

Abstract
The biosynthesis of glycopeptide antibiotics (GPAs) has been an active area of research for decades. Nonetheless, insights into the activity of the cytochrome P450 enzymes required for installing the aromatic crosslinks, which form their cup-shaped topologies and render GPAs bioactive, have only recently emerged. Presently, little is known about the substrate scope and promiscuity of the P450 enzymes. Herein, we report that OxyBvan, the P450 enzyme that installs the first crosslink in vancomycin biosynthesis, is capable of catalyzing the formation of its conventional C-O-D bis-aryl ether bond in non-natural substrates and, furthermore, the formation of a second, novel linkage when D-Trp is incorporated at position 6. HR-MS/MS and isotope labeling studies indicate the second crosslink is formed between rings A and B, resulting in a novel GPA-type scaffold. OxyB is also capable of installing two crosslinks in kistamicin- and complestatin-like substrate peptides. These findings highlight the utility of OxyBvan in creating crosslinked GPA derivatives and provide clues regarding the unusual biosynthesis of kistamicin.
Graphical Abstract
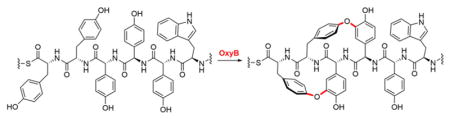
1. Introduction
Vancomycin is one of our most important weapons in the fight against infectious disease and among the WHO’s 100 most essential medicines (1, Fig. 1).1,2 Since its discovery in 1953 as the archetypal glycopeptide antibiotic (GPA), both the biological activity and structural diversity of this compound family has significantly expanded.3 Currently five structural subclasses are recognized, with representative members shown (1–6, Fig. 1). Common to all GPAs is rigid, atropoisomeric topology that is held in place by two to four aromatic crosslinks. The synthesis of a number of GPAs and analogs thereof have already been completed, even those that retain efficacy against drug-resistant pathogens.4 The corresponding biosynthetic pathways, however, remain an area of ongoing investigation that started more than 20 years ago.5 Identification of the biosynthetic gene cluster (BGC) for vancomycin and similar class I GPAs, such as balhimycin, along with pioneering genetic studies suggested that three cytochrome P450 enzymes – named OxyB, OxyA and OxyC – catalyze the oxidative cross-coupling reactions in that order.6–10 Nearly ten years after these initial findings, the enzymatic activity of OxyB from the vancomycin BGC was reconstituted in vitro.11 The discovery of an unusual X-domain, which recruits the P450 enzyme and the pendant 7mer peptide substrate for the crosslinking reaction, and characterization of Oxy enzymes from the teicoplanin (4) BGC have provided additional key insights.12–14 Recently, the activity of OxyA and OxyC have been demonstrated, thus facilitating the in vitro chemo-enzymatic synthesis of vancomycin.15,16
Figure 1.

The glycopeptides antibiotics. Representative members of each subclass are shown. In vancomycin, the rings are labeled and the aromatic crosslinks, installed by OxyB, OxyA, and OxyC in that order, are marked.
With these advances in mind, one can begin to assess the specificities of the P450 enzymes with the goal of creating de novo libraries of vancomycin derivatives that may exhibit enhanced or entirely new biological activities. Toward that end, we herein explore the specificity of the OxyB from the vancomycin BGC (OxyBvan), which installs the first bis-aryl ether crosslink during biosynthesis of the antibiotic. We tested the reaction of OxyBvan with substrate derivatives that, like the class V GPAs (5, 6), contain a Trp residue at position 6. Surprisingly, we find that OxyB doubly crosslinks this substrate to produce a bicyclic product, rather than the usual monocyclic product. HR-MSn and isotope labeling analyses suggest that the product mirrors the kistamicin crosslinks at rings A–B and C–D. Spurred by these results, we investigated the reaction of OxyBvan with the linear 7mer peptide precursors of complestatin (5) and kistamicin (6). Both of these peptides were doubly-crosslinked as well, indicating a significant degree of substrate promiscuity by OxyBvan. Our results carry two major implications: They suggest that the substrate tolerance of OxyBvan can be exploited to chemo-enzymatically generate new vancomycin derivatives with novel crosslinks. They also imply that evolution of new GPAs is possible by simple modulation of the 7mer peptide substrate, even in the absence of major primary sequence changes to the P450 enzymes.
