

Abstract
Aging and diabetes are two well known risk factors for cardiovascular disease (CVD). Over the past 50 years there has been an dramatic increase in life expectancy with a simultaneous increase in the prevalence of diabetes in the older population. This large number of older individuals with diabetes is problematic, given that CVD risk associated with aging and diabetes. In this review, we summarize epidemiological data relating to diabetes and CVD, with an emphasis on the aging population. We then present data on hyperglycemia as a risk factor for CVD and review the current knowledge of age-related changes in glucose metabolism. Next, we review the role of obesity in the pathogenesis of age-related glucose dysregulation, followed by a summary of the results from major randomized controlled trials that focus on cardiovascular risk reduction through glycemic control, with a special emphasis on older adults. We then conclude with our proposed model of aging, that body composition changes and insulin resistance link possible dysregulation of physiological pathways leading to obesity and diabetes—both forms of accelerated aging—and risks for CVD.
Keywords: aging, diabetes, obesity, cardiovascular risk
Aging and diabetes are two well known risk factors for cardiovascular disease (CVD). Aging alone is associated with decline in physiological function leading to chronic diseases such as diabetes, CVD, cognitive impairment and physical disability. Diabetes is a recognized cause of accelerated aging and there is evidence that aging, and diabetes share common pathophysiological pathways. The mechanisms linking advancing age to metabolic dysregulation are multifactorial and complex. Asymptomatic hyperglycemia occurs in many people as they age and creates an insidious and progressive metabolic dysregulation that over many years increases the susceptibility to many age-related chronic diseases, including CVD as well as premature CVD mortality. Altering the trajectory of pathology by lowering mean blood glucose levels may not be possible owing to permanent vascular changes once type 2 diabetes manifested clinically. Thus, there is active research in developing crietria for early diagnosis.
In this report, we summarize the statistics related to diabetes in the context of the current aging population. We review the current knowledge of age-related changes in glucose metabolism, present data on hyperglycemia as a risk factor for CVD, and we attempt to quantify the role of obesity in the pathogenesis of age-related glucose dysregulation. For the purpose of this review, the acronym CVD is meant to include diseases of the heart and blood vessels, with atherosclerosis being the predominant underlying cause, and the main risks factors for CVD, such as obesity, diabetes and aging, acting as prominent players. We conclude with a review of the results from major randomized controlled trials with focus on CVD risk reduction through glycemic control and address a long-time vexing question as to whether reducing blood glucose, independent of other risk factors, also reduces the burden of CVD, with special attention on older adults.
EPIDEMIOLOGY OF AGING, DIABETES AND CARDIOVASCULAR DISEASE
Humans are living longer. Life expectancy has increased dramatically over the past century. In the United States, people older than age 65 increased from 3 million (4% of the population) in 1900 to 40 million (13% of the population) in 2010, and are projected to double to 83 million (21% of the population) in 2050.1 This trend is occurring worldwide, and is even faster in developing countries. In 2015, an estimated 617 million people (8.5%) in the world were age 65 or older and is expected to more than double to about 1.6 billion (17%), in 2050.2
Aging is by far the strongest known risk factor of diabetes and CVD.3 In 1958, 1.6 million Americans, or less than 1% of the US population were diagnosed with diabetes.4 In 2015, an estimated 30.2 million Americans, or 12.2 % of the US population, had diabetes with 7.2 million, or 24%, of them unaware of their diabetes diagnosis.4 Of the 30 million with diabetes, 12 million are over age 65 and represents over 25% of all individuals in this age group.5 If current trend continues, it is projected that by 2050, out of 48 million people with diabetes, nearly 27 million, that is more than half of all people with diabetes, will be in the older than 65 age group. Similar trends are projected for the population worldwide with nearly 253 out of 629 million people with diabetes in the older than 65 age group in 2045.6 Ninety percent of people with diabetes have Type 2 diabetes. For the rest of this review, the term diabetes will be used interchangeably with type 2 diabetes.
Every year, about 1 in 4 deaths in the US are directly due to CVD, with coronary artery disease being the most frequent type of heart disease that kills. In people aged 65–84 years, heart disease, the largest single cause of all deaths in this age group, accounts for at least 25% of all deaths while that number increases to 30% in people age 85 and older, with heart disease again being the single largest cause of death in this age group.7 Viewed through the lens of diabetes, atherosclerotic CVD, defined as coronary heart disease, cerebrovascular disease, or peripheral artery disease, is the leading cause of morbidity and mortality in persons with diabetes.8 In the Framingham study population the presence of diabetes doubled the risk for CVD in men and tripled it in women.9 Similar findings were reported from the Multiple Risk Factor Intervention Study.10 Interestingly, illustrating the impact of circulating glucose levels per se, a study of more than 24,000 non-diabetic individuals found that elevated fasting blood glucose increased the risk of coronary artery disease in this group of apparently healthy adults, after adjusting for other known risk factors.11 Furthermore, cardiologists consider diabetes a risk factor for CVD and and suggest that the screening of diabetes should be an integral part of the CVD evaluation in their assessment guidelines (2013 American College of Cardiology (ACC)/American Heart Association (AHA) risk assessment guidelines).12 However, there is still conflicting information/data about the role of glucose per se in CVD in persons who are not diabetic.
DIABETES DIAGNOSTIC CRITERIA AND THE OLDER ADULTS
According to the latest guidelines by the American Diabetes Association and the World Health organization the diagnosis of diabetes is established according to the following criteria: (1) random plasma glucose ≥ 200 mg/dL (11.1 mmol/L); (2) fasting plasma glucose (FPG) ≥ 126 mg/dL (7 mmol/L); (3) 2hr plasma glucose (2hG) post-75gm oral glucose tolerance test (OGTT) ≥ 200 mg/dL (11.1 mmol/L); or (4) hemoglobin A1c (A1c) ≥ 6.5%.13–15 OGTT is not routinely used in clinical settings for diagnosing diabetes because of the added burden and expense of performing OGTT but continues to be performed in many longitudinal studies, including our own Baltimore Longitudinal Study of Aging (BLSA).
Using FPG only to diagnose diabetes misses a significant proportion of the population with diabetes, especially older adults.16–19 In the Rancho Bernado Study of adults aged 50–89 years, 70% of women and 48% of men with previously undiagnosed diabetes had isolated post-OGTT hyperglycemia.17 The Cardiovascular Health Study reported that 52% of participants newly diagnosed with diabetes had isolated post-OGTT hyperglycemia.19 Results from the DECODE Study Group (Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe) showed that 35% of adults 60–79 years and newly diagnosed with diabetes had isolated post-OGTT hyperglycemia.16 Using our own data collected from the BLSA, we selected all participants with data on A1c and OGTT that had no history of diabetes and were not on antidiabetic medication. Out of 1152 participants, we identified 131 individuals with newly diagnosed type 2 diabetes using either A1c, FPG or 2hG (Figure 1). This sub-cohort has an average age of 70 years. Using FPG alone identified 40 (31%) cases, 2hG alone identified 89 (68%) cases, and A1c alone identified 66 (50%) cases. Clearly, these three measures, FPG, 2hG, and A1c, are each identifying different groups of diabetic individuals. This is especially problematic in older individuals: if only FPG and A1c are used for diagnosis, more than one-third of those with diabetes are missed. The main mechanism underlying age-associated increases in 2hG is the progressive change of body composition due to excessive food consumption. The significance of post-prandial glucose, represented by 2hG in OGTT, in CVD risks will be discussed in detail in this review.
Figure 1:
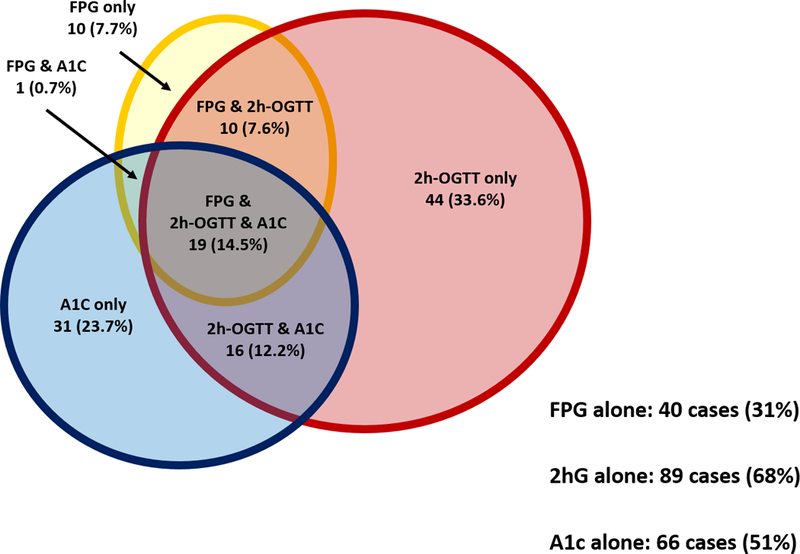
Selected participants from the Baltimore Longitudinal Study of Aging with data on A1c and OGTT that had no history of diabetes and were not on antidiabetic medication. Out of 1152 participants, 131 individuals were newly diagnosed with type 2 diabetes using either A1c, FPG or 2hG. Using FPG (yellow oval) alone identified 40 (31%) cases, 2hrG alone (red circle) identified 89 (68%) cases, and A1c alone (blue circle) identified 66 (51%) cases. If only FPG and A1c are used for diagnosis, more than one-third of those with diabetes would be missed.
The DECODE Study Group reported that 2hG is a better predictor of deaths from all causes, CVD, coronary heart disease and stroke, than FPG.20, 21 The Cardiovascular Health Study found that 2hG was associated with CVD and mortality independent of FPG.22 In a study that combined data from NHANES III (1998–1994) and Continuous NHANES (1999–2004), participants greater than 65 years of age with undiagnosed diabetes and A1c > 6.5% had a 30% greater risk of all-cause mortality compared to those without diabetes and A1c (5.0–5-6%).23 A systematic review (74 published studies) and meta-analyses of (46 studies) reported that in non-diabetic individuals, A1c > 6.5 % has twice the risk of cardiovascular mortality compared to those without diabetes and A1c (5.0–6.0%).24
HYPERGLYCEMIA AND CARDIOVASCULAR RISKS
The worldwide prevalence of impaired glucose tolerance and type 2 diabetes continue to rise, and increasing emphasis, resources, and amounts of GDP of nations are used in the services of lowering circulating plasma glucose levels to prevent CVD and other diabetes-related outcomes. Therefore, the causality of the relationships between elevated plasma glucose, type 2 diabetes and CVD are of great importance to global public health and cannot be overstated.
Despite data accumulated now over several decades, there is still some hesitation in firmly stating that elevated plasma glucose per se is causative for CVD and, if it is not, this would infer that the effects of glucose seen in observational studies of CVD are due to confounding factors, such as hypertension, hyperlipidemia and sleep apnea. There is strong rationale to believe that high glucose may be causative of CVD: (1) advanced glycated end product receptor activation may accelerate atherosclerotic plaque formation by causing increased intima-medial thickening and arterial stiffness; (2) hyperglycemia stimulates the differentiation of monocytes to macrophages leading to formation of foam cells and intracellular accumulation of oxidized lipids; (3) fluctuating glucose levels may promote oxidative stress and activation of protein kinase C and lead to fibrosis and dysfunction of endothelium. Some studies have also reported that variability in blood glucose, especially in the post prandial period, is more detrimental to CVD development than stably high glucose levels that may allow compensatory adaption.25 Of note, if glucose is causative of CVD, then we would expect that increasing glucose levels would be associated with progressively high risk of CVD and overall this does appear to be the case.
Elevated plasma glucose level, without diabetes, does appear to be associated with CVD. Impaired FPG and insulin resistance (without abnormal 2hG) are associated with an increased risk of CVD based on meta-analysis that included over 100 studies.26, 27 Additionally, in a recent retrospective study performed in a Korean Cohort of more than 260,000 individuals free of diabetes or CVD at study entry, change in fasting serum glucose from normal, to impaired to diagnostic for type 2 diabetes resulted in a progressively higher risk of developing myocardial infarction, CVD, and all-cause mortality.28
In the BLSA study population, a combination of FPG and 2hG provides better evaluation of mortality risk than does the FPG alone.29, 30 Categorization of risk based only upon FPG concentration suggests that risk for cardiovascular mortality among men with impaired FPG is intermediate between that of men with normal and diabetic FPG concentrations. Characterization of men by FPG and 2hG concentrations indicates that 1) there is no increased CVD mortality risk for men with impaired FPG who have a normal 2hG, and, 2) CVD risk increases progressively in men with impaired FPG as the 2hG glucose becomes progressively higher.31
Type 2 diabetes, with both diabetic FPG and 2hG levels, is associated with an increased risk of CVD by two to fourfold, according to two observational studies.27, 32 This effect appears to be due to exposure to excessive amount of glucose levels since it is independent of other known type 2 diabetes-associated risk factors such as body mass index (BMI), low-density lipoprotein (LDL) cholesterol, blood pressure, and inflammatory markers and smoking. At least three studies (DECODE study, Japanese cohort study, and Mauritius/Fiji/Nauru study), cross-classified subjects by their FPG and 2hG levels and were followed to determine whether 2hG added to the predictive power of the FPG.33–35 Subjects with normal or impaired FPG were combined into a single nondiabetic group. In the DECODE study, men, but not women, with nondiabetic FPG and a diabetic 2hG did worse than those men with nondiabetic FPG and 2hG concentrations.33 A study performed in the population sampled from Mauritius, Fiji, and Nauru extended the same conclusions to both men and women.34 The Japanese cohort study is difficult to compare to the other reports because participants received a 50-g OGTT, and the cut point for FPG considered normal was 140 mg/dl (7.8 mmol/l), while the cut-point for 2hG was 200 mg/dl (11.1 mmol/l). A more recent study of subjects with genetic variants known to be associated with diabetes suggested that even a small increase in elevated FPG levels in non-diabetic individuals is associated with a trend towards increased CVD risk,36 illustrating that glucose is an important mediator linking rising circulating glucose levels with CVD pathogenesis. These results support the hypothesis that a relationship between circulating glucose and CVD is indeed causal and inextricably linked, and lead to the conclusion, which will be further discussed below, that long-term efforts to lower glucose levels and prevent type 2 diabetes should decrease CVD risk.
