

Abstract
Rationale:
Abnormal SUMOylation has emerged as a characteristic of heart failure (HF) pathology. Previously, we found reduced small ubiquitin-like modifier 1 (SUMO1) expression and sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) SUMOylation in human and animal HF models. SUMO1 gene delivery or small-molecule activation of SUMOylation restored SERCA2a SUMOylation and cardiac function in HF models. Despite the critical role of SUMO1 in HF, the regulatory mechanisms underlying SUMO1 expression are largely unknown.
Objective:
To examine miR-146a-mediated SUMO1 regulation and its consequent effects on cardiac morphology and function.
Methods and Results:
In this study, miR-146a was identified as a SUMO1-targeting microRNA in the heart. A strong correlation was observed between miR-146a and SUMO1 expression in failing mouse and human hearts. miR-146a was manipulated in cardiomyocytes through adeno-associated virus serotype 9 (AAV9)-mediated gene delivery, and cardiac morphology and function were analyzed by echocardiography and hemodynamics. Overexpression of miR-146a reduced SUMO1 expression, SERCA2a SUMOylation, and cardiac contractility in vitro and in vivo. The effects of miR-146a inhibition on HF pathophysiology were examined by transducing a tough decoy of miR-146a into mice subjected to transverse aortic constriction (TAC). miR-146a inhibition improved cardiac contractile function, and normalized SUMO1 expression. The regulatory mechanisms of miR-146a upregulation were elucidated by examining the major miR-146a-producing cell types and transfer mechanisms. Notably, trans-differentiation of fibroblasts triggered miR-146a overexpression and secretion through extracellular vesicles (EVs), and the EV-associated miR-146a transfer was identified as the causative mechanism of miR-146a upregulation in failing cardiomyocytes. Finally, EVs isolated from failing hearts were shown to contain high levels of miR-146a and exerted negative effects on the SUMO1/SERCA2a signaling axis and hence, cardiomyocyte contractility.
Conclusions:
Taken together, our results show that miR-146a is a novel regulator of the SUMOylation machinery in the heart, which can be targeted for therapeutic intervention.
Keywords: Heart failure, SERCA2a, SUMO1, microRNA, extracellular vesicle, miR-146a, posttranscriptional regulation, gene therapy
Keywords: Calcium Cycling/Excitation-Contraction Coupling, Gene Therapy, Heart Failure
INTRODUCTION
Ca2+ cycling abnormalities in cardiomyocytes have been shown to be critical in the pathophysiology of HF.1–3 Experimental studies have demonstrated that stabilization of Ca2+ cycling significantly rescues the HF phenotype and increases animal survival. In this regard, the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) pump and its regulators have been shown to be important targets for intervention.4–9 Previously, our group identified small ubiquitin-like modifier 1 (SUMO1) as a positive regulator of SERCA2a that exhibits reduced levels during HF.10 SERCA2a undergoes SUMOylation at lysine sites 480 and 585 by SUMO1, rendering it more stable and increasing its activity.10 Gene transfer of SUMO1 normalizes SUMO1 expression, enhances SERCA2a SUMOylation, and restores cardiac function in both mouse and swine HF models.10–12 However, the regulatory mechanisms underlying the downregulation of SUMO1 expression have yet to be elucidated.
MicroRNAs (miRs) are short single-stranded noncoding RNAs that evolved to fine-tune biological networks by destabilization and/or translational inhibition of target mRNAs.13 Alterations in the levels of several miRs (i.e., miR-1, 21, and 133) have been observed in human failing myocardium.14 Single miRs tend to regulate multiple targets within the same functional pathway. Furthermore, miR-targeting drugs are unlikely to be limited by immunological issues that commonly accompany virus-mediated gene therapy.15 Therefore, miRs are attractive targets for human diseases including HF. For example, our group found that miR-25 downregulates SERCA2a expression and that miR-25 inhibition enhances Ca2+ handling and cardiac function through the restoration of SERCA2a levels in failing mouse hearts.16 In the present study, we discovered that miR-146a negatively regulates Ca2+ cycling by downregulating SUMO1 expression.
miR-146a was initially reported to act as a negative regulator of innate immune responses by targeting interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6).17 Thus, downregulation of miR-146a is closely associated with autoimmune diseases.18–20 The role of miR-146a in the heart has been shown to vary by heart disease model. Overexpression of miR-146a has exerted beneficial effects by suppressing excessive immune responses in ischemia/reperfusion injury21, septic cardiomyopathy22, and diabetic cardiomyopathy models23. In contrast, miR-146a overexpression has shown to have deleterious effects such as reduced cardiac function and adverse cardiac remodeling in doxorubicin-induced cardiomyopathy24, peripartum cardiomyopathy25, and pressure overload models26 by targeting genes such as Erbb4, Nras, or Dlst. Therefore, the conclusive role of miR-146a during HF has yet to be defined.
Here, we tested the effects of miR-146a modulation in transverse aortic constriction (TAC)-induced HF models and validated the role of miR-146a as a SUMO1-targeting miR. We discovered that miR-146a expression correlates inversely with SUMO1 expression in failing human and mouse hearts. Using genetic techniques, we demonstrated that overexpression of miR-146a attenuates SUMO1 expression, SERCA2a SUMOylation, and cardiac contractile function in vitro and in vivo. In contrast, inhibition of miR-146a normalizes these alternations by the elevated miR-146a levels in the failing conditions. Furthermore, we found that miR-146a is formed in fibroblasts, but not in cardiomyocytes, and then released as an extracellular vesicle (EV)-associated form. The EV-associated miR-146a is transferred into cardiomyocytes and attenuates SUMO1 expression, thereby reducing contractile function. Collectively, we demonstrate a novel mechanism underlying the regulation of SUMO1 expression, which provides novel insights into cardiac intercellular communication in HF.
METHODS
All data have been made publicly available at figshare and can be accessed at 10.6084/m9. figshare.6932888 or from the corresponding author on request. Detailed Methods section is available in the Online Data Supplement.
EV treatment.
Our FACS and NTA analyses revealed the number of cardiac cells and EVs from normal and failing hearts to have the ratio of 1:2000 (cells:EVs). Thus, the EVs were treated at the rate of two thousand in EV treatment experiments. The isolated adult cardiomyocytes were treated with purified EVs for 24hrs after which RNA and contractile properties of cardiomyocytes were analyzed.
EV labeling and confocal analysis.
EVs were subjected to PKH26 labeling according to the manufacturer’s recommendations. For confocal laser scanning microscopy experiments, cardiomyocytes were incubated with PKH26-labeled EVs (2000 EVs/cell) for 24 hrs. Cardiomyocytes were washed twice with PBS solution to remove surface associated EVs and were then fixed in 2% (wt/vol) paraformaldehyde for 5 min on ice. Cardiomyocytes were imaged using Leica SP5 CLSM confocal microscope.
Statistical analysis.
Data are expressed as mean ± SEM or SD as indicated. All data were analyzed using the Student’s t-test for comparison of two groups, One-way ANOVA for three groups. Survival testing was performed with the Kaplan-Meier analysis. To assess the correlation between miR-146a expression and SUMO1 expression, we used Spearman correlation and plotted individual relative expressions of miR-146a and SUMO1 that were normalized by 18S RNA expression. For all in vivo experiments, the groups were randomly allocated and examined under blind test conditions. Statistical outliers were not automatically excluded; only technical failures or sudden death were excluded from the datasets. Differences were considered statistically significant when the p-value was < 0.05.
RESULTS
miR-146a directly targets SUMO1 in cardiomyocytes.