2. Results and Discussion
In all GPA biosynthetic gene cluster examined thus far, one P450 enzyme can be identified for each crosslink, with the kistamicin gene cluster (kis) forming the only exception.17 Indeed, the annotation of the kis cluster revealed only two P450 enzymes, suggesting that one of these installs two aromatic crosslinks. To gain insights into this issue and the specificity of the vancomycin OxyB, we explored the reactivity of the enzyme with a substrate that, like kistamicin (6) contains a Trp residue at position 6. We synthesized two vancomycin-like 7mer peptide substrates by solid-phase peptide synthesis (SPPS): a control bearing the native 3-Cl-D-Tyr (7, Fig. 2A), and a second containing D-Trp at residue 6 (8, Fig. 2A, Fig. S1). Because the C-terminal residue has been reported to be prone to racemization, we used L-Tyr in place of L-3,5-dihydroxy-phenylglycine. Upon completion of the 7mer, each peptide was released from the 2-chlorotrityl resin as an acyl hydrazide, which was subsequently activated and converted to a coenzyme A (CoA) thioester conjugate using a native chemical ligation-based scheme (Fig. 2A, Fig. S1).15 The CoA-7mer conjugate was then loaded onto the X-domain-peptidyl carrier protein from the vancomycin pathway (X-PCPvan) using an optimized Bacillus subtilis 4-phosophpantetheinyl transferase (Sfp R4-4, Fig. 2B).12 This di-domain contains the PCP that covalently carries the 7mer-phosphopentetheine on a conserved Ser residue as well as the X-domain, which serves as a scaffold and enables efficient interaction between the peptide substrate and P450 enzyme. Each substrate was then mixed with OxyBvan and the requisite electron transfer protein partners. At defined time points, the reaction was quenched by cleaving the 7mer peptide from X-PCP via propylamine-mediated aminolysis. The product(s) were then characterized by HPLC-Qtof-MS.
Figure 2.

Reaction of OxyB with a GPA class V-type 7mer substrate results in two products. (A) Synthesis of 7mer-CoA conjugates carrying L-Tyr and 3-Cl-D-Tyr (7) or L-Tyr and D-Trp (8) at residues 1 and 6, respectively. The reaction conditions are shown, see Methods for further details. (B) Attachment of (7) and (8) onto X-PCP and reaction with OxyB and associated assay proteins. Fd, ferredoxin; FdR, ferredoxin reductase; G6P-DH, glucose-6-phosphate dehydrogenase. See Methods for further details. Only one product (9) is observed with 7. A singly-crosslinked (10) and a doubly-crosslinked (11 or 12) product is formed with substrate 8, both of which are OxyB-dependent. (C) MS/MS fragmentation pattern for product 10. (D, E) Observed fragmentation pattern for the doubly-crosslinked product and proposed structures. Both a B-O-A (D) and A-O-B (E) crosslinks (in addition to the C-O-D aryl ether bond) are consistent with the HR-MR/MS data.
With the control peptide, we observed a −2 Da product peak with an [M+H]+obs of 1154.41753 ([M+H]+calc 1154.4152), as expected and previously reported (9, Fig. 2B).15 HR-MS/MS analysis revealed all b and y ions for the substrate, while for the product peak, the b4, b5, y3, y4 ions were no longer detected, consistent with C-O-D aryl ether bond formation. In line with this structure were the b6, b7, y5, y6, y7 ions observed for the product, which were 2 Da lighter compared to the corresponding substrate fragment ions. These data are entirely consistent with the results of Woithe et al and our recent reports, all of which demonstrate C-O-D crosslink formation by OxyBvan.11
With the Trp-containing peptide, a different outcome was obtained (8, 10, Fig. 2A, B). Two OxyB-dependent product peaks were observed corresponding to a loss of 2 Da ([M+H]+obs 1143.46487, [M+H]+calc 1143.4701, 47% yield) and 4 Da ([M+H]+obs 1141.45159, [M+H]+calc 1141.4545, 2% yield) relative to substrate (Table S1). HR-MS/MS analysis confirmed that the −2 Da product contained the C-O-D aryl ether linkage, as expected for an OxyB-catalyzed transformation (10, Fig. 2C, Fig. S2, Table S2). The −4 Da product also contained the C-O-D crosslink, but HR-MS/MS analysis revealed disappearance of the b6 and y2 ions, which can be observed in the C-O-D singly cross-linked peptide (10–12, Fig. 2C, Fig. S2, Table S2). Moreover, the y5, y6, y7 ions were now 4 Da lighter than the corresponding linear substrate fragment ions, indicating the formation of a second cross-link involving rings A and B. These results suggest that, in the presence of D-Trp and L-Tyr at residues 6 and 1, OxyB is capable of installing its canonical C-O-D crosslink on a non-native substrate and, additionally, a second unnatural bridge between residues A and B.