AGE-RELATED CHANGES IN GLUCOSE METABOLISM
Glucose metabolism is central to normal physiological function. The liver is a major metabolic regulatory organ and provides 90–95% of the circulating glucose during the post-absorptive state, when the body is no longer absorbing nutrients from the gut. The brain takes up about 50% of the glucose during post-absorptive phase with skeletal muscle using about 15%. In healthy individuals, skeletal muscle is very sensitive to insulin and can increase its uptake up to 85% of circulating plasma glucose with sudden increases in plasma glucose. Insulin secretion from β cells is tightly coupled to the availability of glucose, allowing the fine tuning of blood glucose levels within a normal range.
With age, some people progressively lose the ability to regulate glucose levels as they did when they were younger.37–43 The 2-hr OGTT data from the BLSA, published in the 1990s, illustrated nicely such positive correlation of age with worsening glucose tolerance in over 770 healthy men and women.44, 45 The decline in glucose tolerance from young (17–39 years) to middle age (40–59 years) is globally explained by the secondary influences of body fat and physical fitness.44 However, changes in glucose tolerance that occur between 60–92 years were significant and remained unexplained when body composition and physical activity were accounted for in the analysis. 44 These findings were replicated three decades later with updated OGTT data from over 2700 non-diabetic BLSA participants (Figure 2 top panel). With advancing age, plasma glucose levels in response to OGTT were progressively higher for every decade of age until they peaked at the 7th decade. Men and women have similar patterns, but levels of glucose were higher in men than in women. In individuals without diabetes, FPG levels were progressively 0.7–1.1 mg/dL higher per age decade and 2hG levels were progressively 5.6–6.6 mg/dL higher per age decade. This phenomenon has been observed in other population studies.46–48
Figure 2: Oral glucose tolerance test.
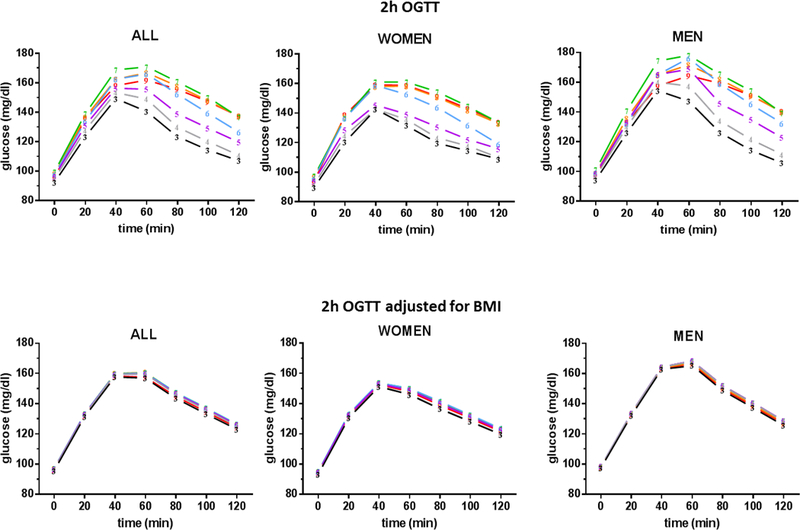
Top panel shows OGTT results in 2,777 nondiabetic participants from the Baltimore Longitudinal Study of Aging. Each line corresponds to one decade. Plasma glucose levels during OGTT rise consecutively for every decade of age until they peaked at the 7th decade. Men and women have similar patterns with men having more severe glucose intolerance. Bottom panel shows the OGTT results adjusted for body mass index (BMI).
After adjusting for BMI, the difference in glucose levels with age is drastically attenuated to 0.01–0.3 mg/dL per age decade for FPG and 0.03–0.5 mg/dL per age decade for 2hG but remains significantly correlated with age (Figure 2 bottom panel). Although BMI is a relatively rough measure of body composition, body fat plays a major role in glucose metabolism which we will discuss in more details later in this review. Other population studies also attributed the decline in glucose tolerance in large part to factors such as obesity, physical activity, dietary habits, and others.46–48
The use of epidemiology data to understand aging and glucose metabolism has limitations. First, some measures may not be precisely assessed. For example, body fat is usually estimated by BMI, and physical activity and diet are assessed by questionnaires that are notoriously biased. Second, the use of statistical methods involves making certain assumptions regarding confounders, linearity of relationships, and interactions among variables that may not be real. To better understand the relationship between aging and glucose metabolism, detailed physiology studies were performed during the 1970s and 1980s period, a large portion of them at the National Institute on Aging (NIA) and used the glucose clamp technique. Box 1 gives a brief description of the two most common clamp procedures, hyperinsulinemic-euglycemic clamp and hyperglycemic clamp. This information may help readers interpreting the clamp results detailed below from different studies.
Box 1: Brief description of glucose clamp technique.
The glucose clamp technique was developed in the 1960s by Andres and colleagues at the National Institute on Aging to conduct in vivo assessment of glucose metabolism in humans.71 Ground breaking studies were done to understand hormonal regulation and pathophysiology of carbohydrate metabolism.54, 202–205 To this day, this clamp technique is still considered the reference standard for studying whole body glucose homeostasis as it is generally considered the most rigorous and reproducible test for studying glucose homeostasis of the whole body or even a specific organ.
Although protocols for clamp come in many forms, most commonly used are the hyperinsulinemic-euglycemic and the hyperglycemic clamps. In all clamp protocols, overnight fast is required for at least 8 hours prior to the procedure. An antecubital intravenous catheter is inserted for infusion of 20% dextrose, insulin, peptides, or isotope. A second catheter is inserted in a retrograde fashion in a dorsal hand or wrist vein for blood sampling. This hand is then placed in a warming box with temperature set at 50°C for about 15 minutes to arterialize the blood prior to starting the clamp. Three to four blood samples are drawn at 10 min intervals to establish basal glucose level. All plasma glucose samples are assayed in real-time.
The hyperinsulinemic-euglycemic clamp procedure is designed to assess in vivo insulin sensitivity and best method for studying hepatic glucose production. This procedure begins with an infusion of priming dose of regular insulin administered as a step function with decreasing level every 2 min for the first 10 minutes then decrease to the desired insulin dose that is maintained for the rest of the clamp procedure. The most common dose use is 6nmol/m2/min (40 mU/m2/min) with insulin doses up to 200 mU/m2/min had been used.206 Four minutes after the start of insulin infusion, glucose infusion is also begun and the glucose infusion rate is adjusted every 5 minutes based on the plasma glucose determined from the sample to maintain the plasma glucose levels at the basal level. Glucose utilization usually reaches a steady state around 80 minutes after the initiation of insulin infusion. At this juncture, the glucose infusion rate is approximately equal to glucose utilization, assuming that hepatic glucose production is completely suppressed by the exogenous hyperinsulinemia. The amount of glucose metabolized is designated as M. For more insulin resistant individuals, hepatic glucose production may not be completely suppressed, and amount of glucose infusion would be lower and hence a lower M value would be obtained.
The hyperglycemic clamp technique is designed to assess β cell function in addition to insulin sensitivity. The procedure begins with an infusion of priming dose of 20% dextrose administered as a step function with decreasing level every 2 min for the first 10 minutes. For the first 10 minutes, blood samples are obtained for glucose measurement every 2 minutes. For the rest of the clamp, plasma glucose samples are obtained every 5 min so that dextrose infusion can be adjusted as to maintain the plasma glucose level at a predetermined level, usually 5.5 mmol/L (98 mg/dL) above basal glucose levels. A range of target glucose levels of 3.0 – 12.8 mmol/L (54–230 mg/dL) above basal glucose levels had been used in studies. Hyperglycemic clamp enables assessment of both β-cell function and peripheral insulin sensitivity. The crucial difference between insulin sensitivity obtained from the hyperglycemic clamp and the hyperinsulinemic-euglycemic clamps is that the insulin sensitivity is in response to variable endogenous insulin release in the former and to stable and reproducible exogenous insulin administration in the later.
Effect of age on hepatic glucose production
The European Group for the Study of Insulin Resistance (EGIR) used the hyperinsulinemic-euglycemic clamp (1mU/kg/min) to study hepatic glucose output in 344 non-diabetic subjects (212 men and 132 women) with age ranged from 18–95 years and BMI from 15–55 kg/m2.49 The basal hepatic glucose output was highly variable; highly correlated with lean body mass and % body fat; 23% higher in men than in women; and was progressively lower at older ages, although the age association was not significant (p=0.10). All these associations disappeared when hepatic glucose output was normalized for lean body mass. Therefore, the variability of basal hepatic glucose output appears to be due to differences of lean body mass which also explains the difference of hepatic glucose output due to age, sex and BMI.
The effect of age on the ability of insulin to suppress hepatic glucose output was also investigated by several other researchers. The EGIR study showed that at insulin levels of 73 uU/mL (510 pmol/L), hepatic glucose output was mostly suppressed in nondiabetic subjects.49 Similarly, two other groups showed that hepatic glucose output was suppressed similarly in nondiabetic young and old persons (~95%) at higher insulin levels (100–110uU/mL).50, 51 Hepatic glucose output at lower physiological plasma insulin levels were also investigated. At insulin levels of 33uU/mL in the young and 61 uU/mL in the old, hepatic glucose output was suppressed more in the young (89%) compare to the old participants (77%).50 However, another group showed that hepatic glucose production was suppressed more rapidly in older individuals at physiologic insulin levels (25–40 uU/mL) with no difference in either the basal hepatic glucose output or in the dose-response curve of its suppression by insulin between young and old individuals.52 The differences in results shown by these studies might be due to small sample size and sex imbalance in their young and old groups. Overall, the available evidence suggests that hepatic glucose output is not affected by age per se but rather by differences in body composition.
Effect of age on peripheral glucose uptake
Glucose uptake occurs by two mechanisms: insulin-mediated glucose uptake (IMGU) and noninsulin-mediated glucose uptake (NIMGU). IMGU occurs primarily in muscle and fat tissues while NIMGU occurs in brain and splanchnic organs. In the fasting state, glucose utilization occurs mostly in NIMGU tissues, specifically brain ~ 50%, splanchnic organs ~20%, skeletal muscle ~15%, adipose tissue ~5%, blood cells ~5%, and other tissues ~5%.53, 54 In post-prandial state, glucose uptake occurs in IMGU tissue with skeletal muscle accounting for the majority of uptake.
Insulin-mediated glucose uptake
Whether age per se causes insulin resistance is controversial. Studies using a variety of techniques (hyperinsulinemic-euglycemic clamp, forearm perfusion, minimal model) have shown decreases in insulin action with advancing age.50, 51, 55, 56. However, reports from some other studies also could not detect any significant association. 57–62 Unfortunately, true comparability between studies is difficult since they used different methods, had relatively small number of subjects and studied mostly men. In addition, some studies included subjects with impaired glucose tolerance, that as explained above, could mean the hepatic glucose output was not fully suppressed while other studies did not take into account body composition.
One of the most comprehensive study of insulin sensitivity was conducted by the EGIR using hyperinsulinemic-euglycemic clamp (1mU/kg/min dose) in 1146 healthy subjects (766 men, 380 women) with normal glucose tolerance and age ranging from 18 to 85 years and BMI ranging from 15 to 55 kg/m2. Insulin sensitivity for the whole group declined slightly with age but this effect of age was no longer significant when differences in BMI were accounted for. Subgroup analysis showed that after adjusting for BMI, insulin sensitivity was lower at older age only in lean women.63
The limitation of the EGIR study is that only a single dose of insulin was tested. It was therefore argued that complete dose-response curves are needed to comprehensively evaluate the effect of age on insulin resistance. Hyperinsulinemic-euglycemic clamps using 3 different insulin doses (20, 80, 200 mU/m2/min) were performed to examine the effect of age on peripheral glucose uptake in 17 young and 10 old nondiabetic non-obese subjects.55 An age-associated decrease in glucose utilization was demonstrated with no change in maximal tissue responsiveness or maximal glucose uptake. In another words, the insulin-glucose dose-response curve shifts to the right with age (Figure 3 left panel). The half-maximal glucose uptake occurred when plasma insulin levels were 54 uU/mL in the young and 113 uU/mL in the old. This association held significant after accounting for lean body mass (Figure 3 right panel). These findings were replicated by other studies that used several insulin doses during hyperinsulinemic-euglycemic clamps.50, 55
Figure 3:
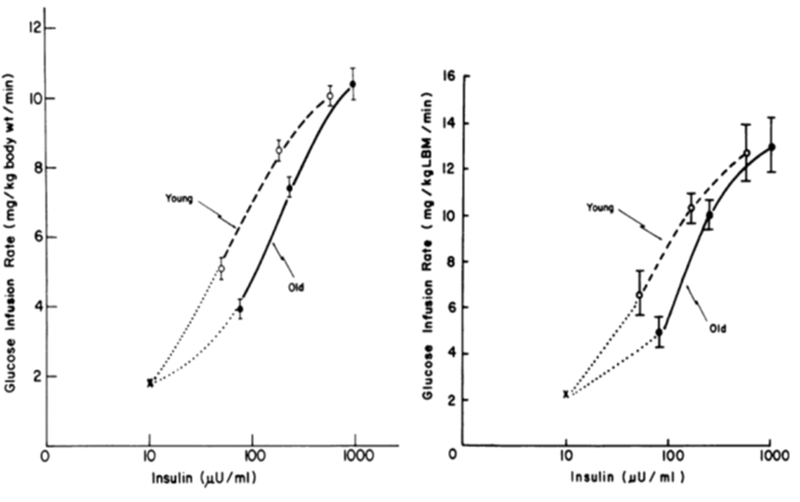
Dose-response curve for insulin-mediated whole-body glucose infusion rate during hyperglycemic clamps in young (---) young and old (—)subjects. Left panel presents glucose infusion rate in mg/kg body weight/min. Right panel presents glucose infusion adjusted for lean body mass. “x” represents basal hepatic production rate (reprint with permission).55 An age-associated decrease in glucose utilization was demonstrated with no change in maximal tissue responsiveness or maximal glucose uptake. The insulin-glucose dose-response curve shifts to the right with age (left panel). The half-maximal glucose uptake occurred when plasma insulin levels were 54 uU/mL in the young and 113 uU/mL in the old. This association held significance after accounting for lean body mass (right panel).