To gain a better understanding of the regulatory mechanisms underlying decreased SUMO1 expression during HF, we sought to identify miRs that affect the expression of SUMO1 using in silico screening methods. Four different prediction programs (microRNA.org, miRSearch, DIANA, and mirdb.org) all yielded miR-146a as a strong candidate (Figure Ia in the Data Supplement). The Sumo1 3′-untranslated region (UTR) contains a putative miR-146a binding site that is highly conserved among selected vertebrates (Figure Ib in the Data Supplement). Intriguingly, miR-146a was previously shown to affect Ca2+ handling in cardiomyocytes.16 Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses revealed that miR-146a levels were significantly elevated in failing human and mouse hearts (Figure 1A and B) and that miR-146a and SUMO1 levels were inversely correlated in these hearts (Figure Id and e in the Data Supplement). miR-146b shares a seed sequence with miR-146a28; however, its expression was not altered in failing hearts (Figure Ic in the Data Supplement). Thus, in this study, we focused predominantly on examining the role of miR-146a. A luciferase reporter assay was used to test whether miR-146a binds directly to the Sumo1 3′-UTR. A miR-146a mimic suppressed the luciferase activity of a reporter containing the Sumo1 mRNA 3′-UTR sequence in a dose-dependent manner (Figure 1C). This suppressive effect was abolished when the putative miR-146a binding site was mutated in the Sumo1 3′-UTR sequence (Figure 1C), indicating that miR-146a binding is specific. Isolated adult mouse cardiomyocytes treated with an adenovirus encoding pre-mir-146a (Ad_pre-mir-146a) exhibited a significant dose-dependent reduction in SUMO1 and SUMO1 conjugate levels, as assessed by qRT-PCR and western blotting (Figure 1D-F and Figure If in the Data Supplement). In striking contrast, treatment with an adenovirus encoding a tough decoy29 for miR-146a (Ad_decoy-146a) led to a significant dose-dependent elevation in SUMO1 and SUMO1 conjugate levels (Figure 1G-I and Figure Ig in the Data Supplement). Taken together, these data suggest that miR-146a regulates SUMO1 expression through specific binding to the 3’-UTR of SUMO1.
Figure 1: MicroRNA-146a (miR-146a) inhibits SUMO1 expression in cardiomyocytes.
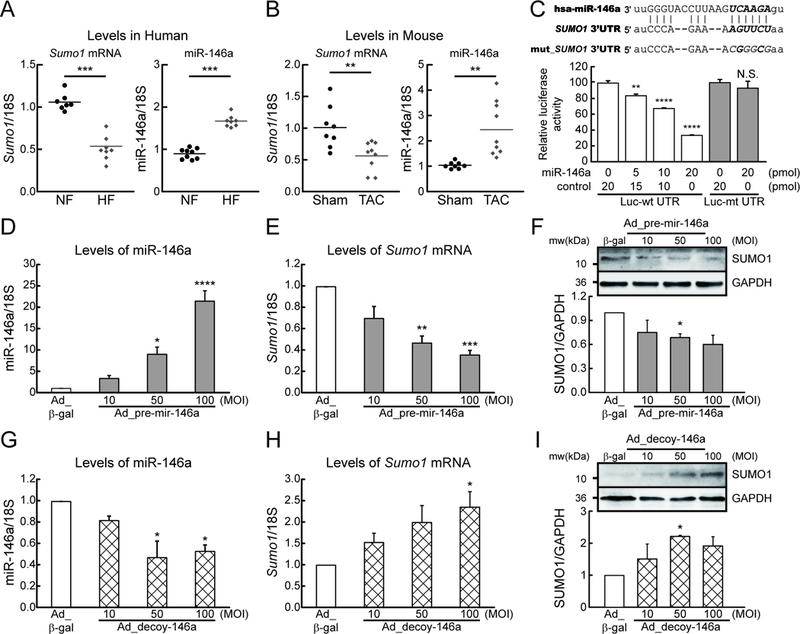
A, Changes in SUMO1 mRNA and miR-146a levels in human hearts of non-failing (NF) and heart failure (HF) patients (n=7–9). B, Changes in SUMO1 mRNA and miR-146a levels in mouse hearts of sham and TAC operated mice (6 weeks after TAC) (n=8–9). C, Sequence alignment of SUMO1 wild-type and mutant 3′ UTRs with miR-146a (top) and luciferase reporter assay (bottom). Luciferase reporters with a putative miR-146a–binding site (Luc-wt UTR) or mutant miR-146a-binding site (Luc-mt UTR) were co-transfected into H9C2 cells with miR-146a mimic or control (cel-miR-39) for 24h, and luciferase activity determined. Values are presented as relative luciferase activity ± s.e.m. (n=3). D and E, The expression of miR-146a (D) and SUMO1 mRNA (E) in isolated adult mouse cardiomyocytes which were infected with an adenovirus encoding pre-mir-146a (Ad_pre-mir-146a) for 48h (n=3). F, Representative blot (top) and quantitative analysis (bottom) showing the expression of SUMO1 protein. G and H, The expression of miR-146a (G) and SUMO1 mRNA (H) in isolated adult mouse cardiomyocytes which were infected with an adenovirus encoding mir-146a decoy (Ad_decoy-146a) for 48h (n=3). I, Representative blot (top) and quantitative analysis (bottom) showing the expression of SUMO1 protein. *, p<0.05, **, p<0.01, ***, p<0.001, ****, p<0.001 versus the respective control, as determined by One-way ANOVA. Data are presented as mean ± s.e.m.
miR-146a regulates Ca2+ cycling in cardiomyocytes.
Reduced SUMO1 levels result in decreased SUMOylation of SERCA2a, which in turn leads to abnormal Ca2+ handling in failing hearts.10 Therefore, we next examined the effect of miR-146a overexpression on SERCA2a SUMOylation and Ca2+ handling in cardiomyocytes. Ad_pre-mir-146a reduced SERCA2a SUMOylation by 30% in cardiomyocytes (Figure 2A). The expression levels of SUMO2, SUMO3, and other SUMOylation-related enzymes were not affected by miR-146a (Figure IIa-g in the Data Supplement). Moreover, Ad_pre-mir-146a reduced cardiomyocyte Ca2+ transient amplitude, SR Ca2+ load, and peak shortening and prolonged the time constant for Ca2+ transient decay (tau) as assessed by a dual-excitation fluorescent photomultiplier system (Figure 2B-F and Table I in the Data Supplement). The effects of miR-146a inhibition were examined in failing cardiomyocytes (Figure IIh in the Data Supplement). Inhibition of miR-146a by Ad_decoy-146a restored SUMO1 expression and SERCA2a SUMOylation (Figure 2G, Figure IIi and j in the Data Supplement) and improved Ca2+ transient and contractility in failing cardiomyocytes (Figure 2H-L and Table I in the Data Supplement). In summary, these data suggest that miR-146a causes abnormal Ca2+ handling in cardiomyocytes through the suppression of SUMO1 expression and the inhibition of miR-146a significantly improves Ca2+ handling in failing cardiomyocytes in vitro.
Figure 2: miR-146a inhibits cardiac contractility via suppression of SERCA2a SUMOylation.