Given that OxyB has been shown to install aryl ether bonds ortho to the phenol-hydroxyl group, rather than biaryl carbon-carbon crosslinks, as is the case for OxyCvan, we surmised that the A–B crosslink could consist either of the Hpg phenol-oxygen bound to the meta position of the Tyr side chain (11, B-O-A), or the Tyr-phenol-oxygen linked to the meta position of Hpg (12, A-O-B, Fig. 2D, 2E). To distinguish between these possibilities, an isotope replacement strategy was envisioned using 3,5-2H2-L-Tyr. In case of formation of 11, we would expect a loss of 5 Da in the final product, whereas a loss of 4 Da would be anticipated with the crosslink shown in 12.
3,5-2H2-L-Tyr was synthesized using acid-mediated exchange of the ortho-hydrogens by deuterons in DCl-D2O (13, Fig. 3A).18 The isotopomer was then Fmoc-protected at the α-amine, incorporated at the C-terminal residue of a 7mer peptide, and loaded onto the X-PCPvan domain as described above to give substrate 14 (Fig. S3). Reaction of OxyB and analysis by HR-MSn revealed a −4 Da product peak, consistent with the loss of four hydrogen atoms only and no deuterons ([M+H]+obs =1143.4643, ([M+H]+calc 1143.4670, Fig. 3B–3D Table S1). In line with this conclusion were HR-MS/MS data, where the y5 ion for the −4 Da product with substrate 14 clearly contained two deuterons, in contrast to the y5 ion of the −4 Da product with substrate 8 (Fig. S4, Table S3). While we cannot exclude a carbon-carbon A–B crosslink, we propose structure 12 as the second product of the OxyB-catalyzed reaction with substrate 8. The A-O-B crosslink ‘mirrors’ the C-O-D crosslink in that a Tyr-OH is anchored to the meta-carbon of an Hpg amnio acid. Interestingly, the A-O-B crosslink only occurs when Trp is incorporated at residue 6, and how this residue potentiates the promiscuous OxyBvan activity remains to be determined. Compound 12 represents a new GPA scaffold, the bioactivity of which will be of interest in future studies.
Figure 3.

OxyB installs an A-O-B crosslink onto a non-native substrate 7mer. (A) Synthesis 3,5-dideutero-L-Tyr (13) and conjugation to CoA to generate 14. (B) Attachment and 14 to X-PCP and reaction with OxyB results in a product with both deuterons intact. (C, D) HPLC-Qtof-MS analysis of the reactions of 8 (C) or 14 (D) with OxyB. Shown are extracted ion chromatograms for substrate (black trace), singly-crosslinked (blue) and doubly-crosslinked (orange) products. Extracted values are within 10 ppm of the calculated substrate/product m/z. No deuterium loss is observed in the product generated from substrate 14.
In light of the unusual substrate tolerance, we wondered whether two crosslinks would also be installed by OxyBvan onto kistamicin- and complestatin-type peptides. We synthesized two peptides to answer this question: the first was identical to the presumed kistamicin 7mer linear substrate, except that it contained D-Tyr at residue 1 (in place D-Hpg, D-Hpg at residue 3 (in place of 3-Cl-D-Hpg), and D-Hpg at residue 5 (in place of D-3,5-dihydroxyphenylglycine, D-Dpg) (15, Fig. 4, Fig. S5). The second peptide was identical to the presumed complestatin precursor, except that it contained D-Tyr at residue 1 (in place of D-Hpg), and D-Hpg at residues 3, 5, and 7 (in place of 3,5-Cl2-D-Hpg) (16, Fig. 4, Fig. S6). These substitutions were chosen to simplify the synthesis of both precursor peptides. The two 7mer substrates were then thioesterified with CoA and appended onto the X-PCPvan di-domain as described above and reacted with OxyBvan.