Noninsulin mediated glucose uptake
The effect of age on noninsulin-mediated glucose uptake (NIMGU) has been relatively understudied. Using a minimal model approach, older subjects were shown to be insulin resistant but glucose effectiveness, a measure of the increase in fractional disappearance of glucose per unit increase in glucose or insulin-independent glucose utilization, was unchanged with aging.56 Another group reported similar results using hyperinsulinemic clamp at four different glucose concentrations.64 In one study, somatostatin infusion was used to suppress endogenous insulin secretion with simultaneous dextrose infusion to evaluate NIMGU. At older ages, NIMGU was impaired at normal FPG levels (~100 mg/dL) but unchanged at physiological hyperglycemia (~200 mg/dL).65
It is clear that besides age, other factors such as body composition (fat mass, fat distribution, lean body mass), physical fitness, dietary composition and genetic factors, influence peripheral insulin sensitivity. When these factors are taken into account, it is not clear that age in and of itself still has an independent effect on peripheral glucose uptake, regardless of whether NIMGU or IMGU are considered.
Effect of age on β-cell function
The balance between insulin secretion from β cells and peripheral insulin sensitivity is what maintains normal glucose homeostasis. Diabetes develops when the ability for β cells to secrete adequate amounts of insulin to maintain normal glycemia falters. Whether aging per se impacts β-cell function has been controversial. With age, β-cell function has been reported to be either unchanged39, 59, 66, 67, or decreased. 56, 68, 69 Again, these studies used different quantification methods, had relatively small number of subjects of mostly men, some studies included subjects with impaired glucose tolerance, and did not take into account body composition.
Perhaps the most robust study on the effect of age on β-cell function was conducted in the BLSA where 230 hyperglycemic clamps were performed in 85 young, 47 middle age and 98 old participants. Four hyperglycemic plateaus were maintained for two hours at 54, 98, 142, 230 mg/dL above basal glucose levels.70 During hyperglycemic clamps, there are several distinct features in the resulting endogenous insulin response. The insulin response to square-wave-induced hyperglycemia is bimodal: there is a first-phase insulin secretion that occurs during the first 10 min with a peak response at approximately 4 min; then, plasma insulin level falls reaching a nadir at approximately 10 min; next, the second insulin secretion phase begins and insulin levels increase linearly until a plateau is reached.71, 72 Figure 4 shows the bimodal insulin response to the four levels of hyperglycemia. Within each age group, higher glucose doses corresponded with statistically higher insulin responses at all levels but differences of insulin responses among the age groups were not significantly different. Similar results were shown in another study using hyperglycemic clamps performed at 125 mg/dL above basal glucose levels.51 In a study of β-cell function of 42 highly trained women athlete aged 18–69 years, insulin secretion did not differ according to age.73
Figure 4:
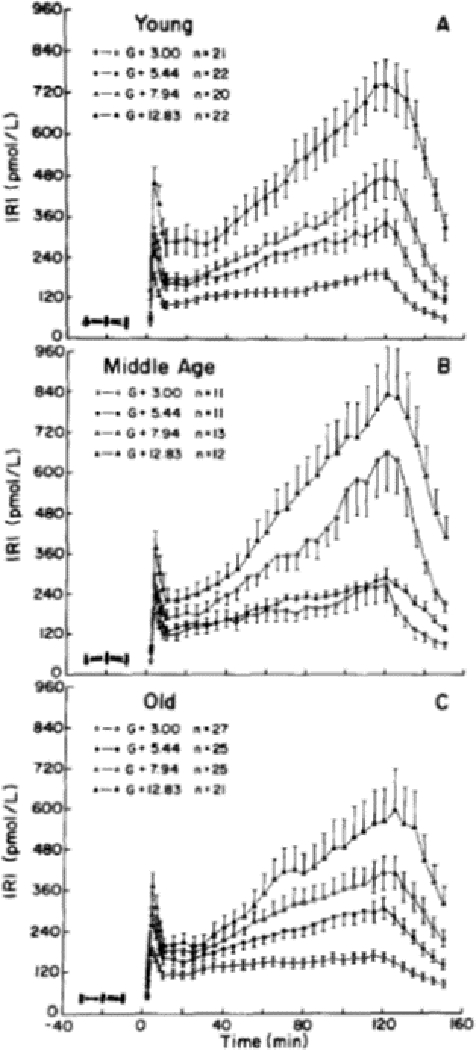
Bimodal time course of the endogenous plasma insulin response during 2h hyperglycemic clamps at four different hyperglycemic plateaus: 54, 98, 142, 230 mg/dL above basal glucose levels. Top panel: young participants; middle panel: middle age participants; bottom panel: old participants (reprint with permission).70 There is a first-phase insulin secretion that occurs during the first 10 min with a peak response at ~4 min; then, plasma insulin level falls reaching a nadir at ~ 10 min; next, the second insulin secretion phase begins and insulin levels increase linearly until a plateau is reached. Within each age group, higher glucose doses corresponded with statistically higher insulin responses at all levels but differences in insulin responses among the age groups were not significantly different.
These dose-response hyperglycemic clamps examined the β-cell response to continuous glucose stimulation at elevated and even supraphysiologic levels for up to two hours. The second phase insulin response has been shown to rise linearly for this two-hour duration, but does this rise ever plateau? And does the continuous glucose stimulation on β cells result in β-cell “fatigue”? Ten-hour hyperglycemic clamps at 98 mg/dL above basal glucose levels were performed in 10 young and 10 old subjects with normal glucose tolerance.74 The second phase insulin responses do reach a plateau during hyperglycemic clamps, around 300 min for the young and 120–150 min for the old group.
Effects of aging on pulsatile insulin secretion
Like most, if not all, hormones, insulin is secreted in a pulsatile fashion. There are two different insulin pulse patterns: the rapid, low amplitude pulses that occur every 8–15 min and the large amplitude ultradian pulses with a periodicity of 60–140 min.75, 76 These two types of pulses have different functions in glucose metabolism: the rapid low amplitude pulses are important in inhibiting hepatic glucose production and preventing insulin receptor downregulation;77–79 while the ultradian pulses are important in stimulating peripheral glucose disposal.80 These insulin pulses are disturbed in individuals with glucose tolerance, obesity, and type 2 diabetes.81–83 In healthy non-obese individuals, aging is associated with a decrease in the amplitude and mass of rapid insulin pulses and a reduction in the frequency of ultradian pulses during the fasted state.84 With stimulation by sustained hyperglycemia for 10 hours, a reduction in mass and amplitude of rapid pulses and a decrease in frequency, amplitude and regularity of ultradian pulses were noted in the old group.74 Whether these age-related changes in insulin pulsatility have any clinically relevant consequences is not known.
The effect of aging on incretins
Incretins are enteroendocrine hormones which augment insulin response from β cells beyond the amount attributed to glucose alone. There are two incretins, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1). Their release from enteroendocrine cells, K cells for GIP and L cells for GLP-1, is stimulated by the presence of nutrients in the gut. Different nutrients have various degree of potency in stimulating incretin secretion: GIP (fatty acids > carbohydrates > amino acids); GLP-1 (carbohydrates > fatty acids > amino acids).85, 86
GIP has been shown to be elevated in type 2 diabetes and obesity in both fasting state and during glucose stimulation. 87–89 Baseline GLP-1 is not affected by type 2 diabetes but its secretion is lower during oral glucose stimulation in obesity.87–89 Fasting GIP levels have been found to be unchanged with aging.90 Compared to young healthy controls, GIP and GLP-1 levels, both fasting and in response to oral glucose have been found to be unchanged in nonobese older adults,89–91 and GIP is increased in obese healthy old people.92
The effect of age on the sensitivity of β cells to endogenous GIP was examined using hyperglycemic clamp in combination with OGTT. Relative to basal state, GIP levels rose by 179% in the young subjects and 265% in the old subjects in response to oral glucose while the rise in insulin levels were comparable between young and old individuals, indicating that β-cell sensitivity to GIP is reduced in advancing age.89 Dose-response of exogenous GIP on insulin secretion was also studied using hyperglycemic clamp at two different glucose levels (198 mg/dL or 330 mg/dL plasma glucose levels) and two different GIP doses (2 or 4 pmol/kg/min). The β-cell sensitivity to exogenous GIP was significantly decreased in the old compared to the young at the lower glycemic levels (198 mg/dL). However, the insulin response to GIP was similar in both young and old at the higher glycemic levels (330 mg/dL).93 The insulinotropic effect of GIP decreases with aging, however, the defect in β cell response to GIP disappear when plasma glucose is increased to higher levels. Taken together, the findings of these studies suggest that the age-related impairment in response to GIP may be an important cause of the glucose intolerance of aging.
The insulinotropic effect of GIP and GLP-1, separately and in combination, were examined during euglycemic or hyperglycemic (198mg/dL) clamps, in healthy subjects and subjects with type 2 diabetes.94 They reported that: 1) GLP-1 is a more potent insulinotropic hormones than GIP; (2) the insulintropic effect of GIP and GLP-1 are additive; (3) GLP-1 has a potent insulinotropic effect while GIP has lost its insulinotropic effect in subjects with type 2 diabetes.94, 95 We, in collaboration with other investigators, studied the insulinotropic effect of exogenous GLP-1 in older type 2 diabetic subjects. We found that GLP-1 not only augments insulin release but also enhances both IMGU and NIMGU in these older type 2 diabetic subjects.96–98 Furthermore, we reported that three months of continuous GLP-1 infusion in the same population was as effective as standard hypoglycemic medications in glycemic control but with a tighter glycemic excursion and less hypoglycemic events.99 To our knowledge, there are no data from humans on the efficacy of GLP-1 as a function of age alone in subjects free of diabetes.
MECHANISMS OF GLUCOSE DYSREGULATION
The detailed mechanistic studies reviewed above clearly show that processes driving metabolic dysregulation in aging are multifaceted, with changes in body composition leading to insulin resistance playing a major role. Insulin resistance is not a disease per se but rather it is a condition where higher insulin levels are required to maintain glucose homeostasis and adequate fatty acid utilization in liver, skeletal muscle, and adipose tissue. With increasing insulin resistance, pancreatic β cells upregulate insulin production and increase secretion to maintain normal glycemia. As long as β cells are able to produce sufficient insulin to overcome the insulin resistance, that is, keep shifting the curve of glucose-insulin secretion to the right, absolute circulating blood glucose concentration stays within a narrow normal range.
Over time, insulin resistance can lead to type 2 diabetes when β cells fail to keep up with the body’s increased need for insulin. It is important to bear in mind that metabolic changes begin to occur many years prior to the diagnosis of diabetes. Two studies have found that 10–13 years prior to diagnosis of diabetes, FPG, 2hG, insulin sensitivity and insulin secretion were already significant different between those who were diagnosed with diabetes and those who were not.100, 101 The evolution of the metabolic characteristics of the groups who eventually developed diabetes compared to the group who remain healthy have never been reported. What causes insulin resistance and ultimately diabetes?
There are many etiologies for insulin resistance – genetic factors, sedentary behaviors, increasing chronological age, medications – but obesity, especially its distribution and content, is by far the single biggest risk factor. As mentioned earlier, the prevalence of diabetes in the US population has increased dramatically over the past half a century. 4, 102 It is impossible not to underline that such increase in prevalence of diabetes is in synchrony with that of obesity, and has generally been attributed to increased food consumption, lack of physical activity and sedentary lifestyles that are facilitated by the proliferation of electronic gadgets and aids. Using data from the US Department of Agriculture (USDA), the daily food consumption for an average American has increased from 2024 calories in 1970 to 2476 calories in 2010, with more than 20% increase in calories.103 Of note, it has been calculated that an extra 500 calories per day roughly translate to one extra pound of weight gain a week! Figure 5 illustrates the prevalence of diagnosed diabetes and obesity together with average daily food consumption per capita over the past 50 years using data from the CDC and USDA. 4, 102,103 It is not at all surprising that the increase prevalence of obesity and diabetes track that of increases in food consumption. This is even more problematic in older persons because aging per se is associated with increased body fatness and declines in lean body mass.
Figure 5:
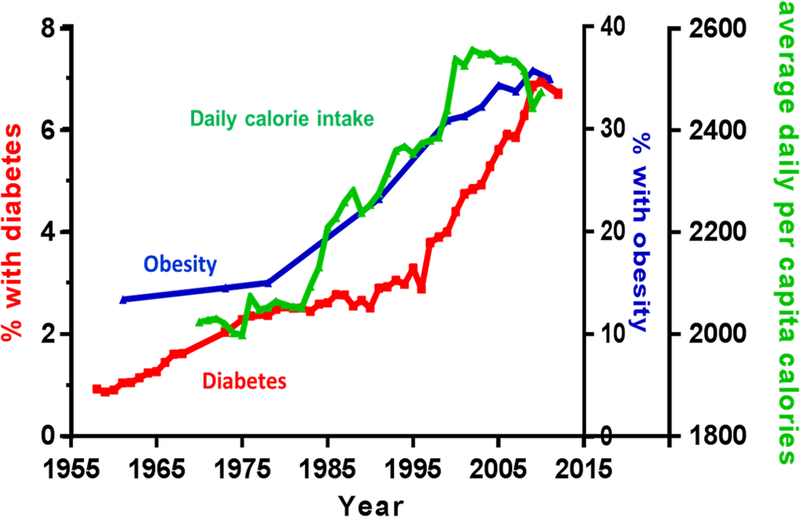
Increase average daily per capita food consumption, prevalence of obesity and diabetes over the past 60 years in the United States (graphs drawn using data from the CDC and USDA). 4, 102,103
In addition to over-consumption of food, older individuals may be more likely than younger individuals to gain weight for many reasons including but not limiting to the following: (1) gradual loss of muscle mass and gain in fat mass starting the 4th decade of life; (2) menopause in women and decline in testosterone with age in men that compound the loss of muscle mass and gain of adipose tissue; and (3) catecholamine-induced lipolysis in adipose tissue blunting in older individuals.104
OBESITY, GLUCOSE DYSREGULATION AND ACCELERATED AGING
Clinically, the definition of obesity is based on crude measure of BMI or waist circumference to gauge total body fat. In prospective studies, increased visceral fat, using waist circumference as a proxy, is an independent risk factor for coronary artery disease and death.105–107 Obesity shares numerous biological similarities with adipose tissue dysfunction of aging such as chronic inflammation, cellular senescence and immune dysfunction. Accordingly, understanding the interplay between accelerated aging related to obesity and adipose tissue dysfunction of aging is critical to gain insight into the aging process and the pathophysiology of obesity resulting in insulin resistance.