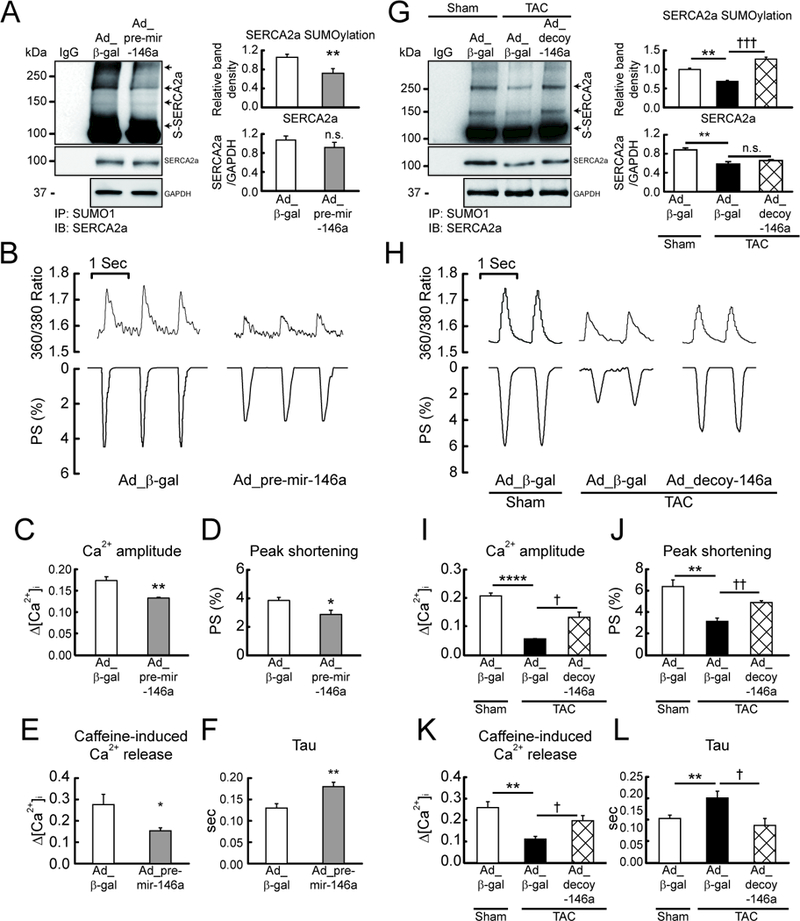
Isolated mouse cardiomyocytes infected with Ad_pre-mir-146a (MOI=50) for 48h. A, Representative blot (left) and quantitative analysis (right) showing the levels of SERCA2a SUMOylation and SERCA2a expression (n=6). B, Representative trace of calcium transients (top) and cardiac contractility (bottom). C and D, Averaged data of calcium transient amplitude (C) and sarcomere length shortening (D) (n=100–150 cells/4 hearts). E, Caffeine-induced Ca2+ transient amplitude at 10 mmol/L Caffeine (n=10 cells/3 hearts). F, Rate constant for the decay of the Ca2+ transient (n=100–150 cells/4 hearts). *, p<0.05, **, p<0.01 versus cardiomyocytes infected with Ad_β-gal, as determined by Student’s t-test. Isolated failing cardiomyocytes from mice subjected to TAC for 6 weeks infected with Ad_decoy-146a (MOI=50) for 48h. G, Representative blot (left) and quantitative analysis (right) showing the levels of SERCA2a SUMOylation and SERCA2a expression (n=3). H, Representative trace of calcium transients (top) and cardiac contractility (bottom). I and J, Averaged data of calcium transient amplitude (I) and sarcomere length shortening (J) (n=100–150 cells/4 hearts). K, Caffeine-induced Ca2+ transient amplitude at 10 mmol/L Caffeine (n=10 cells/3 hearts). L, Rate constant for the decay of the Ca2+ transient (n=100–150 cells/4 hearts). *, p<0.05, **, p<0.01, ****, p<0.0001 versus the sham mouse and †, p<0.05, ††, p<0.01 versus the failing cardiomyocyte treated with Ad_β-gal, as determined by one-way ANOVA. Data are represented as mean ± s.e.m. in all panels.
Overexpression of miR-146a impairs cardiac function in vivo.
These findings led us to investigate the in vivo role of miR-146a in HF. To characterize the physiological consequences of miR-146a overexpression, adeno-associated virus serotype 9 encoding pre-mir-146a (rAAV9_pre-mir-146a) or rAAV9-LacZ (control) were administered to wild-type mice via the tail vein and cardiac morphology and function were monitored at four weeks post gene delivery (Figure 3A). miR-146a levels were significantly increased in cardiomyocytes following injection of rAAV9_pre-mir-146a in a dose-dependent manner (Figure 3B and Figure IIIa-c in the Data Supplement). Hypertrophic indices including heart weight to body weight ratio, left ventricle (LV) wall thickness, the LV cardiomyocyte size, and hypertrophic marker expressions (ANF, BNP) increased at four weeks post-injection of rAAV9_pre-mir-146a, compared to mice injected with the control virus (Figure 3C, D, Figure IIIf, g and Table II in the Data Supplement). Mice with overexpressed miR-146a also exhibited a significant reduction in ventricular systolic function, as evidenced by the dose-dependent reduction in fractional shortening (FS) and ejection fraction (EF) (Figure 3D, E, Figure IIId, e and Table II in the Data Supplement). Moreover, hemodynamic analyses also revealed that LV systolic function was significantly decreased in mice injected with rAAV9-pre-mir-146a, as indicated by the reduced end systolic pressure-volume relationship (ESPVR) and the maximum rate of pressure change (Max dP/dt) (Figure 3F, G and Table III in the Data Supplement). These data suggest that miR-146a overexpression resulted in cardiac systolic dysfunction. Furthermore, the rightward shift of pressure-volume loops observed in mice with overexpressed miR-146a indicated that miR-146a induced ventricular dilation (Figure 3F). SUMO1 expression was significantly decreased in mice injected with rAAV9_pre-mir-146a. SERCA2a expression and SUMOylation levels were also significantly reduced (Figure 3H-K, Figure IIIh and i in the Data Supplement). Overall, these data suggest that elevated miR-146a levels lead to dysregulation of the SUMO1/SERCA2a signaling axis and concomitant deterioration of cardiac morphology and function.
Figure 3: Overexpression of miR-146a promotes cardiac dysfunction in vivo.
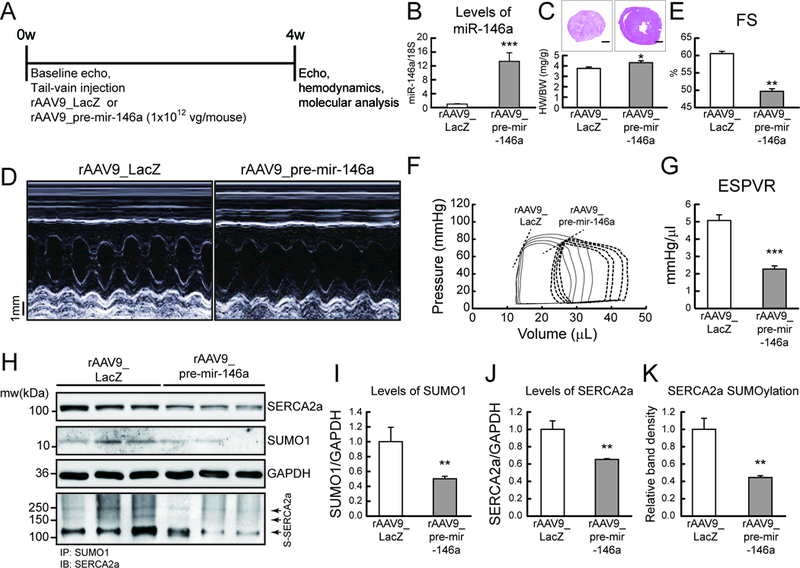
A, Protocol for AAV9-mediated pre-mir-146a gene transfer into normal mice. Six week-old male B6C3/F1 mice received AAV9 carrying pre-mir-146a (rAAV9_pre-mir-146a, 1×1012 vg/mouse) or control virus (rAAV9_LacZ) via tail-vein injection. After 4 weeks of gene delivery, cardiac function was measured by echocardiography and then, further analyzed by hemodynamic measurements. B, levels of miR-146a. C, Cross-section images of a mouse heart (top) and quantification of heart weight/body weight ratio (bottom). Scale bars, 1 mm D and E, Representative LV M-mode images (D) and Factional shortening (FS) (E) (n=8). F and G, Representative pressure-volume loops (F) and ESPVR (G) (n=6). G, Representative blot showing SERCA2a, SUMO1 and total SUMOylation levels after infection with rAAV9_pre-mir-146a. I-K, The protein quantification of SUMO1 (I), SERCA2a (J) and SERCA2a SUMOylation (K) (n=6). *, p<0.05, **, p<0.01, ***, p<0.001 versus mouse infected with rAAV9_LacZ, as determined by Student’s t-test. Data are presented as mean ± s.e.m. in all panels.
Inhibition of miR-146a is cardio-protective in vivo.
