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci Lett. 2004;361(1-3):128–131. doi: 10.1016/j.neulet.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 71.Leker RR, et al. NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke. 2002;33(4):1085–1092. doi: 10.1161/01.STR.0000014207.05597.D7. [DOI] [PubMed] [Google Scholar]
- 72.Richards DA, et al. Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc Natl Acad Sci U S A. 2005;102(17):6166–6171. doi: 10.1073/pnas.0501881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hugel S, Abegg M, de Paola V, Caroni P, Gähwiler BH, McKinney RA. Dendritic spine morphology determines membrane-associated protein exchange between dendritic shafts and spine heads. Cereb Cortex. 2009;19(3):697–702. doi: 10.1093/cercor/bhn118. [DOI] [PubMed] [Google Scholar]
- 74.Verbich D, Prenosil GA, Chang PK, Murai KK, McKinney RA. Glial glutamate transport modulates dendritic spine head protrusions in the hippocampus. Glia. 2012;60(7):1067–1077. doi: 10.1002/glia.22335. [DOI] [PubMed] [Google Scholar]
- 75.Maysinger D, et al. Dendritic polyglycerol sulfate Inhibits microglial activation and reduces hippocampal CA1 dendritic spine morphology deficits. Biomacromolecules. 2015;16(9):3073–3082. doi: 10.1021/acs.biomac.5b00999. [DOI] [PubMed] [Google Scholar]
- 76.Kyriakou EI, et al. Automated quantitative analysis to assess motor function in different rat models of impaired coordination and ataxia. J Neurosci Methods. 2016;268:171–181. doi: 10.1016/j.jneumeth.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 77.Szklarczyk D, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gozes I, Helsmoortel C, Vandeweyer G, Van der Aa N, Kooy F, Sermone SB. The compassionate side of neuroscience: Tony Sermone’s undiagnosed genetic journey--ADNP mutation. J Mol Neurosci. 2015;56(4):751–757. doi: 10.1007/s12031-015-0586-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.