Adipose Tissue Dysfunction
Adipose tissue, once considered an inert, energy storage depot, is now proven to be an active dynamic endocrine organ. It is responsible for energy homeostasis through storing and releasing fat in response to energy-balance needs, synthesizing and secreting peptide hormones by adipocytes to meet energy needs, generating proinflammatory response to bacterial antigens, and other functions.108, 109 Studies have shown that not all adipose tissue is alike and intra-abdominal fat or visceral fat and subcutaneous fat in the upper body and lower extremities may have different functions.110 Furthermore, health risk is associated with location as well as the amount of body fat in which visceral obesity is associated with increased risk for diabetes, hypertension, atherosclerosis, dyslipidemia, cancers, and mortality compared to peripheral obesity.111, 112 Even humans of a normal weight with a high ratio of central-to peripheral fat have an increased likelihood of being insulin resistant.113
Mechanisms of adipose tissue dysfunction in obesity differ from aging in important ways.114 Adipose tissue is composed of primarily adipocytes, preadipocytes, fibroblasts, vascular endothelial cells and a variety of immune cells such as adipose tissue macrophages. Preadipocytes, a progenitor cell, accounts for 15–50% of cells in adipose tissue, and can differentiate into new adipocytes, macrophages, and other cell types.110 The main role of preadipocytes is to give rise to new adipocytes. In addition, preadipocytes secrete cytokines and can undergo cellular senescence, particularly with aging and obesity.114, 115 In vitro, human preadipocytes obtained from abdominal subcutaneous adipose tissue are capable of more extensive replication than those obtained from visceral adipose tissue.116–118
During weight gain, visceral fat generally enlarges through increases in fat cell size, whereas subcutaneous fat enlarges by increasing both fat cell size or number.119, 120 In severe obesity, preadipocytes are driven to become new adipocytes with increase replication cycles, especially those preadipocytes in the subcutaneous adipose tissue, resulting in subcutaneous adipose tissue dysfunction. Subcutaneous adipose tissue dysfunction is associated with: (1) restricted capacity to store excess free fatty acid as triglyceride, thus resulting in dysregulated release of free fatty acids from subcutaneous fat instead of visceral fat; (2) excess free fatty acids from subcutaneous fat contributes to visceral fat enlargement, ectopic lipid accumulation, lipotoxicity resulting in secretion of proinflammatory cytokines and chemokines, immune cell infiltration, accumulation of senescent cells, an increase in senescence-associated secretory phenotype (SASP) components, and insulin resistance.110, 114, 121, 122 With aging, there is a decline in preadipocyte replication, decrease in adipogenesis, increase in susceptibility to lipotoxicity, and increase in pro-inflammatory cytokine and chemokines.114 Furthermore, adipocytes and preadipocytes, instead of macrophages, become the main sources of inflammatory cytokines and chemokines.123–125 All these pathologies associated with adipose tissue dysfunction contribute to insulin resistance.
Cellular senescence
Cellular senescence is a complex process characterized by arrest in replication that is thought to be intrinsically connected with aging. Accumulation of senescent cells occurs during aging and increase in the number of senescent cells are also seen in diseases associated with accelerated aging phenotypes such as obesity and diabetes. Senescent cells are identified by molecular markers such as β-galactosidase (SA-Bgal) and p16INK4a, and other features such as permanent growth and replication arrest and secretion of SASP components.126, 127 SASP components include a large number of cytokines, chemokines, growth factors, and proteases. The physiological role of SASP is not clear, but there is some evidence that SASP is implicated in the mild pro-inflammatory phenotype of aging.
The number of senescent cells in adipose tissue increases with both obesity and aging. Senescent preadipocyte number is higher in obese subjects compared to lean age-matched controls.114 Extremely obese subjects (BMI ~83 kg/m2) have up to 31-fold more senescent preadipocytes than nonobese subjects.114 Higher levels of p53 protein, p21 mRNA expression and SA β-gal activity are found in the adipose tissue from subjects with diabetes.115 Obesity -associated senescent cells may promote chronic, low-grade sterile inflammation through secretion of SASP components thus contributing to development and progression of insulin resistance and type 2 diabetes, although empirical evidence for this association in humans is lacking.128, 129 In a vicious cycle, metabolic and signaling changes seen in diabetes, such as high plasma glucose levels and lipotoxicity, further promote senescent cell formation. Therefore, in diabetes and obesity, senescent cells may be both a cause and consequence of metabolic changes and tissue damage.
There is strong evidence that senescent cells accumulate with aging and, either directly or indirectly through SASPs, contribute to CVD and other chronic diseases. In the context of obesity, the major contribution can probably be ascribed to preadipocytes that acquire senescent attributes, as well as true senescent cells whose origin is still poorly understood. Studies from animal models also suggest that senescence plays a role in atherogenesis, from macrophages with senescent features that transport cholesterol in the arterial intima, to the processing of the intercellular matrix by metalloproteinases, to the fragmentation of the fibrotic cap that destabilizes plaque. However, evidence in humans is sparce. In the context of this review, we have focused more on senescent preadipocytes because their presence in the visceral fat has been associated with insulin resistance, while there is little current evidence that senescent endothelial cells drive this phenotype.
What role, if any, senescent cells play in islets is unclear. β-cell senescence induced by SA β-gal activity in mice fed a high-fat diet was associated with significant decrease in insulin secretion and diet-induced diabetes.130 In another study, levels of p27Kip1, a cell cycle inhibitor and marker of senescence, were reported to be increased in β cells of genetic mouse models of type 2 diabetes. Deletion of p27Kip1 seemed to enhance insulin secretion because of increased number of β cell proliferation.131 However, a recent study showed that in healthy old mice and healthy humans, p16Ink4a-induced senescence of β cells resulted in enhanced insulin secretion compared to β cells from young animals in response to high glucose concentration.132 Clearly, much more research is needed in this area to understand the role of senescent cells as they relate to aging, diabetes, and CVD.
Inflammation
Another crucial factor that intertwine with hyperglycemia and aging is inflammation. Aging is associated with increased in proinflammatory cytokines including interleukin (IL)-1, IL-1 receptor antagonist (IL-1Ra), IL-6, IL-8, IL-13, IL-18, C-reactive protein (CRP), interferons (IFN) α and β, transforming growth factor β (TGF-β), tumor necrosis factor (TNF)-α, its soluble receptors (TNF-R1 and TNF-R2), and serum amyloid. Adipose tissue can produce proinflammatory and chemotactic compounds, such as IL-6, IL-1β, TNF-α and MCP-1, as well as hormones that modulate inflammation, such as adiponectin and leptin.133 Both aging and obesity promote immune cell infiltration into adipose tissue with macrophage infiltration more prominent in obesity and T-regulatory cells more prominent in aging.134, 135 Adipose tissue dysfunction associated with aging or obesity are also linked to increase in number of senescent cells which secrete SASP components including cytokines and chemokines.114 Therefore, chronic inflammation associated with adipose tissue dysfunction, SASPs components, are all intertwined and adding fuel to insulin resistance and accelerated aging phenotype of obesity and diabetes.
A number of clinical trials have clearly demonstrated that treatment with monoclonal antibodies, mostly directed against IL-1β and TNF-α, significantly reduces the risk of developing cardiovascular events. Targeted anti-inflammatory therapy has been suggested for both preventing and treating diabetes and this topic has been extensively reviewed.136 The effects of TNF-α signaling antagonists are still controversial, and the best results have been obtained in patients with overt inflammatory diseases, such as rheumatoid arthritis. The positive effects of anti–IL-1β—including IL-1 receptor antagonist (anakinra) and IL-1β–specific antibody (gevokizumab, canakizumab, and LY21891020)—have been demonstrated in multiple trials, both in terms of reducing inflammation, as well as improving carbohydrate metabolism, with a relatively safe profile. Chronic administration of salsalate, a prodrug of salicylate with fewer adverse reactions than aspirin, has beneficial effects on glycemia and insulin sensitivity, probably by inhibiting the NF-κB pathway. Other anti-inflammatory drugs such as Diacerein (a treatment for arthritis) and chloroquine/hydroxychloroquine (an antimalaric) have also demonstrated benefits in improving glycemia.
Endocannabinoids
Over the last few decades it has become apparent that the endocannabinoid system (ECS) plays a role in the development of obesity, modifies insulin receptor activity in liver, fat and muscle, and is involved in lipid metabolism – all risk factors for CVD. In abdominal obesity the ECS is upregulated both centrally and peripherally.137 The endocannabinoids, anandamide and 2-arachidonoylglycerol, are lipid mediators, whose synthesis is directly influenced by dietary fats and their composition.138 A hyperactive ECS can then, in a vicious cycle, lead to even greater fat accumulation by increasing food intake through activation of cannabinoid 1 receptors (CB1Rs) in brain and favoring reduced energy expenditure in peripheral tissues, such as muscle. In liver, activation of CB1Rs lead to downregulation of insulin receptor activity and worsening hepatic steatosis and steatitis in animals on a high fat diet.139–141 Furthermore, CB1R activation causes endoplasmic reticulum stress that leads to increased ROS and ceramide synthesis and further insulin receptor down-regulation.142 Moreover, islets of Langerhans contain an autonomous ECS system, complete with the enzyme machinery for synthesis and secretion of endocannabinoids and the presence of CB1Rs on β cells.139 Moreover, an overstimulated ECS in islets of Langerhans leads to activation of proapoptotic proteins in β cells and serves as a brake on insulin secretion after β cell depolarization.139, 143, 144 In-depth review of the relationship between the ECS and islets is available.145 Data is now emerging concerning the roles of the ECS directly in cardiovascular functions. Activation of CB1Rs on macrophages enhances their recruitment and their recruitment in turn of other inflammatory-type immune cells to atherosclerotic plaques and sites of injury in the heart and vascular epithelium, and to smooth muscle cells. An activated CB1R also promotes profibrotic pathways in myofibroblasts.146, 147 Research in ECS and aging is lacking however.
Intestinal permeability
In humans, the intestinal epithelium covers a surface of about 32 m2, which is approximately half the size of a badminton court. 148 Such vast surface is the site for interactions with ingested substances, and it serves two important functions: a barrier preventing entry of antigens, toxins and microorganisms into the body; a selective filter facilitating absorption of nutrients, electrolytes, and water; and exchange of molecules between host and environment. Age-dependent intestinal barrier dysfunction or increase in intestinal permeability is associated with metabolic dysfunction, inflammation and death in Drosophila.149 A more permeable gut, in theory, would result in increased entry of antigens such as lipopolysaccharide (LPS), an endotoxin found in the outer membrane of gram-negative bacteria. LPS has been used as a surrogate measure of gut permeability.150
Proinflammatory responses of adipose tissue, primarily preadipocytes, to LPS may be combined with high local concentrations of fatty acids during ensuing cytokine-induced lipolysis to mitigate infection.150 In a population study of 7169 participants, higher endotoxin activity was associated with prevalence and incident of diabetes.151 Plasma LPS concentration was also significantly higher in healthy lean subjects after consuming high-fat, high-carbohydrate meals compared to high-fiber and fruit meal.152 Clearly, much more research is needed in this area to investigate the association between diet (both the amount of calorie consumed and diet quality), gut microbiota, gut permeability and inflammation with aging and age-related diseases.
Mitochondrial dysfunction
The decline of mitochondrial function with age is well described but the relationship between mitochondrial dysfunction and longevity is complex and is an area of active research.153, 154 Mitochondrial dysfunction is associated with decrease in insulin sensitivity and β-cell death in type 2 diabetes.155, 156 Mitochondrial defects of various causes such as mtDNA deletion, and decrease in gene expression of PGC-1α (master control in the regulation of mitochondrial biogenesis) and nuclear respiratory factor 1 (NRF1), have been found in skeletal muscle of diabetic patients.157, 158 Impaired oxidative phosphorylation and fatty acid β-oxidation in skeletal muscle was found in insulin-resistant offspring of patients with type 2 diabetes and older adults with insulin resistance.159, 160 Reduced mitochondrial oxidative capacity measured by 31P-magnetic resonance spectroscopy was associated with the prevalence and duration of insulin resistance in 248 non-diabetic participants in the BLSA.161 Mitochondrial dysfunction observed in obesity is associated with chronic accumulation of nonesterified fatty acids due to excessive caloric intake.162 Excess substrates load such as high-fat diet and hyperglycemia increase reactive oxygen species production in mouse adipocytes.163, 164 In a vicious cycle, reactive oxygen species then reduce oxygen consumption in adipocytes, block fatty acid oxidation, and result in more lipid accumulation.165 Dysfunctional mitochondria that cannot be recycled by mitophagy cause the release of damage-associated molecular species that trigger an inflammatory response by the inflammasome. Both the accumulation of fatty acids and inflammation contribute to insulin resistance.
Altered myocardial mitochondrial metabolism has been observed in aging, as well as in diabetes and obesity. Aging is associated with a decrease in myocardial fatty acid utilization and oxidation, as well as a relative increase in myocardial glucose utilization at rest.166, 167 In response to dobutamine, both younger and older hearts increase myocardial fatty acid metabolism; however, only the younger hearts had increased myocardial glucose utilization.168 In obesity and diabetes, an increase in myocardial fatty acid uptake and oxidation has been observed in addition to a decrease in myocardial glucose metabolism. This increase in fatty acid uptake is coupled to enhanced myocardial oxygen consumption but without a concomitant increase in contractility, thus resulting in reduced cardiac efficiency.169, 170 This age-, obesity-, and diabetes-related mitochondrial dysfunction contributes to the inability of the aging heart to withstand stressors, leading to increased incidence of cardiac failure with aging.
