

Abstract
Background
Defects in protein homeostasis are sufficient to provoke cardiac remodeling and dysfunction. Although post-translational modifications by ubiquitin and ubiquitin-like proteins are emerging as an important regulatory mechanism of protein function, the role of Ufm1, a novel ubiquitin-like protein, has not been explored in either the normal or stressed heart.
Methods and Results
Western blotting revealed that Ufm1 specific ligase 1 (Ufl1), an enzyme essential for Ufm1 modification, was increased in hypertrophic mouse hearts but reduced in the failing hearts of patients with dilated cardiomyopathy. To determine the functional role of Ufl1 in the heart, we generated a cardiac-specific knockout mouse and showed that Ufl1-deficient mice developed age-dependent cardiomyopathy and heart failure, as indicated by elevated cardiac fetal gene expression, increased fibrosis and impaired cardiac contractility. When challenged with pressure overload, Ufl1-deficient hearts exhibited remarkably greater hypertrophy, exacerbated fibrosis and worsened cardiac contractility compared with control counterparts. Transcriptome analysis identified that genes associated with the endoplasmic reticulum (ER) function were dysregulated in Ufl1-deficient hearts. Biochemical analysis revealed that excessive ER stress preceded and deteriorated along with the development of cardiomyopathy in Ufl1-deficient hearts. Mechanistically, Ufl1 depletion impaired PERK signaling and aggravated cardiomyocyte cell death following ER stress. Administration of the chemical ER chaperone tauroursodeoxycholic acid to Ufl1-deficient mice alleviated ER stress and attenuated pressure overload-induced cardiac dysfunction.
Conclusions
Our results advance a novel concept that the Ufm1 system is essential for cardiac homeostasis through regulation of ER function and that upregulation of myocardial Ufl1 could be protective against heart failure.
Introduction
Post-translational protein modifications represent an important control mechanism regulating protein function. Besides small chemical groups like acetyl, methyl and phosphate, proteins can also be covalently modified by a family of small proteins1, 2. Members of this family include ubiquitin and a dozen ubiquitin-like proteins such as SUMO (small ubiquitin-related modifier), NEDD8 (neural precursor cell expressed, developmentally downregulated 8), ISG15 (interferon-stimulated gene 15), Ufm1 (ubiquitin-fold modifier 1), FAT10 (human leukocyte antigen-F associated transcript 10), Urm1 (ubiquitin-related modifier 1)1, 2. Conjugation of these ubiquitin-like proteins to target proteins is mediated by a process analogous to ubiquitination and requires specific E1 (activating), E2 (conjugating) and E3 (ligating) enzymes. Unlike ubiquitination, the modification of proteins with ubiquitin-like proteins generally serve as non-proteolysis signals and regulate various cellular processes through altering the substrate’ structure, stability, localization and/or protein-protein interaction. The fundamental importance of these modifications in both normal cell function and disease is gaining increasing recognition3, 4. Recent studies have begun to unveil crucial roles for SUMO5, 6, NEDD87, 8 and ISG159 in regulating cardiac development and function under physiological conditions and upon pathological insults. However, the biological function of the other ubiquitin-like proteins in the heart remains largely unknown.
Ufm1 (ubiquitin-fold modifier 1) is a novel 9.1-kDa ubiquitin-like protein that is highly conserved in multicellular organisms10. Unlike ubiquitin and other ubiquitin-like proteins such as SUMO and NEDD8, Ufm1 possesses a single active glycine at its C-terminus, which is necessary for its attachment to target proteins. Conjugation of Ufm1 to protein targets (ufmylation) requires the homodimeric Uba5 as an E1 Ufm1 activating enzyme, Ufc1 as an E2 Ufm1 conjugating enzyme and Ufl1 (also known as RCAD) as an E3 Ufm1 ligase11. Ufl1 recruits Ufm1-conjugated Ufc1 to target proteins and transfers Ufm1 to the substrate by generation of an isopeptide bond between the C-terminal glycine of Ufm1 and the lysine residue of the substrate10–12. The Ufm1-specific protease 1 and 2 (Ufsp1 and Ufsp2) can cleave the conjugated Ufm1 from the modified targets 13. To date, only a few proteins have been identified as Ufm1 targets11, 14, 15, with Ufbp1 (Ufm1 binding protein 1, also known as DDRGK1 and C20orf116) being one of the most studied. Ufbp1 is an ER-localized protein and is ubiquitously expressed in different tissues including the heart16. Ufl1 interacts with Ufbp1 and mediates the ufmylation of Ufbp1, which appears to prevent the ubiquitination and degradation of Ufbp111, 17.
Recent studies have begun to uncover the biological functions of the Ufm1 system. Ufmylation has been reported to regulate multiple cellular processes including the endoplasmic reticulum (ER) stress response16, 18–20, fatty acid metabolism21, G-protein-coupled receptor (GPCR) maturation22, cell differentiation15–17 and cell cycle control17, 23. Polymorphisms and dysregulation of the Ufm1 system have been linked to a number of human diseases. Polymorphisms of Ufm1 have been found to associate with von Willebrand disease and atrophic gastritis24, 25. Mutations of Ufsp2 were identified in a family with Beukes familiar hip dysplasia26, 27. The expression of Ufl1 is elevated in human lung adenocarcinoma tissues and regulates estrogen receptor signaling in breast cancer through Ufm1 modification of ASC114, 28. Loss-of-function mutations of Uba5 have been linked to early-onset encephalopathy29, 30, which highlight a pivotal role of ufmylation in central nervous system development and function. Furthermore, loss of Uba5, Ufl1 or Ufbp1 in mice have consistently demonstrated an important role of ufmylation in embryonic development and hematopoiesis19, 20, 31. However, whether and how protein ufmylation regulates cardiac development and function has not yet been investigated.
Here we report that Ufl1 expression is downregulated in human failing hearts and ischemic hearts of mouse models, but upregulated in pressure overloaded murine hearts. To understand the role of ufmylation in the heart, we created cardiomyocyte-restricted Ufl1 knockout (Ufl1CKO) mice. Ufl1CKO mice developed cardiomyopathy during ageing and showed a remarkably increased propensity to develop heart failure in response to hemodynamic stress. RNA sequencing analysis and biochemical assays revealed the implication of prevalent ER stress in Ufl1-deficient hearts. Loss-of-function experiments in cultured cardiomyocytes (CMs) indicate that Ufl1 is required for initiation of adaptive ER stress signaling. Lastly, pharmacological inhibition of ER stress attenuated pressure overload-induced heart failure in Ufl1-deficient hearts. Thus, our data demonstrate an essential role of Ufm1 modification in the heart under physiological and pathological conditions and reveals a critical role of Ufl1 in mediating adaptive ER stress response in CMs.
Materials and Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. This would be accomplished on reasonable and direct request with the corresponding author by e-mail.
Animal models
Generation of a transgenic mouse line bearing a Ufl1 “conditional-knockout” allele (Ufl1flox) was described previously19. The mutant mice were bred to αMHCCre mice32 (The Jackson Laboratory, #011038) to generate cardiomyocyte-restricted Ufl1 knockout (Ufl1CKO) mice. These mice were maintained in the C57BL/6J inbred background for our studies. All animal experiments were approved by the Augusta University Institutional Animal Care and Use Committee.
Some of the animals were treated daily with vehicle (0.15M NaHCO3, pH7.4) or vehicle containing tauroursodeoxycholic acid (TUDCA, 250 mg/kg, EMD Chemicals) through gavage feeding for 2 weeks, starting immediately after TAC surgery.
Human samples
Human heart tissue biopsies from patients with end-stage heart failure due to dilated cardiomyopathy and non-failing controls were retrieved from the archive of the Department of Cardiac Pathology and Laboratory of Electrophysiology at Texas Heart Institute. Control samples were obtained from donors without a history of cardiovascular disorders, whose hearts could not be used for transplantation due to medical or technical reasons. The protocol for the use of human heart samples was reviewed and exempted by the Institutional Review Board at St. Luke’s Episcopal Hospital.
Surgical procedures
To generate a mouse model of myocardial ischemia reperfusion, mice were anesthetized using 1% isoflurane, intubated through the mouth, and ventilated at normal rate. The left anterior descending coronary artery was ligated against a PE10 tubing with an 8–10 nylon suture. After 30 minutes of ligation, the ligature was released to allow reperfusion for another 24 hours. Sham-operated animals were subjected to the same procedure without ligation of the suture.
To create a mouse model of pressure overload, mice were subjected to transverse aortic constriction (TAC). Briefly, mice were anesthetized and ventilated as described above. The transverse aorta was exposed and constricted by banding a 27-gauge needle to the aorta with a suture. The needle served as a uniform model for the constriction and was removed immediately after the ligation of the suture.