To evaluate the consequences of miR-146a inhibition, we examined the cardiac function of wild-type and TAC mice injected with either decoy-146a (rAAV9_decoy-146a) or rAAV9-LacZ (control). Wild-type mice were subjected to TAC surgery to induce pressure overload. The rAAV9_decoy-146a or control virus was injected at two weeks post-TAC and cardiac morphology and function were monitored bi-weekly (Figure 4A). miR-146a levels were elevated in hearts injected with the control virus at six weeks post-TAC; however, miR-146a levels were completely normalized in hearts injected with the rAAV9_decoy-146a (Figure 4B and Figure IVa in the Data Supplement). Various hypertrophic markers were prominently increased in TAC mice injected with the control virus. In contrast, the hypertrophic response was blunted in TAC mice injected with the rAAV9_decoy-146a (Figure 4C, D, Figure IVc-e and Table IX in the Data Supplement). Adverse cardiac remodeling and functional deterioration were evident in mice treated with the control virus under pressure overload, but significantly attenuated in mice injected with the rAAV9_decoy-146a, as determined by FS and EF (Figure 4D, E, Figure IVb and Table IV in the Data Supplement). Hemodynamic analyses revealed that the LV systolic function index (ESPVR and Max dP/dt) was restored to normal levels and ventricular dilation was normalized in TAC mice injected with the rAAV9_decoy-146a (Figure 4F, G and Table V in the Data Supplement). Western blotting showed that SUMO1, SUMO1 conjugates, SERCA2a, and SERCA2a SUMOylation levels were reduced under pressure overload, but were all normalized by rAAV9_decoy-146a under the same conditions (Figure 4H-K and Figure IVf-k in the Data Supplement). Taken together, these results demonstrate that inhibition of miR-146a improves cardiac function through the restoration of SUMO1 expression, which could potentially serve as a therapeutic target for HF.
Figure 4: Inhibition of miR-146a prevents cardiac dysfunction under pressure overload in vivo.
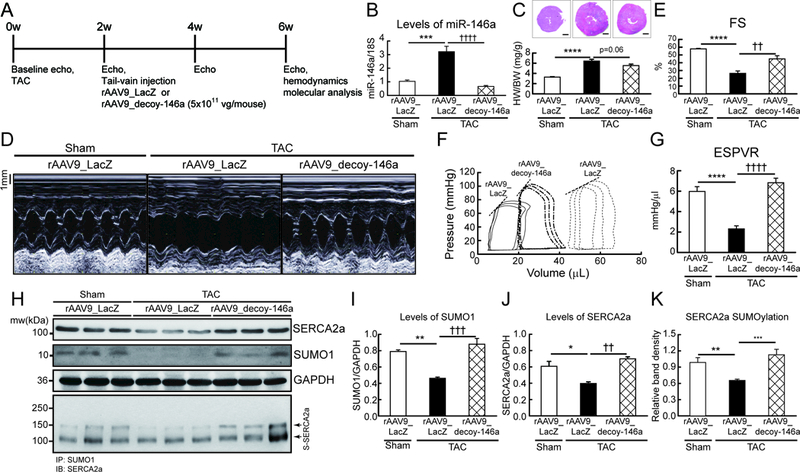
A, Protocol for AAV9-decoy-146a gene transfer in mouse model of pressure overload. Six week-old male B6C3/F1 mice were subjected to TAC and received AAV9 carrying miR-146a decoy (rAAV9_decoy-146a, 5×1011 vg/mouse) or LacZ (rAAV9_LacZ) via tail-vein injection 2 weeks post-TAC surgery. Cardiac function was measured by echocardiography and analyzed by hemodynamic measurements 4 weeks after gene delivery. B, levels of miR-146a. C, Cross-section images of a mouse heart (top) and quantification of heart weight/body weight ratio (bottom). Scale bars, 1 mm D and E, Representative LV M-mode images (D) and Factional shortening (FS) (E) (n=8–10). F and G, Representative pressure-volume loops (F) and ESPVR (G) (n=6–8). H, Representative blot showing SERCA2a, SUMO1 and total SUMOylation after infection of rAAV9_decoy-146a. I-K, Protein quantification of SUMO1 (I), SERCA2a (J) and SERCA2a SUMOylation (K) (n=6). *, p<0.05, **, p<0.01, ***, p<0.001 versus sham operated mice and ††, p<0.01, †††, p<0.001 versus the TAC operated mice infected with rAAV9_LacZ, as determined by one-way ANOVA. Data are represented as mean ± s.e.m. in all panels.
Normalization of SUMO1 levels alleviates miR-146a-mediated cardiac dysfunction.
To confirm our hypothesis that elevated levels of miR-146a induce cardiac dysfunction by downregulating SUMO1, we used the strategy of restoring SUMO1 expression in a miR-146a overexpression model. Cardiac-specific Cre/loxP-conditional Sumo1-transgenic mice (SUMO1 TG) and their negative littermates (NL) were injected with rAAV9_pre-mir-146a and tamoxifen was administrated at two weeks post-viral injection to induce SUMO1 transgene expression. Cardiac function was analyzed at six weeks post-tamoxifen administration (Figure 5A). Consistent with the data shown in Figures 3 and 4, SUMO1 levels were significantly reduced in NL mice, but remained at near-normal levels in SUMO1 TG mice under conditions of miR-146a overexpression (Figure 5B, C, Figure Va and c in the Data Supplement). The SUMOylation assay revealed that reduced SERCA2a SUMOylation under miR-146a overexpression was normalized in SUMO1 TG mice (Figure 5B and D). Normalization of SUMO1 levels attenuated miR-146a-mediated adverse cardiac remodeling and dysfunction (Figure 5E, F, Figure Vb and Table VI in the Data Supplement). Finally, overexpression of miR-146a dramatically increased mortality in NL mice; however, this miR-146a-mediated fatality was significantly attenuated in SUMO1 TG mice, as shown by Kaplan-Meier analysis (Figure 5G). Collectively, these observations suggest that the detrimental effects of miR-146a are mainly mediated through downregulation of SUMO1 in the heart.
Figure 5: Complementation of SUMO1 expression attenuates miR-146a-induced cardiac dysfunction in vivo.
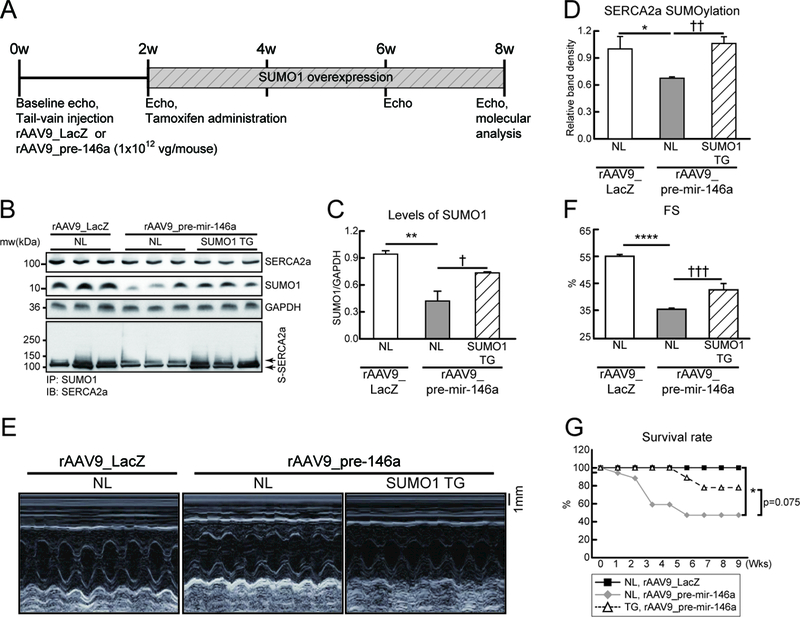
A, Protocol for SUMO1 re-expression in mouse model injected with rAAV9_pre-mir-146a. Six week-old male B6C3/F1 mice were received AAV9 carrying pre-miR-146a (rAAV9_pre-mir-146a, 1×1012 vg/mouse) or LacZ (rAAV9_LacZ) via tail-vein injection. Tamoxifen was administered to induce SUMO1 expression in the SUMO1 TG mice at 2 weeks post- rAAV9_pre-mir-146a injection. Cardiac function was measured by echocardiography and analyzed by hemodynamic measurements 8 weeks after gene delivery. B, Representative blot showing SERCA2a, SUMO1 and total SUMOylation after infection of rAAV9_decoy-146a. C and D, The protein quantification of SUMO1 (C) and SERCA2a SUMOylation (D) protein levels (n=6). E and F, Representative LV M-mode images (E) and Factional shortening (FS) (F) (n=5–8). G, Survival of animals after rAAV9_pre-mir-146a in negative littermate (NL) or SUMO1 TG. The Kaplan-Meier method was used to analyze animal lifespan (NL with rAAV9_LacZ, n=10; NL with rAAV9_pre-mir-146a, n=17; TG with rAAV9_pre-mir-146a, n=9). *, p<0.05, **, p<0.01, ***, p<0.001 versus NL mice injected with rAAV9_LacZ and ††, p<0.01, †††, p<0.001 versus NL mice injected with rAAV9_pre-mir-146a, as determined by one-way ANOVA. Data are represented as mean ± s.e.m. in all panels.