It is apparent that all these factors described above – adipose tissue dysfunction, cellular senescence, inflammation, endocannabinoids, intestinal permeability, and mitochondrial dysfunction – are all interconnected and work in a feedback loop of worsening metabolic control leading to obesity, insulin resistance, diabetes, CVD and associated morbidity and mortality. However, as we started earlier, the biggest single factor contributing to declining whole-body insulin sensitivity with age, is obesity especially when fat is accumulated outside the natural subcutaneous compartment. Researchers at the Mayo Clinic recently published data collected from 116 men and women ages 19–78 and attempted to account for the many factors that we mentioned above, which are supposed to impair insulin action with age. They used hyperinsulinemic-euglycemic clamps to measure M (rate of whole body glucose metabolism; see Box 1), delivered isotopes to measure hepatic glucose suppression, performed magnetic imaging and spectroscopy to quantify subcutaneous, visceral fat deposits, and intrahepatic/intramyocellular lipid amounts, while also using DEXA to measure total fat mass, fat-free mass and % body fat. They also performed muscle biopsies for measuring mitochondrial respiration capacity and reactive oxygen species. In summary the study was, all things considered, a very comprehensive study of liver, muscle, fat depots, and mitochondrial function using state-of-the-art techniques across a spectrum of ages. The relevant findings were: (1) total-body insulin sensitivity negatively correlated with visceral fat and hepatic lipid accumulation (no surprise); (2) hepatic insulin sensitivity was negatively correlated with total body and visceral fat (again, no surprise); (3) however, skeletal muscle mitochondrial function, measured in the biopsy from vastus lateralis muscles, was not a predictor of peripheral insulin sensitivity; and (4) age itself, at least up to age 78, did not appear to be an independent risk factor for reduced insulin sensitivity.171 Another intriguing finding from the study was that intramyocellular lipid amounts, measured in the anterior tibialis muscle, were actually positively predictive of peripheral insulin sensitivity. One can hypothesize that this may be because the fat is fated to go towards ß-oxidation, which may cause increasing mitochondrial turnover and capacity. However, this theory remains to be confirmed. Additionally, the findings from the Mayo group may not be outlier findings because there are hints from other investigators that actual lipid droplets in muscle are neutral or even positive with respect to insulin sensitivity.172, 173
BRIEF SUMMARY OF INTERVENTION TRIALS OF GLUCOSE LOWERING THERAPIES IN CVD PREVENTION
Despite the evidence linking exposure high glucose to CVD, randomized control trials (RCTs) of glucose lowering therapies in patients with type 2 diabetes have not been successful in CVD prevention or mitigation, though studies using the new classes medications, GLP-1 receptor agonists and sodium/glucose cotransporter 2 (SGLT2) inhibitors, appear to be more promising in this direction. The search for therapeutic strategies for preventing CVD by lowering blood glucose has been ongoing for more than 50 years. The University Group Diabetes Program (UGDP), a multicenter randomized controlled trial funded by the National Institutes of Health (NIH) in the 1960s, first tested the hypothesis that lowering glucose would prevent CVD in type 2 diabetic patients.174 One thousand and twenty-seven participants (mean age of 53 years; age>=55 years (45%)) were randomized to 1 of 5 treatment arms: (1) tolbutamide sodium, a first-generation sulfonylurea that was the most frequently prescribed glucose-lowering drug in the US at that time, (2) phenformin hydrochloride, a first-generation biguanide that was another of the most frequently prescribed glucose-lowering drugs at the time, (3) a variable amount of insulin aimed at keeping the blood glucose level close to ‘normal’, (4) a small, fixed dose of insulin based on body size, or (5) a placebo.175
Surprisingly, compared to placebo the UGDP found a 2.9 times higher death rate from CVD in the tolbutamide group (P = 0.005 vs placebo) and a 4 times higher death rate from CVD in the phenformin group (P = 0.04 vs insulin plus placebo); while no differences were seen in microvascular complications compared with placebo in these 2 groups.176–182 The tolbutamide and phenformin arms of the trial were stopped after 8 years because of the increased death rates.176, 178 The study was continued in the two insulin groups because their macrovascular or microvascular complications and mortality were no different from placebo, however results remained the same after 13 years of follow-up with no obvious benefit to insulin treatment in either insulin groups.181, 182
In the 2000s thiazoledienadiones, a new class of glucose lowering agents, became available. The major glucose-lowering action of this new class was to increase insulin sensitivity resulting in a lowering of A1c on average by at least 1%. The expectation would be that lowering insulin resistance would prove beneficial in CVD. However, troglitazone, the first-in-class to become widely used, was withdrawn because of liver toxicity and, in many cases, resulting deaths. In 2007, a meta-analysis of 42 eligible studies (mean age of 56 years) of rosiglitazone compared to placebo/other glucose-lowering agents in treating type 2 diabetes, with pre-specified end points of heart attacks and deaths from cardiovascular disease. They found that rosiglitazone was actually associated with a significant increase in heart attacks and borderline increase in deaths from CVD.183 With these negative effects of two thiazoledienadiones in a row, things did not look good for this class of compounds especially with regards to CVD. However, the third drug in this class, pioglitazone, based on a very recent meta-analysis of nine trials with about 12,000 participants (mean age 62 years), seems to have cardiovascular benefits with regards to major cardiovascular events in people with insulin resistance, glucose impairment and type 2 diabetes, but it increases the risk of heart failure, without increasing CVD deaths, likely because of its side-effect of fluid retention that is common to drugs in this class.184 A meta-analysis of 19 RCTs that involved pioglitazone versus placebo/other glucose-lowering agents in the treatment of type 2 diabetes reported similar results.185 The differences in the two compounds, rosiglitazone and pioglitazone, with regards to CVD are likely independent of any glucose-lowering effect but rather due, at least in part, to differences in lipid handling, with pioglitazone having favorable effects on the LDL particle size in particular.
More recent RCTs have reported inconclusive results on the effect of intensive glucose-lowering therapies regardless of the method, including increasing amounts of insulin to achieve as close to A1c of 7% or below, in the prevention of CVD in patients with type 2 diabetes. Four large-scale RCTs, three of which enrolled over 10,000 patients (mean age ranges from 60 to 66 years), shown no benefit of intensive glucose-lowering therapy, including in subgroup analysis of those older than 65 years of age. Indeed, one of these studies was stopped prematurely because of increased mortality in the treatment arm.186–189
In spite of the negative results of previous trials, a recent meta-analysis of five RCTs of intensive glucose-lowering therapy in type 2 diabetic patients (33, 040 patients, mean age 62 years), indicated a modest benefit to glucose lowering on CVD outcomes.190 Furthermore, a follow-up observational study of patients who had been enrolled in the ACCORD RCT showed some evidence of benefit of intensive glucose-lowering therapy on cardiovascular outcomes.186 And, of great interest, after mining the data from the UKPDS trial, early lowering of blood glucose, by any means, seemed tied into lower CVD at later times, in what is termed the legacy effect.191 However, further follow-up for 2.5 years of the ORIGIN TRIAL participants to look for a legacy effect turned out to be CVD neutral.192
A class of compounds with glucose-lowering effects and minimal systemic or off-target effects, the alpha-glucosidase inhibitors, has fallen out of use despite very favorable CVD outcomes. As mentioned earlier, the DECODE study generated evidence that the 2hG has a strong link to cardiovascular and all-cause mortality: the higher the 2hG glucose, the greater the risk of CVD mortality.193, 194 Acarbose is an alpha glucosidase inhibitor that reduces post-prandial elevations in glucose by reducing absorption of carbohydrates from the small bowel, and, unlike tolbutamide, it carries no risk of hypoglycemia. The STOP-NIDDM study (1,429 participants, mean age 54 years) investigated the effects of acarbose versus placebo on the development of diabetes over 5 years in patients with impaired glucose tolerance. The study also assessed the effect of acarbose on development of major CVD events. The risk of progression to diabetes was reduced by 36% and the risk of developing a CVD event was halved: there was 13 heart attacks in the participants, only one of which was in the acarbose-treated group.195 Because the trial was in people who did not yet have diabetes it can be easily argued that CV pathology was not long-standing or yet irreversible, therefore making it more likely that beneficial effects could be seen. Nonetheless, it shows that CVD can be impacted by lowering post-prandial glucose, especially if done during the diabetes trajectory of increasing blood glucose levels. Use of acarbose in the US fell out of favor, despite the obvious beneficial effects on glucose homeostasis and CVD, because of the gastrointestinal effects of flatulence, bloating and stool changes.
Two new classes of glucose-lowering agents have become available: GLP-1 receptor agonists (since 2005) and sodium/glucose cotransporter (SGLT2) inhibitors. New RCTs were designed and conducted to test the hypothesis that these new compounds, which are more expensive than existing glucose-lowering drugs, were beneficial to CVD versus placebo or compared to existing glucose-lowering agents. The two large RCTs LEADER (9,340 patients, mean age 64 years) and SUSTAIN-6 (3,297 patients, mean age 65 years) found that two GLP-1 agonists, liraglutide and semaglutide, favourably affect the risk of developing CVD in type 2 diabetes.196, 197 Liraglutide reduced CVD death and all-cause mortality compared to placebo, and even resulted in less hospitalizations because of heart failure, with differences becoming even more apparent after 24 months.196 When subgroup analysis was conducted in patients older than 60 years of age in the LEADER study however, the effect was only marginally significant. Participants on semaglutide treatment had fewer heart attacks and nonfatal strokes compared to those on placebo.197 However, another agent in this class, lixisenatide, when studied in the ELIXA trial (6,068 patients, mean age 60 years old) over two years, did not show CVD benefit.198
The EMPA-REG OUTCOME trial (7,020 patients, mean age 63 years), evaluated CV safety and efficacy of empagliflozin (SGLT-2 inhibitor) versus placebo, in a population with established CVD. Subjects with diabetes and known CVD were followed for three years. Empagliflozin use lead to a 38% relative risk reduction in CVD—an impressive result. When subgroup analysis was done in patients older than 65 years of age, the beneficial effect persisted. Additionally, similar to use of GLP-1 agonists, hospitalizations for heart failure were reduced by 35% using empagliflozin. Similar results are appearing for canagliflozin, especially in the older than 65 years subgroup (CANVAS trial with 10142 patients, mean age of 63 years), and therefore point to a class effect.199, 200
A MODEL OF AGING, BODY COMPOSITION CHANGES AND INSULIN RESISTANCE
Based on this review of the literature and our own work, we propose a model (Figure 6) that links the possible dysregulation of physiological pathways leading to obesity and diabetes, both a form of accelerated aging and risk factors for CVD, as a consequence of chronic excessive food consumption. The changes in body composition that are intrinsically connected with the biology of aging together with over indulgence in food and the types of food modify gut microbiota. Changes in gut microbiota, together with nutrients alter intestinal permeability which lead to absorption/infiltration of different types of molecules from the gut lumen including lipopolysaccharides (LPS), large molecules from the outer membrane of gram-negative bacteria. The excess calories and LPS may then lead to upregulated ECS, mitochondrial dysfunction, and chronic inflammation and, over time, increased cellular senescence and adipose tissue dysfunction. Excessive adipose accumulation then causes insulin resistance and eventually, in some people, diabetes. Diabetes and obesity are both forms of accelerated aging and independent risk factors for CVD. Based on this model, diet and exercise should be essential component of treatment of insulin resistance in older persons in preventing development of diabetes. This concept has already been eloquently studied in the Diabetes Prevention Program which showed that diet and exercise were more effective then metformin in preventing development of diabetes.201
Figure 6:
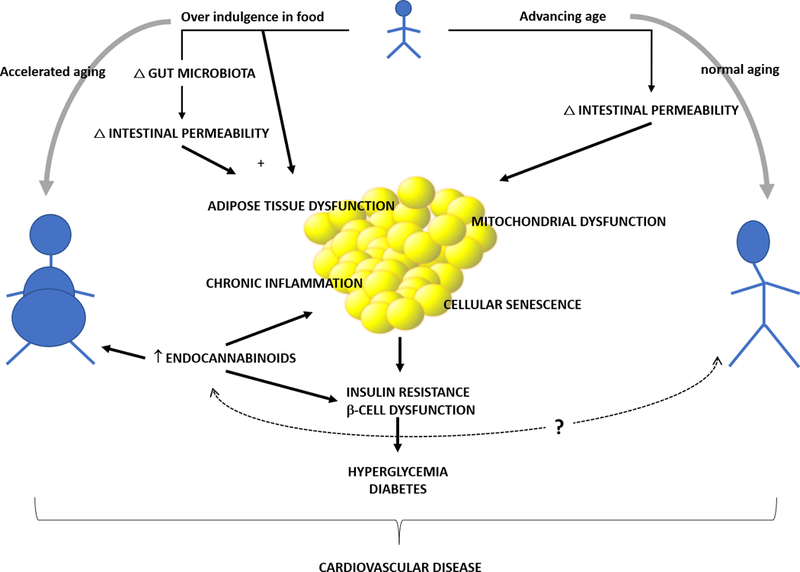
A model of aging, body composition changes and insulin resistance that links the possible dysregulation of physiological pathways leading to obesity and diabetes, both a form of accelerated aging and risk factors for CVD, to chronic excessive food consumption. The changes in body composition that are intrinsically connected with the biology of aging together with over indulgence in food and the types of food modify gut microbiota. Changes in gut microbiota, together with excess food intake alter intestinal permeability which lead to absorption/infiltration of different types of molecules from the gut lumen including lipopolysaccharides (LPS), large molecules from the outer membrane of gram-negative bacteria. The excess calories and LPS may then lead to chronic inflammation and an upregulated endocannabinoid system, and, over time, increasing the amount of senescent cells and adipose tissue dysfunction. Excessive adipose accumulation then leads to insulin resistance and, in some people, eventually to diabetes. Diabetes and obesity are both forms of accelerated aging and independent risk factors for CVD. (Illustration Credit: Ben Smith).
SUMMARY
Over the past 60 years, tremendous progress has been made in understanding the pathogenesis of age-related glucose dysregulation and cardiovascular disease. Despite knowing that excessive food consumption is the predominant cause of obesity, diabetes, and cardiovascular disease, no progress has been made in reversing the prevalence of these diseases. More research, basic, translational and clinical, is needed to better understand interaction of gut microbiota, inflammation, endocannabinoids and whole-body energy consumption in the genesis of diabetes and other metabolic diseases in humans. This knowledge can help us better reinforce selection in “quality” food consumption. Collaborative efforts across disciplines such as behavioral science, mental health, and nutrition are needed so we can get a better understanding of human behavior in overeating and finetune public policies in curbing the obesity and diabetes epidemics. We need to tackle the root cause of the obesity epidemic, which is excessive calorie intake, before we can reverse the tide of ever increasing prevalence of diabetes and cardiovascular disease. Based on our review of the topic, it also appears self-evident that we need to intervene earlier in the glucose trajectory, especially in the post-prandial period, to maintain glucose homeostasis as close to ‘normal’ as possible and before people are given a biochemical diagnosis of diabetes. Blood glucose is a continuum of risk for CVD and it is likely that changes in the vascular epithelium that cannot be reversed simply by lowering blood glucose have already occurred by the time of diabetes diagnosis.
Acknowledgments
SOURCES OF FUNDING
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Non-standard abbriviations:
- 2hG
2hr plasma glucose
- BLSA
Baltimore Longitudinal Study of Aging
- CDC
Centers for Disease Control and Prevention
- CVD
Cardiovascular disease
- FPG
Fasting plasma glucose
- GIP
glucose-dependent insulinotropic polypeptide
- GLP-1
glucagon-like peptide-1
- IMGU
Insulin-mediated glucose uptake
- NIA
National Institute on Aging
- NIH
National Institutes of Health
- NIMGU
noninsulin-mediated glucose uptake
- OGTT
oral glucose tolerance test
- RCT
randomized control trial
- SGLT2
sodium/glucose cotransporter 2
- USDA
US Department of Agriculture
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.West LA, Cole S, Goodkind D and He W. 65+ in the United States: 2010. 2014.