Echocardiography
Transthoracic echocardiography was performed with a linear 30MHz transducer using a VEVO 2100 echocardiography system (VisualSonics). The LV morphometric and functional parameters were analyzed off-line using VEVO 2100 software.
RNASeq and KEGG analysis
RNAs for four control and three Ufl1CKO mice at 2 months of age were extracted using RNeasy Fibrous Tissue Mini Kit (Qiagen). RNA-Seq libraries were prepared using the Illumina TruSeq RNA Kit v2 with 500 ng of total RNA. Library sequencing was run on an Illumina HiSeq 2500 at the Genome Technology Access Center at Washington University. Approximately 20 million reads per sample were generated. Sequencing results were demultiplexed and converted to FASTQ format using Illumina Bcl2FastQ software, and aligned to the mouse genome (build mm10/GRCm38) with Spliced Transcripts Alignment to a Reference (STAR)33. Counts for each transcript were generated using HTSeq package34, normalized to the length of the individual transcript and to the total mapped read counts in each sample, and expressed in counts per millions. For each gene, we compared the expression levels between CTL and Ufl1CKO RNAs. Gene expression differences were evaluated using Fisher’s exact test after normalizing by the total number of mapped reads in each lane. The resulting p-values were corrected via the Benjamini and Hochberg method. Differentially expressed genes were defined as those with log2 changes of at least 1.3 fold between a pair of samples at FDR of 0.05 for genes with a count above 10. The functional annotation of these genes was analyzed using DAVID35. Differentially expressed genes were used as input gene list, and all mouse genes that were expressed in the heart were used as the background. We looked for enrichment for genetic association with KEGG pathways. Dataset was analyzed using R software version 3 and ad hoc packages.
Statistical analysis.
Results are shown as mean ± SD. Paired data were evaluated using a two-tailed Student’s t-test. For multiple comparisons, one-way analysis of variance (ANOVA) or when appropriate, two-way ANOVA, followed by post hoc test was performed. A p-value <0.05 was considered statistically significant.
Detailed methods on real-time PCR, histology and immunohistochemistry analysis, cell culture and Western blot analysis are described in the supplement.
Results
Protein ufmylation in cardiac development and disease
The role of protein ufmylation in cardiac development and disease has not yet been determined. We first confirmed that Ufm1 is expressed in the adult mouse hearts by quantitative real-time PCR (qRT-PCR) analysis (data not shown), and levels were found to be comparable to SUMO 1/2/3, a relatively well-studied ubiquitin-like protein in the heart. During mouse development (day 1 to 1 year old), the abundance of Ufm1-conjugated (ufmylated) proteins were gradually increased, however, the expression of Ufm1 E3 ligase Ufl1 was reduced in ageing hearts (Fig. 1A), indicating that a low level of Ufl1 protein is sufficient to mediate protein ufmylation in adult CMs. We next created mouse models of cardiac hypertrophy by transverse aortic constriction (TAC). Western blot analysis showed that two weeks of pressure overload resulted in a concomitant increase of ufmylated proteins (~1.4 fold) and Ufl1 proteins (~1.6 fold) in mouse hearts (Fig. 1B), suggesting that hemodynamic stress may activate protein ufmylation in the heart in a Ufl1-dependent manner. In mouse hearts subjected to ischemia reperfusion (I/R) surgery, Ufl1 expression was significantly reduced in the ischemic and border zones of the infarct area (Fig. 1C). The reduction of Ufl1 expression in ischemic myocardium was further confirmed by immunostaining and was evident in necrotic CMs as revealed by an Evans Blue Dye (EBD) infiltration assay (Fig. 1D), implicating the potential role of Ufl1 in the regulation of cardiomyocyte survival. Moreover, Western blot analysis of human left ventricular samples from 4 patients with dilated cardiomyopathy and 4 control donors revealed the decreased expression of Ufl1 (Fig. 1E). Thus, protein ufmylation may have an important role in cardiac development and disease.
Figure 1. Expression of Ufm1 and Ufl1 in developing and diseased hearts.
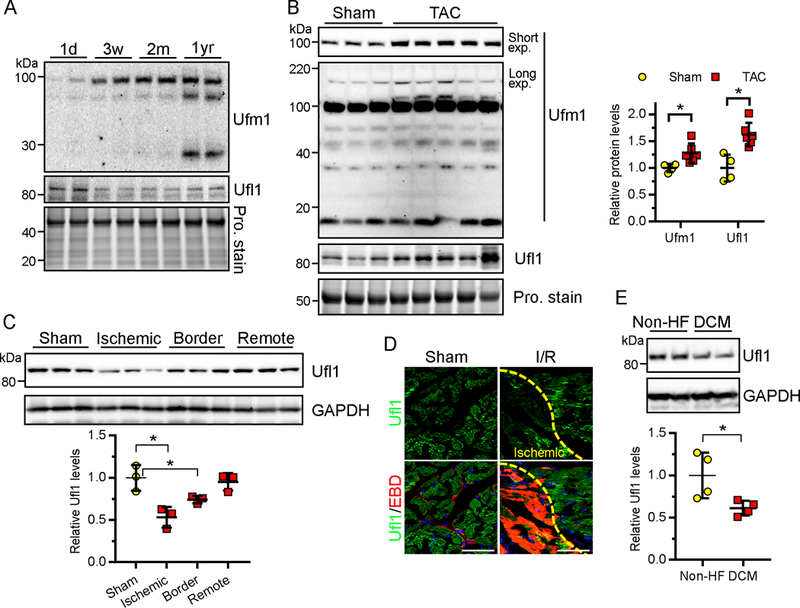
A, Western blots of Ufm1 and Ufl1 in the hearts of C57bl/6 mice at 1 day (1d), 3 weeks (3w), 2 months (2m) and 1 year (1yr) of age. Total protein (Pro.) stain serves as loading control. B, Representative Western blot (left) and densitometric quantification (right) of ufmylated and Ufl1 proteins in mouse hearts at 2 weeks post sham (n=4)- or transverse aortic constriction (TAC, n=6)-operation. C, Western blots (top) and quantification (bottom) of Ufl1 in mouse hearts subject to sham or ischemia-reperfusion (I/R) surgery (30min ischemia followed by 24hr reperfusion. n=3 per group). D, Immunostaining of Ufl1 (green) in I/R mouse hearts. Evans blue dye (EBD, red) was injected into the mice 24 hrs prior to the surgery to identify ischemic CMs that lost membrane integrity following I/R. Bar, 50 μm. E, Representative Western blots of Ufl1 in the hearts of patients with dilated cardiomyopathy (DCM, n=4) or without cardiovascular disorders (non-HF, n=4). * P<0.05.
Creation of cardiomyocyte-restricted Ufl1 knockout (Ufl1CKO) mice
We have prevously generated mice carrying Ufl1flox alleles, wherein exon 7 was flanked by loxP sites19. We then crossed these Ufl1flox mice with α-myosin heavy chain promoter-driven Cre transgenic (αMHCCre) mice32 to achieve cardiomyocyte-restricted knockout of Ufl1 (Fig. 2A). In comparison to littermate control (CTL, Ufl1flox/flox or Ufl1flox/+) mice, we determined that the transcript and protein levels of Ufl1 were dose-dependently reduced in heterozygous and homozygous Ufl1CKO hearts (Fig. 2B–2C), but not in other tissues (Supplemental Fig.1). Western blot analysis revealed that Ufl1 deficiency significantly reduced the abundance of a subset of ufmylated proteins (~80 kDa) in mouse hearts (Fig. 2D). These data collectively indicate that Ufl1 in part regulates ufmylation in CMs.
Figure 2. Generation of Ufl1CKO mice.
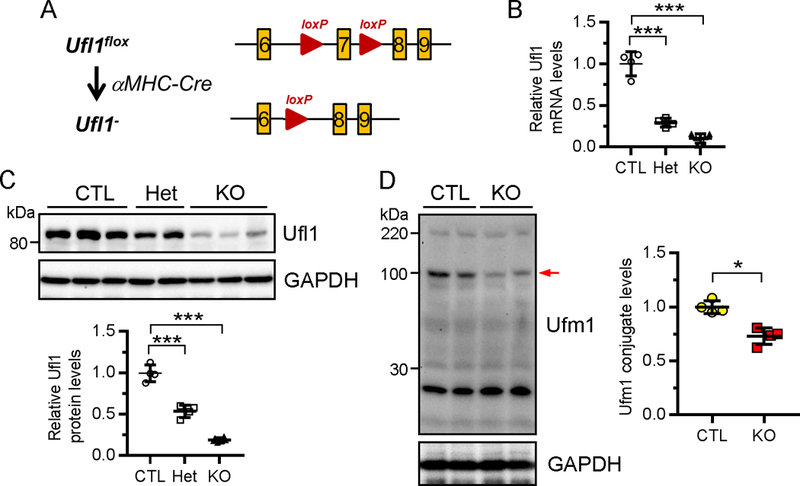
A , Targeting strategy of genomic region of Ufl1 gene. Cardiac-specific knockout of Ufl1 was achieved by intercrossing of Ufl1flox mice with αMHC-Cre transgenic (αMHCCre) mice. B-C, Detection of Ufl1 mRNA (B) and protein levels (C) in the hearts of 2-month-old CTL (Ufl1flox/+ or Ufl1f/f, n=4), heterozygous (αMHCCre:Ufl1f/+, Het, n=4) and Ufl1CKO (αMHCCre:Ufl1f/f, KO, n=6) mice by qRT-PCR and Western blot, respectively. The quantification of (C) is shown under the blots. D, Representative Western blot (top) and the quantification (bottom) of ufmylated proteins in CTL (n=4) and KO (n=4) hearts. Arrow, a significantly reduced ufmylated band. * P<0.05, *** P<0.001 versus CTL.