Figure 4.

OxyB installs A-O-B crosslinks onto kistamicin- and complestatin-like precursors. Reaction scheme for products observed with kistamicin-like (15) and complestatin-like (16) substrates. The ratio of the singly vs. doubly-crosslinked product for both kistamicin- and complestatin-like substrates was ~23:1. In both cases, the monocyclic product was obtained in ~46% yield.
To our surprise, reaction with OxyBvan again showed formation of products with a loss of 2 and 4 Da with both substrates 15 and 16 (Table S1). HR-MS/MS analyses verified that the crosslinks occurred between rings C and D, as well as A and B, similar to the result obtained for substrate 8 (Fig. S7, S8, Table S4, S5). Given the deuteration results in Fig. 3, we propose structures 17 and 18 as the doubly-crosslinked products of OxyBvan with substrates 15 and 16, respectively. In addition to incorporating two aryl ether bonds in the vancomycin 7mer, OxyBvan appears to catalyze a similar reaction with substrates that mirror the 7mers for class V GPAs. The inclusion of Trp at residue 6 and Tyr at residue 1 appears to alter the specificity and enhance the catalytic capabilities of OxyB.
The results with the kistamicin- and complestatin-like precursors are especially remarkable in light of the disparate stereochemical configurations: the vancomycin peptide sequence consists of L-L-D-D-L-D-D amino acids (from C- to N-terminus), whereas the configuration of the kistamicin and complestatin precursors is D-L-D-D-D-D-D. Our results indicate that OxyBvan can install its canonical crosslink between amino acids 2 and 4 in all peptides examined here and a linkage between rings A and B, whether they bear the L-D or D-D configurations at residues 1 and 3, suggesting an unusually non-specific enzyme active site.
The biosynthesis of kistamicin has not yet been examined. Based on sequence comparisons with previously characterized GPA P450 enzymes and the results herein reported, we predict that OxyBkis is capable of catalyzing the C-O-D and B-O-A crosslinks, which are observed in the final product, and that OxyAkis installs the unusual phenol-indole bond. Although the structures of kistamicin and vancomycin are fairly divergent, the P450 enzymes that catalyze the first crosslink exhibit 66% similarity and 52% identity. These insights have implications for the combinatorial chemo-enzymatic preparation of vancomycin derivatives as well as the evolution of new GPAs.
3. Conclusions
We have herein examined the substrate tolerance of OxyBvan and discovered that it is unusually non-specific with respect to the number of crosslinks it catalyzes as well as the stereochemical nature of the 7mer substrate. Specifically, replacements of residues 1 and 6 appear to have an unusual impact on the reaction carried out by the enzyme. Instead of a single crosslink observed in the native peptide, two crosslinks are now found, consisting of C-O-D and A-O-B aryl ether bonds. The molecular basis for this promiscuous enzymatic activity warrants further investigation.
Our results provide some possible clues regarding the biosynthesis of kistamicin, in which – as mentioned above – three crosslinks have been proposed to result from the action of two P450 enzymes. It is fair to assume that the diversity observed in the GPAs is the result of evolution of both the crosslinking P450 enzymes and the sequence of the peptide substrate. Our results, however, suggest that simple modifications to the substrate peptide, even in the absence of modulation or evolution of the P450 enzymes, can result in new GPAs. As such, a primarily substrate-mediated natural product diversification, at least in the first P450 enzyme, could explain the evolution of new GPAs. Additional studies are required to test this idea. Notwithstanding, the alternative reactivity observed might be used to synthesize doubly crosslinked vancomycin derivatives with different stereochemistries, the biological activities of which can now be investigated.