- 2.He W, Goodkind D and Kowal P. An Aging World: 2015; International Population Reports. 2016.
- 3.Savji N, Rockman CB, Skolnick AH, Guo Y, Adelman MA, Riles T and Berger JS. Association between advanced age and vascular disease in different arterial territories: a population database of over 3.6 million subjects. Journal of the American College of Cardiology. 2013;61:1736–43. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. Long-term Trends in Diabetes, 2017 Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services; 2017 2017. [Google Scholar]
- 5.Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017 Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services; 2017. [Google Scholar]
- 6.International Diabetes Federation. IDF Diabetes Atlas, 8th edn. Brussels, Belgium: International Diabetes Federation, 2017. http://www.diabetesatlas.org. [Google Scholar]
- 7.Centers for Disease Control and Prevention. Multiple Cause of Death 1999–2015 Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services;. 2016. [Google Scholar]
- 8.Chamberlain JJ, Johnson EL, Leal S, Rhinehart AS, Shubrook JH and Peterson L. Cardiovascular Disease and Risk Management: Review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640–650. [DOI] [PubMed] [Google Scholar]
- 9.Kannel WB and McGee DL. Diabetes and cardiovascular disease. The Framingham study. Jama. 1979;241:2035–8. [DOI] [PubMed] [Google Scholar]
- 10.Stamler J, Vaccaro O, Neaton JD and Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16:434–44. [DOI] [PubMed] [Google Scholar]
- 11.Nielson C, Lange T and Hadjokas N. Blood glucose and coronary artery disease in nondiabetic patients. Diabetes Care. 2006;29:998–1001. [DOI] [PubMed] [Google Scholar]
- 12.Goff DC Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB Sr, R Gibbons, Greenland P, Lackland DT, Levy D, O’Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr, Sorlie P, Stone NJ and Wilson PW. 2013. ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology. 2014;63:2935–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41:S13–s27. [DOI] [PubMed] [Google Scholar]
- 14.World Health Organization. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia. http://www.who.int/diabetes/publications/diagnosis_diabetes2006/en/ 2006.
- 15.World Health Organization. Use of Glycated Haemoglobin (HbA1c) in the Diagnosis of Diabetes Mellitus. http://www.who.int/diabetes/publications/diagnosis_diabetes2011/en/. 2011. [PubMed]
- 16.Consequences of the new diagnostic criteria for diabetes in older men and women. DECODE Study (Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe). Diabetes Care. 1999;22:1667–71. [DOI] [PubMed] [Google Scholar]
- 17.Barrett-Connor E and Ferrara A. Isolated postchallenge hyperglycemia and the risk of fatal cardiovascular disease in older women and men. The Rancho Bernardo Study. Diabetes Care. 1998;21:1236–9. [DOI] [PubMed] [Google Scholar]
- 18.Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, Little RR, Wiedmeyer HM and Byrd-Holt DD. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes Care. 1998;21:518–24. [DOI] [PubMed] [Google Scholar]
- 19.Wahl PW, Savage PJ, Psaty BM, Orchard TJ, Robbins JA and Tracy RP. Diabetes in older adults: comparison of 1997 American Diabetes Association classification of diabetes mellitus with 1985 WHO classification. Lancet. 1998;352:1012–5. [DOI] [PubMed] [Google Scholar]
- 20.Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour diagnostic criteria. Arch Intern Med. 2001;161:397–405. [DOI] [PubMed] [Google Scholar]
- 21.Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. The DECODE study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative analysis Of Diagnostic criteria in Europe. Lancet. 1999;354:617–21. [PubMed] [Google Scholar]
- 22.Brutsaert EF, Shitole S, Biggs ML, Mukamal KJ, deBoer IH, Thacker EL, Barzilay JI, Djousse L, Ix JH, Smith NL, Kaplan RC, Siscovick DS, Psaty BM and Kizer JR. Relations of Postload and Fasting Glucose With Incident Cardiovascular Disease and Mortality Late in Life: The Cardiovascular Health Study. J Gerontol A Biol Sci Med Sci. 2016;71:370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palta P, Huang ES, Kalyani RR, Golden SH and Yeh HC. Hemoglobin A1c and Mortality in Older Adults With and Without Diabetes: Results From the National Health and Nutrition Examination Surveys (1988–2011). Diabetes Care. 2017;40:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavero-Redondo I, Peleteiro B, Alvarez-Bueno C, Rodriguez-Artalejo F and Martinez-Vizcaino V. Glycated haemoglobin A1c as a risk factor of cardiovascular outcomes and all-cause mortality in diabetic and non-diabetic populations: a systematic review and meta-analysis. BMJ open. 2017;7:e015949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saisho Y Glycemic variability and oxidative stress: a link between diabetes and cardiovascular disease? International journal of molecular sciences. 2014;15:18381–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coutinho M, Gerstein HC, Wang Y and Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care. 1999;22:233–40. [DOI] [PubMed] [Google Scholar]
- 27.Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, Ingelsson E, Lawlor DA, Selvin E, Stampfer M, Stehouwer CD, Lewington S, Pennells L, Thompson A, Sattar N, White IR, Ray KK and Danesh J. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee G, Kim SM, Choi S, Kim K, Jeong SM, Son JS, Yun JM and Park SM. The effect of change in fasting glucose on the risk of myocardial infarction, stroke, and all-cause mortality: a nationwide cohort study. Cardiovascular diabetology. 2018;17:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blake DR, Meigs JB, Muller DC, Najjar SS, Andres R and Nathan DM. Impaired glucose tolerance, but not impaired fasting glucose, is associated with increased levels of coronary heart disease risk factors: results from the Baltimore Longitudinal Study on Aging. Diabetes. 2004;53:2095–100. [DOI] [PubMed] [Google Scholar]
- 30.Metter EJ, Windham BG, Maggio M, Simonsick EM, Ling SM, Egan JM and Ferrucci L. Glucose and insulin measurements from the oral glucose tolerance test and mortality prediction. Diabetes Care. 2008;31:1026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barzilay JI, Spiekerman CF, Wahl PW, Kuller LH, Cushman M, Furberg CD, Dobs A, Polak JF and Savage PJ. Cardiovascular disease in older adults with glucose disorders: comparison of American Diabetes Association criteria for diabetes mellitus with WHO criteria. Lancet. 1999;354:622–5. [DOI] [PubMed] [Google Scholar]
- 32.Bhattacharyya OK, Shah BR and Booth GL. Management of cardiovascular disease in patients with diabetes: the 2008 Canadian Diabetes Association guidelines. CMAJ : Canadian Medical Association journal = journal de l’Association medicale canadienne. 2008;179:920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balkau B New diagnostic criteria for diabetes and mortality in older adults. DECODE Study Group. European Diabetes Epidemiology Group. Lancet. 1999;353:68–9. [DOI] [PubMed] [Google Scholar]
- 34.Shaw JE, Hodge AM, de Courten M, Chitson P and Zimmet PZ. Isolated post-challenge hyperglycaemia confirmed as a risk factor for mortality. Diabetologia. 1999;42:1050–4. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki A Assessment of the new criteria for diabetes mellitus according to 10-year relative survival rates. Diabetologia. 1981;20:195–8. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad OS, Morris JA, Mujammami M, Forgetta V, Leong A, Li R, Turgeon M, Greenwood CM, Thanassoulis G, Meigs JB, Sladek R and Richards JB. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nature communications. 2015;6:7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aging Andres R. and diabetes. Med Clin North Am. 1971;55:835–46. [DOI] [PubMed] [Google Scholar]
- 38.Davidson MB. The effect of aging on carbohydrate metabolism: a review of the English literature and a practical approach to the diagnosis of diabetes mellitus in the elderly. Metabolism. 1979;28:688–705. [DOI] [PubMed] [Google Scholar]
- 39.DeFronzo RA. Glucose intolerance and aging. Diabetes Care. 1981;4:493–501. [DOI] [PubMed] [Google Scholar]
- 40.Broughton DL and Taylor R. Review: deterioration of glucose tolerance with age: the role of insulin resistance. Age and ageing. 1991;20:221–5. [DOI] [PubMed] [Google Scholar]
- 41.Reaven GM, Chen N, Hollenbeck C and Chen YD. Effect of age on glucose tolerance and glucose uptake in healthy individuals. Journal of the American Geriatrics Society. 1989;37:735–40. [DOI] [PubMed] [Google Scholar]
- 42.Elahi D and Muller DC. Carbohydrate metabolism in the elderly. Eur J Clin Nutr. 2000;54 Suppl 3:S112–20. [DOI] [PubMed] [Google Scholar]
- 43.Barrett-Connor E Factors associated with the distribution of fasting plasma glucose in an adult community. American journal of epidemiology. 1980;112:518–23. [DOI] [PubMed] [Google Scholar]
- 44.Shimokata H, Muller DC, Fleg JL, Sorkin J, Ziemba AW and Andres R. Age as independent determinant of glucose tolerance. Diabetes. 1991;40:44–51. [DOI] [PubMed] [Google Scholar]
- 45.Muller DC, Elahi D, Tobin JD and Andres R. Insulin response during the oral glucose tolerance test: the role of age, sex, body fat and the pattern of fat distribution. Aging (Milano). 1996;8:13–21. [DOI] [PubMed] [Google Scholar]
- 46.Zavaroni I, Dall’Aglio E, Bruschi F, Bonora E, Alpi O, Pezzarossa A and Butturini U. Effect of age and environmental factors on glucose tolerance and insulin secretion in a worker population. Journal of the American Geriatrics Society. 1986;34:271–5. [DOI] [PubMed] [Google Scholar]
- 47.Maneatis T, Condie R and Reaven G. Effect of age on plasma glucose and insulin responses to a test mixed meal. Journal of the American Geriatrics Society. 1982;30:178–82. [DOI] [PubMed] [Google Scholar]
- 48.Zamboni M, Armellini F, Harris T, Turcato E, Micciolo R, Bergamo-Andreis IA and Bosello O. Effects of age on body fat distribution and cardiovascular risk factors in women. Am J Clin Nutr. 1997;66:111–5. [DOI] [PubMed] [Google Scholar]
- 49.Natali A, Toschi E, Camastra S, Gastaldelli A, Groop L and Ferrannini E. Determinants of postabsorptive endogenous glucose output in non-diabetic subjects. European Group for the Study of Insulin Resistance (EGIR). Diabetologia. 2000;43:1266–72. [DOI] [PubMed] [Google Scholar]
- 50.Fink RI, Kolterman OG, Griffin J and Olefsky JM. Mechanisms of insulin resistance in aging. J Clin Invest. 1983;71:1523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Defronzo RA. Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes. 1979;28:1095–101. [DOI] [PubMed] [Google Scholar]
- 52.Meneilly GS, Minaker KL, Elahi D and Rowe JW. Insulin action in aging man: evidence for tissue-specific differences at low physiologic insulin levels. J Gerontol. 1987;42:196–201. [DOI] [PubMed] [Google Scholar]
- 53.Brehm A and Roden M. Glucose Clamp Techniques In: Roden M, ed. Clinical Diabetes Research: Methods and Techniques West Sussex, England: John Wiley & Sons, Ltd; 2007: 43–76. [Google Scholar]
- 54.DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–87. [DOI] [PubMed] [Google Scholar]
- 55.Rowe JW, Minaker KL, Pallotta JA and Flier JS. Characterization of the insulin resistance of aging. J Clin Invest. 1983;71:1581–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen M, Bergman RN, Pacini G and Porte D Jr, Pathogenesis of age-related glucose intolerance in man: insulin resistance and decreased beta-cell function. J Clin Endocrinol Metab. 1985;60:13–20. [DOI] [PubMed] [Google Scholar]
- 57.Kimmerling G, Javorski WC and Reaven GM. Aging and insulin resistance in a group of nonobese male volunteers. Journal of the American Geriatrics Society. 1977;25:349–53. [DOI] [PubMed] [Google Scholar]
- 58.Kalant N, Leibovici D, Leibovici T and Fukushima N. Effect of age on glucose utilization and responsiveness to insulin in forearm muscle. Journal of the American Geriatrics Society. 1980;28:304–7. [DOI] [PubMed] [Google Scholar]
- 59.Pacini G, Valerio A, Beccaro F, Nosadini R, Cobelli C and Crepaldi G. Insulin sensitivity and beta-cell responsivity are not decreased in elderly subjects with normal OGTT. Journal of the American Geriatrics Society. 1988;36:317–23. [DOI] [PubMed] [Google Scholar]
- 60.Kahn SE, Larson VG, Schwartz RS, Beard JC, Cain KC, Fellingham GW, Stratton JR, Cerqueira MD and Abrass IB. Exercise training delineates the importance of B-cell dysfunction to the glucose intolerance of human aging. J Clin Endocrinol Metab. 1992;74:1336–42. [DOI] [PubMed] [Google Scholar]
- 61.Coon PJ, Rogus EM, Drinkwater D, Muller DC and Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. J Clin Endocrinol Metab. 1992;75:1125–32. [DOI] [PubMed] [Google Scholar]
- 62.Kohrt WM, Kirwan JP, Staten MA, Bourey RE, King DS and Holloszy JO. Insulin resistance in aging is related to abdominal obesity. Diabetes. 1993;42:273–81. [PubMed] [Google Scholar]
- 63.Ferrannini E, Vichi S, Beck-Nielsen H, Laakso M, Paolisso G and Smith U. Insulin action and age. European Group for the Study of Insulin Resistance (EGIR). Diabetes. 1996;45:947–53. [DOI] [PubMed] [Google Scholar]
- 64.Fink RI, Wallace P and Olefsky JM. Effects of aging on glucose-mediated glucose disposal and glucose transport. J Clin Invest. 1986;77:2034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meneilly GS, Elahi D, Minaker KL, Sclater AL and Rowe JW. Impairment of noninsulin-mediated glucose disposal in the elderly. J Clin Endocrinol Metab. 1989;68:566–71. [DOI] [PubMed] [Google Scholar]
- 66.Palmer JP and Ensinck JW. Acute-phase insulin secretion and glucose tolerance in young and aged normal men and diabetic patients. J Clin Endocrinol Metab. 1975;41:498–503. [DOI] [PubMed] [Google Scholar]
- 67.Dudl RJ and Ensinck JW. Insulin and glucagon relationship during aging in man. Metabolism. 1977;26:33–41. [DOI] [PubMed] [Google Scholar]
- 68.Crockford PM, Harbeck RJ and Williams RH. Influence of age on intravenous glucose tolerance and serum immunoreactive insulin. Lancet. 1966;1:465–7. [DOI] [PubMed] [Google Scholar]
- 69.Barbagallo-Sangiorgi G, Laudicina E, Bompiani GD and Durante F. The pancreatic beta-cell response to intravenous administration of glucose in elderly subjects. Journal of the American Geriatrics Society. 1970;18:529–38. [DOI] [PubMed] [Google Scholar]