Ufl1 is required for physiological cardiac growth and function
To determine if Ufl1 regulates physiological growth of the heart, we characterized the cardiac phenotypes of Ufl1CKO mice over time. Ufl1CKO mice were born at the expected Mendelian ratio and Ufl1CKO hearts were morphologically indistinguishable from CTL hearts at birth. At 2 months of age, Ufl1CKO hearts began to show signs of pathological cardiac remodeling, as evidenced by slightly but significantly reduced heart weight to tibial length (TL) ratios and the upregulation of Bnp, Myh7 and α skeletal actin, fetal genes that are often associated with pathological cardiac remodeling36 (Fig. 3A–3B). From 2 to 6 months after birth, temporal echocardiography analysis demonstrated a progressive deterioration of cardiac function in Ufl1CKO mice (Fig. 3C–3D, Supplemental Table 1). At 2 months of age, Ufl1CKO hearts were functionally comparable to littermate controls. However, at 4 months of age, Ufl1CKO mice displayed significant cardiac chamber dilation and deterioration in cardiac contractility, indicated by a decrease in interventricular septum thickness, an increase in left ventricular internal diameters and a reduction in left ventricle (LV) ejection fraction (EF) and fractional shortening (FS) (Supplemental Table 1). By 6 months of age, LV EF (50.6%±5.9% vs 71.9%±7.2% in controls) and FS (25.3%±3.7% vs 40.3%±3.5% in controls) were profoundly diminished, along with LV wall thinning and chamber dilatation (Fig. 3C–3D, Supplemental Table 1). This was accompanied by decreased heart weight and increased cardiac fetal gene expression in Ufl1CKO mice (Fig. 3A–3B). Histopathology confirmed decreased heart size, LV wall thinning and LV chamber dilatation (Fig. 3E–3F). Masson’s trichrome staining (Fig. 3G–3H) revealed that there was accompanying interstitial fibrosis in the free wall of Ufl1CKO left ventricles. These data indicate that cardiomyocyte-specific knockout of Ufl1 induced severe cardiomyopathy, suggesting an critical role of ufmylation in regulating physiological cardiac growth and maintaining cardiac function.
Figure 3. Ufl1CKO causes dilated cardiomyopathy and heart failure.
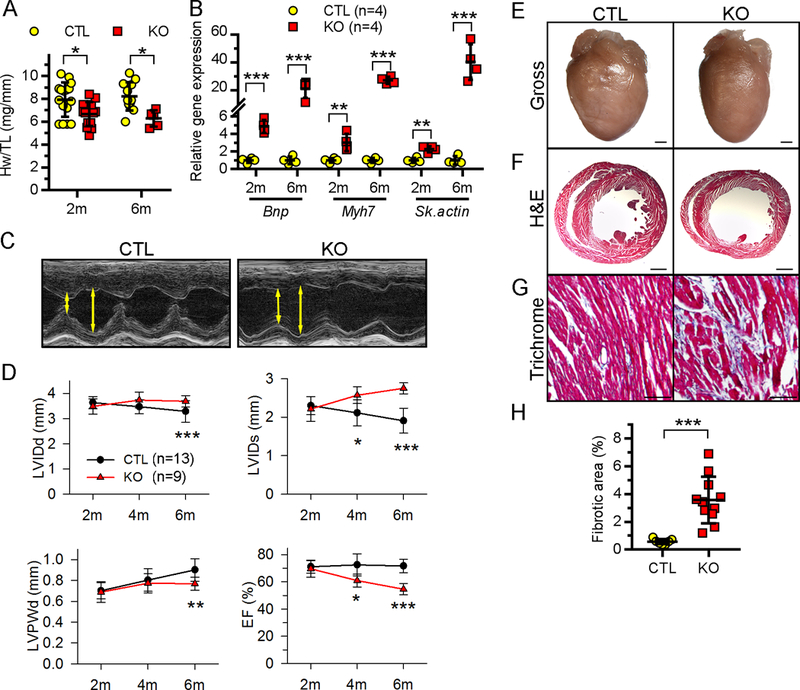
A, Heart weight (Hw) to tibial length (TL) ratio. B, qRT-PCR detection of the indicated fetal genes. C, Representative M-mode echocardiographs of 6-month-old CTL and KO mice. Left ventricle (LV) chamber dimensions at systole and diastole (double head arrows) are marked, respectively. D, Temporal echocardiography analysis of CTL and KO mice at the indicated ages. LV systolic or diastolic internal diameter (LVIDs or LVIDd), LV diastolic posterior wall thickness (LVPWd), and LV ejection fractions (EF) are shown. E-G, Gross morphology (E) of the hearts from 6-month-old CTL and Ufl1CKO (KO) mice and hematoxylin and eosin (H&E) (F) and Masson’s trichrome (G) staining of the myocardium sections. Bars in (E-G) represent 1 mm, 1 mm and 100 μm, respectively. H, Quantification of myocardial fibrotic area in 6-month-old mice. *P<0.05, **P<0.01, ***P<0.001 vs CTL at the respective age.
Ufl1 protects against pressure overload-induced heart failure
Ufl1 expression is upregulated in hypertrophic mouse hearts (Fig. 1B). We hypothesized that myocardial upregulation of Ufl1 is cardioprotective against pathological cardiac remodeling induced by pressure overload. To assess this, transverse aortic constriction (TAC) was applied to CTL and Ufl1CKO mice at two months of age, a time when their cardiac function is comparable (Fig. 3D, Supplemental Table 1). As expected, pressure overload provoked compensatory hypertrophy in CTL mice at two weeks post-TAC, as indicated by increased LV wall thickness, the absence of LV chamber dilation and preserved LV contractility (Fig. 4A–4B, Supplemental Table 2), along with increased heart weight as compared to sham-operated mice (Fig. 4F). However, despite the equivalent pressure overload as revealed by measurement of the pressure gradient across the constricted aortas (Fig. 3B), Ufl1CKO mice subjected to TAC bypassed the compensatory hypertrophy stage and quickly developed heart failure. This is demonstrated by absence of an increase in LV wall thickness, drastic LV chamber dilatation, and significantly impaired contractility when compared with CTL+TAC mice (EF: 46.5%±17.2% vs 66.1%±11.3% of CTL+TAC; FS: 21.1%±8.9% vs 36.3%±7.9% of CTL+TAC) (Fig. 4A–4B, Supplemental Table 2). Histopathology further confirmed LV chamber dilatation of Ufl1CKO hearts post TAC (Fig. 4C). Gravimetric analysis showed that Ufl1CKO mice had a greater increase in heart weight to body weight ratio and a significant increase in lung weight to TL ratio after TAC (Fig. 4E), indicative of LV failure. Moreover, TAC-induced myocardial interstitial fibrosis and reactivation of fetal genes (Myh7) became more pronounced in the Ufl1CKO hearts (Fig. 3D–3E, 3G–3H). Thus, the rapid-onset dilated cardiomyopathy and heart failure in pressure overloaded-Ufl1CKO mice demonstrates the importance of Ufl1 in regulating adaptive cardiac remodeling in response to hemodynamic stress.
Figure 4. Ufl1CKO hearts were more susceptible to TAC-induced pathological cardiac remodeling and heart failure.