Fibroblasts secrete and transfer miR-146a into cardiomyocytes via extracellular vehicles.
To further elucidate the regulatory mechanisms of miR-146a during HF, we examined mature miR-146a and primary transcript (pri-mir-146a) levels in two different heart cell fractions (cardiomyocytes and non-myocytes). An approximately 2.7-fold upregulation of miR-146a was observed in cardiomyocytes isolated from failing hearts, compared to wild-type cardiomyocytes (Figure 6A). However, pri-mir-146a was upregulated exclusively in the non-myocyte fraction of failing hearts (Figure 6B). These observations led us to hypothesize that miR-146a is produced in non-myocyte cells and then delivered to the cardiomyocytes. Intriguingly, recent studies have shown that cardiomyocytes do not induce miR-146a expression by direct stimulation, but rather receive it from other cardiac cells via EVs.25–27 To identify the miR-146a-expressing cell type in the heart, the expression of mature miR-146a and pri-mir-146a was analyzed in the four major cardiac cell types (cardiomyocytes, fibroblasts, endothelial cells, and leukocytes) isolated by fluorescence-activated cell sorting (FACS) (Figure VIa-d and Table VII in the Data Supplement). Although mature miR-146a levels were increased in failing cardiomyocytes, pri-mir-146a levels did not alter under failing conditions, consistent with previous findings (Figure 6A and B). In contrast, a significant elevation in pri-mir-146a levels was observed predominantly in the fibroblast and leukocyte fractions in HF (Figure 6C and D). To compare the level of miR-146a secretion in fibroblasts and leukocytes, EVs were isolated from fibroblast and leukocyte cultures and then analyzed for miR-146a expression. The level of secreted miR-146a was approximately 4-fold higher in EVs secreted from fibroblasts than from leukocytes (Figure 6E). Combined with the fact that fibroblasts are >14-fold more abundant than leukocytes in the heart (Table VII in the Data Supplement), this observation suggests that fibroblasts are the major source of secreted miR-146a in the heart. During HF, fibroblasts trans-differentiate into myofibroblasts, which induces dynamic changes in functional properties and gene expression.30, 31 To mimic pathogenic conditions, fibroblast cultures were treated with transforming growth factor-β (TGF-β), which is known as the primary factor that drives trans-differentiation of fibroblasts.32, 33 miR-146a expression was significantly increased following TGF-β treatment, paralleling the increase in trans-differentiation markers (Figure IVe and f in the Data Supplement). TGF-β treatment also enhanced EV-mediated miR-146a secretion by approximately 20-fold, suggesting that TGF-β triggers the release of miR-146a from fibroblasts (Figure 6F). To confirm the uptake of released miR-146a into cardiomyocytes, EVs isolated from fibroblast cultures (Fib-EV) were labeled with a fluorescent dye (PKH-26) and added to isolated cardiomyocytes. Confocal microscopy revealed the presence of Fib-EVs in cardiomyocytes (Figure 6G and Figure VII in the Data Supplement). qRT-PCR analysis further confirmed that mature miR-146a levels, but not pri-mir-146a levels, increased in cardiomyocytes following Fib-EV treatment. These results imply that the transfer of miR-146a via EVs is the main mechanism by which levels of miR-146a increase in cardiomyocytes and also excludes the possibility of EV treatment in promoting miR-146a synthesis (Figure 6H). The increase of miR-146a levels in cardiomyocytes was accompanied by a decrease in SUMO1 levels and reduced Ca2+ transient amplitude and cardiac contractility (Figure 6I, J and Table VIII in the Data Supplement). These findings indicate that trans-differentiation of fibroblasts, and hence miR-146a secretion, are the causative mechanism of miR-146a upregulation in cardiomyocytes during HF.
Figure 6: miR-146a is transferred from fibroblasts into cardiomyocytes through an EV-mediated mechanism.
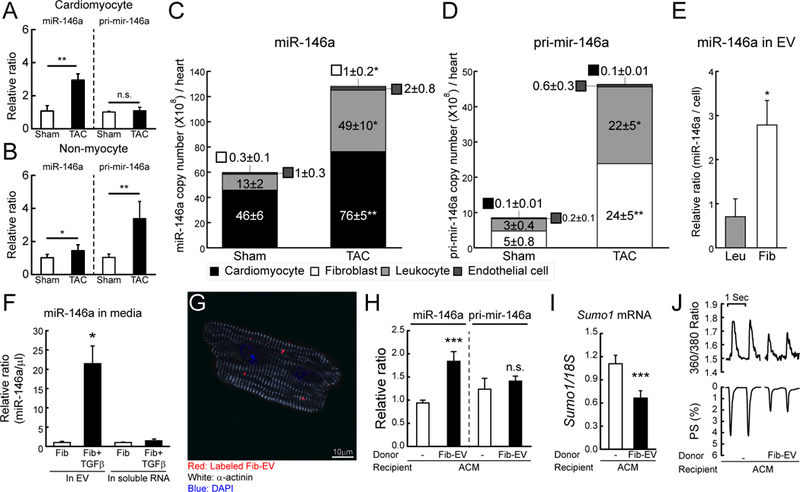
A and B, Changes in miR-146a and pri-mir-146a levels in isolated cardiomyocyte (A) or non-myocyte (B) fractions from mouse hearts of sham and TAC operated mice (TAC, 6 weeks after TAC) (n=3). The RNA levels were normalized to 18S rRNA. C and D, Copy number of miR-146a (C) and pri-mir-146a (D) in sorted cardiac cells from sham and TAC operated mice (6 weeks after TAC) (n=5). E, Levels of miR-146a in isolated EVs from leukocyte (Leu) or fibroblast (Fib)-cultured media. F, Levels of miR-146a in isolated EVs from fibroblast-cultured media with/without TGF-β. G, Representative confocal microscopic image of isolated cardiomyocytes treated with labeled EVs for 24h. H and I, Levels of miR-146a, pri-mir-146a (H) and SUMO1 mRNA (I) in isolated cardiomyocytes treated with/without fibroblast-derived EVs for 24h (n=3). J, Representative trace of calcium transient (top) and cardiac contractility (bottom) (n=60–80 cells/3 hearts). *, p<0.05, **, p<0.01, ***, p<0.001 versus the indicated control, as determined by Student’s t-test. Data are represented as mean ± s.e.m. in all panels.
EV-mediated miR-146a transfer is a critical mechanism of SUMO1 regulation and cardiac function in cardiomyocytes.
To further confirm the role of EV-associated miR-146a on cardiomyocyte function, we examined the effect of either miR-146a-enriched or -depleted EVs on cardiomyocyte function. We generated Fib-EVs in which miR-146a was either enriched (miR-146a-enriched EV) or depleted (miR-146a-depleted EV) by infection with either Ad_pre-mir-146a or Ad_decoy-146a, respectively. The effect of these EVs on cardiomyocyte function was then examined (Figure 7A and Figure VIIIa in the Data Supplement). miR-146a-enriched EVs contained an approximately 2-fold higher level of miR-146a, whereas miR-146a-depleted EVs contained approximately 20% of the miR-146a level in wild-type Fib-EVs (Figure 7B). Cardiomyocytes treated with miR-146a-enriched EVs exhibited increased miR-146a levels, decreased SUMO1 levels, and impaired Ca2+ transients and contractility, whereas cardiomyocytes treated with miR-146a-depleted EVs exhibited slightly decreased miR-146a levels, increased SUMO1 levels, and improved Ca2+ transients and contractility, compared to cells treated with wild-type Fib-EVs (Figure 7C-E and Table VIII in the Data Supplement).
Figure 7: EV-mediated miR-146a transfer affects cardiac function.