Moreover, the lack of specificity for the 7mer peptide, the requirement of a PCP domain and the presence of the X-domain compels us to draw analogies with RiPPs (ribosomally synthesized and post-translationally modified peptides) and their associated RREs (RiPP recognition elements).19–21 The biosynthesis of ribosomally-synthesized natural products begins with a multi-partite precursor peptide, which contains ‘leader’ and ‘core’ sequences. The core is modified by the tailoring enzyme during biosynthesis of the mature natural product. The leader serves to provide specificity by facilitating binding of the peptide substrate to one or more tailoring enzymes, before it is removed in the final stages of maturation. Because specificity has been ‘outsourced’ to the leader portion, RiPP tailoring enzymes are surprisingly tolerant of changes to the core region. Might an analogous model explain our results with OxyBvan? It is possible that specificity during the crosslinking reaction is provided by the interaction between the X-domain, a possible non-ribosomal version of an RRE, and OxyBvan. This arrangement then would confer a fair degree of tolerance toward the ‘core’ region, that is, the 7mer peptide where aromatic cross-coupling takes place. Additional studies are needed to test this hypothesis experimentally, to provide a molecular basis for the extreme promiscuity observed with OxyBvan, and to harness its catalytic potential to create novel GPA-type scaffolds.
4. Experimental
4.1 Materials, strains, and general procedures
Amycolatopsis orientalis DSM40040 was obtained from the American Type Culture Collection (ATCC). LB broth, Terrific Broth and LB agar were purchased from Becton Dickinson. All antibiotics, IPTG, PMSF, lysozyme, β-mercaptoethanol, Sephadex G-25, COMU, NEt3, DIPEA, TIS, DBU, TFA, DCl/D2O, Fmoc-OSu, hydrazine monohydrate, NaNO2, coenzyme A trilithium salt, glucose-6-phosphate dehydrogenase, glucose-6-phosphate, and other components necessary for biochemical assays were obtained from Sigma-Aldrich. Nickel affinity resin and DNase I were purchased from Clontech. Restriction enzymes, T4 DNA ligase, proofreading Q5 DNA polymerase, and the corresponding buffers were purchased from New England Biolabs. PCR reactions were routinely carried out in Failsafe buffer G (Epicentre). Commercially available Fmoc- and side chain-protected amino acids, 2-chlorotrityl chloride resin and other components for solid-phase peptide synthesis were purchased from EMD Millipore and Sigma-Aldrich.
1H NMR spectra were recorded at the Princeton Chemistry NMR facility on a Bruker 500 (500 MHz) and referenced relative to residual acetonitrile (in CD3CN) proton signals at δ 1.94 ppm. Data for 1H spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad, ap = apparent), integration, coupling constant (Hz) and assignment. 13C NMR spectra were recorded on a Bruker 500 (126 MHz) and are referenced relative to residual acetonitrile (in CD3CN) at δ 118.26 and 1.32 ppm. Data for 13C NMR spectra are reported in terms of chemical shift and multiplicity where appropriate.
4.2 Expression and purification of OxyB, OxyA, PCP7-X, Fd, FdR, Sfp R4-4
OxyB and PCP7-X from Amycolatopsis orientalis DSM 40040 were expressed and purified as previously described.15 OxyB exhibited an A420nm/A280nm ratio of 2:1, which is consistent with nearly complete heme loading. Ferredoxin from spinach, ferredoxin reductase from E. coli, Spf R4-4 from B. subtilis (codon-optimized, K28E/T44E/C77Y triple mutant of phosphopantetheinyl transferase) were expressed and purified as previously described.11,12,15
4.3 Synthesis of N-Fmoc-L-3,5-2H2-tyrosine (13)
According to Kanska et al., L-Tyr (600 mg, 3.31 mmol) was dissolved in 18 mL of DCl-D2O (15 ml of D2O + 3 ml conc. DCl/D2O) in a 100-mL round-bottomed flask.18 Inert atmosphere was established in the flask by evacuating and refilling with N2 three times. The reaction mixture was refluxed for 24 h, cooled to RT, and concentrated in vacuo. 1H and 13C NMR spectra show near 100% deuterium incorporation into 3- and 5-positions of L-tyrosine. 1H NMR (500 MHz, D2O): δ 7.19 (s, 2H), 4.20 (dd, J = 7.7, 5.4 Hz, 1H), 3.25 (dd, J = 14.7, 5.4 Hz, 1H), 3.12 (dd, J = 14.7, 7.7 Hz, 1H). 13C NMR (126 MHz, D2O): δ 172.09, 154.93, 130.66, 125.85, 115.61 (t, J = 24.1), 54.61, 34.88. MS-ESI m/z 184.0939 ([M + H]+, C9H10D2NO3, calc. 184.0943).