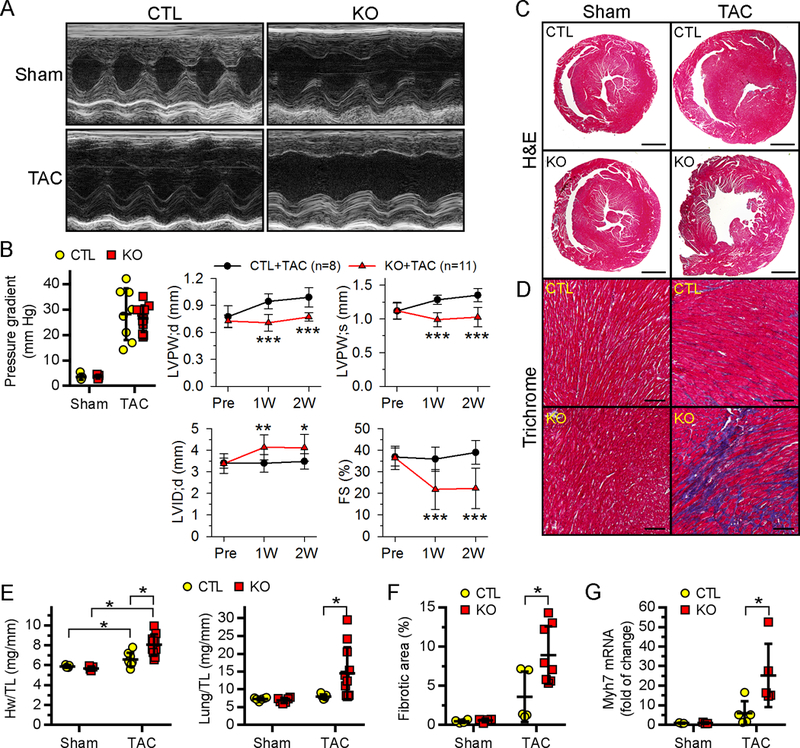
Two-month-old CTL and Ufl1CKO mice were subjected to either sham (n=5 for CTL and 5 for KO) or TAC (n=8 for CTL and 11 for KO) operation for 2 weeks. A-B, Temporal echocardiography analysis of LV functions. Representative M-mode images at 2 weeks post operation (A), the pressure gradients across constricted aorta and the temporal changes of LV morphometry and function (B) are shown. *P<0.05, **P<0.01, ***P<0.001 vs CTL+TAC at indicated times. C-E, Representative images of H&E (C) and Masson’s trichrome (D) stained myocardium sections. Bars in C-D are 1 mm, and 100 μm, respectively. E, Ratios of heart weight (Hw) to tibial length (TL) and lung weight to TL. F-G, The quantification of fibrotic area (F) and Mhy7 mRNA expression (G). *P<0.05.
Differential transcriptome of Ufl1CKO hearts
To identify the cellular pathways that are specifically regulated by Ufl1, we performed RNA sequencing to measure global gene expression in CTL and Ufl1CKO hearts at 2 months of age, when their function is comparable. A total of 41,388 annotated genes from the Ensembl reference database were tested for differential expression. Stringent threshold criteria (count>10, false discovery rate, log2FC±0.3, FDR<0.05) resulted in 967 genes differentially expressed between CTL and Ufl1CKO hearts; 319 genes were downregulated and 648 genes were upregulated in the Ufl1CKO hearts when compared with controls (Fig. 5A, Supplemental Table 1). A heatmap revealed a high level of homogeneity and excellent consistency of the changes within groups (Fig. 5B).
Figure 5. Global transcriptome analysis of Ufl1CKO hearts.
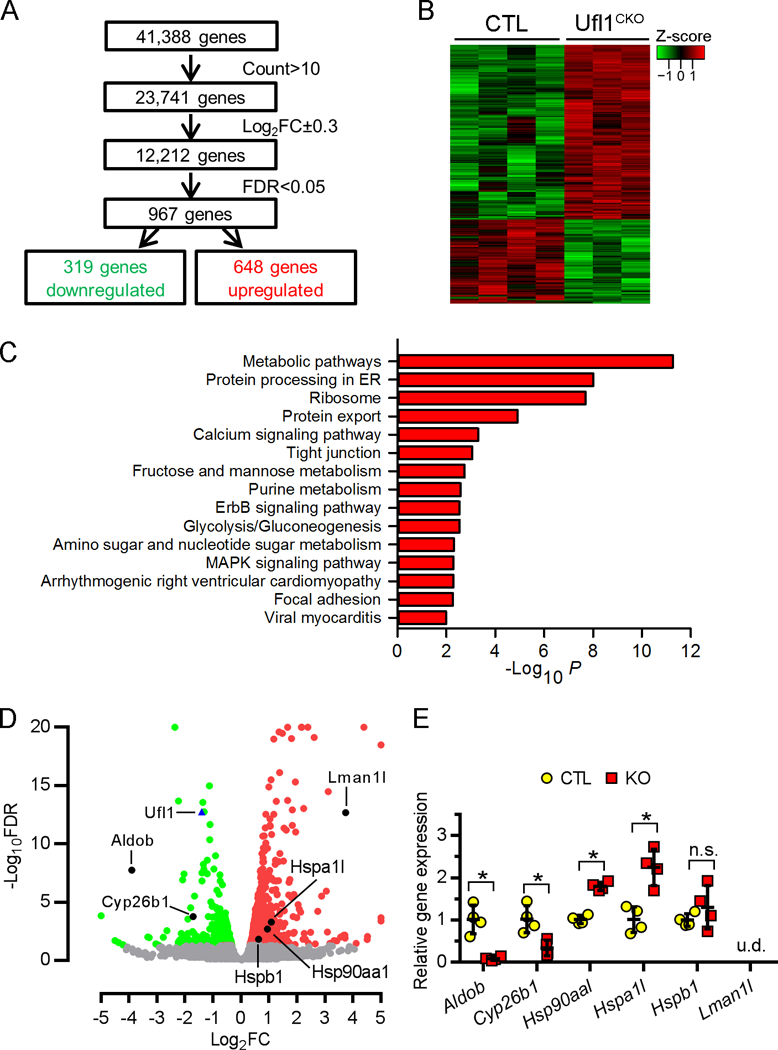
RNAs isolated from 2-month CTL (n=4) and Ufl1CKO (n=3) hearts were used for RNA sequencing analysis. A, Illustration of RNA sequencing analysis. B, Gene clustering by Z-score showing consistency within groups. C, KEGG (Kyoto Encyclopedia Genes and Genomes) analysis shows significantly enriched pathways in Ufl1CKO hearts. Gene set related to ER homeostasis is highlighted. D, Volcano plot of downregulated (green) and upregulated (red) transcripts in Ufl1CKO hearts. ER-related, differentially expressed transcripts, as well as Ufl1 transcript, are marked by dark dots and blue triangle, respectively. Transcripts that were not differentially expressed are in gray. E, qPCR confirmation of mRNA expression of ER-related, differentially expressed genes (n=4). *P <0.05 vs CTL. n.s., not significant. u.d., not detected.
A Kyoto Encyclopedia of Genes and Genomes (KEGG) functional network analysis of the 970 dysregulated genes indicated that the highest significance was in the “Metabolic pathway” and the second highest significance was in “Protein processing in ER” (Fig. 5C). Pathways related to ribosome, protein export, calcium signaling pathway and tight junction, among others, were also prominent, emphasizing the potential roles of Ufl1 in the regulation of these processes. Interestingly, “Arrhythmogenic right ventricular cardiomyopathy” and “Viral myocarditis” also scored with high significance, consistent with the aforementioned critical role of Ufl1 in the maintenance of cardiac function. Since protein ufmylation has been closely linked to ER homeostasis19, 20, 37, we focused on the “Protein processing in ER” pathways. Several ER-localized metabolic enzymes, such as Aldob (coding for adolase B) and Cyp26b1 (coding for cytochrome P450, family 26, subfamily b), were significantly downregulated in Ufl1CKO hearts, whereas ER-resident protein transporter Lman1l and heat shock proteins (Hspa1l, Hsp90aa1 and Hspb1) were significantly upregulated. The positions of these genes in the volcano plot are depicted in Fig. 5D. QPCR with gene-specific primers further confirmed the dysregulation of these genes in 2-month-old Ufl1CKO hearts (Fig. 5E). Of note, Aldob belongs to a family of glycolytic enzymes that associate with cardiac ATP-sensitive K+ channel38, while Cyp26b1 negatively regulates Ca2+ in ER by degrading retinoic acid (RA)39, which mediates signaling essential for heart development40 and adaptive cardiac remodeling41. As normal ER function is essential for Ca2+ cycling and the quality control of secretory proteins, dysregulation of ER-associated regulators of intracellular ion exchange and heat shock proteins in Ufl1CKO hearts implicates a possible role of Ufl1 in the regulation of ER homeostasis.