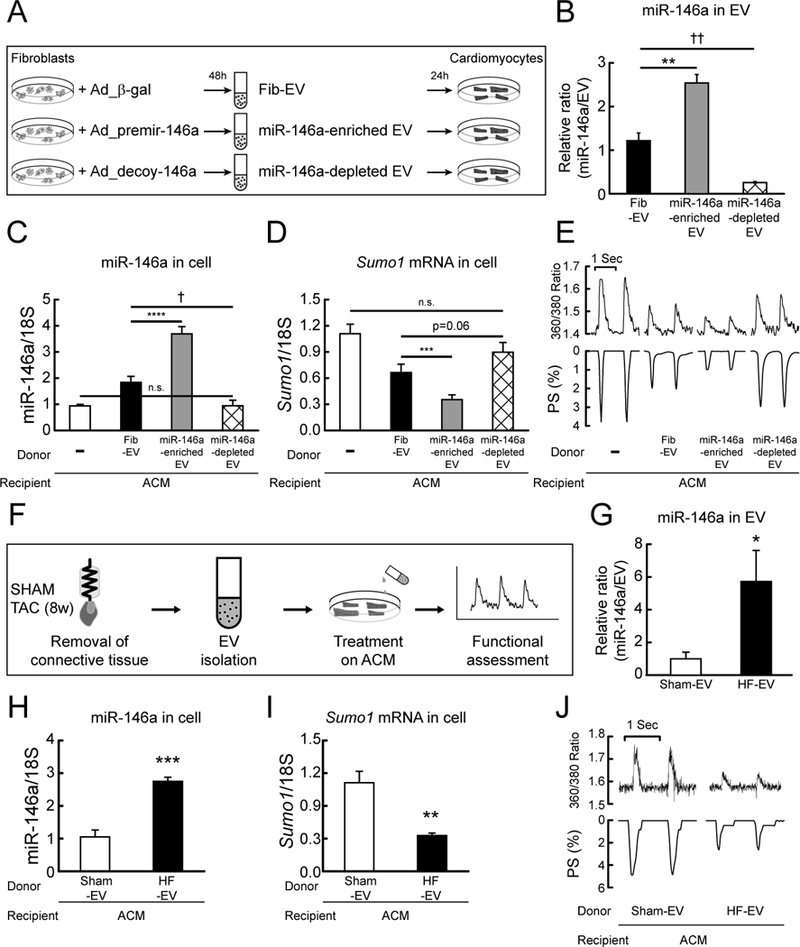
A, Scheme depicting a protocol used for the production of miR-146a enriched or depleted EVs. B, Levels of miR-146a in fibroblast-derived (Fib EV), miR-146a-enriched fibroblast-derived (miR-146a enriched EV), and miR-146a-depleted fibroblast-derived (miR-146a depleted EV) EVs. C and D, Levels of miR-146a (C) and SUMO1 mRNA (D) in isolated cardiomyocytes non-treated or treated with Fib-EV, miR-146a-enriched EV or miR-146a-depleted EV for 24h (n=4). E, Representative trace of calcium transients (top) and cardiac contractility (bottom) (n=60–80 cells/3 hearts). F, Scheme depicting a protocol used for the isolation of EVs from normal and failing hearts. G, Levels of miR-146a in EVs from normal (Sham-EV) and failing mouse hearts (HF-EV) (6 weeks after TAC, n=3). H and I, Levels of miR-146a (H) and SUMO1 mRNA (I) in isolated cardiomyocytes treated with Sham-EVs or HF-EVs for 24h (n=3). J, Representative trace of calcium transient (top) and cardiac contractility (bottom) (n=60–80 cells/3 hearts). *, p<0.05, **, p<0.01, ***, p<0.001, †, p<0.05, ††, p<0.01 versus the indicated control, as determined by Student’s t-test. Data are represented as mean ± s.e.m. in all panels.
Additionally, to clarify the pivotal role of miR-146a in EV delivery in cardiomyocyte functions, the effects of miR-146a inhibition against Fib-EVs were tested. The isolated cardiomyocytes were pre-treated with Ad_decoy-146aand then treated with Fib-EV for 24h after which calcium transient and cardiomyocyte contractility were measured (Figure VIIIb in the Data Supplement). As expected, Ad_decoy-146a pre-treatment blocked the negative inotropic effects that were brought up by upregulation of miR-146a due to Fib-EV uptake (Figure VIIIc-e in the Data Supplement).
Finally, to address the physiological relevance of EV-mediated miR-146a transfer in vivo, isolated cardiomyocytes were treated with EVs isolated from wild-type (Sham-EV) or failing mouse hearts (HF-EV) (Figure 7F and Figure VIIIf in the Data Supplement). The level of miR-146a in EVs isolated from failing hearts was approximately 6-fold higher than in the wild-type control (Figure 7G, Figure VIIIg and h in the Data Supplement). Treatment of wild-type cardiomyocytes with EVs isolated from failing hearts resulted in increased miR-146a levels and decreased SUMO1 expression and significantly impaired Ca2+ transients and cardiac contractility, compared to cardiomyocytes treated with EVs isolated from wild-type hearts (Figure 7H-J and Table VIII in the Data Supplement). Collectively, these data suggest that EV-mediated delivery of miR-146a to cardiomyocytes plays a pivotal role in SUMO1 regulation during HF.
DISCUSSION
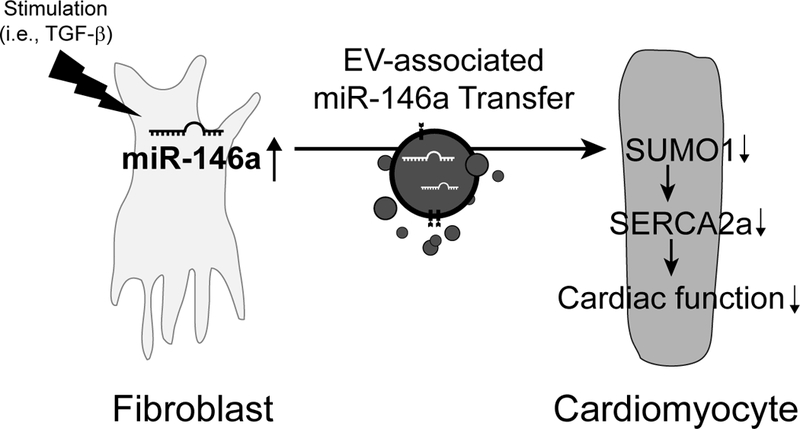
To the best of our knowledge, this study is the first to demonstrate that the upregulation of miR-146a has a direct correlation with the reduction of SUMO1 levels in failing hearts. The reduction of SUMO1 expression by miR-146a overexpression resulted in decreased SERCA2a SUMOylation and contractile function in cardiomyocytes. Moreover, inhibition of miR-146a reversed contractile dysfunction in failing hearts by restoring SUMO1 levels. In failing hearts, miR-146a was secreted from trans-differentiated fibroblasts and then transferred into cardiomyocytes by EV-mediated trafficking. This intracellular communication promoted the inhibition of SUMO1 expression in cardiomyocytes, resulting in cardiac dysfunction during HF. A schematic summary of our findings is shown in Figure 8.
Figure 8: Schematic model depicting miR-146a regulation of SUMOylation in heart failure. During heart failure, miR-146a is expressed and processed in fibroblasts. The mature miR-146a is secreted as an EV-associated form from the activated fibroblasts and then transferred into cardiomyocytes. The fibroblast-derived miR-146a targets SUMO1 and attenuates SERCA2a SUMOylation, thus reducing cardiomyocyte function.
SUMOylation is a post-translational modification process in which SUMO proteins are conjugated to their targets via enzymatic cascade reactions.34 This process has been shown to regulate proteins by altering their structure, enzymatic activity, stability, sub-cellular localization, and protein-protein interactions.34 Recent studies have demonstrated that the SUMO pathway is central in regulating heart development, with defective SUMOylation leading to congenital heart defects.35 Global SUMO1 knockout mice develop congenital heart disease, including atrial and ventricular septal defects, and cardiac-specific SUMO1 knockdown mice show a progression of cardiac dysfunction and sudden death.35 The absence of SENP2, a deSUMOylating enzyme, leads to embryonic lethality with defects in the embryonic mouse heart.36 In addition, cardiac-specific SENP2 overexpression mice exert dilated cardiomyopathy, further underlining the importance of maintaining proper SUMOylation in the heart.37 Our group has previously shown that SERCA2a SUMOylation occurring at lysine 480 and lysine 585 is essential for correct SERCA2a ATPase activity and protein stability in human and mouse cells.10 SUMO1 and SUMOylated SERCA2a levels are reduced in failing human and mouse hearts. However, AAV-mediated SUMO1 gene transfer has exhibited significant beneficial effects in HF animal models.10–12 Recently, our group showed that the small molecule N106 directly activates the SUMO-activating enzyme, SUMO E1 ligase, and induces intrinsic SERCA2a SUMOylation.38 Previous studies have demonstrated that N106 improves ventricular function in HF mice models, suggesting that SUMO1 targeting could serve as the basis for the design of new therapies for countering HF.