L-3,5-2H2-tyrosine was dissolved in 30 ml dioxane/water (1:1) and NaHCO3 (695 mg, 8.28 mmol) was added. The solution was cooled in an ice bath and Fmoc-OSu (1.14 g, 3.38 mmol) was added in portions. The reaction mixture was stirred overnight at room temperature. 20 ml water was added and the solution was extracted into EtOAc (3 × 30 ml). The combined organic layers were washed with brine (20 mL), then dried over anhydrous Na2SO4, and concentrated in vacuo. The resulting white solid was recrystallized from hot Et2O/hexane (1.26 g, 94%). 1H NMR (500 MHz, CD3CN): δ 7.83 (d, J = 7.6 Hz, 2H), 7.60 (dd, J = 7.6, 3.2 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.32 (tq, J = 6.3, 3.8 Hz, 2H), 7.05 (s, 2H), 5.91 (d, J = 8.2 Hz, 1H), 4.36 – 4.22 (m, 3H), 4.18 (t, J = 7.0 Hz, 1H), 3.07 (dd, J = 14.1, 5.0 Hz, 1H), 2.84 (dd, J = 14.1, 9.1 Hz, 1H). 13C NMR (126 MHz, CD3CN): δ 173.35, 156.79, 156.55, 145.00, 144.94, 142.03, 131.18, 129.04, 128.62, 128.03, 126.15, 126.08, 120.90, 67.19, 56.25, 47.88, 37.05. MS-ESI m/z 406.1618 ([M + H]+, C24H20D2NO5, calc. 406.1624).
4.4 Synthesis of and purification of heptapeptide substrates 7, 8, 14, 15, 16
7mer peptides were synthesized and purified as previously described.15 See supporting data for further details (Figs. S1, S3, S5, S6).
4.5 Synthesis and purification of Coenzyme A adducts of 7mer peptides
A 4 mM solution of 7mer peptide in 0.2 M sodium phosphate acidic solution containing 6 M guanidinium chloride at pH 3 was added to an Eppendorf tube equipped with a stir bar. The solution was cooled to −10°C in an ice/salt bath. S odium nitrite (25 equiv.) was added to the reaction mixture, which was stirred for 20 minutes at −10°C. Coenzyme A trilithium salt (20 equiv.) was added to the reaction mixture to a final concentration of 100 mM. Subsequently, the pH was adjusted to 6.8–7.0 with a micro-pH probe and the reaction was allowed to warm to room temperature and stir for 1 h. The reaction mixture was diluted with MeCN (+ 0.1% FA) to a final volume of 0.8 mL. Purification of peptide-CoA was achieved by repeated injections onto an analytical Phenomenex Luna C18 column (5 μm, 250 × 4.6 mm) that had been equilibrated with 10% MeCN in H2O (+ 0.1% FA). The peptide-CoA adducts were eluted with a gradient of 10–55% MeCN in H2O (+ 0.1% FA) over 17 minutes. The purified material was verified by HPLC-MS, aliquoted, and lyophilized (see Figs. S1, S3, S5, S6).
4.6 Activity assays of OxyB
Assay conditions were based on previously described protocols.15 Reactions were carried out on a 100 μL scale. Loading buffer (50 mM HEPES, 20 mM KCl, 10 mM MgCl2, pH 7.0) was added to an Eppendorf tube containing 20 nmol of lyophilized peptide-CoA adduct, to a final concentration of 400 μM. Subsequently, final concentrations of 400 μM PCP7-X and 40 μM of Sfp R4-4 were added to the reaction mixture, which was placed in a 30°C incubator for one hour. In standard reactions, final concentrations of the following reagents were added to the reaction mixture, in this order: 2 mM glucose-6-phosphate, 4 units of glucose-6-phosphate dehydrogenase, 14.4 μM spinach ferredoxin, 3 μM E. coli flavodoxin reductase, 5 μM OxyB, 7.5 μM OxyA. Finally, the oxidative crosslinking reaction was initiated by the addition of 2 mM NADPH. Typical assays were carried out at room temperature for 2 hours in the dark. In order to remove the peptide from the carrier domain, 20,000 equivalents of propylamine were added and the reaction mixture incubated for 15 minutes. Proteins were precipitated by adding 15 μL of formic acid and 50 μL of MeCN (+0.1% FA). Denatured proteins were pelleted and the supernatant was analyzed by HR-HPLC-MS and MS/MS (Figs. S2, S4, S7, S8, Table S1–S5).