Loss of Ufl1 induces ER stress in the heart
Perturbation of ER-associated functions can result in the accumulation of unfolded or misfolded proteins in the ER lumen, a condition termed as ER stress. ER stress is implicated in and contributes to the pathogenesis of various forms of cardiac disease42, 43. Accordingly, we next determined whether the loss of Ufl1 induces ER stress in the heart. BiP (immunoglobulin binding protein, also known as Grp78) and PDI (protein disulfide isomerase) are two ER stress-responsive chaperone proteins that are critical for the activation of the unfolded protein response (UPR) and protein folding, respectively44, 45. Western blot analysis of myocardial lysates from 2-month-old CTL and Ufl1CKO mice showed a significant increase in PDI, as well as the non-specific chaperones Hsp40 and Hsp90, in Ufl1CKO hearts (Fig. 6A). The upregulation of these proteins, along with the levels of Bip and Grp94 proteins, was further augmented in Ufl1CKO hearts with established cardiomyopathy (at 6 months of age) (Fig. 6A–6B). Moreover, CHOP, a key mediator of ER stress-induced cell death,46, 47 was also drastically upregulated in Ufl1CKO hearts at 6 months of age. We also observed an upregulation of PDI and BiP in Ufl1CKO+TAC hearts compared with CTL+TAC hearts and a greater upregulation of PDI in Ufl1CKO+TAC hearts compared with their sham counterparts (Fig. 6C–6D). Moreover, ultrastructural analysis of the ER using electron microscopy revealed massive proliferative and dilated ERs, both hallmarks of ER stress, in Ufl1-deficient CMs (Fig. 6E). In agreement with the notion that persistent ER stress results in cell death, there was a significant increase in TUNEL+ CMs in Ufl1CKO hearts subjected to 2 weeks of TAC (Fig. 6F–6G). Our results suggest that excessive ER stress precedes the onset of cardiomyopathy and then deteriorates along with disease progression in Ufl1CKO hearts, providing evidence in support of a potential causative role.
Figure 6. Elevated ER stress in the Ufl1CKO hearts.
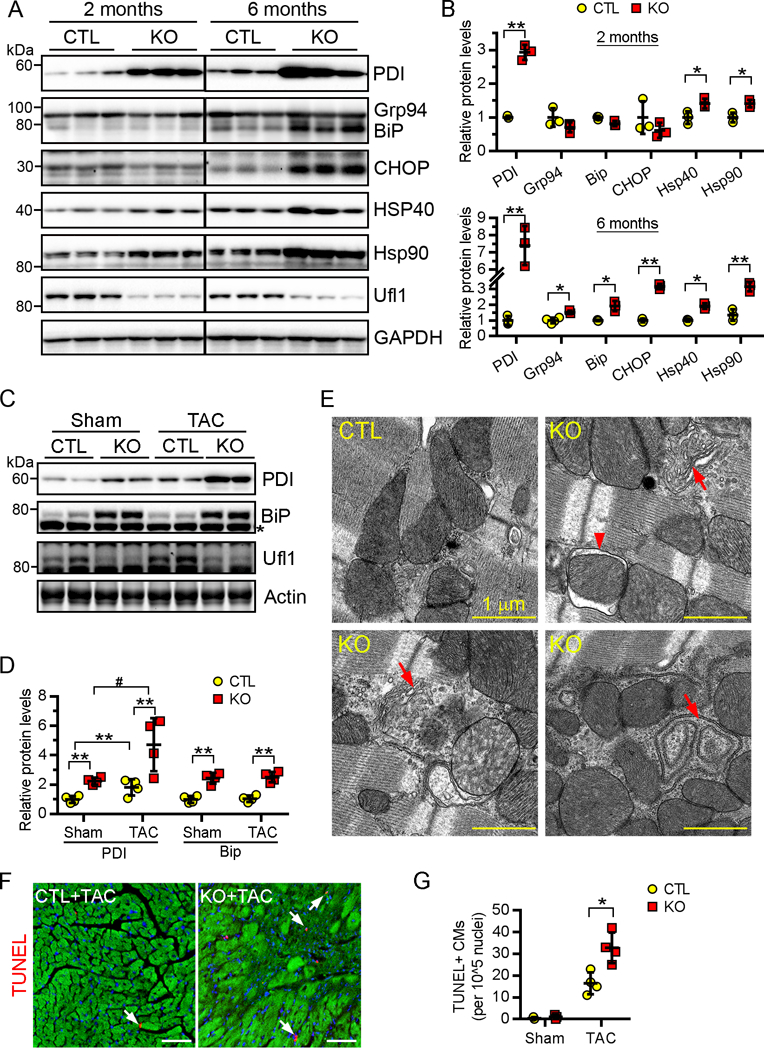
A-D, Western blots (A, C) and the quantification (B, D) of the indicated ER stress-responsive proteins in heart extracts from 2- and 6-month-old mice (A, B. n=3) or 2-month-old mice subjected to sham or TAC for 2 weeks (C, D. n=4). In (A), an anti-KDEL antibody (Santa Cruz, #58774) was used to simultaneously detect Grp94 and Bip. In (C), an anti-Bip antibody (Cell Signal, #3183S) was used to specifically detect Bip.*, a non-specific band. E, Electron micrographs of ventricular sections from 4-month-old mice. Abnormal ER cisternae (arrows) are shown. Three mice per group were analyzed. F-G, Representative images (F) and the quantification (G) of TUNEL+ (red, arrows) cardiomyocytes in myocardium sections (n=3 for sham group, n=4 for TAC group). Cardiomyocytes and nuclei were counterstained by phalloidin (green) and DAPI, respectively. Bar, 50 μm.
Ufl1 is necessary for PERK activation and protects against ER stress-induced cell death in CMs
ER stress triggers a complex signaling network known as the unfolded protein response (UPR) with the goal of maintaining ER homeostasis. The UPR is mediated by three ER membrane-located transducers, PERK, IRE1α and ATF6, and collectively functions to reduce protein synthesis, enhance refolding, increase proteolysis of misfolded proteins, and selectively upregulate the expression of protective proteins42, 43. To understand the role of Ufl1 in regulating UPR, we silenced Ufl1 in neonatal rat ventricular CMs (NRVCs) and treated the cells with the ER stress-inducers thapsigargin (TG) or tunicamycin (TM). Ufl1 silencing effectively diminished TM-induced increase in phosphorylation of PERK and eIF2α, as well as the increase in the PERK downstream target ATF4 (Fig. 7A–7B), indicating defects in activating PERK signaling. In contrast, silencing of Ufl1 did not affect the expression of BiP, calreticulin, PDI or ERp44, which are downstream of ATF6 signaling42. Moreover, silencing of Ufl1 enhanced TG- and TM-induced CM apoptosis, and this is evidenced by the increased cleavage of caspase 12, a major mediator of ER stress-specific apoptosis48, as well as by the increased cleavage of the general apoptosis executor caspase 3 (Fig. 7C). Consistently, silencing of Ufl1 reduced the viability of NRVCs upon TG- and TM-treatment (Fig. 7D). Thus, our results indicate that the loss of Ufl1 inhibits PERK signaling and renders CMs more susceptible to ER stress-induced cell death.
Figure 7. Ufl1 modulates unfolded protein response and protects against ER stress-induced apoptosis.
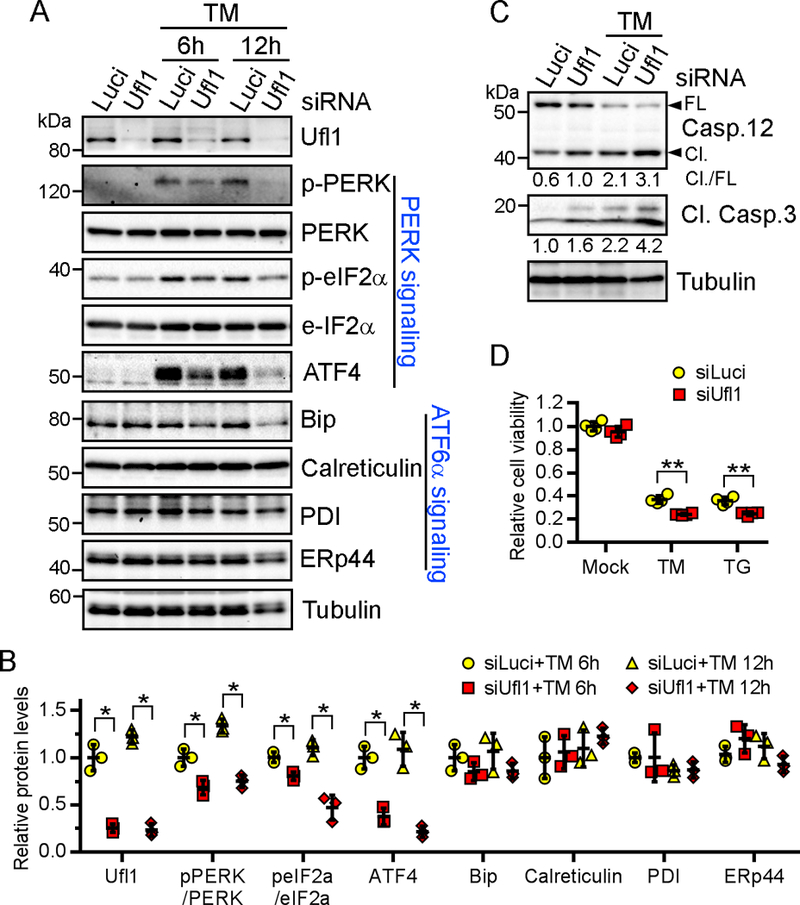
Neonatal cardiomyocytes were transfected with siRNAs against luciferase or Ufl1 for 48 hr and then treated with indicated ER stress inducers for another 6 or 12 (A-B) and 48 hr (C-D). TM, tunicamycin (1 μg/mL). TG, thapsigargin (1 μM). Shown are representative results of three repeats. A-B, Western blots of indicated UPR transducers and downstream targets (A) and the quantification (B). N=3 per group for each repeat. C, Western blots of indicated apoptosis markers. Densitometric quantifications are denoted under the blots. N=2 per group for each repeat. D, Cell viability measured by CellTiter-Glo assay. N=4 per group for each repeat. *P<0.05, **P<0.01 vs siLuci at respective time points.