In this study, we set out to identify miRs that target and regulate SUMO1 expression in HF. We identified miR-146a as a potential SUMO1-targeting miR using in silico prediction tools. Indeed, miR-146a directly binds a site approximately 20~50 bp downstream of the 3′-UTR of the Sumo1 gene, as determined by our luciferase assay. miR-146a overexpression significantly reduced a monomeric form of SUMO1 and SUMO1 conjugates in vitro and in vivo. The reduction in global SUMOylation significantly decreased SUMOylation of SERCA2a. In contrast, miR-146a inhibition attenuated SUMO1 reductions and restored SERCA2a SUMOylation. We also tested other SUMOylation targets in the same experimental condition, but these proteins (Sp1, GATA4) were not susceptible to the changes of SUMO1 expression as much as SERCA2a (Figure IIIj and IVh in the Data Supplement). The features of SERCA2a SUMOylation, that is SUMOylated only by the SUMO1 isoform and does not require a specific SUMO E3 enzyme, seem to determine the susceptibility of SERCA2a SUMOylation to the levels of SUMO1 expression. The miR-146a-mediated reduction in SERCA2a SUMOylation certainly affected cardiac function and sudden death in mice, which is consistent with our previous findings regarding SUMO1 reduction in the heart.10 In contrast, miR-146a inhibition preserved adverse remodeling and dysfunction in TAC mice. Therefore, our findings may provide another attractive therapeutic target for the intervention of HF.
miR-146a was originally discovered as an innate immune suppressor in the THP-1 human monocytic cell line in which it targets IRAK1 and TRAF6.17 Recent studies have reported that overexpression of miR-146a reduced the immune response in myocardial ischemia/reperfusion (I/R) injury21, septic cardiomyopathy22, diabetic cardiomyopathy23, and myocardial infarction (MI).39 Another study using a human cardiac cell line (AC16) showed that miR-146a inhibition enhanced the expression of mRNAs related to inflammation.40 Therefore, we were concerned regarding the excessive inflammation caused by miR-146a inhibition in the present study. To test whether miR-146a inhibition affects cardiac inflammation under pressure overload, representative inflammatory markers were quantified by qRT-PCR and immunohistochemistry (IHC). Based on our qRT-PCR results, although miR-146a levels were elevated, pressure overload stimulated the expression of inflammatory markers such as TNFα, CD11c, CD19, CD3, and F4/80 (Figure IXa-e in the Data Supplement). However, rAAV9_decoy-146a-mediated inhibition of miR-146a in failing hearts abolished inflammatory responses (Figure IXa-e in the Data Supplement). In addition, IHC revealed that CD45 and TNFα expression were significantly increased in failing hearts but decreased in decoy-146a-treated failing hearts (Figure IXf-h in the Data Supplement). These results imply that pressure overload drives the heart to induce inflammatory responses, regardless of the increase in miR-146a levels. In contrast, normalization of miR-146a levels exerted protective effects against inflammation under pressure overload. Thus, although previous reports have provided important evidence that miR-146a attenuates inflammatory response by suppressing its targets, under our HF model settings, we did not observe excessive inflammation and miR-146a inhibition showed beneficial effects on inflammatory responses. Further studies aimed at elucidating the reciprocal relationship between miR-146a and cardiac inflammation are currently underway.
Currently, two conflicting views exist regarding the role of miR-146a in the heart: beneficial versus deleterious effects. The supporting argument for the positive effect of miR-146a in the heart is based on evidence that miR-146a can act as an innate immune suppressor. On the contrary, reports have also shown that miR-146a suppresses the expression of critical proteins (dihydrolipoyl succinyltransferase (DLST), Erb-B2 Receptor Tyrosine Kinase 4 (ERBB4) and neuroblastoma RAS viral oncogene homolog (NRAS)), which have been shown to play crucial roles in cardiac metabolism or angiogenesis.24–26, 41 These studies have demonstrated that inhibition of miR-146a results in reduced cardiac damage in doxorubicin-induced, pressure-overload, and peripartum cardiomyopathy (PPCM) animal models.24–26 In agreement with these reports, our current study also shows beneficial effects of miR-146a inhibition on cardiac function. Taken together, the effects of miR-146a could vary on different physiological settings of disease. The factors that determine if miR-146a is beneficial or deleterious remain to be elucidated. One potential possibility to explain the contradictory results related to miR-146a is the binary effect of an immune response in disease states. Immunologists believe that the relative balance between pathological inflammatory pathways and tissue reparative processes define the trajectory of HF development.42 Suppression of immune response could be beneficial in disease models where pathological inflammation is strongly activated. However, if physiological inflammation is activated in tissue reparative processes, immune suppression is not the correct approach to protect the heart from damage. The role of miR-146a could differ depending on the involvement of pathologic immune response. Therefore, the effects of miR-146a on cardiac function need further exploration in different settings of disease and caution needs to be taken when moving this study into clinical trials.
EVs are heterogeneously sized (20–2000 nm) particles released into biological fluids including blood, urine, tears, and pericardial fluid, as well as into conditioned media from cultured cells.43, 44 Recently, EVs have emerged as biological carriers, transferring their protein, lipid, mRNA, long non-coding RNA and microRNA content to target cells.43, 44 In the heart, EVs are known to play important roles in intercellular communication between cardiac cells. Bang et al. suggested that cardiac fibroblast-derived EVs mediate cardiac hypertrophy through the pro-hypertrophic microRNA, miR-21*45, and Wang et al. showed that diabetic cardiomyocyte-derived EVs reduce angiogenesis by transferring anti-angiogenic miR-320 into endothelial cells.46 Recent studies have shown that miR-146a is also secreted in an EV-associated form.25–27 miR-146a in endothelial cell-derived EVs exerts negative effects on cardiac function in PPCM and pressure overload models.25, 26 Intriguingly, these studies found that endothelial cell-derived EVs play a primary role in regulating miR-146a levels in cardiomyocytes. Because these previous studies used a single cell culture model, we examined the overall changes in miR-146a expression in four major cardiac cell types during HF. According to our FACS analysis, cardiac fibroblasts produced the majority of miR-146a (Figure 6C and D). Consistent with previous studies, endothelial cells also exhibited upregulated expression of miR-146a in failing hearts; however, the level of miR-146a production was much lower than in fibroblasts (Figure 6C and D). Globally, however, fibroblasts seem to be the predominant source of miR-146a in the heart during HF. Another compelling finding was that the ratio between the number of EVs and cells was maintained at 2000:1 (EVs:cells) during HF, although the total number of EVs was doubled, as determined by nanoparticle tracking analysis (Figure VIIIf-h in the Data Supplement). We defined this ratio as physiologically-relevant and tested the functional consequences of different EV to cell ratios. Therefore, our finding may serve as a standard ratio of the effects of EVs on cardiac cells in future studies.
In this study, we demonstrate a novel regulatory mechanism of cardiac function whereby cardiac fibroblasts influence cardiac contractility by diminishing the expression of SUMO1 via EV-associated delivery of miR-146a. In addition, the beneficial effects of miR-146a inhibition via decoy-146a on cardiac function suggest that targeting miR-146a may provide a novel therapeutic strategy for the treatment of HF.
Supplementary Material
NOVELTY AND SIGNIFICANCE
What Is Known?
Small ubiquitin-like modifier 1 (SUMO1) regulates the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) pump and has emerged as a validated molecular target for heart failure.
SUMO1 and SUMOylated SERCA2a levels are reduced in failing hearts and enhancing SUMO1 by gene delivery or pharmacological activation rescues the heart failure phenotype in animal models.