- 70.Elahi D, Muller DC, McAloon-Dyke M, Tobin JD and Andres R. The effect of age on insulin response and glucose utilization during four hyperglycemic plateaus. Exp Gerontol. 1993;28:393–409. [DOI] [PubMed] [Google Scholar]
- 71.DeFronzo RA, Tobin JD and Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–23. [DOI] [PubMed] [Google Scholar]
- 72.Elahi D In praise of the hyperglycemic clamp. A method for assessment of beta-cell sensitivity and insulin resistance. Diabetes Care. 1996;19:278–86. [DOI] [PubMed] [Google Scholar]
- 73.Ryan AS, Muller DC and Elahi D. Sequential hyperglycemic-euglycemic clamp to assess beta-cell and peripheral tissue: studies in female athletes. Journal of applied physiology (Bethesda, Md : 1985). 2001;91:872–81. [DOI] [PubMed] [Google Scholar]
- 74.Meneilly GS, Veldhuis JD and Elahi D. Disruption of the pulsatile and entropic modes of insulin release during an unvarying glucose stimulus in elderly individuals. J Clin Endocrinol Metab. 1999;84:1938–43. [DOI] [PubMed] [Google Scholar]
- 75.Lang DA, Matthews DR, Peto J and Turner RC. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N Engl J Med. 1979;301:1023–7. [DOI] [PubMed] [Google Scholar]
- 76.Polonsky KS, Given BD and Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81:442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paolisso G, Scheen AJ, Giugliano D, Sgambato S, Albert A, Varricchio M, D’Onofrio F and Lefebvre PJ. Pulsatile insulin delivery has greater metabolic effects than continuous hormone administration in man: importance of pulse frequency. J Clin Endocrinol Metab. 1991;72:607–15. [DOI] [PubMed] [Google Scholar]
- 78.Komjati M, Bratusch-Marrain P and Waldhausl W. Superior efficacy of pulsatile versus continuous hormone exposure on hepatic glucose production in vitro. Endocrinology. 1986;118:312–9. [DOI] [PubMed] [Google Scholar]
- 79.Bratusch-Marrain PR, Komjati M and Waldhausl WK. Efficacy of pulsatile versus continuous insulin administration on hepatic glucose production and glucose utilization in type I diabetic humans. Diabetes. 1986;35:922–6. [DOI] [PubMed] [Google Scholar]
- 80.Sturis J, Scheen AJ, Leproult R, Polonsky KS and van Cauter E. 24-hour glucose profiles during continuous or oscillatory insulin infusion. Demonstration of the functional significance of ultradian insulin oscillations. J Clin Invest. 1995;95:1464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polonsky KS, Given BD, Hirsch LJ, Tillil H, Shapiro ET, Beebe C, Frank BH, Galloway JA and Van Cauter E. Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med. 1988;318:1231–9. [DOI] [PubMed] [Google Scholar]
- 82.Lang DA, Matthews DR, Burnett M and Turner RC. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes. 1981;30:435–9. [DOI] [PubMed] [Google Scholar]
- 83.O’Meara NM, Sturis J, Van Cauter E and Polonsky KS. Lack of control by glucose of ultradian insulin secretory oscillations in impaired glucose tolerance and in non-insulin-dependent diabetes mellitus. J Clin Invest. 1993;92:262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meneilly GS, Ryan AS, Veldhuis JD and Elahi D. Increased disorderliness of basal insulin release, attenuated insulin secretory burst mass, and reduced ultradian rhythmicity of insulin secretion in older individuals. J Clin Endocrinol Metab. 1997;82:4088–93. [DOI] [PubMed] [Google Scholar]
- 85.McIntosh CHS, Widenmaier S and Kim SJ. Chapter 15 Glucose‐Dependent Insulinotropic Polypeptide (Gastric Inhibitory Polypeptide; GIP) Vitamins & Hormones: Academic Press; 2009(Volume 80): 409–471. [DOI] [PubMed] [Google Scholar]
- 86.Cho YM, Kieffer TJ and Gerald L. Chapter Four - K-cells and Glucose-Dependent Insulinotropic Polypeptide in Health and Disease Vitamins & Hormones: Academic Press; (Volume 84): 111–150. [DOI] [PubMed] [Google Scholar]
- 87.Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L and Egan JM. GIP Contributes to Islet Trihormonal Abnormalities in Type 2 Diabetes. The Journal of Clinical Endocrinology & Metabolism. 2014;99:2477–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chia CW, Carlson OD, Liu DD, Gonzalez-Mariscal I, Santa-Cruz Calvo S and Egan JM. Incretin secretion in humans is under the influence of cannabinoid receptors. Am J Physiol Endocrinol Metab. 2017;313:E359–E366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elahi D, Andersen DK, Muller DC, Tobin JD, Brown JC and Andres R. The enteric enhancement of glucose-stimulated insulin release. The role of GIP in aging, obesity, and non-insulin-dependent diabetes mellitus. Diabetes. 1984;33:950–7. [DOI] [PubMed] [Google Scholar]
- 90.McConnell JG, Alam MJ, O’Hare MM, Buchanan KD and Stout RW. The effect of age and sex on the response of enteropancreatic polypeptides to oral glucose. Age and ageing. 1983;12:54–62. [DOI] [PubMed] [Google Scholar]
- 91.Korosi J, McIntosh CH, Pederson RA, Demuth HU, Habener JF, Gingerich R, Egan JM, Elahi D and Meneilly GS. Effect of aging and diabetes on the enteroinsular axis. J Gerontol A Biol Sci Med Sci. 2001;56:M575–9. [DOI] [PubMed] [Google Scholar]
- 92.Meneilly GS, Demuth HU, McIntosh CH and Pederson RA. Effect of ageing and diabetes on glucose-dependent insulinotropic polypeptide and dipeptidyl peptidase IV responses to oral glucose. Diabet Med. 2000;17:346–50. [DOI] [PubMed] [Google Scholar]
- 93.Meneilly GS, Ryan AS, Minaker KL and Elahi D. The effect of age and glycemic level on the response of the beta-cell to glucose-dependent insulinotropic polypeptide and peripheral tissue sensitivity to endogenously released insulin. J Clin Endocrinol Metab. 1998;83:2925–32. [DOI] [PubMed] [Google Scholar]
- 94.Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, Habener JF and Andersen DK. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regul Pept. 1994;51:63–74. [DOI] [PubMed] [Google Scholar]
- 95.Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS, Melvin DL and Egan JM. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes. 2009;58:1342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meneilly GS, McIntosh CH, Pederson RA, Habener JF, Gingerich R, Egan JM, Finegood DT and Elahi D. Effect of glucagon-like peptide 1 on non-insulin-mediated glucose uptake in the elderly patient with diabetes. Diabetes Care. 2001;24:1951–6. [DOI] [PubMed] [Google Scholar]
- 97.Meneilly GS, McIntosh CH, Pederson RA, Habener JF, Gingerich R, Egan JM and Elahi D. Glucagon-like peptide-1 (7–37) augments insulin release in elderly patients with diabetes. Diabetes Care. 2001;24:964–5. [DOI] [PubMed] [Google Scholar]
- 98.Meneilly GS, McIntosh CH, Pederson RA, Habener JF, Gingerich R, Egan JM and Elahi D. Glucagon-like peptide-1 (7–37) augments insulin-mediated glucose uptake in elderly patients with diabetes. J Gerontol A Biol Sci Med Sci. 2001;56:M681–5. [DOI] [PubMed] [Google Scholar]
- 99.Meneilly GS, Greig N, Tildesley H, Habener JF, Egan JM and Elahi D. Effects of 3 months of continuous subcutaneous administration of glucagon-like peptide 1 in elderly patients with type 2 diabetes. Diabetes Care. 2003;26:2835–41. [DOI] [PubMed] [Google Scholar]
- 100.Tabak AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimaki M and Witte DR. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. The Lancet. 2009;373:2215–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ohn JH, Kwak SH, Cho YM, Lim S, Jang HC, Park KS and Cho NH. 10-year trajectory of beta-cell function and insulin sensitivity in the development of type 2 diabetes: a community-based prospective cohort study. The lancet Diabetes & endocrinology. 2016;4:27–34. [DOI] [PubMed] [Google Scholar]
- 102.Prevalence of overweight, obesity and extreme obesity among adults: United States, trends 1976–80 through 2005–2006. National Center for Health Statistics, Health E-Stats. December 2008. [Google Scholar]
- 103.Food Availability (Per Capita) Data System. 1970–2015.
- 104.Lonnqvist F, Nyberg B, Wahrenberg H and Arner P. Catecholamine-induced lipolysis in adipose tissue of the elderly. J Clin Invest. 1990;85:1614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Folsom AR, Kaye SA, Sellers TA, Hong CP, Cerhan JR, Potter JD and Prineas RJ. Body fat distribution and 5-year risk of death in older women. Jama. 1993;269:483–7. [PubMed] [Google Scholar]
- 106.Rexrode KM, Carey VJ, Hennekens CH, Walters EE, Colditz GA, Stampfer MJ, Willett WC and Manson JE. Abdominal adiposity and coronary heart disease in women. Jama. 1998;280:1843–8. [DOI] [PubMed] [Google Scholar]
- 107.Huang B, Rodreiguez BL, Burchfiel CM, Chyou PH, Curb JD and Sharp DS. Associations of adiposity with prevalent coronary heart disease among elderly men: the Honolulu Heart Program. Int J Obes Relat Metab Disord. 1997;21:340–8. [DOI] [PubMed] [Google Scholar]
- 108.Rosen ED and Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stout MB, Tchkonia T and Kirkland JL. The Aging Adipose Organ: Lipid Redistribution, Inflammation, and Cellular Senescence In: Fantuzzi G and Braunschweig C, eds. Adipose Tissue and Adipokines in Health and Disease Totowa, NJ: Humana Press; 2014: 69–80. [Google Scholar]
- 110.Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD and Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell metabolism. 2013;17:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arner P, Pollare T and Lithell H. Different aetiologies of type 2 (non-insulin-dependent) diabetes mellitus in obese and non-obese subjects. Diabetologia. 1991;34:483–7. [DOI] [PubMed] [Google Scholar]
- 112.Shuster A, Patlas M, Pinthus JH and Mourtzakis M. The clinical importance of visceral adiposity: a critical review of methods for visceral adipose tissue analysis. The British journal of radiology. 2012;85:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kahn SE, Prigeon RL, Schwartz RS, Fujimoto WY, Knopp RH, Brunzell JD and Porte D Jr. Obesity, body fat distribution, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. The Journal of nutrition. 2001;131:354s–60s. [DOI] [PubMed] [Google Scholar]
- 114.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD and Kirkland JL. Fat tissue, aging, and cellular senescence. Aging cell. 2010;9:667–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F and Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–7. [DOI] [PubMed] [Google Scholar]
- 116.Tchkonia T, Tchoukalova YD, Giorgadze N, Pirtskhalava T, Karagiannides I, Forse RA, Koo A, Stevenson M, Chinnappan D, Cartwright A, Jensen MD and Kirkland JL. Abundance of two human preadipocyte subtypes with distinct capacities for replication, adipogenesis, and apoptosis varies among fat depots. Am J Physiol Endocrinol Metab. 2005;288:E267–77. [DOI] [PubMed] [Google Scholar]
- 117.Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, Forse RA, Chinnappan D, Martin-Ruiz C, von Zglinicki T and Kirkland JL. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–8. [DOI] [PubMed] [Google Scholar]
- 118.Van Harmelen V, Rohrig K and Hauner H. Comparison of proliferation and differentiation capacity of human adipocyte precursor cells from the omental and subcutaneous adipose tissue depot of obese subjects. Metabolism. 2004;53:632–7. [DOI] [PubMed] [Google Scholar]
- 119.DiGirolamo M, Fine JB, Tagra K and Rossmanith R. Qualitative regional differences in adipose tissue growth and cellularity in male Wistar rats fed ad libitum. Am J Physiol. 1998;274:R1460–7. [DOI] [PubMed] [Google Scholar]
- 120.Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL and Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci U S A. 2010;107:18226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tchkonia T, Corkey BE and Kirkland JL. Current Views of the Fat Cell as an Endocrine Cell: Lipotoxicity In: Bray GA and Ryan DH, eds. Endocrine Updates: Overweight and the Metabolic Syndrome New York: Springer; 2006(26): 105–118. [Google Scholar]
- 122.Stout MB, Justice JN, Nicklas BJ and Kirkland JL . Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology (Bethesda, Md). 2017;32:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tchkonia T, Pirtskhalava T, Thomou T, Cartwright MJ, Wise B, Karagiannides I, Shpilman A, Lash TL, Becherer JD and Kirkland JL. Increased TNFalpha and CCAAT/enhancer-binding protein homologous protein with aging predispose preadipocytes to resist adipogenesis. Am J Physiol Endocrinol Metab. 2007;293:E1810–9. [DOI] [PubMed] [Google Scholar]
- 124.Cartwright MJ, Schlauch K, Lenburg ME, Tchkonia T, Pirtskhalava T, Cartwright A, Thomou T and Kirkland JL. Aging, depot origin, and preadipocyte gene expression. J Gerontol A Biol Sci Med Sci. 2010;65:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu D, Ren Z, Pae M, Guo W, Cui X, Merrill AH and Meydani SN. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J Immunol. 2007;179:4829–39. [DOI] [PubMed] [Google Scholar]
- 126.Aging Campisi J., Senescence Cellular, and Cancer. Annual Review of Physiology. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.He S and Sharpless NE. Senescence in Health and Disease. Cell. 2017;169:1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dandona P, Aljada A and Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends in immunology. 2004;25:4–7. [DOI] [PubMed] [Google Scholar]
- 129.Esser N, Legrand-Poels S, Piette J, Scheen AJ and Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105:141–50. [DOI] [PubMed] [Google Scholar]
- 130.Sone H and Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. [DOI] [PubMed] [Google Scholar]
- 131.Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, Kido Y, Hayashi Y, Nakayama KI, White MF and Kasuga M. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med. 2005;11:175–82. [DOI] [PubMed] [Google Scholar]
- 132.Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, Tornovsky-Babeay S, Dai C, Glaser B, Powers AC, Shapiro AM, Magnuson MA, Dor Y and Ben-Porath I. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016;22:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garg SK, Delaney C, Shi H and Yung R. Changes in adipose tissue macrophages and T cells during aging. Critical reviews in immunology. 2014;34:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, Oatmen K, Martinez-Santibanez G, Julius A, Garg S and Yung RL. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J Immunol. 2011;187:6208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13:465–76. [DOI] [PubMed] [Google Scholar]