TUDCA administration ameliorates TAC-induced cardiac phenotype of Ufl1CKO mice
TUDCA (tauroursodeoxycholic acid) is a classic chemical chaperone that has been shown to alleviate ER stress in vitro and in vivo by enhancing ER folding capacity49. To determine whether excessive ER stress is the driving force that sensitizes Ufl1-deficient hearts to hemodynamic stress, we subjected 2-month-old CTL and Ufl1CKO mice to TAC surgery and administered TUDCA (250 mg/kg/day) through daily intraperitoneal injections for 2 weeks. As shown in Fig. 8A, TUDCA markedly attenuated TAC-induced enlargement of Ufl1CKO hearts. This is confirmed by a decreased heart weight to tibial length ratio in Ufl1CKO+TAC hearts after TUDCA treatment (Fig. 8B). Moreover, there was also a significant decrease in the lung weight to tibial length ratio in Ufl1CKO+TAC+TUDCA hearts when compared with Ufl1CKO+TAC hearts (Fig. 8C). We also performed echocardiography to determine heart function. While TUDCA administration did not have any impact on the function of CTL+TAC hearts due to preserved heart function at this stage (Fig. 8D, Supplemental Table 3), it significantly suppressed LV dilatation and improved LV contractility in Ufl1CKO+TAC hearts. This is indicated by a reduction in LV internal chamber diameter at both systolic and diastolic phase as well as an increase in LV EF in Ufl1CKO+TAC+TUDCA hearts compared with their vehicle-treated counterparts (Fig. 8D, Supplemental Table 3). TUDCA treatment also significantly reduced TAC-induced CM apoptosis in the Ufl1CKO hearts (Fig. 8E). Lastly, Western blot analysis demonstrated that TUDCA blunted the upregulation of PDI in Ufl1CKO hearts, validating the effectiveness of TUDCA on alleviation of ER stress (Fig. 8F–8G). Together, our data suggest that alleviation of ER stress mitigates TAC-induced cardiac dysfunction in Ufl1CKO hearts.
Figure 8. Inhibition of ER stress attenuates cardiac dysfunction in Ufl1CKO hearts after pressure overload.
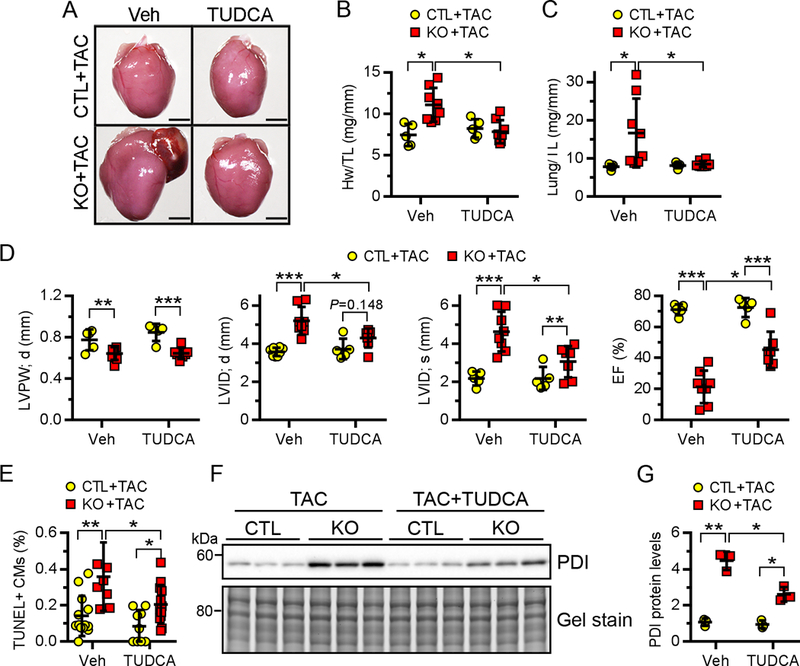
Mice at 2 months of age were subjected to TAC surgery and immediately received TUDCA (250 mg/kg/day, n=5 for CTL and 7 for KO) or vehicle (n=5 for CTL and 8 for KO) through daily intraperitoneal injections for 2 weeks. A-C, Gross morphology (A), heart weight to body weight (Hw/Bw) ratio (B) and lung weight to body weight (Lung/Bw) ratio (C) of indicated mice. D, Echocardiography analysis of cardiac function. E, Quantification of TUNEL+ cardiomyocytes. n=3 mice per group. F-G, Western blot analysis of PDI in heart extracts (F) and the quantification (G). n=3 mice per group. *P<0.05, **P<0.01, ***P<0.001.
Discussion
In summary, our findings demonstrate for the first time that Ufl1, an E3 ligase for a novel ubiquitin-like protein Ufm1, is required for maintenance of normal cardiac structure and function and protects against pressure overload-induced pathological cardiac remodeling. Our results support a role for Ufl1 in regulating ER homeostasis via sustaining PERK signaling in the heart (Fig. 9). These findings establish the importance of protein ufmylation in the heart and warrant further experiments to clarify the molecular/cellular actions of Ufl1-mediated ufmylation in the heart and to assess the potential of upregulating myocardial Ufl1 levels as a therapeutic option in heart failure.
Figure 9. A model illustrating the role of ufmylation in the heart.
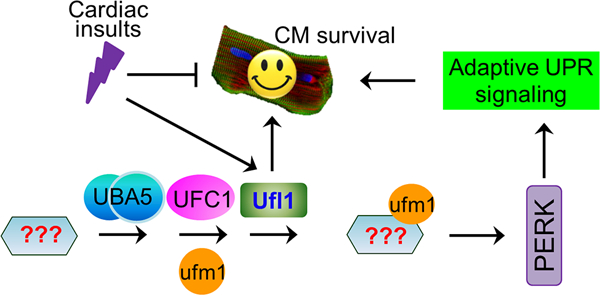
Protein ufmylation is mediated by the Ufm1-specific enzymes UBA5, UFC1 and Ufl1, and is dysregulated under pathological conditions. Myocardial Ufl1 is indispensable for the maintenance of normal heart function and protect against pressure overload-induced cardiac remodeling and heart failure, which is in part through ensuring ER homeostasis by sustaining adaptive PERK signaling.
In this current study, we have identified Ufl1 as a novel regulator of cardiac homeostasis. We present evidence that implicates dysregulated ufmylation in hypertrophic, ischemic and dilated cardiomyopathy in both animal models and human patients (Fig. 1). We further show that mice lacking Ufl1 develop age-dependent dilated cardiomyopathy and are more susceptible to pressure overload-induced heart failure (Fig. 3 and Fig. 4). Interestingly, the loss of Ufl1 in CMs leads to a reduced heart size during development (Fig. 3A) but augmented cardiac hypertrophy in response to pressure overload (Fig. 4E), suggesting that Ufl1 has a dichotomous role in regulating physiological and pathological cardiac growth. The E3 Ufm1 ligase Ufl1 has no obvious sequence similarity to any known ubiquitin E3 ligases and has been shown to promote the ufmylation of Ufbp1 (an ER-resident protein)11, ASC1 (a transcriptional coactivator of estrogen receptor α)14 and ribosomal proteins50. In agreement with its role as the E3 Ufm1 ligase, we observed a reduction of ufmylated proteins in Ufl1-deficient hearts. The lack of a global change in protein ufmylation in Ufl1CKO hearts could be due to the presence of other unknown Ufm1 E3 ligases, compensatory activation of protein de-ufmylation, and/or enduring ufmylation in non-cardiomyocytes in the hearts. While it is unknown whether ufmylation always requires a dedicated E3 ligase, it is conceivable that an E3 ligase could enhance the Ufm1 modification of cellular targets, as seen in the case of SUMO modification51. Thus far, there is no evidence suggesting that Ufl1 has an uncharacterized molecular function beyond ufmylation. Therefore, we propose that Ufl1-mediated protein ufmylation strongly contributes to the maintenance of normal cardiac function.