The regulatory mechanisms underlying SUMO1 expression in the heart are unknown.
What New Information Does This Article Contribute?
Micro RNA-146a regulates SUMO1/SERCA2a axis in cardiomyocytes and its upregulation contributes to the contractile dysfunction in heart failure.
Mir-146a is secreted from trans-differentiated fibroblasts and then transferred into cardiomyocytes through extracellular vehicles-mediated trafficking and inhibits the SUMO1 expression.
Inhibition of miR-146a reverses contractile dysfunction by restoring SUMO1 levels in mice with pathologic cardiac hypertrophy.
SUMO1 is identified, as a new therapeutic target molecule for HF, however, its regulatory mechanism has not been explored. For the first time, we report that miR-146a induction plays a critical role in the abnormal regulation of SUMO1 pathway. In addition, we validated the delivery of miR-146a from cardiac fibroblasts into cardiomyocytes through extracellular vehicles. The beneficial effects of miR-146a inhibition on cardiac function suggest that targeting miR-146a may provide a novel therapeutic strategy for the treatment of heart failure.
Acknowledgments
SOURCES OF FUNDING
This work is supported by NIH R00 HL116645 (C. K.). R.J.H. was supported by R01 HL119046, R01 HL128099, R01 HL 117505, R01 HL129814, R01 HL131404, P50 HL112324, T32 HL007824, and a Transatlantic Leducq Foundation grant. We would like to acknowledge the Gene Therapy Resource Program (GTRP) of the National Heart, Lung, and Blood Institute, National Institutes of Health for providing the gene vectors used in this study. J.G.O was funded by AHA 17POST33410877.
Nonstandard Abbreviations and Acronyms:
- HF
heart failure
- SERCA2a
sarcoplasmic reticulum Ca2+-ATPase
- SUMO1
small ubiquitin-like modifier 1
- AAV9
adeno-associated virus serotype 9
- miR
microRNA
- IRAK1
interleukin-1 receptor-associated kinase 1
- TRAF6
TNF receptor-associated factor 6
- TAC
transverse aortic constriction
- EV
extracellular vesicle
- UTR
untranslated region
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- LV
left ventricle
- FS
fractional shortening
- EF
ejection fraction
- ESPVR
end systolic pressure-volume relationship
- Max dP/dt
maximum rate of pressure change
- SUMO1 TG
cardiac-specific Cre/loxP-conditional Sumo1-transgenic mice
- NL
negative littermates
- FACS
fluorescence-activated cell sorting
- TGF-β
transforming growth factor-β
- Fib-EV
EVs isolated from fibroblast cultures
- I/R
ischemia/reperfusion
- MI
myocardial infarction
- IHC
immunohistochemistry
- PPCM
peripartum cardiomyopathy
- DLST
dihydrolipoyl succinyltransferase
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2012;123(1):46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ Res. 2013;113(6):690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97(2):793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.del Monte F, Hajjar RJ, Harding SE. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ Res. 2001;88(11):E66–67. [DOI] [PubMed] [Google Scholar]
- 6.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, Nakajima-Takenaka C, Tsuji T, Konishi N, del Monte F, Hajjar RJ, Takaki M. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol. 2007;292(2):H1204–1207. [DOI] [PubMed] [Google Scholar]
- 7.Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif JC, Tanguay JF, Hajjar RJ. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51(11):1112–1119. [DOI] [PubMed] [Google Scholar]
- 8.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15(3):171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477(7366):601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med. 2013;5(211):211ra159. [DOI] [PubMed] [Google Scholar]
- 12.Lee A, Jeong D, Mitsuyama S, Oh JG, Liang L, Ikeda Y, Sadoshima J, Hajjar RJ, Kho C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal. 2014;21(14):1986–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melman YF, Shah R, Das S. MicroRNAs in heart failure: is the picture becoming less miRky? Circ Heart Fail. 2014;7(1):203–214. [DOI] [PubMed] [Google Scholar]
- 15.van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res. 2008;103(9):919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, Hajjar RJ, Mercola M. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508(7497):531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo M, Mao X, Ji Q, Lang M, Li S, Peng Y, Zhou W, Xiong B, Zeng Q. miR-146a in PBMCs modulates Th1 function in patients with acute coronary syndrome. Immunol Cell Biol. 2010;88(5):555–564. [DOI] [PubMed] [Google Scholar]
- 19.Pauley KM, Stewart CM, Gauna AE, Dupre LC, Kuklani R, Chan AL, Pauley BA, Reeves WH, Chan EK, Cha S. Altered miR-146a expression in Sjogren’s syndrome and its functional role in innate immunity. Eur J Immunol. 2011;41(7):2029–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu WD, Lu MM, Pan HF, Ye DQ. Association of MicroRNA-146a with autoimmune diseases. Inflammation. 2012;35(4):1525–1529. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013;97(3):432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL, Li C. Attenuation of Cardiac Dysfunction in Polymicrobial Sepsis by MicroRNA-146a Is Mediated via Targeting of IRAK1 and TRAF6 Expression. J Immunol. 2015;195(2):672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng B, Chen S, Gordon AD, Chakrabarti S. miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol. 2017;105:70–76. [DOI] [PubMed] [Google Scholar]
- 24.Horie T, Ono K, Nishi H, Nagao K, Kinoshita M, Watanabe S, Kuwabara Y, Nakashima Y, Takanabe-Mori R, Nishi E, Hasegawa K, Kita T, Kimura T. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res. 2010;87(4):656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halkein J, Tabruyn SP, Ricke-Hoch M, Haghikia A, Nguyen NQ, Scherr M, Castermans K, Malvaux L, Lambert V, Thiry M, Sliwa K, Noel A, Martial JA, Hilfiker-Kleiner D, Struman I. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013;123(5):2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heggermont WA, Papageorgiou AP, Quaegebeur A, Deckx S, Carai P, Verhesen W, Eelen G, Schoors S, van Leeuwen R, Alekseev S, Elzenaar I, Vinckier S, Pokreisz P, Walravens AS, Gijsbers R, Van Den Haute C, Nickel A, Schroen B, van Bilsen M, Janssens S, Maack C, Pinto Y, Carmeliet P, Heymans S. Inhibition of MicroRNA-146a and Overexpression of Its Target Dihydrolipoyl Succinyltransferase Protect Against Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction. Circulation. 2017;136(8):747–761. [DOI] [PubMed] [Google Scholar]
- 27.Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, Seabra MC, Round JL, Ward DM, O’Connell RM. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun. 2015;6:7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paterson MR, Kriegel AJ. MiR-146a/b: a family with shared seeds and different roots. Physiol Genomics. 2017;49(4):243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9(8):840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118(6):1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gyongyosi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, Gonzalez A, Thum T, Diez J, Jaisser F, Pizard A, Zannad F. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19(2):177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51(4):600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8(12):947–956. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Chen L, Wen S, Zhu H, Yu W, Moskowitz IP, Shaw GM, Finnell RH, Schwartz RJ. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res A Clin Mol Teratol. 2011;91(6):468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, Cheng J, Yeh ET. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell. 2010;38(2):191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim EY, Chen L, Ma Y, Yu W, Chang J, Moskowitz IP, Wang J. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J Mol Cell Cardiol. 2012;52(3):638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K, Sanchez R, DeVita RJ, Christensen G, Dahl R, Hajjar RJ. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun. 2015;6:7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang W, Tian SS, Hang PZ, Sun C, Guo J, Du ZM. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol Ther Nucleic Acids. 2015;5:e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palomer X, Capdevila-Busquets E, Botteri G, Davidson MM, Rodriguez C, Martinez-Gonzalez J, Vidal F, Barroso E, Chan TO, Feldman AM, Vazquez-Carrera M. miR-146a targets Fos expression in human cardiac cells. Dis Model Mech. 2015;8(9):1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. 2012;111(10):1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick SA, Epelman S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res. 2016;119(1):159–176. [DOI] [PubMed] [Google Scholar]
- 43.Kosaka N, Yoshioka Y, Hagiwara K, Tominaga N, Katsuda T, Ochiya T. Trash or Treasure: extracellular microRNAs and cell-to-cell communication. Front Genet. 2013;4:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan GC. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol. 2014;74:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.