- 137.Bluher M, Engeli S, Kloting N, Berndt J, Fasshauer M, Batkai S, Pacher P, Schon MR, Jordan J and Stumvoll M. Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes. 2006;55:3053–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alvheim AR, Torstensen BE, Lin YH, Lillefosse HH, Lock EJ, Madsen L, Froyland L, Hibbeln JR and Malde MK. Dietary linoleic acid elevates the endocannabinoids 2-AG and anandamide and promotes weight gain in mice fed a low fat diet. Lipids. 2014;49:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim W, Doyle ME, Liu Z, Lao Q, Shin YK, Carlson OD, Kim HS, Thomas S, Napora JK, Lee EK, Moaddel R, Wang Y, Maudsley S, Martin B, Kulkarni RN and Egan JM. Cannabinoids Inhibit Insulin Receptor Signaling in Pancreatic {beta}-Cells. Diabetes. 2011;60:1198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gonzalez-Mariscal I, Krzysik-Walker SM, Doyle ME, Liu QR, Cimbro R, Santa-Cruz Calvo S, Ghosh S, Ciesla L, Moaddel R, Carlson OD, Witek RP, O’Connell JF and Egan JM. Human CB1 Receptor Isoforms, present in Hepatocytes and beta-cells, are Involved in Regulating Metabolism. Scientific reports. 2016;6:33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, Yin S, Gao P, Shan X, Pickel J, Bataller R, O’Hare J, Scherer T, Buettner C and Kunos G. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–1228.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cinar R, Godlewski G, Liu J, Tam J, Jourdan T, Mukhopadhyay B, Harvey-White J and Kunos G. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology (Baltimore, Md. 2014;59:143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shin H, Han JH, Yoon J, Sim HJ, Park TJ, Yang S, Lee EK, Kulkarni RN, Egan JM and Kim W. Blockade of cannabinoid 1 receptor improves glucose responsiveness in pancreatic beta cells. Journal of cellular and molecular medicine. 2018;22:2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gonzalez-Mariscal I, Montoro RA, Doyle ME, Liu QR, Rouse M, O’Connell JF, Santa-Cruz Calvo S, Krzysik-Walker SM, Ghosh S, Carlson OD, Lehrmann E, Zhang Y, Becker KG, Chia CW, Ghosh P and Egan JM. Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia. 2018;61:1470–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gonzalez-Mariscal I and Egan JM. Endocannabinoids in Islets of Langerhans: The Ugly, the Bad and the Good Facts. Am J Physiol Endocrinol Metab. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stanley C and O’Sullivan SE. Vascular targets for cannabinoids: animal and human studies. Br J Pharmacol. 2014;171:1361–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tiyerili V, Zimmer S, Jung S, Wassmann K, Naehle CP, Lutjohann D, Zimmer A, Nickenig G and Wassmann S. CB1 receptor inhibition leads to decreased vascular AT1 receptor expression, inhibition of oxidative stress and improved endothelial function. Basic research in cardiology. 2010;105:465–77. [DOI] [PubMed] [Google Scholar]
- 148.Helander HF and Fandriks L. Surface area of the digestive tract - revisited. Scandinavian journal of gastroenterology. 2014;49:681–9. [DOI] [PubMed] [Google Scholar]
- 149.Rera M, Clark RI and Walker DW. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A. 2012;109:21528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A and Wells JM. Intestinal permeability--a new target for disease prevention and therapy. BMC gastroenterology. 2014;14:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pussinen PJ, Havulinna AS, Lehto M, Sundvall J and Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. 2011;34:392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM and Dandona P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. 2009;32:2281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang Y and Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science. 2015;350:1204–7. [DOI] [PubMed] [Google Scholar]
- 155.Supale S, Li N, Brun T and Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends in endocrinology and metabolism: TEM. 2012;23:477–87. [DOI] [PubMed] [Google Scholar]
- 156.Wang CH, Wang CC and Wei YH. Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Ann N Y Acad Sci. 2010;1201:157–65. [DOI] [PubMed] [Google Scholar]
- 157.van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, van de Kamp JJ and Maassen JA. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nature genetics. 1992;1:368–71. [DOI] [PubMed] [Google Scholar]
- 158.Liang P, Hughes V and Fukagawa NK. Increased prevalence of mitochondrial DNA deletions in skeletal muscle of older individuals with impaired glucose tolerance: possible marker of glycemic stress. Diabetes. 1997;46:920–3. [DOI] [PubMed] [Google Scholar]
- 159.Petersen KF, Dufour S, Befroy D, Garcia R and Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW and Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fabbri E, Chia CW, Spencer RG, Fishbein KW, Reiter DA, Cameron D, Zane AC, Moore ZA, Gonzalez-Freire M, Zoli M, Studenski SA, Kalyani RR, Egan JM and Ferrucci L. Insulin Resistance Is Associated With Reduced Mitochondrial Oxidative Capacity Measured by 31P-Magnetic Resonance Spectroscopy in Participants Without Diabetes From the Baltimore Longitudinal Study of Aging. Diabetes. 2017;66:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hirabara SM, Curi R and Maechler P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. Journal of cellular physiology. 2010;222:187–94. [DOI] [PubMed] [Google Scholar]
- 163.Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, Hawkins M, Barzilai N, Rhodes CJ, Fantus IG, Brownlee M and Scherer PE. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem. 2005;280:4617–26. [DOI] [PubMed] [Google Scholar]
- 164.Talior I, Yarkoni M, Bashan N and Eldar-Finkelman H. Increased glucose uptake promotes oxidative stress and PKC-delta activation in adipocytes of obese, insulin-resistant mice. Am J Physiol Endocrinol Metab. 2003;285:E295–302. [DOI] [PubMed] [Google Scholar]
- 165.Wang T, Si Y, Shirihai OS, Si H, Schultz V, Corkey RF, Hu L, Deeney JT, Guo W and Corkey BE. Respiration in adipocytes is inhibited by reactive oxygen species. Obesity (Silver Spring, Md. 2010;18:1493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lesnefsky EJ, Chen Q and Hoppel CL . Mitochondrial Metabolism in Aging Heart . Circulation research. 2016;118:1593–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kates AM, Herrero P, Dence C, Soto P, Srinivasan M, Delano DG, Ehsani A and Gropler RJ. Impact of aging on substrate metabolism by the human heart. Journal of the American College of Cardiology. 2003;41:293–9. [DOI] [PubMed] [Google Scholar]
- 168.Soto PF, Herrero P, Kates AM, Dence CS, Ehsani AA, Davila-Roman V, Schechtman KB and Gropler RJ. Impact of aging on myocardial metabolic response to dobutamine. Am J Physiol Heart Circ Physiol. 2003;285:H2158–64. [DOI] [PubMed] [Google Scholar]
- 169.Rider OJ, Cox P, Tyler D, Clarke K and Neubauer S. Myocardial substrate metabolism in obesity. International journal of obesity (2005). 2013;37:972–9. [DOI] [PubMed] [Google Scholar]
- 170.Konig A, Bode C and Bugger H. Diabetes mellitus and myocardial mitochondrial dysfunction: bench to bedside . Heart failure clinics. 2012;8:551–61. [DOI] [PubMed] [Google Scholar]
- 171.Lalia AZ, Dasari S, Johnson ML, Robinson MM, Konopka AR, Distelmaier K, Port JD, Glavin MT, Esponda RR, Nair KS and Lanza IR. Predictors of Whole-Body Insulin Sensitivity Across Ages and Adiposity in Adult Humans. J Clin Endocrinol Metab. 2016;101:626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF and Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–4. [DOI] [PubMed] [Google Scholar]
- 173.Toledo FG, Menshikova EV, Azuma K, Radikova Z, Kelley CA, Ritov VB and Kelley DE. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes. 2008;57:987–94. [DOI] [PubMed] [Google Scholar]
- 174.Blackburn H and Jacobs DR Jr. The University Group Diabetes Program 1961–1978: pioneering randomized controlled trial. International journal of epidemiology. 2017;46:1354–1364. [DOI] [PubMed] [Google Scholar]
- 175.Klimt CR, Knatterud GL, Meinert CL and Prout TE. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes, I: Design, methods and baseline characteristics. Diabetes. 1970;19:747–783. [PubMed] [Google Scholar]
- 176.Meinert CL, Knatterud GL, Prout TE and Klimt CR. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. II. Mortality results. Diabetes. 1970;19:Suppl: 789–830. [PubMed] [Google Scholar]
- 177.Goldner MG, Knatterud GL and Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes: III. clinical implications of ugdp results. JAMA. 1971;218:1400–1410. [PubMed] [Google Scholar]
- 178.Knatterud GL, Meinert CL, Klimt CR, Osborne RK and Martin DB. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes: IV. a preliminary report on phenformin results. JAMA. 1971;217:777–784. [PubMed] [Google Scholar]
- 179.Knatterud GL, Klimt CR, Goldner MG, Hawkins BS, Weisenfeld S, Kreines K and Haddock L. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes.V. Evaluation of phenformin therapy. Diabetes. 1975;24:65–184.1090475 [Google Scholar]
- 180.Knatterud GL, Meinert CL, Klimt CR, Osborne RK and Martin DB. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VI. Supplementary report on nonfatal events in patients treated with tolbutamide. Diabetes. 1976;25:1129–53.992232 [Google Scholar]
- 181.Knatterud GL, Klimt CR, Levin ME, Jacobson ME and Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978;240:37–42. [PubMed] [Google Scholar]
- 182.VIII UGDP. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VIII. Evaluation of insulin therapy: final report. Diabetes. 1982;31:1–81. [PubMed] [Google Scholar]
- 183.Nissen SE and Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. [DOI] [PubMed] [Google Scholar]
- 184.Liao HW, Saver JL, Wu YL, Chen TH, Lee M and Ovbiagele B. Pioglitazone and cardiovascular outcomes in patients with insulin resistance, pre-diabetes and type 2 diabetes: a systematic review and meta-analysis. BMJ open. 2017;7:e013927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lincoff AM, Wolski K, Nicholls SJ and Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. Jama. 2007;298:1180–8. [DOI] [PubMed] [Google Scholar]
- 186.Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH Jr, Probstfield JL, Simons-Morton DG and Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, Warren SR, Goldman S, McCarren M, Vitek ME, Henderson WG and Huang GD. Glucose Control and Vascular Complications in Veterans with Type 2 Diabetes. New England Journal of Medicine. 2009;360:129–139. [DOI] [PubMed] [Google Scholar]
- 188.Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R and Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72. [DOI] [PubMed] [Google Scholar]
- 189.Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, Pogue J, Probstfield J, Ramachandran A, Riddle MC, Ryden LE and Yusuf S. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367:319–28. [DOI] [PubMed] [Google Scholar]
- 190.Ray KK, Seshasai SR, Wijesuriya S, Sivakumaran R, Nethercott S, Preiss D, Erqou S and Sattar N. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet. 2009;373:1765–72. [DOI] [PubMed] [Google Scholar]
- 191.Holman RR, Paul SK, Bethel MA, Matthews DR and Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89. [DOI] [PubMed] [Google Scholar]
- 192.Cardiovascular and Other Outcomes Postintervention With Insulin Glargine and Omega-3 Fatty Acids (ORIGINALE). Diabetes Care. 2016;39:709–16. [DOI] [PubMed] [Google Scholar]
- 193.Is fasting glucose sufficient to define diabetes? Epidemiological data from 20 European studies. The DECODE-study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative analysis of Diagnostic Criteria in Europe. Diabetologia. 1999;42:647–54. [DOI] [PubMed] [Google Scholar]
- 194.Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care. 2003;26:688–96. [DOI] [PubMed] [Google Scholar]
- 195.Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A and Laakso M. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. Jama. 2003;290:486–94. [DOI] [PubMed] [Google Scholar]
- 196.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM and Buse JB. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. New England Journal of Medicine. 2016;375:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J and Vilsbøll T. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. New England Journal of Medicine. 2016;375:1834–1844. [DOI] [PubMed] [Google Scholar]
- 198.Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Køber LV, Lawson FC, Ping L, Wei X, Lewis EF, Maggioni AP, McMurray JJV, Probstfield JL, Riddle MC, Solomon SD and Tardif J-C. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. New England Journal of Medicine. 2015;373:2247–2257. [DOI] [PubMed] [Google Scholar]
- 199.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC and Inzucchi SE. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. New England Journal of Medicine. 2015;373:2117–2128. [DOI] [PubMed] [Google Scholar]
- 200.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M and Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. New England Journal of Medicine. 2017;377:644–657. [DOI] [PubMed] [Google Scholar]
- 201.Crandall J, Schade D, Ma Y, Fujimoto WY, Barrett-Connor E, Fowler S, Dagogo-Jack S and Andres R. The influence of age on the effects of lifestyle modification and metformin in prevention of diabetes. J Gerontol A Biol Sci Med Sci. 2006;61:1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Andres R, Swerdloff R, Pozefsky T and Coleman D. Manual feedback technique for the control of blood glucose concentration In: Skeggs JLT, ed. Automation in Analytical Chemistry New York: Mediad; 1966: 486–491. [Google Scholar]
- 203.Sherwin RS, Kramer KJ, Tobin JD, Insel PA, Liljenquist JE, Berman M and Andres R. A model of the kinetics of insulin in man. J Clin Invest. 1974;53:1481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Insel PA, Liljenquist JE, Tobin JD, Sherwin RS, Watkins P, Andres R and Berman M. Insulin control of glucose metabolism in man: a new kinetic analysis. J Clin Invest. 1975;55:1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–214. [DOI] [PubMed] [Google Scholar]
- 206.Bergman RN, Finegood DT and Ader M. Assessment of insulin sensitivity in vivo. Endocr Rev. 1985;6:45–86. [DOI] [PubMed] [Google Scholar]