The endoplasmic reticulum (ER) is a multifunctional subcellular organelle that coordinates the synthesis, folding, assembly, trafficking and degradation of proteins in eukaryotic cells. Disruption of ER function due to various cellular stressors such as ischemia, hypoxia, heat shock, oxidative stress and increased protein synthesis results in ER stress, which has been recognized as an important mechanism underlying the pathogenesis of heart failure42, 43. However, the regulatory control of ER stress in CMs is incompletely understood. Our study identifies ufmylation as a novel regulator of ER homeostasis in CMs. Ufl1 deficiency results in excessive ER stress as evidenced by the dysregulation of ER stress genes, significant increases in ER stress-responsive proteins and abnormal ER ultrastructure in the heart (Fig. 5D and Fig. 6). The excessive ER stress precedes discernable cardiac dysfunction in Ufl1-deficient hearts, and attenuation of ER stress ameliorates TAC-induced heart failure (Fig. 8), indicating that persistent ER stress plays a causative role in cardiac dysfunction in Ufl1-deficient hearts. These findings are in line with previous reports that links ufmylation with ER homeostasis. Ufl1 depletion in bone marrow cells has been shown to induce ER stress and impair hematopoiesis19. Silencing different components of the ufmylation pathway also enhanced ER stress in a beta-cell line16. We found that Ufl1 depletion impaired PERK signaling and sensitized CMs to ER stressed-induced cell death (Fig. 7). Cardiac-specific inhibition of PERK signaling exacerbates pressure overload-induced heart failure52, supporting the concept that Ufl1-conferred cardioprotection may be mediated by sustained PERK signaling in CMs. Interestingly, Ufl1 depletion in bone marrow cells appeared to stimulate both the Xbp-1s and PERK branches of UPR, suggesting that Ufl1 may have cell-type specific effects in initiating the UPR19. Nevertheless, more details remain to be understood with regard to how ufmylation controls different branches of ER stress signaling in cardiomyocytes.
Despite the established role of ufmylation in regulation of ER function, exactly how ufmylation regulates ER homeostasis remains unclear, largely due to the elusive identities of bona fide Ufm1 targets. Evidence from several reports suggest that Ufbp1, a putative ER-localized Ufm1 substrate17, may be a Ufl1 downstream effector in regulating ER stress. Ufbp1 forms a protein complex with Ufl1 and C53 (also known as Cdk5rap3), a tumor suppressor that regulates NFκB signaling53, at the cytosolic site of ER54. Ufl1 catalyzes Ufm1 conjugation to the lysine 267 of Ufbp1 and stabilizes Ufbp154. Ufbp1 depletion leads to ER stress and elevated PERK signaling in both bone marrow cells and hepatocyte cell lines20, 37, possibly due to destabilization of IRE1α25. Moreover, germline and somatic deletion of Ufbp1 also causes embryonic lethality and defective hematopoiesis, phenocopying mice deficient in Ufl120. Whether and how Ufbp1 mediates cardioprotective effects downstream of Ufl1 awaits further investigation.
It is worth noting that attenuation of ER stress failed to restore the cardiac function of TAC-operated Ufl1-deficient hearts to the level of CTL+TAC hearts (Fig. 8), indicating that other ER-independent mechanisms contribute. Indeed, our transcriptome analysis reveals that ufmylation may potentially regulate calcium cycling, ribosome function and metabolism, along with other pathways, in CMs (Fig. 5C), all of which are known to be crucial for the maintenance of cardiomyocyte contractility55. As fine-tuning of the calcium concentration in ER is critical for proper protein folding and transport, it will be intriguing to investigate whether dysregulation of Ca2+ cycling in Ufl1-deficient hearts is the proximal cause of ER stress and heart failure in future studies. Additionally, NFκB signaling, through cooperation with the NFAT pathway, has been shown to potentiate TAC-induced pathological cardiac remodeling56. It was previously found that silencing Ufl1 or its interacting partner C53, promotes NFκB activity54. Thus, Ufl1 may restrain NFkB activation to protect against pathological cardiac hypertrophy.
Our findings suggest that myocardial upregulation of Ufl1 is an adaptive response to pathological insults such as pressure overload. Hence, a better understanding of how Ufl1 expression is regulated in CMs may be helpful to develop new approaches for the treatment of heart failure. Previous studies have reported that the Ufm1 system can be transcriptionally induced by ER stress in mouse embryonic fibroblasts18. Specifically, Ufm1 seems to be a potential target of XBP-1 and depletion of XBP-1 can blunt ER stress-induced Ufm1 expression. Interestingly, ER stress-induced upregulation of Ufl1 is not dependent on XBP-1s and PERK signaling18, raising the possibility of Ufl1 being the downstream target of ATF6. Activation of ATF6 signaling has been shown to protect against myocardial ischemia-reperfusion damage57, 58, which is in part mediated by the induction of two of its direct targets in the heart: RCAN1 and Derl359, 60. Thus, it will be interesting to test whether Ufl1 acts as another ATF6 direct target to mediate its cardioprotective effect.
Supplementary Material
Novelty and Significance
What Is New?
The expression of Ufm1 specific E3 ligase 1 (Ufl1) is upregulated in murine hypertrophied hearts and downregulated in human failing hearts.
Ufl1 deficiency induces age-dependent cardiomyopathy and exacerbates pressure overload-induced cardiac hypertrophy, fibrosis and dysfunction.
Ufl1 deficiency impairs myocardial endoplasmic reticulum (ER) homeostasis and inhibits PERK signaling.
What Are the Clinical Implications?
Inadequate protein ufmylation may contribute to the development of cardiomyopathy.
Upregulation of myocardial Ufl1 levels could be a future therapeutic option in heart failure.
Targeting ufmylation could be a new strategy for treatment of diseases associating with ER stress.
Acknowledgments
We would like to thank Dr. David Fulton, Vascular Biology Center at Medical College of Georgia, Augusta University, for critically reading the manuscript and suggesting substantial improvements.
Sources of Funding
This study was in part supported by the US National Institutes of Health grants (R01HL124248 to H.S.) and the American Heart Association grants (16SDG30940002 to J.L. and 17POST33410592 to J.Z.).
Footnotes
Disclosures
None.
References
- 1.Hochstrasser M Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annu Rev Biochem. 2012;81:323–357. [DOI] [PubMed] [Google Scholar]
- 3.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Johnson JA, Su H. Ubiquitin and ubiquitin-like proteins in cardiac disease and protection. Curr Drug Targets. 2018;19:989–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. Sumo1-dependent modulation of serca2a in heart failure. Nature. 2011;477:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendler L, Braun T, Muller S. The ubiquitin-like sumo system and heart function: From development to disease. Circ Res. 2016;118:132–144. [DOI] [PubMed] [Google Scholar]
- 7.Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, Wang X. Perturbation of cullin deneddylation via conditional csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su H, Li J, Osinska H, Li F, Robbins J, Liu J, Wei N, Wang X. The cop9 signalosome is required for autophagy, proteasome-mediated proteolysis, and cardiomyocyte survival in adult mice. Circ Heart Fail. 2013;6:1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahnefeld A, Klingel K, Schuermann A, Diny NL, Althof N, Lindner A, Bleienheuft P, Savvatis K, Respondek D, Opitz E, Ketscher L, Sauter M, Seifert U, Tschope C, Poller W, Knobeloch KP, Voigt A. Ubiquitin-like protein isg15 (interferon-stimulated gene of 15 kda) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation. 2014;130:1589–1600. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K. A novel protein-conjugating system for ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatsumi K, Sou YS, Tada N, Nakamura E, Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E, Yamamoto M, Tanaka K, Komatsu M. A novel type of e3 ligase for the ufm1 conjugation system. J Biol Chem. 2010;285:5417–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniel J, Liebau E. The ufm1 cascade. Cells. 2014;3:627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K, Chung CH. Two novel ubiquitin-fold modifier 1 (ufm1)-specific proteases, ufsp1 and ufsp2. J Biol Chem. 2007;282:5256–5262. [DOI] [PubMed] [Google Scholar]
- 14.Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW, Ka SH, Sou YS, Komatsu M, Tanaka K, Lee ST, Noh DY, Baek SH, Jeon YJ, Chung CH. Modification of asc1 by ufm1 is crucial for eralpha transactivation and breast cancer development. Mol Cell. 2014;56:261–274. [DOI] [PubMed] [Google Scholar]