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (grants GM065483 to E.J.S. and DP2-AI-124786 to M.R.S.) for support of this work. C.C.F. was supported by an Eli Lilly-Edward C. Taylor Fellowship in Chemistry.
Footnotes
Supplementary data related to this article can be found at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butler MS, Hansford KA, Blaskovich MAT, Halai R, Cooper MA. J Antibiot. 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- 2.WHO. Model List of Essential Medicines, 19th List. Apr, 2015. [Google Scholar]
- 3.Nicolaou KC, Boddy CNC, Bräse S, Winssinger N. Angew Chem Int Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 4.Okano A, Isley NA, Boger DL. Chem Rev. 2017;117:11952–11993. doi: 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbard BK, Walsh CT. Angew Chem Int Ed. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 6.Pelzer S, Süssmuth RD, Heckmann D, Recktenwald J, Huber P, Jung G, Wohlleben W. Antimicrob Agents Chemother. 1999;43:1565–1573. doi: 10.1128/aac.43.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bischoff D, Pelzer S, Höltzel A, Nicholson GJ, Stockert S, Wohlleben W, Jung G, Süssmuth RD. Angew Chem Int Ed. 2001;40:1693–1696. doi: 10.1002/1521-3773(20010504)40:9<1693::aid-anie16930>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Bischoff D, Pelzer S, Bister B, Nicholson GJ, Stocker S, Schirle M, Wohlleben W, Jung G, Süssmuth RD. Angew Chem Int Ed. 2001;40:4688–4691. doi: 10.1002/1521-3773(20011217)40:24<4688::aid-anie4688>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Stegmann E, Pelzer S, Bischoff D, Puk O, Stockert S, Butz D, Zerbe K, Robinson J, Süssmuth RD, Wohlleben W. J Biotechnol. 2006;124:640–653. doi: 10.1016/j.jbiotec.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 10.van Wageningen AM, Kirkpatrick PN, Williams DH, Harris BR, Kershaw JK, Lennard NJ, Jones M, Jones SJ, Solenberg PJ. Chem Biol. 1998;5:155–162. doi: 10.1016/s1074-5521(98)90060-6. [DOI] [PubMed] [Google Scholar]
- 11.Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K, Robinson JA. J Am Chem Soc. 2007;129:6887–6895. doi: 10.1021/ja071038f. [DOI] [PubMed] [Google Scholar]
- 12.Haslinger K, Maximowitsch E, Brieke C, Koch A, Cryle MJ. ChemBioChem. 2014;15:2719–2728. doi: 10.1002/cbic.201402441. [DOI] [PubMed] [Google Scholar]
- 13.Peschke M, Brieke C, Cryle MJ. Sci Rep. 2016;6:35584. doi: 10.1038/srep35584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haslinger K, Peschke M, Brieke C, Maximowitsch E, Cryle MJ. X-domain of peptide synthetases recruits oxygenases crucial for glycopeptide biosynthesis. Nature. 2015;521:105–109. doi: 10.1038/nature14141. [DOI] [PubMed] [Google Scholar]
- 15.Forneris CC, Ozturk S, Gibson MI, Sorensen EJ, Seyedsayamdost MR. ACS Chem Biol. 2017;12:2248–2253. doi: 10.1021/acschembio.7b00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forneris CC, Seyedsayamdost MR. Under review. [Google Scholar]
- 17.Nazari B, Forneris CC, Gibson MI, Moon K, Schramma KR, Seyedsayamdost MR. Med Chem Commun. 2017;8:780–788. doi: 10.1039/c6md00637j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanska M, Drabarek S. Radiochem Radioanal Lett. 1980;44:207–210. [Google Scholar]
- 19.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Goeransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Mueller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl H-G, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Suessmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh CT. ACS Chem Biol. 2014;9:1653–1661. doi: 10.1021/cb5003587. [DOI] [PubMed] [Google Scholar]
- 21.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564–570. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.