- 15.Pirone L, Xolalpa W, Sigurethsson JO, Ramirez J, Perez C, Gonzalez M, de Sabando AR, Elortza F, Rodriguez MS, Mayor U, Olsen JV, Barrio R, Sutherland JD. A comprehensive platform for the analysis of ubiquitin-like protein modifications using in vivo biotinylation. Sci Rep. 2017;7:40756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemaire K, Moura RF, Granvik M, Igoillo-Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E, Cnop M, Schuit F. Ubiquitin fold modifier 1 (ufm1) and its target ufbp1 protect pancreatic beta cells from er stress-induced apoptosis. PLoS One. 2011;6:e18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Lei G, Mei M, Tang Y, Li H. A novel c53/lzap-interacting protein regulates stability of c53/lzap and ddrgk domain-containing protein 1 (ddrgk1) and modulates nf-kappab signaling. J Biol Chem. 2010;285:15126–15136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Zhang M, Wu J, Lei G, Li H. Transcriptional regulation of the ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS One. 2012;7:e48587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, Pace B, Tuan-Lo D, Ganapathy V, Singh N, Li H. Rcad/ufl1, a ufm1 e3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015;22:1922–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang M, Chen J, Makala L, Lu C, Wu J, Teng Y, Pace B, Tuan D, Singh N, Li H. Ufbp1, a key component of the ufm1 conjugation system, is essential for ufmylation-mediated regulation of erythroid development. PLoS Genet. 2015;11:e1005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gannavaram S, Connelly PS, Daniels MP, Duncan R, Salotra P, Nakhasi HL. Deletion of mitochondrial associated ubiquitin fold modifier protein ufm1 in leishmania donovani results in loss of beta-oxidation of fatty acids and blocks cell division in the amastigote stage. Mol Microbiol. 2012;86:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Itakura E, Weber KP, Hegde RS, de Bono M. An er complex of odr-4 and odr-8/ufm1 specific protease 2 promotes gpcr maturation by a ufm1-independent mechanism. PLoS Genet. 2014;10:e1004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon J, Cho HJ, Han SH, No JG, Kwon JY, Kim H. A novel lzap-binding protein, nlbp, inhibits cell invasion. J Biol Chem. 2010;285:12232–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maran S, Lee YY, Xu S, Rajab NS, Hasan N, Syed Abdul Aziz SH, Majid NA, Zilfalil BA. Gastric precancerous lesions are associated with gene variants in helicobacter pylori-susceptible ethnic malays. World J Gastroenterol. 2013;19:3615–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Loon J, Dehghan A, Weihong T, Trompet S, McArdle WL, Asselbergs FW, Chen MH, Lopez LM, Huffman JE, Leebeek FW, Basu S, Stott DJ, Rumley A, Gansevoort RT, Davies G, Wilson JJ, Witteman JC, Cao X, de Craen AJ, Bakker SJ, Psaty BM, Starr JM, Hofman A, Wouter Jukema J, Deary IJ, Hayward C, van der Harst P, Lowe GD, Folsom AR, Strachan DP, Smith N, de Maat MP, O’Donnell C. Genome-wide association studies identify genetic loci for low von willebrand factor levels. Eur J Hum Genet. 2016;24:1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson CM, Crinnion LA, Gleghorn L, Newman WG, Ramesar R, Beighton P, Wallis GA. Identification of a mutation in the ubiquitin-fold modifier 1-specific peptidase 2 gene, ufsp2, in an extended south african family with beukes hip dysplasia. S Afr Med J. 2015;105:558–563. [DOI] [PubMed] [Google Scholar]
- 27.Di Rocco M, Rusmini M, Caroli F, Madeo A, Bertamino M, Marre-Brunenghi G, Ceccherini I. Novel spondyloepimetaphyseal dysplasia due to ufsp2 gene mutation. Clin Genet. 2018;93:671–674. [DOI] [PubMed] [Google Scholar]
- 28.Kim CH, Nam HS, Lee EH, Han SH, Cho HJ, Chung HJ, Lee NS, Choi SJ, Kim H, Ryu JS, Kwon J. Overexpression of a novel regulator of p120 catenin, nlbp, promotes lung adenocarcinoma proliferation. Cell Cycle. 2013;12:2443–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muona M, Ishimura R, Laari A, Ichimura Y, Linnankivi T, Keski-Filppula R, Herva R, Rantala H, Paetau A, Poyhonen M, Obata M, Uemura T, Karhu T, Bizen N, Takebayashi H, McKee S, Parker MJ, Akawi N, McRae J, Hurles ME, Study DDD, Kuismin O, Kurki MI, Anttonen AK, Tanaka K, Palotie A, Waguri S, Lehesjoki AE, Komatsu M. Biallelic variants in uba5 link dysfunctional ufm1 ubiquitin-like modifier pathway to severe infantile-onset encephalopathy. Am J Hum Genet. 2016;99:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colin E, Daniel J, Ziegler A, Wakim J, Scrivo A, Haack TB, Khiati S, Denomme AS, Amati-Bonneau P, Charif M, Procaccio V, Reynier P, Aleck KA, Botto LD, Herper CL, Kaiser CS, Nabbout R, N’Guyen S, Mora-Lorca JA, Assmann B, Christ S, Meitinger T, Strom TM, Prokisch H, Consortium F, Miranda-Vizuete A, Hoffmann GF, Lenaers G, Bomont P, Liebau E, Bonneau D. Biallelic variants in uba5 reveal that disruption of the ufm1 cascade can result in early-onset encephalopathy. Am J Hum Genet. 2016;99:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatsumi K, Yamamoto-Mukai H, Shimizu R, Waguri S, Sou YS, Sakamoto A, Taya C, Shitara H, Hara T, Chung CH, Tanaka K, Yamamoto M, Komatsu M. The ufm1-activating enzyme uba5 is indispensable for erythroid differentiation in mice. Nat Commun. 2011;2:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. Star: Ultrafast universal rna-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT, Huber W. Htseq--a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 36.Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–2424. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Wang Y, Song L, Zeng L, Yi W, Liu T, Chen H, Wang M, Ju Z, Cong YS. A critical role of ddrgk1 in endoplasmic reticulum homoeostasis via regulation of ire1alpha stability. Nat Commun. 2017;8:14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong M, Kefaloyianni E, Bao L, Malester B, Delaroche D, Neubert TA, Coetzee WA. Cardiac atp-sensitive k+ channel associates with the glycolytic enzyme complex. FASEB J. 2011;25:2456–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross AC, Zolfaghari R. Cytochrome p450s in the regulation of cellular retinoic acid metabolism. Annu Rev Nutr. 2011;31:65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xavier-Neto J, Sousa Costa AM, Figueira AC, Caiaffa CD, Amaral FN, Peres LM, da Silva BS, Santos LN, Moise AR, Castillo HA. Signaling through retinoic acid receptors in cardiac development: Doing the right things at the right times. Biochim Biophys Acta. 2015;1849:94–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu S, Guleria RS, Thomas CM, Roth A, Gerilechaogetu F, Kumar R, Dostal DE, Baker KM, Pan J. Loss of myocardial retinoic acid receptor alpha induces diastolic dysfunction by promoting intracellular oxidative stress and calcium mishandling in adult mice. J Mol Cell Cardiol. 2016;99:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groenendyk J, Sreenivasaiah PK, Kim do H, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–1197. [DOI] [PubMed] [Google Scholar]
- 43.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutkevich LA, Cohen-Doyle MF, Brockmeier U, Williams DB. Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol Biol Cell. 2010;21:3093–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. [DOI] [PubMed] [Google Scholar]
- 46.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishitoh H Chop is a multifunctional transcription factor in the er stress response. J Biochem. 2012;151:217–219. [DOI] [PubMed] [Google Scholar]
- 48.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. [DOI] [PubMed] [Google Scholar]
- 49.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce er stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, Barna M. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell. 2017;169:1051–1065 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melchior F Sumo--nonclassical ubiquitin. Annu Rev Cell Dev Biol. 2000;16:591–626. [DOI] [PubMed] [Google Scholar]
- 52.Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, Wei Y, Cavener DR, Hu X, Hall J, Bache RJ, Chen Y. Endoplasmic reticulum stress sensor protein kinase r-like endoplasmic reticulum kinase (perk) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64:738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG. Lzap, a putative tumor suppressor, selectively inhibits nf-kappab. Cancer Cell. 2007;12:239–251. [DOI] [PubMed] [Google Scholar]
- 54.Wu J, Jiang H, Luo S, Zhang M, Zhang Y, Sun F, Huang S, Li H. Caspase-mediated cleavage of c53/lzap protein causes abnormal microtubule bundling and rupture of the nuclear envelope. Cell Res. 2013;23:691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. [DOI] [PubMed] [Google Scholar]
- 56.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between nfkappab and nfat coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC. Atf6 decreases myocardial ischemia/reperfusion damage and links er stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120:862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of atf6. Circ Res. 2006;98:1186–1193. [DOI] [PubMed] [Google Scholar]
- 59. Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (rcan1), a novel atf6-inducible gene. J Biol Chem. 2008;283:14012–14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belmont PJ, Chen WJ, San Pedro MN, Thuerauf DJ, Gellings Lowe N, Gude N, Hilton B, Wolkowicz R, Sussman MA, Glembotski CC. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene derlin-3 in the ischemic heart. Circ Res. 2010;106:307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.